

Abstract
The determination of single nucleotide polymorphisms (SNPs) has become faster and more cost effective since the advent of short read data from next generation sequencing platforms such as Roche's 454 Sequencer, Illumina's Solexa platform, and Applied Biosystems SOLiD sequencer. The SOLiD sequencing platform, which is capable of producing more than 6 GB of sequence data in a single run, uses a unique encoding scheme where color reads represent transitions between adjacent nucleotides. The determination of SNPs from color reads usually involves the translation of color alignments to likely nucleotide strings to facilitate the use of tools designed for nucleotide reads. This technique results in the loss of significant information in the color read, producing many incorrect SNP calls, especially if regions exist with dense or adjacent polymorphism. Additionally, color reads align ambiguously and incorrectly more often than nucleotide reads making integrated SNP calling a difficult challenge. We have developed ComB, a SNP calling tool which operates directly in color space, using a Bayesian model to incorporate unique and ambiguous reads to iteratively determine SNP identity. ComB is capable of accurately calling short consecutive nucleotide polymorphisms and densely clustered SNPs; both of which other SNP calling tools fail to identify. ComB, which is capable of using billions of short reads to accurately and efficiently perform whole human genome SNP calling in parallel, is also capable of using sequence data or even integrating sequence and color space data sets. We use real and simulated data to demonstrate that ComB's iterative strategy and recalibration of quality scores allow it to discover more true SNPs while calling fewer false positives than tools which use only color alignments as well as tools which translate color reads to nucleotide strings.
Key words: algorithms, genome analysis, SNPs, next generation sequencing, statistics
1. Introduction
The completion of the human genome project in 2001 (Venter et al., 2001; Lander et al., 2001) marked a watershed moment in biomedical research and bioinformatics. Since the first genome was sequenced using traditional Sanger sequencing, there have been several attempts to duplicate the feat at a fraction of the cost using massively parallel next generation sequencing technologies (Pushkarev et al., 2009). Single nucleotide polymorphisms (SNPs) and short block nucleotide polymorphisms (BNPs) are thought to play a major role in human phenotypic variation, disease development, and drug response (Shastry, 2007; Yue and Moult, 2006). The identification of such sites, a necessary step in human genome resequencing, may be key for the realization of personal medicine (Mancinelli et al., 2000) and disease prevention (Manolio et al., 2009). There exist many SNP calling algorithms dedicated to the next generation sequencing platforms which produce nucleotide reads such as Illumina's Solexa sequencer and Roche's 454 sequencing platform (Li et al., 2009b; Brockman et al., 2008; Quinlan et al., 2008). In contrast, Applied Biosystems' high-throughput (Ondov et al., 2008) ligation mediated SOLiD sequencer produces reads composed of colors that represent nucleotide transitions (Fig. 1), a departure from other sequencing machines that provides both new benefits and challenges to SNP calling. In color space, sequencing errors are rarely mistaken for SNPs, whose color signatures exist only on a subset of consecutive color mismatches (Ondov et al., 2008). Many mapping programs (Homer et al., 2009; Langmead et al., 2009; Li and Durbin, 2009) attempt to take advantage of this by translating color reads to their most likely nucleotide string after alignment. SNPs can then be identified through a “pileup” or count of converted nucleotides which cover each position (Li et al., 2009a). While this strategy provides an output format identical to that produced from sequence data, the conversion required cannot adequately maintain all of the information contained in the original color read and quality sequence. In addition, sequencing errors will obscure otherwise obvious SNP signatures creating incomplete SNP information that cannot be maintained if reads are translated to nucleotide sequences (Fig. 2). ComB handles dependencies associated with SNP loci in color space by considering the likelihood of all supported polymorphisms with the read data in aggregate, which produces significantly more accurate estimation of the genome from which the reads were sequenced.
FIG. 1.

Consider reads which map to a location with genome sequence AAAAAAAAA, which in color space is BBBBBBBB. The reads may differ from the color space genome sequence, because of either SNPs or sequencing errors. The goal of identifying likely sets of sequencing errors and SNPs that would produce the given read is complicated by the dependence between adjacent colors. Valid color substitutions are restricted to those which change the read sequence locally, while sequencing errors are not restricted. Above are a set of reads that map to the genome location in color space, along with a set of possible interpretations of the read as results of sequencing errors and SNPs. Determining the combination of SNPs and sequencing errors which affected any single read is difficult, ComBs statistical model allows the likely interpretation to be made using the entire collection of reads.
FIG. 2.

SOLiD colors represent the transition between the nucleotides. This unique encoding scheme results in each nucleotide being sequenced twice, decreasing the probability of sequencing errors being interpreted as SNPs. However, color signals are not independent, making necessary new methods to accurately determine polymorphism.
ComB's SNP calling strategy involves two parts:
Maximize available information regarding each potential polymorphism loci by staying in color space, including ambiguous reads, and iteratively updating the target genome.
Minimize error by recalibrating quality scores and applying a Bayesian model to calculate the posterior probability of polymorphism conditioned on the observed read set and subsequent alignments.
