

Abstract
Histidine-rich protein II (HRPII) is an abundant protein released into the bloodstream by Plasmodium falciparum, the parasite that causes the most severe form of human malaria. Here, we report that HRPII binds tightly and selectively to coagulation-active glycosaminoglycans (dermatan sulfate, heparan sulfate, and heparin) and inhibits antithrombin (AT). In purified systems, recombinant HRPII neutralized the heparin-catalyzed inhibition of factor Xa and thrombin by AT in a Zn2+-dependent manner. The observed 50% inhibitory concentration (IC50) for the HRPII neutralization of AT activity is approximately 30nM for factor Xa inhibition and 90nM for thrombin inhibition. Zn2+ was required for these reactions with a distribution coefficient (Kd) of approximately 7μM. Substituting Zn2+ with Cu2+, but not with Ca2+, Mg2+, or Fe2+, maintained the HRPII effect. HRPII attenuated the prolongation in plasma clotting time induced by heparin, suggesting that HRPII inhibits AT activity by preventing its stimulation by heparin. In the microvasculature, where erythrocytes infected with P falciparum are sequestered, high levels of released HRPII may bind cellular glycosaminoglycans, prevent their interaction with AT, and thereby contribute to the procoagulant state associated with P falciparum infection.
Introduction
Malaria is an ancient parasitic disease that continues to take an enormous toll on persons and communities, as well as on global economies. In humans, the disease is caused by 5 protozoan parasites of the genus Plasmodium, including P ovale, P malariae, P vivax, P knowlesi, and P falciparum. Current infection rates are estimated at 250 million annually, with approximately 1 million of those cases resulting in fatalities.1,2 P falciparum infection is the most dreaded, accounting for almost all malaria deaths. Morbidity and mortality from P falciparum malaria result from serious complications that arise during the course of the infection, including severe anemia, neurologic impairment, acute renal failure, and coagulopathy. Coagulation disturbances may contribute to the inflammation and organ failure associated with severe disease.3,4 Patients with P falciparum infections have prolonged prothrombin time and activated partial thromboplastin time (aPTT),5,6 reduced levels of clotting factors, including antithrombin (AT),7,8 and elevated levels of fibrin degradation products.9,10 Although uncommon, frank disseminated intravascular coagulation is observed in severe cases.5,9,11 Activation of the intrinsic coagulation cascade and induction of tissue factor expression have been proposed,12–14 but the precise mechanisms leading to hemostatic alterations in P falciparum infection remain poorly understood. Whether secreted parasite products are involved has not been established. Products released by intraerythrocytic parasites include a pair of proteins with unusually high histidine content: histidine-rich protein II (HRPII) and HRPIII.15 Of these 2 homologous proteins, HRPII has been best studied.
HRPII is a 277-amino acid protein with 35% histidine content and contains 51 repeats of the tripeptide His-His-Ala. It is a particularly interesting protein in that it is only produced by P falciparum and not by any other Plasmodium species that infect humans. HRPII is released by late-stage and rupturing parasitized erythrocytes16 and circulates at high levels (up to high-nanomolar/low-micromolar concentrations).17 The protein is relatively stable in plasma, where it remains detectable for several days or weeks after treatment.18 HRPII detection methods are thus widely used for the rapid diagnosis of P falciparum malaria.19 Despite its abundance, the precise function of HRPII is not clear. It is reported to be involved in the formation of hemozoin,20 heme binding,21 suppression of the immune response,22 and Zn2+ binding.23 As a secreted protein that is unique to P falciparum, the question arises whether HRPII could play a role in the more severe clinical course of P falciparum malaria.
Zinc ion binding and high histidine content are properties of HRPII that are shared with a well-studied human plasma glycoprotein called histidine rich glycoprotein (HRG). HRG binds to heparin and less tightly to some other glycosaminoglycans (GAGs, heparan sulfate, dermatan sulfate, chondroitin sulfate A) in vitro.24 The binding of HRG to heparin has been shown to inhibit heparin's interaction with the serpins AT25,26 and heparin cofactor II (HCII).27 Several studies have also implicated HRG in the regulation of coagulation in vivo.28–30 Although no sequence or structural homology has been found between HRG and HRPII, it is conceivable that HRPII may mimic HRG functions. In this report, we provide evidence that HRPII has a procoagulant effect by binding to heparin and abrogating its acceleration of AT activity. The implications of this finding in the context of the coagulopathy observed in P falciparum malaria are discussed.
Methods
Expression and purification of HRPII
The recombinant expression and purification of HRPII constructed in pET 15b vector and using Escherichia coli BL 21/DE3 as the host cells have been previously described.20 This clone was used for the present studies, following a similar protocol for expression and purification, with minor modifications.31 Briefly, bacterial starter cultures (2 mL) grown at 37°C for 16 hours were seeded into 2 L of LB broth and grown at 37°C to an optical density of 0.6 at 600 nm. Isopropyl β-D-1-thiogalactopyranoside (0.4mM final concentration) was then added and the culture incubated at room temperature for 16 hours. The bacteria were harvested by centrifugation and lysed by sonication in binding buffer (20mM Tris, 500mM NaCl, pH 8.0, containing 50mM imidazole). The lysate was centrifuged at 16 000g for 30 minutes, and the supernatant was filtered through a 0.45-μm filter (Millipore) and then passed by gravity flow over an activated Ni2+ column (2 mL; Novagen) pre-equilibrated with binding buffer. The column was washed with 5 bed volumes of binding buffer, followed by 5 bed volumes of wash buffer (binding buffer containing 200mM imidazole), and then eluted with 1M imidazole in binding buffer. The eluted protein was dialyzed for 48 hours in 100mM sodium acetate, pH 4.8, with 5 changes of dialysate, followed by a final dialysis in Tris-buffered saline (TBS; 50mM Tris, 150mM NaCl, pH 7.4). Because the protein was purified from the soluble fraction of bacterial lysate and because the purification was performed under nondenaturing conditions, no refolding procedure was used. The HRPII concentration was estimated using the biuret assay-based Micro BCA Protein Assay Kit (Thermo Scientific), which has been shown to be accurate for measuring HRPII concentration.21 After analyzing its purity by SDS-PAGE, the purified protein was stored at −80°C until use.
