

Abstract
17(R),18(S)-Epoxyeicosatetraenoic acid [17(R),18(S)-EETeTr], a cytochrome P450 epoxygenase metabolite of eicosapentaenoic acid (EPA), exerts negative chronotropic effects and protects neonatal rat cardiomyocytes against Ca2+-overload with an EC50 ~1–2 nM. Structure-activity studies revealed a cis-Δ11,12- or Δ14,15-olefin and a 17(R),18(S)-epoxide are minimal structural elements for anti-arrhythmic activity whereas antagonist activity was often associated with the combination of a Δ14,15-olefin and a 17(S),18(R)-epoxide. Compared with natural material, the agonist and antagonist analogs are chemically and metabolically more robust and several show promise as templates for future development of clinical candidates.
Introduction
The intake of omega-3 polyunsaturated fatty acids (ω-3 PUFAsa) such as eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA) is associated with manifold health benefits,1 especially for cardiovascular disease,2 e.g., lower triglyceride levels, amelioration of ischemic injury, and reduction of sudden cardiac death resulting from fatal arrhythmia.3 However, the mechanism(s) of action as well as the molecular specie(s) or metabolite(s) responsible for the protective effects of the ω-3 PUFAs remain largely obscure.4 A leading hypothesis is that EPA and its congeners compete with arachidonic acid (AA) for enzymatic transformations and that the ω-3 PUFA metabolites have physiological consequences that differ from those of AA.5
In concert with this proposal, a series of in vitro studies demonstrated that many major cytochrome P450 (CYP) epoxygenase isoforms preferentially metabolize EPA to the 17,18-epoxide 1 (17,18-EETeTr)6 and often favor the 17(R),18(S)-enantiomer 2.6b,c,7 A recent in vivo study revealed that dietary ω-3 PUFA supplementation causes a profound shift in the endogenous CYP-eicosanoid profile from AA- to EPA/DHA-derived metabolites.8 In fact, 1 represented the predominant epoxy-metabolite in the heart, lung, kidney and several other organs of rats fed an EPA/DHA-rich diet, whereas this metabolite was produced only in trace amounts when the animals received normal chow rich in ω-6 PUFAs.8 There is also initial evidence for the in vivo formation of EETeTrs in humans based on studies showing the occurrence of these EPA metabolites and/or their hydrolysis products in urine and plasma of volunteers ingesting increased amounts of ω-3 PUFAs.9,10 Epoxide 2 is a potent vasodilator and stimulates large-conductance K+ (BK) channels in rat cerebral arteries.11 The vasodilator effects of epoxides 1 and 2 are not blocked by the epoxyeicosatrienoic acid (EET) antagonist 14,15-epoxyeicosa-5(Z)-enoic acid (EEZE; 10 μM), suggesting a recognition or binding site independent of the putative EET receptor.12,13 In the human lung, 1 also hyperpolarizes and relaxes pulmonary artery and bronchial smooth muscle cells and exerts anti-inflammatory effects.14,15
A major open question concerning the whole field of CYP-eicosanoid research is the heretofore unidentified, primary targets of CYP-eicosanoid action. Although some studies indicate direct interactions with certain ion channels and transcription factors, most of the observed CYP-eicosanoid actions apparently rely on the ability of these metabolites to trigger distinct intracellular signaling pathways. These pathways, depending upon the specific CYP metabolite and physiological context, can involve protein kinase A, guanine nucleotide binding proteins, tyrosine kinases, mitogen-activated kinases, phosphoinositol-3 kinase, IkappaB kinase, Rho-kinase or the epidermal growth factor and a series of other well-known key components of intracellular signaling.16 In general, these signaling components become specifically engaged in pathways that are primarily elicited by membrane receptors. Recent studies indeed revealed the presence of a high affinity EET-binding site in U937 cells, which is probably a G-protein coupled receptor.17 These findings suggest that the various CYP-dependent metabolites of ω-6 and ω-3-PUFAs may exert their specific functions via a family of G-protein coupled receptors analogous with the cyclooxygenase- and lipoxygenase-generated eicosanoids.
Neonatal rat cardiomyocytes (NRCMs) have been introduced as an in vitro model to analyze the anti-arrhythmic mechanisms of ω-3 PUFAs.3a,18 In this model, EPA (3–10 μM) reduces the contraction rate of the spontaneously beating heart cells, i.e., it exerts a strong negative chronotropic effect. Furthermore, EPA reduces the response of NRCMs to β-adrenergic agonists as well as increases extracellular Ca2+-concentrations and terminates the arrhythmias induced by these agents.18 We have shown previously that 1 and 2 (Table 1) exert the same effects as EPA on NRCMs, but in contrast are active in the low nanomolar range suggesting that these metabolites may function as mediators of the anti-arrhythmic mechanism of ω-3 PUFAs.8 In the present study, we used the chronotropic effect on NRCMs as a bioassay to evaluate the relative “anti-arrhythmic potencies” of a series of synthetic analogs based on the structure of the natural EPA epoxy-metabolites. This SAR study was performed to (i) identify the structural determinants that mediate the unique biological activities of 1, (ii) to find selective agonists19 and antagonists as required for future mechanistic studies and for isolation of the putative receptor, and (iii) to generate agonists with improved chemical and metabolic stability as a first step toward the development of novel anti-arrhythmic drugs.
Table 1.
Chronotropic Effects of 17,18-EETeTr and Analogs on NRCMsa
| Compd | Analog | Change (beats/min) | nb |
|---|---|---|---|
| 1 |
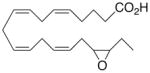
|
−22.5 ± 0.8 | 60 |
| 2 |
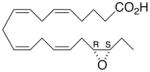
|
−21.3 ±0.9 | 12 |
| 3 |
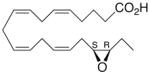
|
+1.3 ±1.6 | 12 |
| 4 |
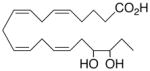
|
0.0 ±0.0 | 10 |
| 5 |
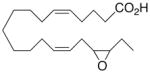
|
0.0 ±1.2 | 14 |
| 6 |
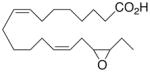
|
−2.7 ±1.4 | 22 |
| 7 |
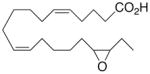
|
−19.8 ±0.8 | 28 |
| 8 |
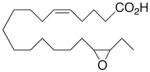
|
−0.6 ±1.0 | 14 |
| 9 |
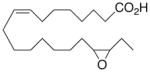
|
−0.6 ±1.3 | 14 |
| 10 |
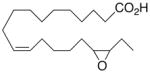
|
−18.3 ±1.5 | 21 |
| 11 |
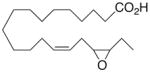
|
−1.2 ±1.3 | 14 |
| 12 |
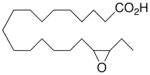
|
+0.8 ±1.6 | 14 |
| 13 |
|
−20.3 ±1.2 | 27 |
| 14 |
|
+3.3 ±1.1 | 29 |
| 15 |
|
−21.1 ±0.8 | 18 |
| 16 |
|
+0.7 ±0.9 | 18 |
| 17 |
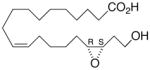
|
−4.8 ± 0.26 | 18 |
| 18 |
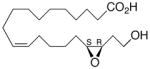
|
−1.4 ±0.34 | 18 |
All compounds were tested at a final concentration of 30 nM at 37 °C (control NRCM: 120–150 beats/min).
n = number of determinations.
Results and Discussion
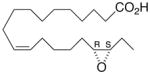
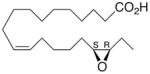
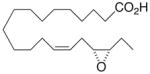
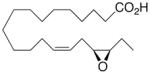
Whilst EPA required prolonged incubation and a ~1000-fold higher concentration, epoxide 2 was almost immediately effective with an EC50 ~1–2 nM (Fig. 1). Notably, the negative chronotropic activity resides with the 17(R),18(S)-enantiomer 2 while its antipode 3 had no significant effect (Table 1). The soluble epoxide hydrolase (sEH) metabolite of 1, i. e., diol 4, was completely inactive. To capitalize on these observations for the development of more efficacious anti-arrhythmic therapies,20 we initiated a systematic structure-activity relationship study to characterize this pharmacophore and to simultaneously address the chemical and metabolic labilities of 1 and 2 that restrict their wider utility.21,22 Thus, our first priority was the partial saturation of the polyenoic backbone with the intention of strategically disrupting the three 1,4-dienyl combinations. This would obviate both autooxidation and metabolism by lipoxygenases and cycloxygenases.4c Of the three possible tetrahydro-variants 5–7 that satisfied this criteria, only 7 was still competent to significantly reduce the beating rate. The mono-olefinic series 8–11 was consistent with the preceding observations and clearly established the presence of a Δ11,12-olefin, as found in analog 10, confers strong negative chronotropic activity. The fully saturated analog 12 proved to be largely inactive. As might be anticipated from the differences between the 17,18-EETeTr enantiomers 2 and 3, analog 13 was a powerful negative chronotrope whereas its mirror image, analog 14, had the opposite, albeit more modest activity. The enantiomeric pair 15 and 16 presented a similar pharmacological profile that suggests that the absolute configuration of the epoxide can trump the influence of olefin position. Terminal hydroxylation is a common degradation pathway for fatty acids and eicosanoids,23 so it was no surprise that this modification mostly abrogated biological activity in the arrhythmia assay, e.g., 17 and 18.