ComB was designed to perform SNP calling on the SAM alignment (Li et al., 2009a) output produced by many popular alignment programs, or the “mapping” output format produced by the alignment program PerM (Chen et al., 2009). The Methods section below contains a detailed description of ComB's statistical model and its iterative design, which produces efficient and accurate identification of polymorphism.
2. Methods
2.1. Motivation for a statistical model
The design of ComB was motivated in part by the need for a consistent statistical model to call genome variants in color space. Working in color space allows more information about SNPs to be preserved, but also requires the dependencies and mapping biases, which do not exist in nucleotide space, be addressed. For this reason, ComB's methods are described for reads in color space, although each feature of ComB generalizes to perform the same task for sequence data.
Initially, the model is described for the simplest case: haploid SNP loci are isolated and spanned only by uniquely mapping reads. Modifications to deal with additional complexities are discussed after the model introduction.
2.1.1. Bayesian statistical model
To calculate the posterior probability for the identity of the target base tj, ComB considers both the base on the reference genome gj as well as the set of reads R(tj), whose mappings span tj. ComB assumes a unique alignment constitutes a correct alignment. The read set R(tj) is composed of reads which map to unique regions spanning tj. The posterior probability of the true nucleotide identity tj = λ for 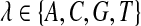 , conditioned on the observed mapping data, can be written using Bayes theorem as
, conditioned on the observed mapping data, can be written using Bayes theorem as
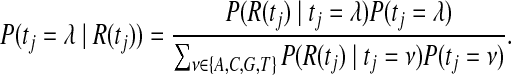 |
(1) |
where P(tj = ν) is the prior distribution of base being considered, and P(R(tj)∣tj = λ) is the conditional distribution of the read set, which can be written in terms of the read alignment scores P(r∣s, tj = λ).
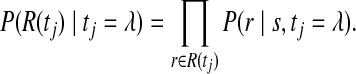 |
(2) |
The read alignment scores and SNP priors are described in the next two sections.
2.1.2. Quality calibration and alignment scores
The error distribution resulting from the sequencing process has been shown to vary depending on the sequencing platform, sequence motifs, and read position (Shendure and Ji, 2008). The SOLiD error rate, which increases on later cycles and at the tails of reads, is estimated to be quite high. In many datasets fewer than half the reads map with fewer than three mismatches (Ondov et al., 2008). The raw quality scores provided by sequencing machines reflect signal intensity and fail to capture many sources of error [Li et al., 2004]. To recalibrate the quality scores to more accurately reflect the true error rate, ComB calculates color call rates with consideration to color, reference dinucleotides, quality scores, and read position. Letting ∣Q∣ represent the number of bins spanning the space of quality scores, ComB uses uniquely mapped reads to build a matrix with dimensions 4 × 4 × 4 × ∣Q∣ for each position in the sequence. Each entry in the matrix represents the probability of seeing a color (4×), between a pair of bases (4 × 4), at a quality score (∣Q∣).
Thus, the entry in the matrix describing the probability of observing color x with quality q between reference dinucleotides b1 and b2 at the ith position in the read is
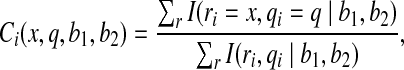 |
(3) |
where ri and qi represent the color and quality for a given read at its ith position, and I() is the indicator function, and the sum is over all uniquely mapped reads (r). This recalibration of the call rates serves primarily to identify sequence specific errors and reduce the false positive rate.
The call rates matrix described by Eqn. (3) can also be used to measure the alignment between a read r and it's quality q and a genome substring s. This read alignment score, P(r∣s), is calculated by multiplying the observation probabilities from the call rates matrix for each color and quality score along the length of the read:
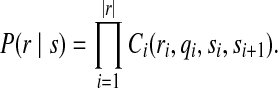 |
(4) |
This is the probability that sequencing the genome ∣r∣-mer represented by s would produce the read r.
2.1.3. Prior SNP probability
The prior SNP probability or global mutation rate P(tj = λ∣gj), depends on the genetic distance between the reference and target genomes. For the human genome, if a global GC rate of 43% (Karro et al., 2008) as well as homozygous and heterozygous SNP rates of 0.048% and 0.054% (Levy et al., 2007) are assumed, then the probability that a given reference nucleotide has mutated into one of nine possible target SNPs can be estimated using the SNP frequencies from the approximately 20 million base mutations annotated in the dbSNP database build 131 (Database of Single Nucleotide Polymorphisms [dbSNP]) (dbSNP, 2001). For example, the prior probability of the event that the reference base gj has mutated from A to homozygous C is
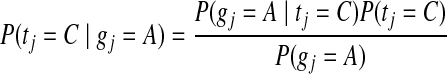 |
(5) |
Here the marginal probability of nucleotide A is estimated from the genomic GC rate:
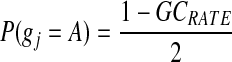 |
(6) |
and the marginal probability of a mutation leading to nucleotide C (P(tj = C)) and conditional probability of reference A in the event of a mutation (P(gj = A∣tj = C)) are estimated from the frequencies in dbSNP. For non-model organisms where SNP data is not available estimated priors can be provided, or the prior parameters can be calculated iteratively.
