

Abstract
Bone morphogenetic protein 10 (BMP10) is a member of the TGF-β superfamily and plays a critical role in heart development. In the postnatal heart, BMP10 is restricted to the right atrium. The inactive pro-BMP10 (∼60 kDa) is processed into active BMP10 (∼14 kDa) by an unknown protease. Proteolytic cleavage occurs at the RIRR316↓ site (human), suggesting the involvement of proprotein convertase(s) (PCs). In vitro digestion of a 12-mer peptide encompassing the predicted cleavage site with furin, PACE4, PC5/6, and PC7, showed that furin cleaves the best, whereas PC7 is inactive on this peptide. Ex vivo studies in COS-1 cells, a cell line lacking PC5/6, revealed efficient processing of pro-BMP10 by endogenous PCs other than PC5/6. The lack of processing of overexpressed pro-BMP10 in the furin- and PACE4-deficient cell line, CHO-FD11, and in furin-deficient LoVo cells, was restored by stable (CHO-FD11/Fur cells) or transient (LoVo cells) expression of furin. Use of cell-permeable and cell surface inhibitors suggested that endogenous PCs process pro-BMP10 mostly intracellularly, but also at the cell surface. Ex vivo experiments in mouse primary hepatocytes (wild type, PC5/6 knock-out, and furin knock-out) corroborated the above findings that pro-BMP10 is a substrate for endogenous furin. Western blot analyses of heart right atria extracts from wild type and PACE4 knock-out adult mice showed no significant difference in the processing of pro-BMP10, implying no in vivo role of PACE4. Overall, our in vitro, ex vivo, and in vivo data suggest that furin is the major convertase responsible for the generation of BMP10.
Keywords: Bone Morphogenetic Protein (BMP), Convertases, Enzyme Processing, Heart, Transforming Growth Factor β (TGFbeta), BMP10, Cardiac-specific Growth Factor, Furin, Proprotein Convertase, Proprotein Convertase Inhibitor
Introduction
The heart is the first organ to be formed during embryogenesis and it develops via a complex and sequential process that involves simultaneous morphogenesis and differentiation. Cardiac morphogenesis is under the control of a large variety of regulatory proteins, including TGF-β-related family members (1). TGF-β-like proteins and other substrates like neuropeptides, receptors, or viral glycoproteins are known targets for members of the secretory basic amino acid (aa)2-specific proprotein convertase(s) (PCs) (2, 3). In mammals, seven members of the basic aa-specific PC family have been characterized (mouse genes Pcsk1 to Pcsk7) that cleave various precursors at the consensus motif K/R-Xn-K/R↓ (n = 0, 2, 4, or 6 and X is any aa, except Cys) (2). Four of them, furin, PC5/6, PACE4, and PC7, are ubiquitously or widely expressed and are responsible for the majority of processing events occurring in the constitutive secretory pathway, at the cell surface and/or in the extracellular matrix (3).
Despite their functional redundancy ex vivo, these PCs fulfill key processing events in vivo, as revealed by the specific phenotypes resulting from their respective gene inactivation in mice. Pcsk3 (furin gene) knock-out (KO) in mice causes early death by embryonic day 10.5 (E10.5) due to hemodynamic insufficiency and cardiac ventral closure defects translated into failure of the heart tube to fuse and undergo looping morphogenesis (4). These phenotypes emphasize the critical involvement of furin in cardiac development. Pcsk5 (PC5/6 gene) KO leads to death at birth with an altered antero-posterior patterning, including extra vertebrae, lack of tail, kidney agenesis, hemorrhages, collapsed alveoli, and retarded ossification, as well as heart ventricular-septal defects (5, 6). Mice lacking Pcsk6 (PACE4 gene) KO survive to adulthood, and some develop incompletely penetrant left-right patterning defects combined with cyclopia, craniofacial, and cardiac malformations (7, 8). Pcsk7 (PC7 gene) KO mice exhibit no overt abnormalities (9).3 Thus, heart defects are a common phenotype associated with the single KO of the mouse genes coding for furin, PC5/6, or PACE4, but not PC7.
Bone morphogenetic protein 10 (BMP10) is a newly identified cardiac-specific growth factor that is a member of the TGF-β superfamily and is known to play a critical role in heart development. BMP10 expression is most abundant in the developing and postnatal heart and weaker in the adult liver and lung (10). During mouse cardiogenesis, after completion of embryonic cardiac patterning and looping and at the onset of trabeculation and chamber maturation, BMP10 is transiently expressed in the ventricular trabecular myocardium (E9–E13.5). By E16.5–E18.5, BMP-10 is only expressed in the atria, and is restricted to the right atrium (RA) in the postnatal heart, where it promotes increased cardiomyocyte and heart size (10–12). Homozygous BMP10 KO embryos die in utero between E10 and E10.5 due to arrested cardiac development. Compared with wild type (WT) embryos, KO embryos appear normal at E8.5, but display cardiac dysgenesis at E9–E9.5 with profound hypoplastic ventricular walls, absence of ventricular trabeculae, and a significantly lower heart rate (11).