Figure 1.

Dose dependency of EPA-epoxide 2 and analogs 13, 19, 21, and 22 on NRCM beating rates.
In recognition of the central role of the sEH enzyme in the metabolism of fatty epoxanoids,24 replacement of the epoxide with an achiral epoxide bioisostere21 was deemed crucial to the development of any robust, long-lived analogs intended for in vivo applications. Hence, we were delighted to find 19,21 a 1,3-disubstituted urea version of 10, decisively suppressed the beating rate (Table 2). In contrast to prior experience with 14,15-EET analogs,21 the introduction of N-methyls on to the urea, i.e., analog 20, strongly dampened the anti-arrhythmic effect. 1,4-Disubstituted oxamide 21 proved to be the most efficacious of all the bioisosteres and, thus, was subjected to additional scrutiny. Amongst these, the trans-olefinic and acetylenic versions 22 and 23, respectively, were the most informative. The strict stereochemical requirement for this portion of the molecule suggests the recognition or binding site contains a narrow pocket that best accommodates the hairpin configuration of a cis-olefin. The related amides 24–26 were disappointing; however, shifting the amide functionality just one position away from the ω-terminus as in 27 restored most of the negative chronotropic influence. Repositioning the olefin to the Δ14,15-position consistently reversed or annulled any negative chronotropic behavior irrespective of the epoxide surrogate: 28 vs 25, 29 vs 21, and 30 vs 19.
Table 2.
Chronotropic Effects of Epoxide Bioisosteric Analogs on NRCMsa
| Compd | Analog | Change (beats/min) | nb |
|---|---|---|---|
| 19 |
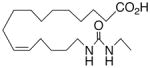
|
−27.0 ± 1.2 | 27 |
| 20 |
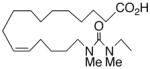
|
−5.8 ± 0.4 | 6 |
| 21 |
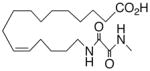
|
−33.7 ±1.3 | 24 |
| 22 |
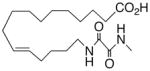
|
−4.7 ± 0.45 | 18 |
| 23 |
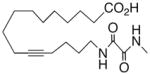
|
−2.6 ± 0.35 | 18 |
| 24 |
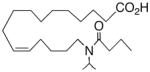
|
+0.2 ±1.3 | 18 |
| 25 |
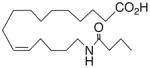
|
−1.6 ±0.9 | 18 |
| 26 |
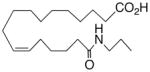
|
−4.7 ±1.0 | 18 |
| 27 |
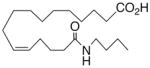
|
−22.4 ±1.7 | 18 |
| 28 |
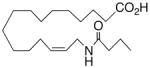
|
+12.8 ±1.5 | 18 |
| 29 |
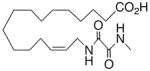
|
−1.3 ±1.0 | 18 |
| 30 |
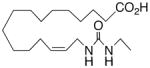
|
+17.8 ±1.4 | 18 |
All compounds were tested at a final concentration of 30 nM at 37 °C (control NRCM: 120–150 beats/min)
n = number of determinations.
Having identified several potent analogs containing both epoxide and bioisosteric replacements, it was of interest to evaluate the dose response profile of select examples versus 17(R),18(S)-EETeTr (2) on NRCM beating rates. The weakly active trans-olefinic analog 22 was included as a control. As evident in Figure 1, urea 19 and oxamide 21 outperformed 2 at most concentrations. Above ca. 5 nM, all of the test compounds began to plateau, except for analog 22 which did not display any significant efficacy until the low μM range.
In recognition of the ability of analog 16 to block the negative chronotropic effects of its antipode 15, we sought to identify additional antagonists amongst the analogs in Table 1 that were themselves largely devoid of activity. To this end, NRCMs were preincubated with the test antagonist (30 nM final concentration) for 5 min at 37 °C before the addition of analog 10 (30 nM final concentration). Only analogs 6 and 11 resulted in any useful antagonism (Figure 2). While both share a Δ14,15-olefin, this is not sufficient for antagonism since analog 5 also contains a Δ14,15-olefin, yet was not distinguishable from vehicle.
Figure 2.

Antagonism of 10 by Other Analogs.
While analog 6 emerged as the most powerful antagonist in the foregoing study, there was no certainty that it could function as a universal antagonist against other negative chronotropes. Consequently, NRCMs were preincubated with several of the most efficacious negative chronotropes, inter alia, EPA, 1, 7 and oxamide 21, and then challenged with an equimolar amount of 6 (Figure 3). In all cases, blockades of 90% or better were achieved. Of particular note, antagonist 6 (30 nM) blocked not only the effects of epoxide 1 (30 nM) and its synthetic agonists, but also that of EPA (3.3 μM) suggesting that EPA and its natural metabolite act via the same mechanism.
Figure 3.

Antagonism of Negative Chronotropes by 6.
To demonstrate the chemical modifications described above resulted in agonists with improved metabolic stability, we analyzed the metabolism of 1, 12, 13, 19 and 21 by rat liver homogenates. The results are shown in detail in the Supporting Information section. As expected, epoxide 1 was rapidly hydrolyzed to vic-diol 4 and further attacked by epoxidation and hydroxylation reactions. In total, about 80% of 1 was metabolized within 30 min. Analog 21, carrying an oxamide moiety instead of the epoxy-group and only the functionally essential 11,12-double bond, provided the currently best solution to overcome the metabolic instability of the natural compound. LC-MS analysis indicated that analog 21 was only slowly metabolized to regioisomeric hydroxy-metabolites and more than 85% of the original compound remained intact after 30 min incubation with liver homogenate (see Supporting Information).
It was also of interest to understand the consequences, if any, of the structural modifications on sEH activity. Other laboratories25 have developed highly potent sEH-inhibitors that utilize ureas as the epoxide bioisostere. Similar functionality appears in some of our 17,18-EETeTr agonists, e.g., 19 (Table 2). Accordingly, the biological activity of our analogs might be attributable in part or entirely to inhibition of sEH and subsequent accumulation of endogenous epoxy-eicosanoids in the cardiomyocytes. To test this possibility, we analyzed selected compounds containing the urea (analog 19) or oxamide (analogs 21 and 29) moieties for their capacity to inhibit sEH. Analog 19 exerted weak dose-dependent sEH inhibition at 1 and 5 μM, i.e., concentrations 1000-fold higher than required for reducing the contractility of cardiomyocytes (see Supporting Information, Figure 2). Analogs 21 and 29 lacked any inhibitory effects up to 5 μM. Under the same conditions, AUDA (12-(3-adamantan-1-yl-ureido)dodecanoic acid), a representative of the urea-class of highly potent sEH-inhibitors, almost completely blocked sEH activity at 100 nM.25 AUDA itself had no effect on the spontaneous beating rate of cultured cardiomyocytes. However, AUDA partially inhibited the negative chronotropic effect of 1 (see Supporting Information, Figure 3). Furthermore, combined administration of 19 or 21 with 17,18-EETeTr did not result in additive or synergistic effects (see Supporting Information, Figure 3). Taken together, these results show that our synthetic analogs do not or only weakly target sEH; hence, sEH inhibition can be excluded as a significant contributor to their effects on cardiomyocytes.
Chemistry
Syntheses of 7, 8, 10, and 12 are summarized in Scheme 1 and are representative of the methodology used to prepare the other epoxide-containing analogs. Following literature precedent,26 commercial octa-1,7-diyne (31) was alkylated with a limiting amount of 2-(4-bromobutoxy)-tetrahydro-2H-pyran27 in THF/HMPA to give 32. A second alkylation using 8-bromo-oct-3(Z)-ene28 followed by acidic cleavage of the THP and Jones oxidation furnished acid 33 that was then subjected to Fischer esterification and selective epoxidation of the Δ17,18-olefin. The resultant epoxide 34 smoothly led to dienoate 35 via semi-hydrogenation of over P-2 nickel/hydrogen gas. Random hydrogenation of 35 using diimide generated in situ gave rise to a chromatographically separable mixture of unreacted 35, mono-olefins 36 and 37, and tetrahydro-epoxide 38. LiOH hydrolysis of the individual methyl esters yielded the corresponding free acids 7, 8, 10, and 12.
Scheme 1.

Synthesis of 7, 8, 10, and 12a
aReagents and conditions: (a) n-BuLi (0.9 equiv), THF/HMPA (6:1), −78 °C to −10 °C, 2 h; Br(CH2)4OTHP, −78 °C to rt, 3 h; rt, 12 h, 65%; (b) n-BuLi, THF/HMPA (6:1), −78 °C to −10 °C, 2 h; Br(CH2)4CH=CHCH2CH3, −78 °C to rt, 3 h; rt, 12 h, 65%; (c) p-TSA, MeOH, rt, 4 h, 92%; (d) Jones reagent, acetone, −40 °C to −10 °C, 88%; (e) p-TSA, MeOH, rt, 10 h, 82%; (f) m-CPBA, CH2Cl2, rt, 2 h, 82%; (g) P-2 Ni/(H2NCH2)2, H2 (1 atm), EtOH, 98%; (h) (H2N)2, CuSO4, O2, EtOH, rt, 12 h; (i) LiOH, THF/H2O (4:1), rt 10 h, 90–93%.