2.1.4. Ambiguous reads
Reads which align to multiple loci in a reference genome within a certain mismatch threshold are referred to as ambiguous. In such cases, the assumption is made that every alignment but one is spurious and may cause us to incorrectly infer the probability of a genome variant. Error in the selection of the correct alignment can lead to incorrect mapping and false determination of genome variants. For this reason, ambiguous reads are often discarded. Unfortunately, for large genomes, large fractions of mapped reads are often ambiguous. ComB preserves this data by weighting each ambiguous mapping by its relative alignment score to each reference location. Formally, if a read r maps to a set of genomic subsequences S(r), then the conditional probability of observing r given the identity at locus tj is
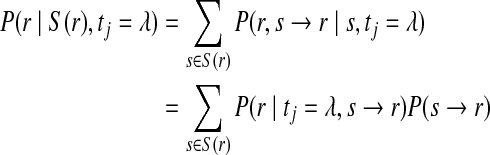 |
(7) |
where s → r represents the event that r is sequenced from s (correct alignment). The expression P(r∣S(r), tj = λ) replaces the single alignment score used in the unique read case. Since 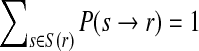 , then for each
, then for each 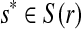 ,
,
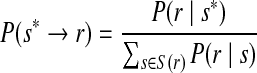 |
(8) |
where P(r∣s) is the alignment score defined in Eqn. (4).
2.1.5. Consecutive nucleotide polymorphism dependency
In color space, the positions in consecutive nucleotide polymorphisms are not independent and result in different signatures than those of isolated SNPs. Short block nucleotide polymorphism (BNPs) (defined as polymorphism between 2 and 8 bp in length) are not uncommon in the human genome. Of the approximately 20.5 million positions annotated as either single or multiple nucleotide polymorphism in dbSNP build 131, 11.07% are adjacent to another nucleotide polymorphism (dbSNP, 2001). The majority (>60%) of these positions are members of blocks of length two or three. Unlike isolated SNPs, which always change two consecutive colors, BNPs can result in a change to two or more color mismatches that are not necessarily consecutive (Fig. 1). Thus, consecutive SNP candidate blocks must be evaluated for all possible combinations of polymorphisms along the length of the block. The read alignment score can be conditioned on multiple events, 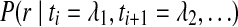 and Eqn. 1 remains applicable.
and Eqn. 1 remains applicable.
2.1.6. Diploid and multiploid organisms
If the target genome sequence comes from an organism with multiple copies of chromosomes, the parameter λ should represent the space of all possible nucleotide groupings on the chromosomes. For simplicity, consider a diploid organism where λ = W, i.e., heterozygous for nucleotides A and T. In this event, the observation score should consider the event that the read is sequenced from either chromosome. Thus, the observation score for a read aligned to such a locus is expressed as
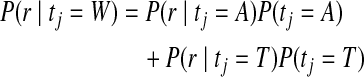 |
(9) |
where
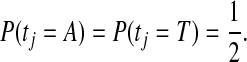 |
(10) |
When considering diploid space, 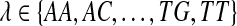 , accuracy is markedly improved through the use of iterative SNP calling. The iterative method will first call homozygous nucleotide polymorphisms (NPs), update the target genome, and then call potential diploid locations to reduce bias to the reference genome. This bias otherwise causes homozygous SNPs to be interpreted as heterozygous.
, accuracy is markedly improved through the use of iterative SNP calling. The iterative method will first call homozygous nucleotide polymorphisms (NPs), update the target genome, and then call potential diploid locations to reduce bias to the reference genome. This bias otherwise causes homozygous SNPs to be interpreted as heterozygous.
2.1.7. Iterative mapping
Dense polymorphism often produces reads with too many mismatches to map to their correct location. This is further exacerbated in color space where SNPs cause multiple color mismatches. Unfortunately, allowing more mismatches may further bias the mapping results as many reads map incorrectly to positions of relative similarity.
This bias is alleviated through ComB's iterative mapping strategy which updates the target genome to increase the number of reads that cover SNP loci.
Assuming a set of short reads 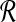 , sequenced from an unknown target genome
, sequenced from an unknown target genome  , being mapped to a similar reference genome
, being mapped to a similar reference genome 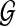 , each iteration consists of the following four steps:
, each iteration consists of the following four steps:
Initially
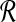 is mapped to
is mapped to 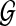 .
.The machine quality scores are recalibrated and call rates are estimated (Eqn. (3)).
The genome is scanned and SNP candidate blocks are located and tested using Eqn. (1).
The target genome is updated at positions with high posterior SNP probability, and the mapping is repeated.