Partial purification of native HRPII from conditioned medium of parasitized erythrocytes
P falciparum clone 3D7 was grown in O+ human erythrocytes at 2% hematocrit, using RPMI 1640 supplemented with 0.5% albumax I, 30mM N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid, 30mM sodium bicarbonate, 100μM hypoxanthine, and 1mM sodium pyruvate. Extracellular medium was collected from asynchronous cultures after 24 hours of incubation. The conditioned medium (1L) was centrifuged at 6000g for 1 hour and filtered through a 0.22-μm filter. Zinc acetate (100μM final concentration) was added, and the cleared medium was passed by gravity flow over a 0.8 × 4 cm Poly-Prep chromatography column (Bio-Rad), which contained a suspension of heparin-agarose (Sigma-Aldrich, 1-mL bed volume) preequilibrated with TBS containing 100μM zinc acetate. The column was washed with equilibration buffer containing 2M NaCl and then eluted with 10mM ethylenediaminetetraacetic acid (EDTA). Fractions were analyzed by Western blotting, and those containing HRPII were pooled and concentrated to 20 μL using a Microcon YM-10 centrifugal filter device (Millipore). Buffer exchange was carried out by diluting the concentrate with 0.5 mL of TBS and repeating the concentration procedure 5 times to remove EDTA. The HRPII concentration was estimated by quantitative Western blotting with a monoclonal antibody against HRPII (mAB2G12),32 using known amounts of recombinant HRPII expressed in E coli as standards. The protein concentrate in 150 μL was stored at −80°C until used.
Mass spectroscopic analysis of HRPII
The intact molecular weight of HRPII was determined by matrix-assisted laser desorption/ionization time-of-flight mass spectrometry at the Stanford University PAN Facility. Briefly, 1 μL of a desalted protein solution was spotted onto a silver matrix-assisted laser desorption/ionization plate containing 1 μL of sinapinic acid matrix (10 mg/mL in 50% acetonitrile: 50% trifluoroacetic acid, 0.1%). The sample was analyzed on an ABI Voyager-DE RP matrix-assisted laser desorption/ionization time-of-flight mass spectrometer in linear mode (accelerating voltage 25 kV, grid voltage 92%, guide wire 0.15%, delay time 350 nsec).
SDS-PAGE and Western blot
SDS-PAGE was performed according to the method of Laemmli.33 The acrylamide concentration was 12%, and the gel was stained with the Coomassie-based Simply blue dye (Invitrogen). Samples were reduced with 5% 2-mercaptoethanol before loading. For Western blotting, the protein sample was similarly run on a 12% gel and transferred to a nitrocellulose membrane. The membrane was blocked with 5% nonfat dry milk in TBS containing 0.1% Tween 20 for 1 hour at room temperature. After three 10-minute washes with TBS containing 0.1% Tween 20, the membrane was probed with mAB2G12. A HRP-linked anti-mouse IgG (Sigma-Aldrich) and chemiluminescence with the Lumi-LightPlus Western blotting detection kit (Roche Diagnostics) were then used to detect the primary antibody.
Binding assay
Heparin binding to HRPII was performed at room temperature as described by Kluszynski et al,34 with minor modifications. In brief, a Poly-Prep chromatography column (0.8 × 4 cm; Bio-Rad) was packed with a suspension of heparin-agarose (Sigma-Aldrich, 1-mL bed volume). After washing extensively with distilled water, the column was equilibrated with buffer (20mM Tris, 10mM NaCl, pH 7.4). HRPII (450nM) in equilibration buffer (500 μL) was then applied to the column by gravity flow, followed by 3 bed volumes of equilibration buffer to wash off any unbound protein. To evaluate HRPII binding to the heparin, the column was eluted stepwise with 1 mL equilibration buffer containing increasing amounts of NaCl (0.1-3M). After a first set of 0.1 to 3M salt elutions (fractions 1-5), a second set of elutions followed with a similar series of elution buffers to which 1mM EDTA (final concentration) had been added (fractions 6-10). In a separate experiment, the same procedure was followed except that all the elution buffers contained 15μM zinc acetate. To detect HRPII in each eluted fraction, a 10-μL aliquot was spotted onto a nitrocellulose membrane and allowed to dry. The HRPII was then detected with mAB2G12 as described for the detection of HRPII by Western blotting under “SDS-PAGE and Western blot.”
Competition assays
The binding of heparan sulfate, dermatan sulfate, keratan sulfate, and chondroitin sulfates A and C (all purchased from Sigma-Aldrich) was assessed by their abilities to compete with tritiated heparin for binding to HRPII. The competition assays were performed as described by Chakravarty et al35 with minor modifications. All steps in this procedure, including dilutions, were done in phosphate-buffered saline, pH 7.0. First, 200 μCi of tritiated heparin (New England Nuclear, 0.51 mCi/mg) was fractionated by applying it over a preequilibrated Sephadex G75 column (0.7 cm × 28 cm), and collecting 52 500-μL fractions; 1 μL of each fraction was dissolved in 5 mL scintillation fluid and measured in a scintillation counter (Beckman LS-230). Fractions outside the void volume that measured more than 15 000 cpm/μL were pooled, and the volume adjusted with buffer to obtain a working stock of purified tritiated heparin of 20 000 cpm/μL. For competition assays, reaction mixtures (25 μL) were constructed in wells of a 96-well round-bottom tissue culture test plate (Midwest Scientific), by mixing 5 μL of purified tritiated heparin stock, 5 μL of zinc acetate, and 10 μL of HRPII with 5 μL of various concentrations of unlabeled GAG. The plate was incubated at 37°C for 1 hour, after which the mixtures were transferred to FilterMAT glass fiber filters (Skatron Instruments), washed with deionized water using the model 11000 Classic Cell Harvester (Skatron Instruments), and dried. The dried filters were then counted in 5 mL of scintillation fluid to determine the amount of tritiated heparin bound to HRPII. Final concentrations in the 25-μL reactions were as follows: tritiated heparin (4 μg/mL), zinc acetate (18 μg/mL), HRPII (0.4 mg/mL), and unlabeled GAGs (0-5 mg/mL). For each unlabeled GAG, each reaction condition was tested in triplicate.