Access to oxamide 21 (Scheme 2) began with alkylation of 12-(tert-utyldiphenylsilyloxy)dodec-1-yne29 (39) with 2-(4-bromobutoxy)-tetrahydro-2H-pyran.30 Subsequent desilylation evolved 40 which was advanced to 41 via P-2 nickel mediated semi-hydrogenation and Jones oxidation. Exposure of the product to acidic methanol simultaneously esterified the carboxylic acid and removed the THP ether. Replacement of the terminal alcohol with azide using diphenylphosphoryl azide (DPPA) under Mitsunobu-type conditions completed the transformation to 42. Sequential Staudinger reduction, EDCI amidation with 2-(methylamino)-2-oxoacetic acid,30 and hydrolysis delivered 21 and could be accomplished without the purification of intermediates.
Scheme 2.

Synthesis of 21a
aReagents and conditions: (a) n-BuLi, THF/HMPA (6:1), −78 °C to −10 °C, 2 h; Br(CH2)4OTHP, −78 °C to rt, 3 h; rt, 12 h, 66%; (b) n-Bu4NF, THF, rt, 5 h, 80%; (c) P-2 Ni/(H2NCH2)2, H2 (1 atm), EtOH, rt, 99%; (d) Jones reagent, acetone, −40 °C to −10 °C, 68%; (e) p-TSA, MeOH, rt, 10 h, 83%; (f) DEAD, Ph3P, THF, −20 °C, 30 min; (PhO)2P(O)N3, 0 °C, 6 h, 78%; (g) Ph3P, THF/H2O, rt, 8 h; (h) EDCI, HOBt, i-PrEt2N, MeNHC(O)C(O)OH, DMF, rt, 12 h, 68% over two steps; (i) LiOH, THF/H2O (4:1), rt 10 h, 89%
Conclusions
The potency and rapid onset of 17(R),18(S)-EETeTr (2) are consistent with a prominent role for this cytochrome P450-dependent eicosanoid in mediating the anti-arrhythmic effects of EPA. Structure-activity correlations helped define the essential pharmacophore as well as led to the development of agonist and antagonist analogs that are more robust than natural material and that provide a sound foundation for the future development of potential clinical candidates. Additionally, these data are also consistent with the existence of a specific ω-3 epoxanoid receptor, albeit other mechanisms of actions cannot be excluded at present.
Experimental Section
General Procedures
Unless stated otherwise, yields refer to purified products and are not optimized. Final compounds were judged ≥95% pure by HPLC using a Zorbax Eclipse C18 (250 × 4.6 mm; Agilent) connected to an Agilent 1200 API/LC-MS with acetonitrile/water combinations as solvent; enantiomers were also analyzed by chiral phase HPLC using a Chiralcel OJ-H column (250 × 4.6 mm; Daicel) with hexane/isopropyl alcohol combinations as solvent and judged to be ≥95% e.e. All oxygen and/or moisture sensitive reactions were performed under an argon atmosphere using oven-dried glassware and anhydrous solvents. Anhydrous solvents were freshly distilled from sodium benzophenone ketyl, except for CH2Cl2, which was distilled from CaH2. Extracts were dried over anhydrous Na2SO4 and filtered prior to removal of all volatiles under reduced pressure. Unless otherwise noted, commercially available materials were used without purification. Flash chromatography (FC) was performed using E Merck silica gel 60 (240–400 mesh). Thin layer chromatography was performed using pre-coated plates purchased from E. Merck (silica gel 60 PF254, 0.25 mm). Nuclear magnetic resonance (NMR) spectra were recorded on Varian 300, 400 or 500 spectrometers at operating frequencies of 300/400/500 MHz (1H) or 75/100/125 MHz (13C). Nuclear magnetic resonance (NMR) splitting patterns are described as singlet (s), doublet (d), triplet (t), quartet (q), and broad (br); the values of chemical shifts (δ) are given in ppm relative to residual solvent (chloroform δ = 7.27 for 1H NMR or δ = 77.23 for proton decoupled 13C NMR), and coupling constants (J) are given in Hertz (Hz). Melting points were determined using an OptiMelt (Stanford Research Systems) and are uncorrected. The Michigan State University Mass Spectroscopy Facility or Prof. Kasem Nithipatikom (Medical College of Wisconsin) kindly provided high-resolution mass spectral analyses.
32. n-BuLi (30 mL of 2.5 M soln in hexanes, 70 mmol) was added dropwise to a −78 °C solution of 31 (9.0 g, 84.9 mmol; G F Smith, Inc.) in dry THF/HMPA (350 mL, 6:1) under an argon atmosphere. After 30 min, the reaction mixture was warmed to −10 °C over 2 h and maintained at this temperature for 20 min, then re-cooled to −78 °C. To this was added a solution of 2-(4-bromobutoxy)-tetrahydro-2H-pyran27 (15 g, 63.68 mmol) in dry THF (15 mL). The resulting mixture was warmed to room temperature over 3 h, maintained at this temperature for 12 h, then quenched with sat. aq. NH4Cl (25 mL). After 20 min, the mixture was extracted with Et2O (2 × 125 mL). The combined ethereal extracts were washed with water (2 × 100 mL), brine (100 mL), dried over Na2SO4 and concentrated under reduced pressure. The residue was purified by SiO2 column chromatography using 5% EtOAc/hexanes as eluent to give 32 (10.85 g, 65%) as a colorless oil. TLC: 10% EtOAc/hexanes, Rf ≈ 0.6; 1H NMR (400 MHz, CDCl3) δ 4.57 (t, J = 2.5 Hz, 1H), 3.82–3.87 (m, 1H), 3.70–3.77 (m, 1H), 3.46–3.51 (m, 1H), 3.36–3.42 (m, 1H), 2.14–2.20 (m, 6H), 1.93 (t, J = 2.5 Hz, 1H), 1.46–1.72 (m, 14H); 13C NMR (100 MHz, CDCl3) δ 98.78, 84.23, 80.31, 79.82, 68.50, 67.07, 62.25, 30.80, 30.76, 28.98, 28.07, 27.58, 25.96, 25.57, 19.68, 18.64, 18.31. HRMS calcd for C16H25O2 [M+1]+ 249.1855, found 249.1852.
33. Compound 32 (4.5 g, 17.2 mmol) was alkylated using n-BuLi (8.3 mL of 2.5 M soln in hexanes, 20.65 mmol) and 8-bromo-oct-3(Z)-ene31 (4.1 g, 21.5 mmol) in THF/HMPA (180 mL, 6:1) as described above for the synthesis of 32 to give 2-[eicosa-17(Z)-en-5,11-diynyloxy]tetrahydro-2H-pyran (4.15 g, 65%) as a colorless oil. TLC: 10% EtOAc/hexanes, Rf ≈ 0.6; 1H NMR (400 MHz, CDCl3) δ 5.26–5.41 (m, 2H), 4.58 (t, J = 2.5 Hz, 1H), 3.82–3.87 (m, 1H), 3.70–3.77 (m, 1H), 3.46–3.51 (m, 1H), 3.36–3.42 (m, 1H), 2.11–2.20 (m, 8H), 1.92–2.04 (m, 4H), 1.62–1.86 (m, 4H), 1.39–1.59 (m, 14H), 0.94 (t, J = 7.5 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 132.54, 128.42, 98.98, 80.47, 79.99, 68.56, 67.25, 62.49, 34.01, 32.49, 30.93, 29.12, 28.37, 28.21, 27.71, 26.34, 26.08, 25.69, 20.72, 19.83, 18.79, 18.46, 18.16, 14.53. HRMS calcd for C24H39O2 [M+1]+ 359.2950, found 359.2951.
A solution of 2-[eicos-17(Z)-en-5,11-diynyloxy]tetrahydro-2H-pyran (1.3 g, 3.49 mmol) and p-TSA (50 mg) in MeOH (50 mL) was stirred at room temperature for 4 h, then concentrated in vacuo. The residue was purified by SiO2 column chromatography using 15% EtOAc/hexanes as eluent to give eicosa-17(Z)-en-5,11-diyn-1-ol (925 mg, 92%) as a colorless oil. TLC: 30% EtOAc/hexanes, Rf ≈ 0.35; 1H NMR (400 MHz, CDCl3) δ 5.27–5.42 (m, 2H), 3.66 (t, J = 6.8 Hz, 2H), 2.00–2.19 (m, 12H), 1.43–1.72 (m, 12H), 0.95 (t, 3H, J = 7.7 Hz); 13C NMR (100 MHz, CDCl3) δ 132.56, 128.45, 80.42, 80.22, 62.69, 34.06, 32.51, 32.07, 28.39, 28.20, 27.73, 26.36, 25.51, 20.74, 18.73, 18.45, 18.16, 14.45. HRMS calcd for C20H33O [M+1]+ 289.2531, found 289.2534.