2.1.8. Parallel implementation and default settings
For small single platform datasets, each iteration of ComB can be run through a single command. To increase speed and save disk space, intermediate information such as the estimated call rates are compressed and stored internally; only the consensus genome and called SNPs are written to disk. As a compromise to memory usage, by default, ComB only considers sites as NP candidates if they are spanned by at least three reads and at least 20% of such reads contain valid polymorphism signatures. BNP lengths are also limited to five positions per iteration and only sites whose posterior SNP probability exceeds 95% are intended to be called as SNPs. These parameters can be changed depending on the expected variation between the reference and target genome. For large or multi-platform datasets, multiple instances of ComB can generate coverage and valid SNP signature counts in parallel. This information is then merged to determine SNP candidate locations, and the posterior probability at each location is updated in parallel and combined to produce ComB's SNP calling output. This implementation drastically reduces running time, cutting an iteration of mapping and NP identification of 2.7 billion 50-bp reads to the unmasked human genome to just over 2 hours. Additionally, this implementation allows information from different platforms and read lengths to be combined. The intermediate files hold only information regarding candidate location and posterior probability allowing information generated from sequence space and color space reads to be merged, which provides greater accuracy for SNP calling and resequencing.
3. Results and Discussion
Tests were performed to compare ComB to Corona Lite (McKernan et al., 2009) ABIs native color space aligner and SNP caller, as well as soapSNP (Li et al., 2009b) and MAQ-consensus (Li et al., 2008), the two popular SNP calling algorithms which operate on SOLiD data after it is transformed to nucleotide based SAM (Li et al., 2009a) format. The mapping program BWA (Li and Durbin, 2009) was used to perform the color-to-nucleotide alignment and translation necessary for SOLiD data, while the alignment program PerM was used for the alignment of sequence data. BWA was run with the parameters “−c −N −k 4 −n 6” which finds all alignments of fewer than four mismatches and many with six or fewer, as this best mimics the sensitivity of PerM which is full sensitive to four mismatch alignments and partially sensitive to alignments which have as many as six mismatches on short (≤64 bp) reads. Corona Lite was also used to identify alignments with fewer than six mismatches. In all cases, alignment files were trimmed such that each SNP caller was evaluated on an equal number of mapped reads to control for small differences in alignment sensitivity. When run with it's default parameters, two iterations of ComB, run on a single CPU, performed faster than all other SNP callers. For this reason, ComB was always run for only two iterations in SNP calling comparisons.
To compare SNP discovery on real sequence and color space data, two similar E. coli datasets and genomes were downloaded from the NCBI short read archive (Archive a, 2010; Archive b, 2010) and the UCSC genome browser (Karolchik et al., 2003). The genome for a third strain (E. coli-REL606) which is moderately diverged from the other two (≤35K SNPs and little rearrangement) was also downloaded from the UCSC genome browser (Karolchik et al., 2003), and the SNPs called between this strain and each of the others were compared to the results of a global alignment generated from the program MuMMER (Kurtz et al., 2004).
To determine the experimental precision of the different algorithms and determine the specific SNP distributions that most affected performance, four simulations were performed using the Drosophila melanogaster genome.
To demonstrate ComB's ability to perform massively parallel human genome SNP calling, 2.705 billion reads from an anonymous individual of African origin were downloaded (Archive c, 2010) and used to call over 2 million NPs. These results were compared to the annotation in the most recent dbSNP database.
3.1. E. coli SNP calling experiments
3.1.1. E. coli SNP-calling in color space
Over 25 million 36-bp SOLiD reads (Archive a; 2010) from a sequencing run of the fragment library of E. coli k12-DH10B to determine SNP calling accuracy between the 4.75-Mbp genome and a similar strain of E. coli, REL606 (Karolchik et al., 2003). A global alignment between the two strains which was performed using the software MuMMER (Kurtz et al., 2004), yielded 31,902 SNPs and little genomic rearrangement. Of the NP sites, 2,314 were members of block polymorphism. Though the global SNP rate was relatively low (0.67%), the SNPs were not uniformly distributed and showed alternating areas of low and high density. The greatest distance between SNPs was 118,088 bp, while there were 19 non-overlapping regions of length 100 with at least 25 SNPs. The most SNP-dense 100-bp stretch included 33 SNPs. Of the SNPs, 20,827 were preceded by a snp than 50 bp away. Additionally, we observed a bias in inter-SNP distance due to codon degeneracy (Fig. 3).
FIG. 3.

E. coli SNP separation bias. The inter-SNP distances in the global alignment between the two E. coli strains, DH10B and REL606, were most frequently multiples of three due to codon degeneracy in protein coding regions. The most common inter-SNP distance of 3 bp is evidence that there were many regions of dense polymorphism.
ComB was compared to soapSNP, MAQ-Consensus and Corona Lite at differing coverage levels. At 25× coverage, ComB identified 10% more snps than any of the other SNP callers. Additionally, at 25× coverage ComB was able to identify 1,382 of the 2,314 sites which were members of Block Nucleotide Polymorphisms. In comparison, soapSNP and MAQ-Consensus were able to identify 795 and 791 of the 2,314 sites at 25× coverage respectively. Corona lite had far less sucess at locating block nucleotide polymorphism. In total, there were only 952 unsupported SNPs that were identified with all four SNP callers. It's likely that these are true SNPs, while others may be false positives. The results of SNP calling with SOLiD data at 25× coverage is summarized in Table 1, while SNP identification at different coverage levels is shown in Figure 4.