As with all members of the TGF-β superfamily, BMP10 is synthesized as an inactive precursor protein (pro-BMP10, ∼60 kDa) that is presumably activated by proteolytic cleavage, likely at the motif RIRR313↓ (mouse nomenclature) releasing the secreted non-glycosylated C-terminal mature peptide of 108 aa (∼14 kDa; BMP10) and an N-terminal prosegment of ∼50 kDa (supplemental Fig. S1). Mature BMP10 exhibits a conserved pattern of 7 cysteines, one of which is thought to be engaged in an interchain disulfide bond (13, 14) (supplemental Fig. S1). BMP10 shares >98% aa sequence identity among human, mouse, and rat orthologs.
Cleavage of pro-BMP10 at the motif RXRR↓ suggests the involvement of basic aa-specific PCs (2). The restricted expression of BMP10 mRNA to the RA of the heart in adult mice, and heart-specific phenotypes observed in mice lacking furin, PC5/6, or PACE4, led us to test herein the implication of these convertases in the generation of mature BMP10.
EXPERIMENTAL PROCEDURES
Bioinformatic Searches
We identified all human and mouse protein sequences with signal peptides in ENSEMBL and screened them for the consensus sequence (R/K)-Xn-R↓[N] (where X = 0, 2, 4, 6 aa) using the FuzzPro program (EMBOSS). Positive hits, where the potential cleavage motif was present in both human and mouse proteins, were screened against the Mouse Genome Informatics data base to identify the proteins where there is also genetic evidence for their relevance in heart development. Data were integrated and queried using a relational data base.
In Vitro Assays
Enzymatic in vitro activities of the purified furin, PC5/6, PACE4, and PC7 (15) were measured at 37 °C in 100 μl of buffer (25 mm Tris-MES, 2 mm CaCl2, pH 7) in the presence of 100 μm of the fluorogenic substrate pyroglutamic acid-RTKR-7-amido-4-methylcoumarin (Pyr-RTKR-MCA; Peptide International). The release of free 7-amino-4-methylcoumarin (AMC) was detected with a Spectra MAX GEMINI EM microplate spectrofluorimeter (Molecular Devices) (excitation, 370 nm; emission, 460 nm; emission cutoff, 435 nm). 1 unit of enzymatic activity was defined as 1 pmol of AMC released from 100 μm Pyr-RTKR-MCA/min at 37 °C. The 12-mer synthetic peptides encompassing the predicted cleavage site of mouse pro-BMP10, DSSARIRR313↓NAKG (WT-mBMP10), and DSSARIRR313↓DAKG (N314D-mBMP10, in which the Asn at the P1′ position was mutated to Asp), were obtained from GenScript (NJ).
To determine the specificity of the PCs for the processing of the above synthetic substrates, peptides were individually incubated at 37 °C for 2 h in vitro (200 μm peptide in 100 μl of buffer: 25 mm Tris-MES, 2 mm CaCl2, pH 7) with 2 units of purified PCs (15). For measurement of the time dependence of in vitro cleavage of WT-mBMP10 peptide by furin, PC5/6, or PACE4, the incubations were carried out as above for 0, 10, 20, 30, and 40 min. For each incubation mixture the cleavage products were separated by reversed-phase HPLC (RP-HPLC) on a Varian C18 column (5 μm, 100 Å, 4.6 × 250 mm) using a 0–50% acetonitrile (+0.1% TFA) gradient over 50 min, at 1 ml/min flow rate. Peptide bonds were detected at 214 nm. The cleavage site RIRR313↓ was confirmed by mass spectrometry using MS/MS.