Jones reagent (5 mL of a 10 N aq. solution) in acetone (10 mL) was added slowly to a stirring, −40 °C solution of eicosa-17(Z)-ene-5,11-diyn-1-ol (1.0 g, 3.47 mmol) in acetone (50 mL). After 1 h, the reaction mixture was warmed to −10 °C and maintained at this temperature for 3 h, then quenched with excess isopropanol. The green chromium salts were removed by filtration, the filter cake was washed with acetone, and the combined filtrates were concentrated in vacuo. The residue was dissolved in EtOAc (100 mL), washed with water (50 mL), and concentrated in vacuo. The residue was purified by SiO2 column chromatography using 15% EtOAc/hexanes as eluent to give 33 (920 mg, 88%) as a colorless oil. TLC: 30% EtOAc/hexanes, Rf ≈ 0.35; 1H NMR (400 MHz, CDCl3) δ 5.24–5.41 (m, 2H), 2.41 (t, J = 6.9 Hz, 3H), 2.10–2.19 (m, 8H), 1.98–2.09 (m, 4H), 1.75–1.81 (m, 2H), 0.96 (t, J = 7.7 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 184.37, 132.54, 128.44, 84.43, 80.45, 80.26, 78.58, 34.08, 32.53, 32.09, 28.36, 28.23, 27.75, 26.38, 25.53, 20.76, 18.77, 18.48, 18.03, 14.59. HRMS calcd for C20H31O2 [M+1]+ 303.2324, found 303.2324.
34. A solution of 33 (0.8 g, 2.63 mmol) and p-TSA (20 mg) in MeOH (30 mL) was stirred at room temperature for 10 h, then concentrated in vacuo and the residue was purified by SiO2 column chromatography using 3% EtOAc/hexanes as eluent to give methyl eicos-17(Z)-en-5,11-diynoate (682 mg, 82%) as a colorless oil. TLC: 10% EtOAc/hexanes, Rf ≈ 0.60; 1H NMR (400 MHz, CDCl3) δ 5.27–5.42 (m, 2H), 3.67 (s, 3H), 2.43 (t, 2H, J = 7.6 Hz), 2.12–2.21 (m, 8H), 1.99–2.09 (m, 4H), 1.76–1.82 (m, 2H), 1.42–1.58 (m, 8H), 0.95 (t, 3H, J = 7.7 Hz); 13C NMR (100 MHz, CDCl3) δ 184.30, 132.57, 128.48, 84.52, 80.46, 80.21, 79.97, 51.19, 33.09, 32.43, 32.19, 28.39, 28.27, 27.65, 26.36, 25.63, 20.73, 18.72, 18.49, 18.07, 13.76. HRMS calcd for C21H33O2 [M+1]+ 317.2481, found 317.2485.
m-CPBA (1.6 g, 4.76 mmol) was added in portions to a 0 °C solution of methyl eicosa-17(Z)-en-5,11-diynoate (1.15 g, 3.66 mmol) in CH2Cl2 (50 mL). After 2 h at room temperature, the reaction mixture was diluted with CH2Cl2 (25 mL), washed with sat. aq. NaHCO3 (2 × 25 mL), brine (2 × 25 mL), water (50 mL), dried over Na2SO4, and concentrated under reduced pressure. The residue was purified by SiO2 column chromatography using 5% EtOAc/hexanes as eluent to give 34 (990 mg, 82%) as a colorless oil. TLC: 10% EtOAc/hexanes, Rf ≈ 0.3; 1H NMR (400 MHz, CDCl3) δ 3.67 (s, 3H), 2.84–2.94 (m, 2H), 2.42 (t, 2H, J = 7.3 Hz), 2.14–2.23 (m, 8H), 1.74–1.83 (m, 2H), 1.42–1.61 (m, 12H), 1.03 (t, 3H, J = 7.6 Hz); 13C NMR (100 MHz, CDCl3) δ 184.21, 83.62, 80.35, 80.08, 62.96, 59.43, 52.32, 34.19, 32.28, 32.20, 29.17, 28.32, 28.25, 27.75, 26.43, 25.72, 20.75, 18.63, 18.28, 18.14, 14.53. HRMS calcd for C21H33O3 [M+1]+ 333.2430, found 333.2434.
35. NaBH4 (33 mg, 0.88 mmol) was added portionwise to a stirring, rt solution of nickel(II) acetate tetrahydrate (190 mg, 0.76 mmol) in absolute ethanol (5 mL) under a hydrogen blanket (1 atm). After 15 min, freshly distilled ethylenediamine (200 mg, 3.24 mmol) was added followed by 34 (250 mg, 0.75 mmol) in absolute ethanol (5 mL) while maintaining the hydrogen blanket (1 atm). The heterogeneous mixture was stirred at rt for 90 min, then diluted with ether (40 mL), and filtered through a short pad of silica gel; the filtration pad was washed with ether (3 × 10 mL). The combined filtrates were dried over anhydrous Na2SO4 and concentrated in vacuo to give 35 (246 mg, 98%) as a colorless oil sufficiently pure to be used directly in the next step. TLC: 20% EtOAc/hexanes, Rf ≈ 0.65; 1H NMR (400 MHz, CDCl3) δ 5.27–5.42 (m, 4H), 3.66 (s, 3H), 2.83–2.93 (m, 2H), 2.30 (t, 2H, J = 7.3 Hz), 1.92–2.09 (m, 8H), 1.63–1.72 (m, 2H), 1.25–1.58 (m, 12H), 1.03 (t, 3H, J = 7.7 Hz); 13C NMR (100 MHz, CDCl3) δ 174.29, 131.12, 130.20, 129.68, 128.64, 58.54, 57.45, 51.63, 33.62, 29.78, 29.53, 29.48, 27.79, 27.28, 26.71, 26.41, 25.05, 21.28, 10.80. HRMS calcd for C21H37O3 [M+1]+ 337.2743, found 337.2741.
7. An aqueous solution of LiOH (1 mL, 2 M soln) was added to a 0 °C solution of 35 (250 mg, 0.74 mmol) in THF (8 mL) and deionized H2O (2 mL). After stirring at room temperature overnight, the reaction mixture was cooled to 0 °C, the pH was adjusted to 4 with 1 M aq. oxalic acid, and extracted with ethyl acetate (2 × 20 mL). The combined extracts were washed with water (30 mL), brine (25 mL), dried over anhydrous Na2SO4, and concentrated in vacuo. The residue was purified by SiO2 column chromatography using 25% EtOAc/hexanes as eluent to give 7 (222 mg, 93%) as a colorless oil. TLC: 30% EtOAc/hexanes, Rf ≈ 0.3; 1H NMR (400 MHz, CDCl3) δ 5.28–5.40 (m, 4H), 2.87–2.97 (m, 2H), 2.34 (t, 3H, J = 7.0 Hz), 1.97–2.12 (m, 8H), 1.63–1.74 (m, 2H), 1.30–1.60 (m, 12H), 1.02 (t, 3H, J = 7.4 Hz); 13C NMR (300 MHz, CDCl3) δ 180.06, 131.75, 130.03, 129.77, 128.66, 58.86, 57.87, 33.93, 29.93, 29.84, 29.81, 27.89, 27.68, 26.41, 26.36, 24.83, 21.26, 10.84. HRMS calcd for C20H35O3 [M+1]+ 323.4901, found 323.4901.
Syntheses of 8, 10, and 12
A stream of ethanol saturated air was passed through a stirring solution of hydrazine hydrate (400 mg, 12 mmol, 20 equiv), 35 (200 mg, 0.60 mmol), and CuSO4•5H2O (10 mg) in ethanol (5 mL).26 After 12 h, the reaction mixture was passed through a short pad of silica gel and the filter cake was washed with dichloromethane (3 × 10 mL). The combined filtrates were dried over anhydrous Na2SO4 and concentrated in vacuo. The residue was resolved into its components by AgNO3-impregnated PTLC using 2% CH2Cl2/benzene: Rf ≈ 0.2, 0.4, 0.55, and 0.85 for 35, 37, 36, and 38, respectively, isolated in a ratio of 2:3:3:2, respectively.
36: 1H NMR (400 MHz, CDCl3) δ 5.27–5.42 (m, 2H), 3.66 (s, 3H), 2.84–2.92 (m, 2H), 2.30 (t, J = 7.4 Hz, 2H), 1.96–2.08 (m, 4H), 1.64–1.71 (m, 2H), 1.45–1.58 (m, 4H), 1.21–1.36 (m, 16H), 1.03 (t, J = 7.3 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 174.45, 131.88, 128.63, 58.64, 57.87, 51.96, 33.88, 29.99, 29.86, 29.74, 29.46, 27.98, 27.76, 26.88, 26.72, 25.88, 21.32, 10.48. HRMS calcd for C21H39O3 [M+1]+ 339.2899, found 339.2896. Hydrolysis of 36 as described above gave 8 (92%) as a colorless oil: TLC, 30% EtOAc/hexanes, Rf ≈ 0.3; 1H NMR (300 MHz, CDCl3) δ 5.27–5.43 (m, 2H), 2.85–2.93 (m, 2H), 2.34 (t, J = 7.6 Hz, 2H), 1.95–2.11 (m, 4H), 1.64–1.72 (m, 2H), 1.49–1.60 (m, 4H), 1.22–1.36 (m, 16H), 1.03 (t, J = 7.4 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ 179.42, 131.54, 128.40, 60.08, 58.75, 57.73, 34.59, 31.86, 29.86, 29.74, 29.71, 29.45, 27.84, 27.42, 26.81, 26.64, 24.85, 21.28, 15.47, 10.81. HRMS calcd for C20H37O3 [M+1]+ 325.2743, found 325.2747.