Table 1.
SNP Calling Performance for 36-BP SOLiD or Illumina Reads at 25× Coverage
| |
E. coli DHB10 vs. Rel606 (31,902 Total SNPs) |
|
|---|---|---|
| SNP-Caller | Valid SNPs | Unsupported |
| ComB | 28,374 | 3,430 |
| soapSNP | 25,305 | 6,500 |
| MAQ-consensus | 25,031 | 6,773 |
| Corona lite | 16,384 | 1,883 |
| |
E. coli MG1665 vs. Rel606 (33,944 Total SNPs) |
|
|---|---|---|
| SNP-Caller | Valid SNPs | Unsupported |
| ComB | 32,095 | 7,530 |
| soapSNP | 31,823 | 16,188 |
| MAQ-consensus | 31,823 | 16,191 |
PerM was used to produce all alignments for Illumina data while BWA was used for color-space alignment and translation in preparation for MAQ-Consensus and soapSNP. Corona Lites results come Corona-Lite's match pipeline. Alignment files were trimmed so that equal coverage was provided for all SNP-callers. The results for each algorithm were compared to the SNPs which exist in a global alignment between each reference and target genome. Unsupported SNPs are not present in the global alignment of the reference sequences but may be present in the individuals sequenced.
FIG. 4.

E. coli experiment: Shown are the validated SNP sites each SNP caller was able to identify at different levels of coverage for 36-bp SOLiD reads.
3.1.2. E. coli SNP-Calling with nucleotide reads
Over 20 million 36-bp Illumina reads from a sequencing run of E. coli k12-MG1665 were downloaded from NCBI (Archive b; 2010) to test the different SNP callers performance in nucleotide space. The global alignment between E. coli K12-MG1655 and REL606 was very similar to that of K12-DHB10 and REL606. In total the global alignement produced 33,994 SNPs including 2469 which were members of block nucleotide polymorphism and 22,377 which were preceded by a SNP fewer than 50 bp away. At 25× coverage, ComB found just slightly more SNPs than were located by MAQ-Consensus and soapSNP. However, ComB found far fewer unsupported SNPs. In nucleotide data, ComB was able to identify 2248 of 2469 block polymorphism sites while soapSNP and MAQ-consensus were able to locate 2019 and 2011 sites, respectively. The results at 25× coverage are summarized in Table 1 and the change in performance at different coverage levels in shown in Figure 5.
FIG. 5.

E. coli experiment: Shown are the validated SNP sites each SNP caller returned at different coverage levels for 36-bp Illumina reads.
3.1.3. Effect of Quality Calibration in E. coli Data
To test the effectiveness of recalibrating call rates we performed SNP calling using E. coli data with and without recalibration of quality score. Using color reads at 25× coverage, quality calibration resulted in the identification of 39 more SNPs and a decrease in the false positive rate of 19% (800 sites). In Illumina data, recalibration of quality score resulted in six fewer SNPs being identified; however, the false discovery rate was decreased by 6% (243 sites). This shows that in both Color and Nucleotide space that the recalibration of quality scores will lead to fewer false SNP calls.
3.2. Drosophila Simulation
To determine the SNP distributions which affect performance, and to accurately assess the algorithms propensity to make false positives, the 22.4 million bp Drosophilia melanogaster X chromosome was used to simulate 50-bp color reads from four different distributions. Each simulation described below included a uniform 1% error rate.
Isolated (22,422 uniformly selected snps, 0.1% SNP rate)
Dense SNPs (2,242,000 uniformly selected SNPs, 10% SNP rate) (Figs. 6 and 7)
Block Polymorphism (10,000 dinucleotide polymorphisms and 10,000 tri-nucleotide polymorphisms 0.2% SNP rate) (Figs. 8 and 9)
Heterozygous SNPs (22,422 SNPs located on only one chromosome, 0.1% SNP rate)
FIG. 6.

Drosophila simulation: The precision of different SNP calling programs for the dense SNP(10% SNP rate) case as a function of coverage. ComB's iterative strategy allows it to use SNP locations to improve mapping accuracy as well as solve ambiguous mappings, leading to improved performance.
FIG. 7.

Drosophila experiment: The recall to dense SNPS (10% SNP rate) for each SNP caller as a function of coverage. ComB shows an approximate increase of 20% recall in comparison to soapSNP and MAQ-consensus.
FIG. 8.

Drosophila simulation: The precision of different algorithms when confronted with adjacent SNPs show the problems with converting the color reads to nucliotide space. Adjacent SNPs generate color signatures which could also be interpreted as color errors by the translation algorithm. Information is lost in the translation, and the SNP calling precision is affected.