For the determination of kinetic parameters Vmax(app) and Km(app), furin samples (2 units) were incubated at 37 °C with increasing concentrations of WT-mBMP10 peptide ranging from 0 to 200 μm in a total volume of 200 μl of buffer as described above. From each reaction mixture, corresponding to one peptide concentration, 25 μl were subjected to RP-HPLC analysis per time point (see above) and 2–3 time points were assayed, varying from 0 to 15 min. The initial rate of substrate cleavage was obtained from the increase over time of the normalized peak area (peak area/number of peptide bonds) corresponding to the C-terminal fragment NAKG. The values were transformed into micromolar/h by a standard curve generated from variable concentrations of intact WT-mBMP10 peptide for which the peak area was normalized to the number of peptide bonds. Vmax(app) and Km(app) were calculated from a fit to Michaelis-Menten equation (vi = (Vmax(app) × [S])/(Km(app) + [S])) of plots of initial rates of cleavage (vi) against the peptide concentration ([S]) using TableCurve2D software (TableCurve2D version 5.01, SYSTAT Software Inc.).
Plasmids and Reagents
The cDNA encoding for human BMP10 (pCMV6 × 15 vector) was obtained from OriGene (MD). Human BMP10 and its mutant cDNAs (R313A, R316A, and R313A/R316A) were subcloned, with a ProtC tag at the N terminus, into pIRES2-EGFP vector (Clontech). Human furin, human PACE4, mouse PC5A, and their mutant cDNAs were cloned without a tag into pIRES-EGFP vector, as previously described (16). All cDNAs were verified by DNA sequencing.
Cell Culture, Transfections, and Cell Treatments
HEK293 and COS-1 cell lines were cultured in Dulbecco's modified Eagle's medium (DMEM; Invitrogen) supplemented with 10% (v/v) fetal bovine serum (FBS; Invitrogen), whereas CHO-K1, furin-deficient CHO-FD11 (17), and furin-deficient LoVo (18, 19) cells were grown in Ham's F-12 medium with 10% (v/v) FBS. All cells were maintained at 37 °C under 5% CO2.
In a typical transfection experiment, at about 80–90% confluence, cells were transiently transfected as follows: COS-1 (total of 4 μg of cDNA), CHO-K1 (total of 3 μg of cDNA), and CHO-FD11 (total of 3 μg of cDNA) cells with Lipofectamine 2000 (Invitrogen); HEK293 cells (total of 0.6 μg of cDNA) with Effectene (Qiagen); LoVo cells (total of 2 μg of cDNA) with FuGENE HD (Roche Diagnostics). In co-transfection experiments with cDNAs coding for (ProtC)-BMP10 and for the prosegment of furin (ppFurin) (20) or PACE4 (ppPACE4) (21), the cDNA ratio of BMP10 to inhibitor was 1:2. The transfection efficiencies were as follows: COS-1, 40–42%; CHO-K1, 27–30%; CHO-FD11, 30–40%; HEK293, 75–80%; LoVo, 10–11%. At 24 h post-transfection, cells were washed for 1 h (at 37 °C) in serum-free medium followed by incubation for an additional 20 h in serum-free medium alone or in combination with 10 μm hexa-d-arginine (D6R; EMD Chemicals), or 25 μm decanoyl-RVKR-chloromethyl ketone (RVKR-cmk; Bachem), respectively, as indicated in the figure legends. Following treatments, cells and/or conditioned media were collected for Western blot analyses (see below).
Enzymatic Digestion of Glycosyl Moieties
Proteins from conditioned media and cell lysates were incubated with endoglycosidase H (Endo H) or N-glycosidase F (PNGase F) for 90 min at 37 °C. Products were separated on 8% Tris glycine SDS-PAGE gels, transferred to a PVDF membrane (0.45 μm; PerkinElmer Life Sciences), and revealed by Western blot (see below).
Isolation, Culture, and Transfection of Mouse Primary Hepatocytes
Mouse primary hepatocytes were isolated from 8–10-week-old male livers using a two-step collagenase perfusion method, as previously described (22). In 3.5-cm Petri dishes coated with fibronectin (0.5 mg/ml, Sigma), 5 × 105 cells were seeded in Williams' medium E supplemented with 10% (v/v) FBS (Invitrogen). After 2 h, the medium was replaced with hepatozyme medium (Invitrogen) for 12 h prior to transfection. Transfections were performed with Effectene (Qiagen) using a total of 5 μg of cDNA, following the manufacturer's instructions (transfection efficiencies of 10–15%). The medium was collected 48 h post-transfection and subjected to immunoprecipitation followed by Western blot analysis (see below).