37: 1H NMR (300 MHz, CDCl3) δ 5.25–5.35 (m, 2H), 3.61 (s, 3H), 2.79–2.89 (m, 2H), 2.25 (t, J = 7.3 Hz, 2H), 1.93–2.04 (m, 4H), 1.19–1.60 (m, 22H), 1.00 (t, J = 7.2 Hz, 3H); 13C NMR (100 MHz, CDCl3) d174.48, 130.41, 129.54, 58.54, 57.45, 51.62, 34.27, 29.92, 29.81, 29.67, 29.63, 29.47, 29.46, 29.34, 27.80, 27.42, 27.27, 26.42, 25.14, 10.82. HRMS calcd for C21H39O3 [M+1]+ 339.2899, found 339.2900. Hydrolysis of 37 as described above gave 10 (92%) as a colorless oil: TLC, SiO2, 30% EtOAc/hexanes, Rf ≈ 0.3; 1H NMR (300 MHz, CDCl3) δ 5.28–5.40 (m, 2H), 2.84–2.94 (m, 2H), 2.31 (t, J = 7.6 Hz, 2H), 1.96–2.04 (m, 4H), 1.02–1.62 (m, 22H), 1.01 (t, 3H, J = 7.4 Hz); 13C NMR (75 MHz, CDCl3) δ 180.10, 130.45, 129.57, 58.74, 57.67, 34.27, 29.92, 29.81, 29.66, 29.60, 29.46, 29.43, 29.25, 27.76, 27.43, 27.28, 26.41, 24.89, 21.27, 10.81. HRMS calcd for C20H37O3 [M+1]+ 325.2743, found 325.2745.
38: 1H NMR (400 MHz, CDCl3) δ 3.67 (s, 3H), 2.84–2.94 (m, 2H), 2.31 (t, 2H, J = 7.4 Hz), 1.42–1.65 (m, 6H), 1.22–1.34 (m, 24H), 1.04 (t, 3H, J = 7.3 Hz). HRMS calcd for C21H41O3 [M+1]+ 341.3056, found 341.3056. Hydrolysis of 38 as described above gave 12 (94%) as white solid, mp 62.1–62.5 °C; TLC, 30% EtOAc/hexanes, Rf ≈ 0.35; 1H NMR (400 MHz, CDCl3) δ 2.86–2.94 (m, 2H), 2.34 (t, 2H, J = 7.3 Hz), 1.46–1.65 (m, 30H), 1.04 (t, 3H, J = 7.35 Hz); 13C NMR (100 MHz, CDCl3) δ 180.04, 58.83, 57.47, 34.24, 30.06, 30.03, 29.92, 29.81, 29.66, 29.60, 29.46, 29.43, 29.25, 27.76, 27.43, 27.28, 26.41, 24.89, 21.27, 10.89. HRMS calcd for C20H39O3 [M+1]+ 327.5219, found 327.5216.
Synthesis of 40
Alkylation of 3929 with 2-(4-bromobutoxy)tetrahydro-2H-pyran-2-yloxy32 as described for 32 produced tert-butyldiphenyl-[16-(tetrahydro-2H-pyran-2-yloxy)hexadec-11-ynyloxy]silane (66%) as a colorless oil which was used without further purification. TLC: 10% EtOAc/hexane, Rf ≈ 0.5.
A solution of the above crude tert-butyldiphenyl-[16-(tetrahydro-2H-pyran-2-yloxy)hexadec-11-ynyloxy]silane (6 g, 10.42 mmol) and tetra-n-butylammonium fluoride (3.14 g, 12.5 mL of a 1 M soln in THF, 12.50 mmol) in THF (150 mL) was stirred at rt under an argon atmosphere. After 5 h, the reaction mixture was quenched with sat. aq. NH4Cl (5 mL), washed with water (100 mL), and brine (75 mL). The aqueous layer was back-extracted with ether (2 × 75 mL). The combined organic extracts were dried over Na2SO4, concentrated under reduced pressure, and the residue was purified by SiO2 column chromatography using 5–10% EtOAc/hexanes as eluent to give 40 (3.17 g, 80% overall) as a colorless oil. TLC: 40% EtOAc/hexanes, Rf ≈ 0.4; 1H NMR (CDCl3, 300 MHz) δ 4.57–4.59 (m, 1H), 3.82–3.90 (m, 1H), 3.71–3.79 (m, 1H), 3.64 (t, 2H, J = 6.8 Hz), 3.46–3.53 (m, 1H), 3.36–3.44 (m, 1H), 2.10–2.22 (m, 4H), 1.20–1.80 (m, 26H); 13C NMR (100 MHz, CDCl3) δ 18.90, 18.95, 21.51, 25.01, 25.63, 25.82, 27.85, 29.03, 29.15, 29.44, 29.61, 29.72, 29.96, 30.42, 63.88, 64.57, 66.65, 80.20, 80.55, 108.24. HRMS calcd for C21H39O3 [M+1]+ 339.2899, found 339.2897.
Synthesis of 41
Semi-hydrogenation of 40 as described above gave 16-(tetrahydro-2H-pyran-2-yloxy)hexadec-11(Z)-en-1-ol (99%) as a colorless oil. TLC: 20% EtOAc/hexane, Rf ≈ 0.30; 1H NMR (CDCl3, 300 MHz) δ 5.33–5.37 (m, 2H), 4.58 (m, 1H), 3.83–3.90 (m, 1H), 3.73–3.77 (m, 1H), 3.65 (t, 2H, J = 6.7 Hz), 3.46–3.53 (m, 1H), 3.34–3.44 (m, 1H), 1.97–2.09 (m, 4H), 1.20–1.83 (m, 26H); 13C NMR (100 MHz, CDCl3) δ 20.70, 23.41, 23.99, 24.90, 24.96, 28.45, 28.47, 29.06, 29.37, 30.13, 30.23, 30.46, 30.51, 31.26, 32.44, 61.81, 62.37, 68.14, 107.45, 130.56, 131.83. HRMS calcd for C21H41O3 [M+1]+ 341.3056, found 341.0360.
Jones oxidation of the preceding 16-(tetrahydro-2H-pyran-2-yloxy)hexadec-11(Z)-en-1-ol as described above gave 41 (68%) as a colorless oil. TLC: SiO2, 40% EtOAc/hexanes, Rf ≈ 0.40; 1H NMR (CDCl3, 300 MHz) δ 5.33–5.37 (m, 2H), 4.56–4.58 (m, 1H), 3.83–3.88 (m, 1H), 3.73–3.78 (m, 1H), 3.49–3.53 (m, 1H), 3.35–3.43 (m, 1H), 2.34 (t, J = 7.0 Hz, 2H) 1.97–2.09 (m, 4H), 1.20–1.84 (m, 24H); 13C NMR (100 MHz, CDCl3) δ 19.54, 23.55, 25.71, 25.98, 26.41, 28.53, 28.67, 29.01, 29.14, 29.18, 29.22, 29.32, 30.04, 30.36, 32.56, 64.63, 67.89, 107.26, 130.85, 130.88, 180.48. HRMS calcd for C21H39O4 [M+1]+ 341.3056, found 341.0360.
Synthesis of 42
A solution of 41 (2.1 g, 5.93 mmol) and p-TSA (50 mg) in MeOH (30 mL) was stirred at room temperature for 10 h, then concentrated in vacuo and the residue was purified by SiO2 column chromatography using 15% EtOAc/hexanes as eluent to give methyl 16-hydroxyhexadec-11(Z)-enoate (1.42 g, 83%) as a colorless oil. TLC: 20% EtOAc/hexanes, Rf ≈ 0.35; 1H NMR (CDCl3, 300 MHz) δ 5.33–5.37 (m, 2H), 3.65 (s, 3H), 3.63 (t, J = 7.3 Hz, 2H), 2.29 (t, J = 7.0 Hz, 2H), 1.97–2.08 (m, 4H), 1.21–1.64 (m, 18H); 13C NMR (75 MHz) δ 174.77, 130.30, 129.70, 62.95, 51.73, 34.28, 32.42, 29.64, 29.45, 29.17, 29.03, 27.29, 27.10, 27.08, 26.03, 25.07. HRMS calcd for C17H33O3 [M+1]+ 285.2430, found 285.2434.