FIG. 9.

Drosophila simulation: The recall to block nucleotide polymorphisms is roughly equal for MAQ-Consensus and soapSNP. Corona-Lite locates very few BNP, while ComB finds close to 95% of BNPs.
For each of the above SNP distributions, 100 million color reads were simulated to assure sufficient coverage after alignment. Similar to the E. coli tests, the reads were mapped with BWA to facilitate color translation for MAQ-Consensus and soapSNP and with PerM for ComB. SNP calling was performed with different size trimmed read files for each algorithm to test performance at different coverage levels. Each SNP caller had the same number of alignments to perform SNP calling. ComB vastly outperformed all other SNP calling algorithms at identification of Block Nucleotide Polymorphism and Dense polymorphism. Performance was roughly equal for ComB, MAQ-Consensus and soapSNP for the isolated and heterozygous SNP distributions. The results are shown in Tables 2–5.
Table 2.
Drosophila Experiment: Isolated SNPs
| Algorithm | TP | FP | Precision | Recall |
|---|---|---|---|---|
| ComB | 21,882 | 26 | 0.999 | 0.976 |
| MAQ-con. | 21,308 | 186 | 0.991 | 0.950 |
| soapSNP | 21,309 | 194 | 0.991 | 0.950 |
| Corona Lite | 18,511 | 6,091 | 0.752 | 0.826 |
SNP Calling was performed for four different Simulated SNP distributions. In each distribution, 50-bp color reads were simulated uniformly with a 1% error rate. In the isolated test, 22,422 SNPs were uniformly selected on the 22.4 million–bp Drosophilia melanogaster X chromosome.
Table 5.
Block Nucleotide Polymorphism
| Algorithm | TP | FP | Precision | Recall |
|---|---|---|---|---|
| ComB | 46,783 | 178 | 0.996 | 0.936 |
| MAQ-con. | 25,509 | 25,509 | 0.494 | 0.500 |
| soapSNP | 25,506 | 25,506 | 0.496 | 0.499 |
| Corona Lite | 4,308 | 4,308 | 0.019 | 0.002 |
10,000 dinucleotide polymorphisms and 10,000 tri-nucleotide polymorphims (50,000 total sites) were uniformly selected.
Table 3.
Drosophila Experiment: Heterozygous SNPs
| Algorithm | TP | FP | Precision | Recall |
|---|---|---|---|---|
| ComB | 18,547 | 8 | 1.000 | 0.827 |
| MAQ-con. | 18,922 | 37 | 0.998 | 0.844 |
| soapSNP | 18,920 | 36 | 0.998 | 0.844 |
| Corona Lite | 3,210 | 1,151 | 0.736 | 0.143 |
22,422 Heterozgous SNPs were uniformly selected.
Table 4.
Dense SNPs
| Algorithm | TP | FP | Precision | Recall |
|---|---|---|---|---|
| ComB | 1,676,675 | 9,310 | 0.994 | 0.748 |
| MAQ-con. | 1,320,543 | 167,894 | 0.887 | 0.589 |
| soapSNP | 1,320,562 | 167,911 | 0.887 | 0.589 |
| Corona Lite | 211,668 | 32,852 | 0.866 | 0.095 |
2,242,000 SNPs were uniformly selected.
3.3. Unmasked Human Genome Alignment
To demonstrate ComB's ability to perform human genome scale SNP calling, we downloaded 1.48 billion single end 50-bp SOLiD reads from the ABI website (Archive c; 2010), a small subset of the reads which were previously used to detect variants with ABI's Corona Lite package (McKernan et al., 2009). 832.07 million reads had at least one mapping (and fewer than 100 mappings) to the non-repeat masked human genome with fewer than five mismatches, providing a mean and median coverage of approximately 13× and 6×, respectively. Only 170.3 million reads (20.4%) mapped without mismatches and the mean number of mismatches was 2.06, which is suggestive of a high error rate. ComB used 96 CPUs to perform two homozygous and one heterozygous mapping and SNP calling iterations in fewer than seven hours (approximately 140 minutes per iteration). The second and third iterations led to an increase of 6.25 and 2.5 million reads mapping with two or fewer mismatches than the previous iteration. After the heterozygous iteration ComB identified 2,185,105 likely polymorphism sites (posterior probability > 0.95). These sites included 56,317 block calls, 974,122 heterozygous calls, and 1,154,666 homozygous calls. Included in these calls were 11,841 dense regions (100 bp region with ten or more NPs). In total, 84.06% of the identified NP sites were annotated as a single SNP or BNP in the latest build of dbSNP 131 (dbSNP, 2001). A small fraction 6,454 (<0.3%) of the sites were annotated with different base substitutions. ComB's SNP calling results are shown in Table 6.
Table 6.