Assays for clotting factor inhibition
Assays were performed at room temperature in TBS containing 0.1% BSA. By titrating heparin against AT, and vice versa in factor Xa (FXa) inhibition assays, it was determined that, in the absence of any divalent cations, when heparin (Baxter Healthcare) (0.01 U/mL, 180 U/mg) and AT (Sigma-Aldrich; 100nM) were incubated for 1 minute with FXa (Enzyme Research Laboratories; 5nM), approximately 50% of the FXa activity was inhibited. Under these conditions, neither heparin alone nor AT alone significantly affected FXa activity. These conditions were thus used for testing the effect of HRPII on the AT-heparin inhibition of FXa. A similar approach was used to develop conditions for assays of thrombin inhibition by AT and HCII.27 All the aforementioned indicated concentrations are final concentrations.
Amidolytic assays
Assays to determine the activity of FXa or thrombin were performed in 96-well plates at room temperature in a final volume of 150 μL. FXa-containing samples (100 μL) were mixed with 50 μL of the synthetic chromogenic substrate S-2222, benzoyl-Ile-Glu-Gly-Arg-p-nitroanilide hydrochloride (Diapharma; 500μM). Similarly, thrombin-containing samples (100 μL) were mixed with 50 μL of substrate S-2238, H-D-Phe-pip-Arg-p-nitroanilide dihydrochloride (Diapharma; 100μM). S-2222 or S-2238 hydrolysis was then measured by following the change in absorbance at 405nM (p-nitroaniline release) on a SpectraMax 340 plate reader (Molecular Devices). All the aforementioned indicated concentrations are final concentrations.
Effect of various divalent cations and zinc counterions on HRPII activity and determination of Kd for Zn2+ interaction with HRPII
Inhibitor mixtures were constructed with AT (100nM) plus heparin (0.01 U/mL) with or without HRPII (300nM) in reaction buffer containing no divalent cation or one of the following: zinc acetate (15μM), ZnCl2 (15μM), ZnSO4 (15μM), CuSO4 (15μM), FeSO4 (15μM), or MgSO4 (2mM), which are approximate plasma concentrations. The mixtures were then added to FXa (5nM) to start the reaction. After 1 minute, the FXa activity remaining was determined as described under “Amidolytic assays.”
To determine the Kd for the binding of Zn2+ to HRPII, reaction mixtures were constructed containing various concentrations of zinc acetate (0-160μM), and a fixed amount of CaCl2 (5mM), AT (100nM), heparin (0.01 U/mL), and HRPII (300nM). The inhibition of FXa (5nM) and measurement of residual FXa activity were then assessed as described in the preceding paragraph. All the aforementioned indicated concentrations are final concentrations in a reaction volume of 100 μL.
Clotting assay
The aPTT assay is a clotting assay often used to monitor the activity of heparin in plasma. In the assay, citrated plasma is mixed with an aPTT reagent (which contains an activator of intrinsic coagulation) for a specified time, followed by the addition CaCl2. Timing is begun from the time of addition of CaCl2, and the time required for the clot to form is the aPTT. The assay indirectly measures the generation and activity of thrombin on fibrinogen to form a clot. Here, the aPTT was used to determine the effect of HRPII on heparin activity in the plasma environment. Assays were performed using the aPTT-Soluble Activator Reagent Kit (Helena Laboratories) in a total volume of 300 μL. All dilutions were carried out in TBS/BSA. The aPTT reagent (100 μL) was premixed with 100 μL of citrated pooled normal plasma (George King Biomedical), before adding zinc acetate (10 μL, 20μM final), with or without varied concentrations of HRPII (20 μL, 0-700nM), with or without varied concentrations of heparin (20 μL, 0-0.1 U/mL). The mixture was allowed to incubate at 37°C for 3 minutes after which CaCl2 (50 μL, 10mM final) prewarmed to 37°C was added and the clot time recorded using a precision coagulation timer (BBL Fibrosystem). The exogenous Zn2+ was necessary to compensate for the plasma dilution in the assay.
Results
Purified HRPII
SDS-PAGE and Western blot analysis of the purified recombinant HRPII are shown in Figure 1. Based on its amino acid sequence, the predicted molecular weight of HRPII is 29 025. Nevertheless, the HRPII produced by P falciparum and the recombinant protein expressed in E coli migrate with a higher apparent molecular weight on SDS-PAGE.32 The reason for this migration pattern is unknown but could be related to the unusual amino acid content of HRPII. Matrix-assisted laser desorption/ionization mass spectroscopic analysis yielded a mass of 29 087, which is well within error of the calculated mass of 29 025. N-terminal sequencing yielded the expected sequence, H2N-AKNAKGLNLN. It has been shown previously that rapid overexpression of HRPII in E coli can lead to amino acid substitutions and protein heterogeneity.31 Electrospray ionization showed low heterogeneity comparable with that described by these authors for slow expression (supplemental Figure 1, available on the Blood Web site; see the Supplemental Materials link at the top of the online article).
Figure 1.
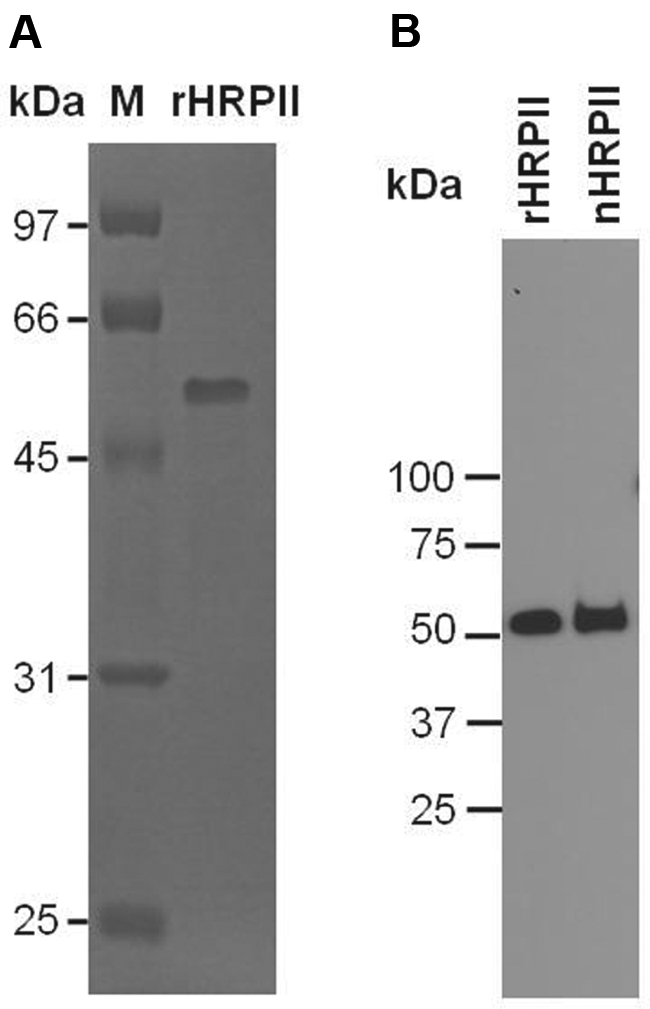
SDS-PAGE and Western blot analysis of purified HRPII. (A) SDS-PAGE. Recombinant HRPII (0.5 μg) was treated with 5% 2-mercaptoethanol and run on 12% SDS-PAGE. The gel was stained with Coomassie brilliant blue. (B) Western blot. Recombinant HRPII (10 ng) and native HRPII (20 ng) were run on SDS-PAGE as in panel A, except that a 10% to 20% gradient gel was used. The gel was blotted onto a nitrocellulose membrane and probed with a monoclonal antibody to HRPII (mAB2G12) as described in “SDS-PAGE and Western blot.” (A-B) Molecular mass markers are listed on the left. rHRPII indicates recombinant HRPII; and nHRPII, native HRPII.