Diisopropyl azodicaboxylate (DIAD; 1.15 g, 5.70 mmol,) was added dropwise to a −20 °C solution of triphenylphosphine (1.49 g, 5.70 mmol) in dry THF (30 mL) under an argon atmosphere. After stirring for 10 min, a solution of methyl 16-hydroxyhexadec-11(Z)-enoate (1.35 g, 4.75 mmol) in anhydrous THF (5 mL) was added dropwise. After 30 min at −20 °C, the reaction mixture was warmed to 0 °C and diphenylphosphoryl azide (DPPA, 1.38 g, 5.70 mmol) was added dropwise. After stirring at room temperature for 6 h, the reaction was quenched with water (3 mL), diluted with ether (50 mL), and washed with brine (40 mL). The aqueous layer was back-extracted with ether (2 × 30 mL). The combined organic extracts were dried over Na2SO4, and concentrated under reduced pressure. The residue was purified by SiO2 column chromatography using 5% EtOAc/hexanes as eluent to give 42 (1.14 g, 78%) as a pale yellow oil. TLC: 10% EtOAc/hexanes, Rf ≈ 0.45; 1H NMR (300 MHz) δ 5.31–5.43 (m, 2H), 3.66 (s, 3H), 3.26 (t, J = 6.7 Hz, 2H), 2.30 (t, J = 7.1 Hz, 2H), 1.97–2.10 (m, 4H), 1.50–1.64 (m, 4H), 1.15–1.48 (m, 14H); 13C NMR (100 MHz) δ 174.32, 130.84, 129.01, 51.09, 51.05, 34.21, 32.58, 32.18, 29.86, 29.61, 29.59, 29.54, 29.42, 29.31, 27.40, 26.94, 25.23; IR (neat) 2985, 2954, 2845, 2106, 1754, 1250, 1104, 1029 cm−1; HRMS calcd for C17H32N3O2 [M+1]+ 310.2495, found 310.2500.
Synthesis of 21
Triphenylphosphine (1.15 g., 4.41 mmol) was added to a room temperature solution of 42 (1.05 g., 3.4 mmol) in THF (25 mL). After 2 h, water (200 mL) was added and the stirring was continued for another 8 h. The reaction mixture was then diluted with EtOAc (20 mL), washed with water (20 mL) and brine (25 mL). Aqueous layers were back-extracted with EtOAc (2 × 30 mL). The combined organic extracts were dried over Na2SO4, concentrated under reduced pressure and further dried under high vacuum for 4 h. The crude methyl 16-aminohexadec-11(Z)-enoate was used in the next step without additional purification.
2-(Methylamino)-2-oxoacetic acid29 (79 mg, 0.77 mmol), 1-hydroxybenzotriazole (101 mg, 0.77 mmol; HOBt) and diisopropylethylamine (105 mg, 0.77 mmol; DIPEA) were added to a stirring solution of crude methyl 16-aminohexadec-11(Z)-enoate (180 mg, 0.64 mmol) from above in anhydrous DMF (20 mL) under an argon atmosphere. After 5 min, 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (147 mg, 0.77 mmol; EDCI) was added as a solid. After stirring for 12 h at room temperature, the reaction mixture was diluted with EtOAc (30 mL), washed with water (3 × 20 mL), and brine (20 mL). The combined aqueous layers were back-extracted with EtOAc (3 × 30 mL). The combined organic extracts were dried over Na2SO4, concentrated under reduced pressure, and the residue was purified by SiO2 column chromatography using EtOAc as eluent to give methyl 16-(2-(methylamino)-2-oxoacetamido)hexadec-11(Z)-enoate (160 mg, 68%). TLC: 100% EtOAc, Rf ≈ 0.4; 1H NMR (CDCl3, 300 MHz) δ 7.45 (br s, 1H), 5.26–5.42 (m, 2H), 3.66 (s, 3H), 3.27–3.35 (m, 2H), 2.90 (d, 3H, J = 5.2 Hz), 2.30 (t, 2H, J = 7.3 Hz), 1.96–2.08 (m, 4H), 1.24–1.66 (m, 18H); 13C NMR (CDCl3, 75 MHz) δ 174.60, 160.81, 159.94, 130.87, 129.08, 51.68, 39.79, 34.33, 29.91, 29.68, 29.63, 29.50, 29.46, 29.36, 29.02, 27.46, 27.08, 26.91, 26.40, 25.17. HRMS calcd for C20H36N2O4 [M+1]+ 368.2675, found 368.2675.
Methyl 16-(2-(methylamino)-2-oxoacetamido)hexadec-11(Z)-enoate (150 mg, 0.40 mmol) was hydrolyzed using LiOH as described above to afford 21 (126 mg, 89%) as a white powder, mp 110.2–110.6 °C. TLC: 5% MeOH/CH2Cl2, Rf ≈ 0.4; 1H NMR (CDCl3, 300 MHz) δ 7.80 (br s, 1H), 7.66 (br s, 1H), 5.26–5.42 (m, 2H), 3.28–3.35 (m, 2H), 2.90 (s, 3H), 2.36 (t, 2H, J = 7.3 Hz), 1.97–2.08 (m, 4H), 1.51–1.64 (m, 4H), 1.22–1.42 (m, 14H); 13C NMR (CDCl3, 75 MHz) δ 177.98, 160.96, 159.93, 130.83, 129.22, 39.91, 33.91, 29.58, 29.25, 29.12, 29.01, 28.95, 27.21, 27.09, 26.93, 26.46, 24.89. HRMS calcd for C19H34N2O4 [M+1]+ 354.2519, found 354.2516.
NRCM Bioassays
The biological activities of the metabolites and synthetic analogs were determined using a bioassay essentially as described previously.33 Briefly, neonatal rat cardiomyocytes (NRCM) were isolated from 1–2 day old Wistar rats and cultured as monolayers on the bottom of Falcon flasks (12.5 cm) in 2.0 mL of Halle SM 20-I medium supplemented with 10% heat-inactivated fetal calf serum and 2 μM fluoro-deoxyuridine. Spontaneously beating cell clusters occurred after 5–7 days (120–150 beats/min, monitored at 37 °C using an inverted microscope). The beating rates were determined for 6 to 8 individual clusters before and five minutes after addition of the test substance(s). Based upon the difference between the basal and compound-induced beating rate of the individual clusters, the chronotropic effects (changes in beats/min) were calculated and are given as mean SE values; n = 18–40 clusters originating from at least three independent NRCM cultures. All test compounds were prepared as 1000-fold stock solutions in ethanol and tested at a final concentration of 30 nM. The exception was EPA that required a final concentration of 3.3 mM and 30 min preincubation.
Supplementary Material
Acknowledgments
Financial support provided by the Robert A. Welch Foundation (GL625910) and NIH (GM31278, DK38226) to JRF and by the Deutsche Forschungsgemeinschaft (SCHU-822/5-1) to AK, WHS and DNM. Prof. Kasem Nithipatikom (Medical College of Wisconsin) kindly provided high-resolution mass spectral analyses.
Footnotes
Abbreviations: ω-3 PUFAs, omega-3 polyunsaturated fatty acids; EPA, eicosa-5(Z),8(Z),11(Z),14(Z),17(Z)-pentaenoic acid; DHA, docosa-5(Z),8(Z),11(Z),14(Z),16(Z),19(Z)-hexaenoic acid; AA, arachidonic acid or eicosa-5(Z),8(Z),11(Z),14(Z)-tetraenoic acid; EETeTr, epoxyeicosatetraenoic acid; NRCM, neonatal rat cardiomyocytes; sEH, soluble epoxide hydrolase; EET, epoxyeicosatrienoic acid; EEZE, 14,15-epoxyeicosa-5(Z)-enoic acid.