Human Variant Calls Produced by ComB
| NP Type | Called | Annotated, n | Annotated, % | Novel |
|---|---|---|---|---|
| Homozygous SNPs | 1,154,666 | 1,095,973 | 94.91 | 58,693 |
| Heterozygous SNPs | 974,122 | 711,413 | 73.03 | 262,709 |
| Block NP sites | 56,317 | 29,519 | 52.41 | 26,789 |
| Total | 2,185,105 | 1,836,905 | 84.06 | 348,200 |
2.71 billion 50-bp reads were iteratively mapped to the unmasked Hg19. The lower annotation rate for heterozygous SNPs may be due to the difficulty of calling heterozygous bases at low or moderate coverage. Additionally, it is likely that SNPs which are not yet fixed in a population would be unannotated.
4. Conclusion
ComB provides an efficient and accurate tool to perform SNP calling for both Illumina and SOLiD data. The demonstration on the human genome shows that ComB can be efficient and accurate even for the largest of datasets. ComB's algorithms were designed to be especially accurate for SOLiD data; features such as the inclusion of ambiguous reads, sensitivity to BNPs, and a Bayesian model that remains in color space allow ComB to take full advantage of SOLiD's novel color encoding and provide a statistically sound method to determine genome variants in color space. The results on the E. coli genomes show that ComB is capable of identifying more SNPs on both SOLiD and Illumina data than MAQ-Consensus, soapSNP, and Corona-Lite. The results on the simulated data show that ComB is more accurate than the other SNP callers especially when SNP loci are dense. That ComB did not significantly outperform other SNP calling tools on a simulated isolated SNP distritbution is evidence that real data has a complicated SNP distribution and that a SNP caller should be robust to dense SNPs as well as BNPs to fully analyze SNP data. ComB is an efficent tool which is capable of providing scientists the ability to find new more accurate information from short color or nucleotide reads.
Acknowledgments
We would like to thank Yangho Chen for helpful discussion. This work was supported by the NIH Center of Excellence in Genomic Sciences (NIH CEGS; grant 2-P50-HG002790-06) and by the National Institute of Mental Health (NIMH; grant 1-RC2-HD064482-01).
Disclosure Statement
No competing financial interests exist.
References
- NCBI Short Read Archive. Solid system ecoli dh10b data set, a. 2010. http://www.ncbi.nlm.nih.gov/sra/SRX000353. [Marc;2011 ]. http://www.ncbi.nlm.nih.gov/sra/SRX000353 Archive a.
- NCBI Short Read Archive. Solexa 36bp ecoli mg1655 data set, b. 2010. http://www.ncbi.nlm.nih.gov/sra/SRX000429. [Mar 15;2011 ]. http://www.ncbi.nlm.nih.gov/sra/SRX000429 Archive b.
- NCBI Short Read Archive. Ab solid sequencing of human hapmap individual na18507, c. 2010. http://www.ncbi.nlm.nih.gov/sra/SRX003963. [Mar 15;2011 ]. http://www.ncbi.nlm.nih.gov/sra/SRX003963 Archive c.
- Brockman W. Alvarez P. Young S., et al. Quality scores and snp detection in sequencing-by-synthesis systems. http://www.hubmed.org/display.cgi?uids=18212088. [Mar 15;2011 ];Genome Res. 2008 18:763–770. doi: 10.1101/gr.070227.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y. Souaiaia T. Chen T. Perm: efficient mapping of short sequencing reads with periodic full sensitive spaced seeds. http://www.hubmed.org/display.cgi?uids=19675096. [Mar 15;2011 ];Bioinformatics. 2009 25:2514–2521. doi: 10.1093/bioinformatics/btp486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- National Library of Medicine. Database of Single Nucleotide Polymorphisms. National Center for Biotechnology Information. dbsnp: the ncbi database of genetic variation. http://www.hubmed.org/display.cgi?uids=11125122. [Mar 15;2011 ];Nucleic Acids Res. 2001 29:308–311. doi: 10.1093/nar/29.1.308. dbSNP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Homer N. Merriman B. Nelson S.F. Bfast: an alignment tool for large-scale genome resequencing. http://www.hubmed.org/display.cgi?uids=19907642. [Mar 15;2011 ];PLoS One. 2009 4:11. doi: 10.1371/journal.pone.0007767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karolchik D. Baertsch R. Diekhans M., et al. The UCSC genome browser database. http://view.ncbi.nlm.nih.gov/pubmed/12519945. [Mar 15;2011 ];Nucleic Acids Res. 2003 31:51–54. doi: 10.1093/nar/gkg129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karro J.E. Peifer M. Hardison R.C., et al. Exponential decay of gc content detected by strand-symmetric substitution rates influences the evolution of isochore structure. http://www.hubmed.org/display.cgi?uids=18042807. [Mar 15;2011 ];Mol. Biol. Evol. 2008 25:362–374. doi: 10.1093/molbev/msm261. [DOI] [PubMed] [Google Scholar]