HRPII binds to heparin and other coagulation-active GAGs
HRPII bound efficiently to immobilized heparin in the absence of Zn2+. The bound HRPII could be eluted with 100 to 200mM NaCl (Figure 2A top left panel). After 500mM NaCl treatment, further salt increases (up to 3M) did not yield any further HRPII elution from the column. On subsequent addition of EDTA with increasing salt (up to 3M) to the same heparin column, no further elution of HRPII was observed (Figure 2A top right panel), confirming complete elution of HRPII from heparin with low concentrations of NaCl when Zn2+ is absent. In contrast, when HRPII was bound to heparin in the presence of Zn2+, the affinity of HRPII for heparin was so greatly enhanced that 3M salt did not release the HRPII from heparin (Figure 2A bottom left panel). In the presence of Zn2+, the bound HRPII could be released from the heparin column only after adding EDTA to chelate the Zn2+ (Figure 2A bottom right panel). As the high salt used in the elution attempt in the absence of EDTA could have carried over to the HRPII elution after the addition of EDTA, it was verified in separate experiments that 100mM NaCl and 1mM EDTA indeed eluted HRPII from heparin while 1mM EDTA alone did not elute bound HRPII from the heparin column without NaCl (supplemental Figure 2).
Figure 2.

Interaction of HRPII with GAGs. (A) Binding of HRPII to immobilized heparin. HRPII in 10mM NaCl binding buffer in the absence of Zn2+ (top) or presence of 15μM Zn2+ (bottom) was applied to a heparin-agarose chromatography column. After washing with binding buffer, fractions were collected with successive applications of increasing amounts of NaCl. Note that, after fraction 5 for each panel, EDTA (1mM) was included in the buffers for the elution of fractions 6-10. Fractions were analyzed for HRPII by immune-dotblots using mAB2G12 as described in “Binding assay.” (B) Competition of various unlabeled GAGs with tritiated heparin for binding to HRPII. A fixed concentration of tritiated heparin was mixed with fixed amounts of HRPII and zinc acetate along with various amounts of unlabeled GAGs in a 25-μL reaction volume in 96-well plate format. Thereafter, reactions were incubated at 37°C for 1 hour before processing to determine the amount of tritiated heparin bound to HRPII as described in “Competition assays.” Data are mean ± SE for triplicate wells.
In binding competition assays where various unlabeled GAGs were allowed to compete with tritiated heparin for binding to HRPII, heparan sulfate and dermatan sulfate effectively competed with heparin for HRPII binding (Figure 2B). In contrast, keratan sulfate, chondroitin sulfate A, and chondroitin sulfate C failed to compete with heparin for binding to HRPII. These data show that HRPII selectively binds to some anticoagulant GAGs.
HRPII neutralizes serpin-heparin inhibition of clotting factor activity in a Zn2+-dependent manner
Data showing that HRPII affects the AT-heparin inhibition of FXa are presented in Figure 3. The assay was designed so that AT and heparin together inhibited approximately 50% of FXa activity in the absence of any divalent ions (Figure 3A). The addition of HRPII to AT-heparin has no effect on FXa inhibition under these conditions (Figure 3A). In the presence of Ca2+ alone, the AT-heparin inhibition of FXa is enhanced, and addition of HRPII to the AT-heparin does not affect the observed FXa inhibition (Figure 3B). However, in the presence of Zn2+ with or without Ca2+, HRPII completely reversed the AT-heparin inhibition of FXa (Figure 3C-D). As anticipated, neither AT alone, nor heparin alone, nor HRPII alone significantly affected FXa activity in these assays (Figure 3A-D). In addition, a combination of HRPII with AT, or with heparin with or without Zn2+, had no effect on FXa activity (data not shown). Native HRPII prepared from P falciparum culture supernatant had a similar effect (Figure 3E).
Figure 3.

Effect of HRPII on the AT-heparin inhibition of FXa. Reaction buffer alone as control or reaction buffer containing indicated combinations of heparin (0.01 U/mL), AT (100nM), and HRPII (300nM) was added to a fixed amount of FXa (5nM). The resulting mixture was incubated for 1 minute at room temperature, and the FXa activity remaining was then determined by amidolytic assay using the chromogenic substrate S-2222 as described in “Amidolytic assays.” Four reaction conditions were used: (A) no divalent cations; (B) 5mM CaCl2 present; (C) 15μM ZnCl2 present; and (D) both 5mM CaCl2 and 15μM ZnCl2 present. (B,D) The AT-heparin inhibition of FXa is enhanced by Ca2+. (E) Same as panel D, except that native HRPII (5nM) was used. Assays were done in triplicate, and data are mean ± SE. Hep indicates heparin; P-HRPII, AT-hep + HRPII.
Substituting Zn2+ with Cu2+ maintains the full HRPII effect, whereas similar to Ca2+, replacement with either Fe2+ or Mg2+ failed to support HRPII inhibition of AT-heparin activity altogether (Figure 4A). Similar to Ca2+, the AT-heparin inhibition of FXa is enhanced by Mg2+ but not by Fe2+, Cu2+, or Zn2+ (Figure 4A). The effect of Zn2+ was independent of the carrier anion as zinc acetate, ZnSO4, and ZnCl2 produced identical results (Figure 4A). Zn2+ titration against the HRPII effect showed that Zn2+ interacts with HRPII with a Kd of approximately 7μM (Figure 4B), which is around the physiologic concentration of Zn2+.36
Figure 4.