Supporting Information Available. Synthetic procedures, analytical data, and NMR spectra for all new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Siddiqui RA, Shaikh SR, Sech LA, Yount HR, Stillwell W, Zaloga GP. Omega 3-fatty acids: Health benefits and cellular mechanisms of action. Mini-Rev Med Chem. 2004;4:859–871. doi: 10.2174/1389557043403431. [DOI] [PubMed] [Google Scholar]; (b) Geleijnse JM, Giltay EJ, Schouten EG, de Goede J, Oude Griep LM, Teitsma-Jansen AM, Katan MB, Kromhout D. Effect of low doses of ω-3 fatty acids on cardiovascular diseases in 4,837 post-myocardial infarction patients: Design and baseline characteristics of the Alpha Omega Trial. Am Heart J. 2010;159:539–546. doi: 10.1016/j.ahj.2009.12.033. [DOI] [PubMed] [Google Scholar]
- 2.(a) Holub BJ. Docosahexaenoic acid (DHA) and cardiovascular disease risk factors. Prostag Leukotr Ess. 2009;81:199–204. doi: 10.1016/j.plefa.2009.05.016. [DOI] [PubMed] [Google Scholar]; (b) Lavie CJ, Milani RV, Mehra MR, Ventura HO. Omega-3 polyunsaturated fatty acids and cardiovascular diseases. J Am Coll Cardio. 2009;54:585–594. doi: 10.1016/j.jacc.2009.02.084. [DOI] [PubMed] [Google Scholar]
- 3.(a) Leaf A, Kang JX, Xiao YF, Billman GE. Clinical Prevention of Sudden Cardiac Death by n-3 Polyunsaturated Fatty Acids and Mechanism of Prevention of Arrhythmias by ω-3 Fish Oils. Circulation. 2003;107:2646–2652. doi: 10.1161/01.CIR.0000069566.78305.33. [DOI] [PubMed] [Google Scholar]; (b) von Schacky C. Cardiovascular disease prevention and treatment. Prostag Leukotr Ess. 2009;81:193–198. doi: 10.1016/j.plefa.2009.05.009. [DOI] [PubMed] [Google Scholar]; (c) Fedačko J, Pella D, Mechírová V, Horvath P, Rybár RP, Varjassyová P, Vargová V. ω-3 PUFAs-From dietary supplements to medicines. Pathophysiol. 2007;14:127–132. doi: 10.1016/j.pathophys.2007.04.001. [DOI] [PubMed] [Google Scholar]; (d) Fischer R, Dechend R, Qadri F, Markovic M, Feldt S, Herse F, Park JK, Gapelyuk A, Schwarz I, Zacharzowsky UB, Plehm R, Safak E, Heuser A, Schirdewan A, Luft FC, Schunck WH, Muller DN. Dietary ω-3 Polyunsaturated Fatty Acids and Direct Renin Inhibition Improve Electrical Remodeling in a Model of High Human Renin Hypertension. Hypertension. 2008;51:540–546. doi: 10.1161/HYPERTENSIONAHA.107.103143. [DOI] [PubMed] [Google Scholar]; (e) Marchioli R, Barzi F, Bomba E, Chieffo C, Di Gregorio D, Di Mascio R, Franzosi MG, Geraci E, Levantesi G, Maggioni AP, Mantini L, Marfisi RM, Mastrogiuseppe G, Mininni N, Nicolosi GL, Santini M, Schweiger C, Tavazzi L, Tognoni G, Tucci C, Valagussa F. Early protection against sudden death by ω-3 polyunsaturated fatty acids after myocardial infarction: time-course analysis of the results of the Gruppo Italiano per lo Studio della Sopravvivenza nell’Infarto Miocardico (GISSI)-Prevenzione. Circulation. 2002;105:1897–1903. doi: 10.1161/01.cir.0000014682.14181.f2. [DOI] [PubMed] [Google Scholar]
- 4.(a) Harris WS. Omega-3 fatty acids and cardiovascular disease: A case for omega-3 index as a new risk factor. Pharmacol Res. 2007;55:217–223. doi: 10.1016/j.phrs.2007.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Pignier C, Revenaz C, Rauly-Lestienne I, Cussac D, Delhon A, Gardette J, Le Grand B. Direct protective effects of poly-unsaturated fatty acids, DHA and EPA, against activation of cardiac late sodium current: a mechanism for ischemia selectivity. Basic Res Cardiol. 2007;102:553–564. doi: 10.1007/s00395-007-0676-x. [DOI] [PubMed] [Google Scholar]; (c) Oliw EH. 17R(18S)-epoxyeicosatetraenoic acid, a cytochrome P-450 metabolite of 20:5ω-3 in monkey seminal vesicles, is metabolized to novel prostaglandins. Biochem Biophys Res Commun. 1991;178:1444–1450. doi: 10.1016/0006-291x(91)91055-h. [DOI] [PubMed] [Google Scholar]
- 5.(a) Smith WL. Cyclooxygenases, peroxide tone and the allure of fish oil. Curr Opin Cell Biol. 2005;17:174–182. doi: 10.1016/j.ceb.2005.02.005. [DOI] [PubMed] [Google Scholar]; (b) Li Y, Kang JX, Leaf A. Differential effects of various eicosanoids on the production or prevention of arrhythmias in cultured neonatal rat cardiac myocytes. Prostaglandins. 1997;54:511–530. doi: 10.1016/s0090-6980(97)00122-6. [DOI] [PubMed] [Google Scholar]; (c) Vedin I, Cederholm T, Freund-Levi Y, Basun H, Hjorth E, Irving GF, Eriksdotter-Joenhagen M, Schultzberg M, Wahlund LO, Palmblad J. Reduced prostaglandin F2α release from blood mononuclear leukocytes after oral supplementation of ω-3 fatty acids: the OmegAD study. J Lipid Res. 2010;51:1179–1185. doi: 10.1194/jlr.M002667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.(a) Schwarz D, Kisselev P, Chernogolov A, Schunck WH, Roots I. Human CYP1A1 variants lead to differential eicosapentaenoic acid metabolite patterns. Biochem Biophys Res Commun. 2005;336:779–783. doi: 10.1016/j.bbrc.2005.08.172. [DOI] [PubMed] [Google Scholar]; (b) Barbosa-Sicard E, Markovic M, Honeck H, Christ B, Muller DN, Schunck WH. Eicosapentaenoic acid metabolism by cytochrome P450 enzymes of the CYP2C subfamily. Biochem Biophys Res Commun. 2005;329:1275–1281. doi: 10.1016/j.bbrc.2005.02.103. [DOI] [PubMed] [Google Scholar]; (c) Muller DN, Schmidt C, Barbosa-Sicard E, Wellner M, Gross V, Hercule H, Markovic M, Honeck H, Luft FC, Schunck WH. Mouse Cyp4a isoforms: enzymatic properties, gender- and strain-specific expression, and role in renal 20-hydroxyeicosatetraenoic acid formation. Biochem J. 2007;403:109–118. doi: 10.1042/BJ20061328. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Oliw EH, Sprecher HW. Metabolism of polyunsaturated (ω-3) fatty acids by monkey seminal vesicles: isolation and biosynthesis of omega-3 epoxides. Biochim Biophys Acta. 1991;1086:287–294. doi: 10.1016/0005-2760(91)90172-e. [DOI] [PubMed] [Google Scholar]; (e) VanRollins MJ. Epoxygenase metabolites of docosahexaenoic and eicosapentaenoic acids inhibit platelet aggregation at concentrations below those affecting thromboxane synthesis. Pharmacol Exp Ther. 1995;274:798–804. [PubMed] [Google Scholar]; (f) Fer M, Dreano Y, Lucas D, Corcos L, Salauen JP, Berthou F, Amet Y. Metabolism of eicosapentaenoic and docosahexaenoic acids by recombinant human cytochromes P450. Arch Biochem Biophys. 2008;471:116–125. doi: 10.1016/j.abb.2008.01.002. [DOI] [PubMed] [Google Scholar]
- 7.(a) Schwarz D, Kisselev P, Ericksen SS, Szklarz GD, Chernogolov A, Honeck H, Schunck WH, Roots I. Arachidonic and eicosapentaenoic acid metabolism by human CYP1A1: highly stereoselective formation of 17(R),18(S)-epoxyeicosatetraenoic acid. Biochem Pharmacol. 2004;67:1445–1457. doi: 10.1016/j.bcp.2003.12.023. [DOI] [PubMed] [Google Scholar]; (b) Lucas D, Goulitquer S, Marienhagen J, Fer M, Dreano Y, Schwaneberg U, Amet Y, Corcos L. Stereoselective epoxidation of the last double bond of polyunsaturated fatty acids by human cytochromes P450. J Lipid Res. 2010;51:1125–1133. doi: 10.1194/jlr.M003061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Arnold C, Markovic M, Blossey K, Wallukat G, Fischer R, Dechend R, Konkel A, von Schacky C, Luft FC, Muller DN, Rothe M, Schunck WH. Arachidonic acid-metabolizing cytochrome P450 enzymes are targets of omega-3 fatty acids. J Biol Chem. 2010;285:32720–32733. doi: 10.1074/jbc.M110.118406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Knapp HR, Miller AJ, Lawson JA. Urinary excretion of diols derived from eicosapentaenoic acid during n-3 fatty acid ingestion by man. Prostag. 1991;42:47–54. doi: 10.1016/0090-6980(91)90093-u. [DOI] [PubMed] [Google Scholar]
- 10.Shearer GC, Harris WS, Pedersen TL, Newman JW. Detection of omega-3 oxylipins in human plasma and response to treatment with omega-3 acid ethyl esters. J Lipid Res. 2009;51:2074–2081. doi: 10.1194/jlr.M900193-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.(a) Lauterbach B, Barbosa-Sicard E, Wang MH, Honeck H, Kärgel E, Theuer J, Schwartzman ML, Haller H, Luft FC, Gollasch M, Schunck W-H. Cytochrome P450-dependent eicosapentaenoic acid metabolites are novel BK channel activators. Hypertension. 2002;39:609–613. doi: 10.1161/hy0202.103293. [DOI] [PubMed] [Google Scholar]; (b) Hercule HC, Salanova B, Essin K, Honeck H, Falck JR, Sausbier M, Ruth P, Schunck WH, Luft FC, Gollasch M. The vasodilator 17,18-epoxyeicosatetraenoic acid targets the pore-forming BK alpha channel subunit in rodents. Exp Physiol. 2007;92:1067–1076. doi: 10.1113/expphysiol.2007.038166. [DOI] [PubMed] [Google Scholar]
- 12.Gauthier KM, Deeter C, Krishna UM, Reddy YK, Bondlela M, Falck JR, Campbell WB. 14,15-epoxyeicosa-5(Z)-enoic acid: A selective epoxyeicosatrienoic acid antagonist that inhibits endothelium-dependent hyperpolarization and relaxation in coronary arteries. Circ Res. 2002;90:1028–1036. doi: 10.1161/01.res.0000018162.87285.f8. [DOI] [PubMed] [Google Scholar]
- 13.Yang W, Tuniki VR, Anjaiah S, Falck JR, Hillard CJ, Campbell WB. Characterization of epoxyeicosatrienoic acid binding site in U937 membranes using a novel radiolabeled agonist, 20-125I-14,15-epoxyeicosa-8(Z)-enoic acid. J Pharm Exp Ther. 2008;324:1019–1027. doi: 10.1124/jpet.107.129577. [DOI] [PubMed] [Google Scholar]
- 14.Morin C, Sirois M, Echave V, Rizcallah E, Rousseau E. Relaxing effects of 17(18)-EpETE on arterial and airway smooth muscles in human lung. Am J Physiol Lung Cell Mol Physiol. 2009;296:L130–139. doi: 10.1152/ajplung.90436.2008. [DOI] [PubMed] [Google Scholar]
- 15.Morin C, Sirois M, Echave V, Albadine R, Rousseau E. 17,18-EpETE targets PPAR{gamma} and p38MAPK to mediate its anti-inflammatory effect in lung: role of sEH. Am J Respir Cell Mol Biol. 2009;43:564–575. doi: 10.1165/rcmb.2009-0155OC. [DOI] [PubMed] [Google Scholar]
- 16.Recent review: Spector AA, Norris AW. Action of epoxyeicosatrienoic acids on cellular function. Am J Physiol Cell Physiol. 2007;292:C996–1012. doi: 10.1152/ajpcell.00402.2006.