- Kurtz S. Phillippy A. Delcher A.L., et al. Versatile and open software for comparing large genomes. http://www.hubmed.org/display.cgi?uids=14759262. [Mar 15;2011 ];Genome Biol. 2004 5:2. doi: 10.1186/gb-2004-5-2-r12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lander E.S. Linton L.M. Birren B., et al. Initial sequencing and analysis of the human genome. http://www.hubmed.org/display.cgi?uids=11237011. [Mar 15;2011 ];Nature. 2001 409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- Langmead B. Trapnell C. Pop M., et al. Ultrafast and memory-efficient alignment of short dna sequences to the human genome. http://www.hubmed.org/display.cgi?uids=19261174. [Mar 15;2011 ];Genome Biol. 2009 10:3. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy S. Sutton G. Ng P.C., et al. The diploid genome sequence of an individual human. http://www.hubmed.org/display.cgi?uids=17803354. [Mar 15;2011 ];PLoS Biol. 2007 5:10. doi: 10.1371/journal.pbio.0050254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H. Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. http://www.hubmed.org/display.cgi?uids=19451168. [Mar 15;2011 ];Bioinformatics. 2009 25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H. Ruan J. Durbin R. Mapping short DNA sequencing reads and calling variants using mapping quality scores. http://www.hubmed.org/display.cgi?uids=18714091. [Mar 15;2011 ];Genome Res. 2008 18:1851–1858. doi: 10.1101/gr.078212.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H. Handsaker B. Wysoker A., et al. The sequence alignment/map format and samtools. http://www.hubmed.org/display.cgi?uids=19505943. [Mar 15;2011 ];Bioinformatics. 2009a 25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M. Nordborg M. Li L.M. Adjust quality scores from alignment and improve sequencing accuracy. http://www.hubmed.org/display.cgi?uids=15459287. [Mar 15;2011 ];Nucleic Acids Res. 2004 32:5183–5191. doi: 10.1093/nar/gkh850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li R. Li Y. Fang X., et al. Snp detection for massively parallel whole-genome resequencing. http://www.hubmed.org/display.cgi?uids=19420381. [Mar 15;2011 ];Genome Res. 2009b 19:1124–1132. doi: 10.1101/gr.088013.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mancinelli L. Cronin M. Sadée W. Pharmacogenomics: the promise of personalized medicine. http://www.hubmed.org/display.cgi?uids=11741220. [Mar 15;2011 ];AAPS PharmSci. 2000 2:1. doi: 10.1208/ps020104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manolio T.A. Collins F.S. Cox N.J., et al. Finding the missing heritability of complex diseases. http://www.hubmed.org/display.cgi?uids=19812666. [Mar 15;2011 ];Nature. 2009 461:747–753. doi: 10.1038/nature08494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKernan K.J. Peckham H.E. Costa G.L., et al. Sequence and structural variation in a human genome uncovered by short-read, massively parallel ligation sequencing using two-base encoding. http://www.hubmed.org/display.cgi?uids=19546169. [Mar 15;2011 ];Genome Res. 2009 19:1527–1541. doi: 10.1101/gr.091868.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ondov B.D. Varadarajan A. Passalacqua K.D., et al. Efficient mapping of applied biosystems solid sequence data to a reference genome for functional genomic applications. http://www.hubmed.org/display.cgi?uids=18842598. [Mar 15;2011 ];Bioinformatics. 2008 24:2776–2777. doi: 10.1093/bioinformatics/btn512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pushkarev D. Neff N.F. Quake S.R. Single-molecule sequencing of an individual human genome. http://www.hubmed.org/display.cgi?uids=19668243. [Mar 15;2011 ];Nat. Biotechnol. 2009 27:847–852. doi: 10.1038/nbt.1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan A.R. Stewart D.A. Strömberg M.P., et al. Pyrobayes: an improved base caller for snp discovery in pyrosequences. http://www.hubmed.org/display.cgi?uids=18193056. [Mar 15;2011 ];Nat Methods. 2008 5:179–181. doi: 10.1038/nmeth.1172. [DOI] [PubMed] [Google Scholar]
- Shastry B.S. Snps in disease gene mapping, medicinal drug development and evolution. http://www.hubmed.org/display.cgi?uids=17928948. [Mar 15;2011 ];J. Hum. Genet. 2007 52:871–880. doi: 10.1007/s10038-007-0200-z. [DOI] [PubMed] [Google Scholar]
- Shendure J. Ji H. Next-generation DNA sequencing. http://www.hubmed.org/display.cgi?uids=18846087. [Mar 15;2011 ];Nat. Biotechnol. 2008 26:1135–1145. doi: 10.1038/nbt1486. [DOI] [PubMed] [Google Scholar]
- Venter J.C., et al. The sequence of the human genome. http://www.sciencemag.org/cgi/content/abstract/291/5507/1304. [Mar 15;2011 ];Science. 2001 291:1304–1351. doi: 10.1126/science.1058040. [DOI] [PubMed] [Google Scholar]
- Yue P. Moult J. Identification and analysis of deleterious human snps. http://www.hubmed.org/display.cgi?uids=16412461. [Mar 15;2011 ];J. Mol. Biol. 2006 356:1263–1274. doi: 10.1016/j.jmb.2005.12.025. [DOI] [PubMed] [Google Scholar]