Effect of various divalent cations on the reversal of the AT-heparin inhibition of FXa by HRPII. (A) Effect of various cations and Zn2+ counterions. Heparin (0.01 U/mL), AT (100nM), with or without HRPII (300nM) were added to a fixed amount of FXa (5nM) in buffer without divalent ions, or buffer containing the indicated salt (2mM for MgCl2 and 15μM for the rest). The resulting mixture was incubated for 1 minute at room temperature, and the FXa activity remaining was then determined by amidolytic assay using S-2222 as described in “Amidolytic assays.” Note that AT-heparin inhibition of FXa is enhanced by Mg2+. Assays were done in triplicate, and results are the mean ± SE. (B) Zn2+ titration for determination of Kd for Zn2+ binding to HRPII. The assay was performed as described in panel A in the presence of HRPII, using various concentrations of zinc acetate (0-160μM), at fixed heparin (0.01 U/mL), AT (100nM), HRPII (300nM), FXa (5nM), and CaCl2 (5mM).
Data for the titration of HRPII at fixed AT-heparin for the inhibition of FXa and thrombin are presented in Figure 5. In the presence of Ca2+ and Zn2+, the apparent IC50 value for the HRPII neutralization of FXa inhibition by AT-heparin is approximately 30nM (Figure 5A). For the HRPII neutralization of thrombin inhibition, the apparent IC50 is approximately 90nM (Figure 5B). The titration of HRPII at fixed HCII-heparin for the inhibition of thrombin yields an apparent IC50 value of approximately 50nM (Figure 5C). These HRPII concentrations are well within the range reported in blood of patients infected with P falciparum.17,37 Local concentrations near sequestered infected erythrocytes38 and on the endothelial surface39 could be even higher.
Figure 5.

IC50 for HRPII reversal of AT-heparin and HCII-heparin activities. (A) IC50 for HRPII reversal of the AT-heparin inhibition of FXa. Heparin (0.01 U/mL), AT (100nM), and increasing amounts of HRPII (0-300nM) were added to a fixed amount of FXa (5nM) in the presence of zinc acetate (15μM) and CaCl2 (5mM). The reaction was incubated for 1 minute at room temperature, and the FXa activity remaining was then determined by amidolytic assay using S-2222 as described in “Amidolytic assay.” (B) IC50 for HRPII reversal of the AT-heparin inhibition of thrombin. The assay was performed similarly as described for FXa in panel A. Final concentrations were heparin (0.02 U/mL), AT (100nM), HRPII (0-600nM), thrombin (2.5nM), zinc acetate (15μM), and CaCl2 (5mM). The thrombin activity remaining was determined by amidolytic assay using S-2238 as described in “Amidolytic assay.” (C) IC50 for HRPII neutralization of the HCII-heparin inhibition of thrombin. Reaction mixtures in 100 μL contained HCII (300nM), heparin (0.05 U/mL), thrombin (2.5nM), CaCl2 (5mM), zinc acetate (15μM), and various HRPII (0-400nM). After 1 minute incubation at room temperature, the thrombin activity remaining was determined as in panel B. (A-B) Data are the percentage of activity in the absence of AT-heparin. (C) Data are the percentage of activity in the absence of HCII-heparin.
HRPII reverses the prolongation in plasma clotting time produced by heparin
To test whether the HRPII effect on the activity of heparin observed in purified system is relevant in the plasma environment, aPTT assays were performed using citrated plasma (Figure 6). The addition of heparin to plasma resulted in the prolongation of the aPTT (Figure 6A). When HRPII was included in the assay, the effect of heparin on clotting time was significantly reduced. The effect of HRPII on clotting time in the presence of heparin was concentration dependent (Figure 6B). HRPII had little effect on clotting time in the absence of heparin.
Figure 6.

Effect of HRPII on the heparin-induced prolongation of plasma clot time. aPTT reagent (100 μL) was premixed with citrated pooled normal plasma (100 μL) before adding zinc acetate (10 μL, 20 μM final) with or without varied concentrations of HRPII (20μL) with or without varied concentrations of heparin (20 μL). The mixture was allowed to incubate at 37°C for 3 minutes, after which CaCl2 (50 μL, 10mM final, prewarmed to 37°C) was added and the clot time recorded as described in “Clotting assay.” (A) Effect of increasing amounts of heparin on aPTT in the presence and absence of 700nM HRPII. (B) Effect of increasing amounts of HRPII on aPTT in the presence and absence of 0.1 U/mL heparin. Data points and error bars represent mean ± SE for 2 independent experiments performed in duplicate.
Discussion
Currently, there is no licensed vaccine against P falciparum malaria. Moreover, the development of resistance to currently available antimalarial drugs remains a major impediment to effective treatment. Consequently, enormous effort is being directed at exploring novel treatment alternatives. Discerning the mechanisms leading to hemostatic imbalance in severe P falciparum infection would provide a vital contribution to this effort. The present study focused on investigating whether HRPII, a protein secreted uniquely by P falciparum affects blood coagulation.
Our data show that HRPII binds to several GAGs, including heparin, heparan sulfate, and dermatan sulfate (Figure 2). Indeed, the A-H-H and A-H-H-A-A repeats of HRPII resemble the consensus sequences for GAG recognition.40 The binding of HRPII to heparin is enhanced by physiologic concentrations of Zn2+ (Figure 2A). Because HRPII has a high affinity for Zn2+ (Figure 4B), the modulation of HRPII-heparin binding observed in the presence of Zn2+ (Figure 2A) is probably the result of the binding of Zn2+ to HRPII. Zn2+ has previously been reported to enhance the binding of other proteins to GAGs, including HRG.34 HRPII also has a high affinity for Cu2+, and indeed has been previously shown to bind Cu2+ even more tightly than Zn2+.23 It is therefore not surprising that Cu2+ also supports the HRPII activity against AT (Figure 4A).