- 17.(a) Yang W, Tuniki VR, Anjaiah S, Falck JR, Hillard CJ, Campbell WB. Characterization of epoxyeicosatrienoic acid binding site in U937 membranes using a novel radiolabeled agonist, 20-125I-14,15-epoxyeicosa-8(Z)-enoic acid. J Pharmacol Exp Ther. 2008;324:1019–1027. doi: 10.1124/jpet.107.129577. [DOI] [PubMed] [Google Scholar]; (b) Chen Y, Falck JR, Tuniki VR, Campbell WB. 20-125Iodo-14,15-epoxyeicosa-5(Z)-enoic acid: a high-affinity radioligand used to characterize the epoxyeicosatrienoic acid antagonist binding site. J Pharmacol Exp Ther. 2009;331:1137–1145. doi: 10.1124/jpet.109.157818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.(a) Kang JX, Leaf A. Effects of long-chain polyunsaturated fatty acids on the contraction of neonatal rat cardiac myocytes. Proc Natl Acad Sci U S A. 1994;91:9886–9890. doi: 10.1073/pnas.91.21.9886. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Kang JX, Leaf A. Prevention and termination of β-adrenergic agonist-induced arrhythmias by free polyunsaturated fatty acids in neonatal rat cardiac myocytes. Biochem Biophys Res Commun. 1995;208:629–636. doi: 10.1006/bbrc.1995.1385. [DOI] [PubMed] [Google Scholar]; (c) Kang JX, Xiao YF, Leaf A. Free, long-chain, polyunsaturated fatty acids reduce membrane electrical excitability in neonatal rat cardiac myocytes. Proc Natl Acad Sci U S A. 1995;92:3997–4001. doi: 10.1073/pnas.92.9.3997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.In the context of this anti-arrhythmic study, agonist activity refers to the ability of a species to reduce the compound-induced abnormal or pathogenic beating rate of NRCMs towards the normal, spontaneous beating rate of 120–150 beats/min. Inverse agonist activity refers to the ability of a species to increase the beating rate beyond the normal, spontaneous beating rate, i.e., it has a biological activity opposite to an agonist. An antagonist is a species capable of partially or completely blocking the activity of an agonist or inverse agonist, while having no significant effect of its own on receptor or physiological activity.
- 20.Review: Dobrev D, Nattel S. New antiarrhythmic drugs for treatment of atrial fibrillation. Lancet. 2010;375:1212–1223. doi: 10.1016/S0140-6736(10)60096-7.
- 21.For a discussion of chemical and metabolic restrictions for epoxanoids see, Falck JR, Kodela R, Manne R, Atcha KR, Puli N, Dubasi N, Manthati VL, Capdevila JH, Yi X-Y, Goldman DH, Morisseau C, Hammock BD, Campbell WB. 14,15-Epoxyeicosa-5,8,11-trienoic Acid (14,15-EET) Surrogates Containing Epoxide Bioisosteres: Influence upon Vascular Relaxation and Soluble Epoxide Hydrolase Inhibition. J Med Chem. 2009;52:5069–5075. doi: 10.1021/jm900634w.
- 22.To estimate comparative metabolic stability, EPA-epoxide 1 and several analogs were incubated for 30 min at 37 °C with rat liver homogenate in the presence of NADPH and analyzed by LC/MS (Supporting Information Section).
- 23.Cowart LA, Wei S, Hsu M-H, Johnson EF, Krishna MU, Falck JR, Capdevila JH. The CYP4A Isoforms Hydroxylate Epoxyeicosatrienoic Acids to Form High Affinity Peroxisome Proliferator-activated Receptor Ligands. J Biol Chem. 2002;277:35105–35112. doi: 10.1074/jbc.M201575200. [DOI] [PubMed] [Google Scholar]
- 24.Imig JD. Cardiovascular therapeutic aspects of soluble epoxide hydrolase inhibitors. Cardiovascular Drug Rev. 2006;24:169–188. doi: 10.1111/j.1527-3466.2006.00169.x. [DOI] [PubMed] [Google Scholar]
- 25.(a) Morisseau C, Goodrow MH, Dowdy D, Zheng J, Greene JF, Sanborn JR, Hammock BD. Potent urea and carbamate inhibitors of soluble epoxide hydrolases. Proc Natl Acad Sci U S A. 1999;96:8849–8854. doi: 10.1073/pnas.96.16.8849. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Kowalski JA, Swinamer AD, Muegge I, Eldrup AB, Kukulka A, Cywin CL, De Lombaert S. Rapid synthesis of an array of trisubstituted urea-based soluble epoxide hydrolase inhibitors facilitated by a novel solid-phase method. Bioorg Med Chem Lett. 2010;20:3703–3707. doi: 10.1016/j.bmcl.2010.04.078. [DOI] [PubMed] [Google Scholar]; (c) Shen HC, Ding F-X, Wang S, Deng Q, Zhang X, Chen Y, Zhou G, Xu S, Chen H-s, Tong X, Tong V, Mitra K, Kumar S, Tsai C, Stevenson AS, Pai L-Y, Alonso-Galicia M, Chen X, Soisson SM, Roy S, Zhang B, Tata JR, Berger JP, Colletti SL. Discovery of a highly potent, selective, and bioavailable soluble epoxide hydrolase inhibitor with excellent ex vivo target engagement. J Med Chem. 2009;52:5009–5012. doi: 10.1021/jm900725r. [DOI] [PubMed] [Google Scholar]
- 26.Gunstone FD, Lie Ken Jie M. Fatty acids. 24. Synthesis of ten octadecadiynoic acids and of the related cis,cis- and trans,trans-octadecadienoic acids. Chem Phys Lipids. 1970;4:1–14. [Google Scholar]
- 27.Ochiai M, Sueda T. Tetrahydrofuranylation of alcohols catalyzed by alkylperoxy-λ 3-iodane and carbon tetrachloride. Tetrahedron Lett. 2004;45:3557–3559. [Google Scholar]
- 28.Seifert RM. Synthesis, spectra, odor properties, and structural relationship of (Z)-6-Nonenal and (Z)-5-octen-1-ol to fruit fly attractants. J Agric Food Chem. 1981;29:647–649. [Google Scholar]
- 29.Harcken C, Bruckner R. Elucidation of the stereostructure of the annonaceous acetogenin (+)-montecristin through total synthesis. New J Chem. 2001;25:40–54. [Google Scholar]
- 30.Wessel JC, Matyja M, Neugebauer M, Kiefer H, Daldrup T, Tarbah FA, Weber H. Characterization of oxalic acid derivatives as new metabolites of metamizol (dipyrone) in incubated hen’s egg and human. Eur J Pharm Sci. 2006;28:15–25. doi: 10.1016/j.ejps.2005.11.010. [DOI] [PubMed] [Google Scholar]
- 31.Molander GA, Figueroa R. cis-Dihydroxylation of Unsaturated Potassium Alkyl- and Aryltrifluoroborates. Org Lett. 2006;8:75–78. doi: 10.1021/ol052549e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Corey EJ, Eckrich TM. A method for the stereospecific synthesis of chiral cis-2-alkylcyclopropyllithium reagents. Tetrahedron Lett. 1984;25:2415–2418. [Google Scholar]
- 33.Wallukat G, Muñoz Saravia SG, Haberland A, Bartel S, Araujo R, Valda G, Duchen D, Diaz Ramirez I, Borges AC, Schimke I. Distinct patterns of autoantibodies against G-protein-coupled receptors in Chaga’s cardiomyopathy and megacolon: their potential impact for early risk assessment in asymptomatic Chagas’ patients. J Am Coll Cardiol. 2010;55:463–468. doi: 10.1016/j.jacc.2009.06.064. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.