The binding of HRPII to heparin appears to be stronger than the binding of HRG to heparin. This is evident in the fact that, in the presence of Zn2+, HRPII could not be eluted from heparin with 3M NaCl (Figure 2A). Under similar conditions, HRG will elute from immobilized heparin in the presence of 1M NaCl.34 This is significant in that, because HRG is reported to bind more tightly to heparin than any other plasma protein,41,42 conceivably HRPII could out-compete all other proteins for binding to GAGs, such as heparan sulfate and dermatan sulfate on the endothelial surface during P falciparum infection. HRPII has been shown to coat the vascular endothelium in humans with falciparum malaria.39 With organ failure being a frequent cause of death from P falciparum infection, it can be envisioned that parasite sequestration in the microvasculature, tissue factor induction, and assembly of macromolecular coagulation complexes,14 and the inhibition of AT activity by HRPII would all contribute to the microvascular blockades observed in severe P falciparum infection and precipitate organ failure.
GAGs in the blood vessels are involved in maintaining the anticoagulant and antithrombotic surface of the endothelium, and small changes to the integrity of the GAGs lining blood vessels appear to have profound procoagulant effects.43,44 Although GAGs have some procoagulant functions, their most prominent role on the endothelium is in inhibiting coagulation. GAGs on the surface of cells that circulate in the blood also play an anticoagulant role,45 and the binding of HRPII to GAGs could therefore have consequences for blood coagulation beyond the endothelial surface.
AT regulates the activities of several coagulation factors, including thrombin, FVIIa-tissue factor complex, FXa, FIXa, FXIa, FXIIa, and kallikrein, with FXa and thrombin being its primary targets in blood. GAGs can increase the rates of protease inhibition by AT by up to 10 000 times,46 achieved in 2 ways: (1) by providing a bridge or template on which the serpin and protease can encounter each other; and (2) GAG binding results in a conformational change in AT that increases its affinity for thrombin and FXa.47 In the present study, HRPII prevented the heparin-mediated acceleration in the AT inhibitions of both FXa and thrombin, indicating a functional significance for the HRPII-heparin binding. The obvious explanation for this inhibition is that the binding of HRPII to GAGs prevent their interaction with AT and FXa or thrombin. Presumably, HRPII would also affect the AT inhibition of its other coagulation targets. Although other plasma proteins that bind GAGs, including HRG, platelet factor 4, and serum amyloid P, have been reported to neutralize heparin,41,48,49 only HRG has been reported to do so in the plasma environment.26 Consequently, if HRPII affects heparin activity in plasma, it might interfere with the physiologic functions of HRG in blood. We have shown that HRPII can neutralize the anticoagulant effect of heparin in plasma at concentrations comparable with levels of HRPII measured in patients with P falciparum.17 Zinc levels around the concentration present in plasma were shown to be sufficient for this HRPII modulation of the anticoagulant effect of heparin. This study has also revealed that AT might not be the only coagulation inhibitor whose activity would be affected by HRPII in the blood. Heparin increases the rate of inhibition of thrombin by HCII, and HRG abrogates this effect.27 Indeed, HRPII is also capable of neutralizing HCII-heparin activity (Figure 5C). Another example of a coagulation inhibitor whose activity could be affected by HRPII is C1 inhibitor, the main inhibitor of FXIa in plasma when heparin is present.50
In conclusion, we have shown that HRPII possesses coagulant properties capable of affecting the tightly balanced activities of procoagulant and anticoagulant factors that maintain hemostasis. The interaction of HRPII with GAGs and divalent metal ions may have diverse consequences on other physiologic processes unrelated to coagulation; and as an abundant protein that is unique to P falciparum among the 5 species of human malaria parasites, HRPII may play a key role toward the more severe clinical course of this deadly protozoan.
Supplementary Material
Acknowledgments
The authors thank Nina Lasky and Anna Oksman for their technical assistance, Dr Diane Taylor for providing mAB2G12, and Paul Sigala and the Stanford University PAN and mass spectrometry facilities for their assistance with the mass spectral analysis of HRPII.
This work was supported in part by the National Institute of Child Health and Human Development (grant K12-HD000850), the American Academy of Pediatrics, and the American Pediatric Society. A.S.M. was a Fellow of the Pediatric Scientist Development Program.
Footnotes
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: M.N. and O.O.B. designed and performed the research and wrote the manuscript; A.S.M. performed the research and wrote the manuscript; D.M.T. provided advice on experimental design; G.J.B. provided advice on experimental design and wrote the manuscript; D.E.G. conceived the project, provided advice, and wrote the manuscript; and all authors read and reviewed the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
The current affiliation for O.O.B. is Department of Pathology, Brigham and Women's Hospital, Harvard University School of Medicine, Boston, MA.
Correspondence: Daniel E. Goldberg, Howard Hughes Medical Institute, Departments of Medicine and Molecular Microbiology, Washington University School of Medicine, PO Box 8230, 660 S Euclid Ave, St Louis, MO 63110; e-mail: goldberg@borcim.wustl.edu.
References
- 1.World Health Organization. World Malaria Report 2009. [Accessed May 4, 2011]. http://www.who.int/malaria/publications/atoz/9789241563901/en/index.html.
- 2.Sachs J, Malaney P. The economic and social burden of malaria. Nature. 2002;415(6872):680–685. doi: 10.1038/415680a. [DOI] [PubMed] [Google Scholar]
- 3.Francischetti IM, Seydel KB, Monteiro RQ. Blood coagulation, inflammation, and malaria. Microcirculation. 2008;15(2):81–107. doi: 10.1080/10739680701451516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Francischetti IM. Does activation of the blood coagulation cascade have a role in malaria pathogenesis? Trends Parasitol. 2008;24(6):258–263. doi: 10.1016/j.pt.2008.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rojanasthien S, Surakamolleart V, Boonpucknavig S, Isarangkura P. Hematological and coagulation studies in malaria. J Med Assoc Thai. 1992;75(suppl 1):190–194. [PubMed] [Google Scholar]
- 6.Prasad R, Das BK, Pengoria R, Mishra OP, Shukla J, Singh TB. Coagulation status and platelet functions in children with severe falciparum malaria and their correlation of outcome. J Trop Pediatr. 2009;55(6):374–378. doi: 10.1093/tropej/fmp028. [DOI] [PubMed] [Google Scholar]
- 7.Dennis LH, Eichelberger JW, Inman MM, Conrad ME. Depletion of coagulation factors in drug-resistant Plasmodium falciparum malaria. Blood. 1967;29(5):713–721. [PubMed] [Google Scholar]
- 8.Vogetseder A, Ospelt C, Reindl M, Schober M, Schmutzhard E. Time course of coagulation parameters, cytokines and adhesion molecules in Plasmodium falciparum malaria. Trop Med Int Health. 2004;9(7):767–773. doi: 10.1111/j.1365-3156.2004.01265.x. [DOI] [PubMed] [Google Scholar]
- 9.Liechti ME, Zumsteg V, Hatz CF, Herren T. Plasmodium falciparum cerebral malaria complicated by disseminated intravascular coagulation and symmetrical peripheral gangrene: case report and review. Eur J Clin Microbiol Infect Dis. 2003;22(9):551–554. doi: 10.1007/s10096-003-0984-5. [DOI] [PubMed] [Google Scholar]
- 10.Jaroonvesama N, Harinasuta T, Muangmanee L, Asawapokee N. Coagulation studies in falciparum and vivax malaria. Southeast Asian J Trop Med Public Health. 1975;6(3):419–424. [PubMed] [Google Scholar]
- 11.Edwards IR. Malaria with disseminated intravascular coagulation and peripheral tissue necrosis successfully treated with streptokinase. Br Med J. 1980;280(6226):1252–1253. doi: 10.1136/bmj.280.6226.1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Clemens R, Pramoolsinsap C, Lorenz R, Pukrittayakamee S, Bock HL, White NJ. Activation of the coagulation cascade in severe falciparum malaria through the intrinsic pathway. Br J Haematol. 1994;87(1):100–105. doi: 10.1111/j.1365-2141.1994.tb04877.x. [DOI] [PubMed] [Google Scholar]
- 13.Pernod G, Polack B, Peyron F, et al. Monocyte tissue factor expression induced by Plasmodium falciparum-infected erythrocytes. Thromb Haemost. 1992;68(2):111–114. [PubMed] [Google Scholar]
- 14.Francischetti IM, Seydel KB, Monteiro RQ, et al. Plasmodium falciparum-infected erythrocytes induce tissue factor expression in endothelial cells and support the assembly of multimolecular coagulation complexes. J Thromb Haemost. 2007;5(1):155–165. doi: 10.1111/j.1538-7836.2006.02232.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Howard RJ, Uni S, Aikawa M, et al. Secretion of a malarial histidine-rich protein (Pf HRP II) from Plasmodium falciparum-infected erythrocytes. J Cell Biol. 1986;103(4):1269–1277. doi: 10.1083/jcb.103.4.1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Desakorn V, Dondorp AM, Silamut K, et al. Stage-dependent production and release of histidine-rich protein 2 by Plasmodium falciparum. Trans R Soc Trop Med Hyg. 2005;99(7):517–524. doi: 10.1016/j.trstmh.2004.11.014. [DOI] [PubMed] [Google Scholar]
- 17.Manning L, Laman M, Stanisic D, et al. Plasma Plasmodium falciparum histidine-rich protein-2 concentrations do not reflect severity of malaria in Papua New Guinean children. Clin Infect Dis. 2011;52(4):440–446. doi: 10.1093/cid/ciq105. [DOI] [PubMed] [Google Scholar]
- 18.Iqbal J, Siddique A, Jameel M, Hira PR. Persistent histidine-rich protein 2, parasite lactate dehydrogenase, and panmalarial antigen reactivity after clearance of Plasmodium falciparum monoinfection. J Clin Microbiol. 2004;42(9):4237–4241. doi: 10.1128/JCM.42.9.4237-4241.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Garcia M, Kirimoama S, Marlborough D, Leafasia J, Rieckmann KH. Immunochromatographic test for malaria diagnosis [letter]. Lancet. 1996;347(9014):1549. doi: 10.1016/s0140-6736(96)90700-x. [DOI] [PubMed] [Google Scholar]
- 20.Sullivan DJ, Jr, Gluzman IY, Goldberg DE. Plasmodium hemozoin formation mediated by histidine-rich proteins. Science. 1996;271(5246):219–222. doi: 10.1126/science.271.5246.219. [DOI] [PubMed] [Google Scholar]
- 21.Schneider EL, Marletta MA. Heme binding to the histidine-rich protein II from Plasmodium falciparum. Biochemistry. 2005;44(3):979–986. doi: 10.1021/bi048570p. [DOI] [PubMed] [Google Scholar]
- 22.Das P, Grewal JS, Chauhan VS. Interaction of Plasmodium falciparum histidine-rich protein II with human lymphocytes leads to suppression of proliferation, IFN-gamma release, and CD69 expression. Parasitol Res. 2006;100(1):39–50. doi: 10.1007/s00436-006-0228-6. [DOI] [PubMed] [Google Scholar]
- 23.Panton LJ, McPhie P, Maloy WL, Wellems TE, Taylor DW, Howard RJ. Purification and partial characterization of an unusual protein of Plasmodium falciparum: histidine-rich protein II. Mol Biochem Parasitol. 1989;35(2):149–160. doi: 10.1016/0166-6851(89)90117-5. [DOI] [PubMed] [Google Scholar]
- 24.Borza DB, Morgan WT. Histidine-proline-rich glycoprotein as a plasma pH sensor: modulation of its interaction with glycosaminoglycans by pH and metals. J Biol Chem. 1998;273(10):5493–5499. doi: 10.1074/jbc.273.10.5493. [DOI] [PubMed] [Google Scholar]
- 25.Lijnen HR, van Hoef B, Collen D. Interaction of heparin with histidine-rich glycoprotein and with antithrombin III. Thromb Haemost. 1983;50(2):560–562. [PubMed] [Google Scholar]
- 26.Lijnen HR, Van Hoef B, Collen D. Histidine-rich glycoprotein modulates the anticoagulant activity of heparin in human plasma. Thromb Haemost. 1984;51(2):266–268. [PubMed] [Google Scholar]
- 27.Tollefsen DM, Pestka CA. Modulation of heparin cofactor II activity by histidine-rich glycoprotein and platelet factor 4. J Clin Invest. 1985;75(2):496–501. doi: 10.1172/JCI111725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tsuchida-Straeten N, Ensslen S, Schäfer C, et al. Enhanced blood coagulation and fibrinolysis in mice lacking histidine-rich glycoprotein (HRG). J Thromb Haemost. 2005;3(5):865–872. doi: 10.1111/j.1538-7836.2005.01238.x. [DOI] [PubMed] [Google Scholar]
- 29.Kuhli C, Scharrer I, Koch F, Hattenbach LO. Recurrent retinal vein occlusion in a patient with increased plasma levels of histidine-rich glycoprotein. Am J Ophthalmol. 2003;135(2):232–234. doi: 10.1016/s0002-9394(02)01940-2. [DOI] [PubMed] [Google Scholar]
- 30.Souto JC, Garí M, Falkon L, Fontcuberta J. A new case of hereditary histidine-rich glycoprotein deficiency with familial thrombophilia. Thromb Haemost. 1996;75(2):374–375. [PubMed] [Google Scholar]
- 31.Schneider EL, King DS, Marletta MA. Amino acid substitution and modification resulting from Escherichia coli expression of recombinant Plasmodium falciparum histidine-rich protein II. Biochemistry. 2005;44:987–995. doi: 10.1021/bi048571h. [DOI] [PubMed] [Google Scholar]
- 32.Rock EP, Marsh K, Saul AJ, et al. Comparative analysis of the Plasmodium falciparum histidine-rich proteins HRP-I, HRP-II and HRP-III in malaria parasites of diverse origin. Parasitology. 1987;95(2):209–227. doi: 10.1017/s0031182000057681. [DOI] [PubMed] [Google Scholar]
- 33.Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227(5259):680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 34.Kluszynski BA, Kim C, Faulk WP. Zinc as a cofactor for heparin neutralization by histidine-rich glycoprotein. J Biol Chem. 1997;272(21):13541–13547. doi: 10.1074/jbc.272.21.13541. [DOI] [PubMed] [Google Scholar]
- 35.Chakravarty L, Rogers L, Quach T, Breckenridge S, Kolattukudy PE. Lysine 58 and histidine 66 at the C-terminal alpha-helix of monocyte chemoattractant protein-1 are essential for glycosaminoglycan binding. J Biol Chem. 1998;273(45):29641–29647. doi: 10.1074/jbc.273.45.29641. [DOI] [PubMed] [Google Scholar]
- 36.Davies IJ, Musa M, Dormandy TL. Measurements of plasma zinc: I. In health and disease. J Clin Pathol. 1968;21(3):359–363. doi: 10.1136/jcp.21.3.359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dondorp AM, Desakorn V, Pongtavornpinyo W, et al. Estimation of the total parasite biomass in acute falciparum malaria from plasma PfHRP2. PLoS Med. 2005;2(8):e204. doi: 10.1371/journal.pmed.0020204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen Q, Schlichtherle M, Wahlgren M. Molecular aspects of severe malaria. Clin Microb Rev. 2000;13(3):439–450. doi: 10.1128/cmr.13.3.439-450.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Aikawa M, Iseki M, Barnwell JW, Taylor D, Oo MM, Howard RJ. The pathology of human cerebral malaria. Am J Trop Med Hyg. 1990;43(2):30–37. doi: 10.4269/ajtmh.1990.43.30. [DOI] [PubMed] [Google Scholar]
- 40.Verrecchio A, Germann MW, Schick BP, Kung B, Twardowski T, San Antonio JD. Design of peptides with high affinities for heparin and endothelial cell proteoglycans. J Biol Chem. 2000;275(11):7701–7707. doi: 10.1074/jbc.275.11.7701. [DOI] [PubMed] [Google Scholar]
- 41.Fu CL, Horn MK., III Histidine-rich glycoprotein plus zinc to neutralize heparin. J Lab Clin Med. 2002;139(4):211–217. doi: 10.1067/mlc.2002.121854. [DOI] [PubMed] [Google Scholar]
- 42.Lijnen HR, Collen D. Interaction of heparin with histidine-rich glycoprotein. Ann N Y Acad Sci. 1989;556:181–185. doi: 10.1111/j.1749-6632.1989.tb22502.x. [DOI] [PubMed] [Google Scholar]
- 43.Ihrcke NS, Wrenshall LE, Lindman BJ, Platt JL. Role of heparan sulfate in immune system-blood vessel interactions. Immunol Today. 1993;14(10):500–505. doi: 10.1016/0167-5699(93)90265-M. [DOI] [PubMed] [Google Scholar]
- 44.McGee M, Wagner WD. Chondroitin sulfate anticoagulant activity is linked to water transfer: relevance to proteoglycan structure in atherosclerosis. Arterioscler Thromb Vasc Biol. 2003;23(10):1921–1927. doi: 10.1161/01.ATV.0000090673.96120.67. [DOI] [PubMed] [Google Scholar]
- 45.Leung L, Saigo K, Grant D. Heparin binds to human monocytes and modulates their procoagulant activities and secretory phenotypes: effects of histidine-rich glycoprotein. Blood. 1989;73(1):177–184. [PubMed] [Google Scholar]
- 46.Olson ST, Björk I, Shore JD. Kinetic characterization of heparin-catalyzed and uncatalyzed inhibition of blood coagulation proteases by antithrombin. Methods Enzymol. 1993;222:525–560. doi: 10.1016/0076-6879(93)22033-c. [DOI] [PubMed] [Google Scholar]
- 47.Huntington JA. Mechanisms of glycosaminoglycan activation of the serpins in hemostasis. J Thromb Haemost. 2003;1(7):1535–1549. doi: 10.1046/j.1538-7836.2003.00305.x. [DOI] [PubMed] [Google Scholar]
- 48.Lane DA, Pejler G, Flynn AM, Thompson EA, Lindahl U. Neutralization of heparin-related saccharides by histidine-rich glycoprotein and platelet factor 4. J Biol Chem. 1986;261(9):3980–3986. [PubMed] [Google Scholar]
- 49.Peterson CB, Morgan WT, Blackburn MN. Histidine-rich glycoprotein modulation of the anticoagulant activity of heparin: evidence for a mechanism involving competition with both antithrombin and thrombin for heparin binding. J Biol Chem. 1987;262(16):7567–7574. [PubMed] [Google Scholar]
- 50.Wuillemin WA, Eldering E, Citarella F, de Ruig CP, ten Cate H, Hack CE. Modulation of contact system proteases by glycosaminoglycans: selective enhancement of the inhibition of factor XIa. J Biol Chem. 1996;271(22):12913–12918. doi: 10.1074/jbc.271.22.12913. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.