Quantitative RT-PCR (QPCR) Analyses
RNA from isolated mouse atria (right and left) were extracted using TRIzol, as recommended by the manufacturer (Invitrogen). Primers from neighboring exons were used to measure BMP10, furin, PACE4, PC5/6, and mouse TATA-box binding protein. cDNA synthesis and QPCR were performed as described previously (23). The sets of primers were as follows: BMP10, 5′-CATCATCCGGAGCTTCAAGAAC versus 5′-TCCGGAGCCCATTAAAAGTG; furin, 5′-CATGACTACTCTGCTGATGG versus 5′-GAACGAGAGTGAACTTGGTC; PACE4, 5′-GCTGGCTAAACAAGCTTTCGA versus 5′-CAAAAATGGAGCCCAGACCTT; PC5/6, 5′-ACTCTTCAGAGGGTGGCTA versus 5′-GCTGGAACAGTTCTTGAATC; TATA-box binding protein, 5′-GCTGAATATAATCCCAAGCGATTT versus 5′-GCAGTTGTCCGTGGCTCTCT.
Western Blotting and Antibodies
Media from COS-1 cells (transfected with non-tagged pro-BMP10) or mouse atria protein extracts (50 μg) were subjected to non-reducing (8% Tricine) SDS-PAGE analyses, followed by transfer to a 0.2-μm PVDF membrane (Millipore) and BMP10 detection using a BMP10 antibody (BMP10 Ab; under non-reducing conditions) (1:500; R&D Systems) and the corresponding secondary antibody conjugated to horseradish peroxidase (HRP) (1:10,000; Invitrogen).
Proteins from medium of cultured cells (transfected with ProtC-tagged pro-BMP10) were analyzed by 8% Tris glycine SDS-PAGE, followed by Western blotting using a ProtC-Ab (1:1000; GenScript) and a rabbit HRP-conjugated secondary Ab (1:10,000; Invitrogen). Pro-BMP10 and its prosegment tagged with ProtC at the N terminus were immunoprecipitated from mouse primary hepatocytes culture medium (200 μl) using a ProtC-Ab (2 μg; Abgent), resolved by 8% Tris glycine SDS-PAGE gels, and immunodetected using the same ProtC-Ab (1:1,000) and anti-rabbit IgG-HRP (1:2,000, TrueBlot).
The antigen-antibody complexes were visualized using the enhanced chemiluminescence kit (ECL; Pierce). Quantitation was performed using ImageJ software (National Institutes of Health), and normalization was reported to β-actin. When media were analyzed by Western blotting, the same volumes were loaded on SDS-PAGE gels.
Biosynthetic Analyses
HEK293 cells (1 × 106 cells) were transiently transfected in 60-mm dishes using Effectene (Qiagen) and a total of 1 μg of DNA. Two days post-transfection, the cells were washed and pulse-labeled with 250 μCi/ml of [32S]Met/Cys for either 15 min (pulse-chase experiments) or 2 h. When pulse-labeled for 2 h, the cells were also incubated with brefeldin A (2.5 μg/ml), tunicamycin (5 μg/ml), D6R (10 μm), cell permeable RVKR-CMK (50 μm), or dimethyl sulfoxide. For the pulse-chase experiments, following the 15-min pulse, the cells were chased for various periods in a medium containing excess unlabeled Met/Cys. The cells were lysed in modified radioimmune precipitation assay buffer (150 mm NaCl and 50 mm Tris-HCl, pH 7.5), 1% Nonidet P-40, 0.5% deoxycholate, 0.1% SDS, and proteinase inhibitor mixture (Mixture Set I; Calbiochem). Cell lysates and media were immunoprecipitated with BMP10 Ab (2 μg) recognizing the mature form. Immunoprecipitates were resolved by SDS-PAGE on 8% Tricine gels and autoradiographed.
RESULTS
Bioinformatic Analysis of Potential Secretory PC Substrates Exhibiting Asn at P1′, and Whose Gene Deletion Is Associated with Heart Defects
Genetic evidence indicated that the proprotein convertases play key roles in cardiogenesis and left-right (furin and PACE4) or antero-posterior patterning (PC5/6) (5–8). Indeed, deletion of the PC5/6 gene (Pcsk5) led to death at birth, with mice exhibiting multiple bone morphogenic defects and heart abnormalities, including ventricular-septal defects (5, 6). One of the in vivo specific PC5/6 substrates associated with patterning defects was identified as growth differentiating factor 11 (Gdf11) (5, 6). The PC5/6-specific processing of NTKRSRR295↓NLGLD in pro-Gdf11 required the presence of Asn at the P1′ position, as its replacement by Asp led to the loss of PC5/6 specificity (5).
We therefore hypothesized that the motifs (R/K)X(R/K)R↓N, (R/K)XXR↓N, or (R/K)XXXXR↓N may represent specific PC5/6 recognition sequences, as they fit the general basic aa-specific PC consensus motif (R/K)XnR↓, where Xn = 0, 2, or 4 aa (2). Consequently, we undertook a bioinformatic analysis of all the genes coding for secretory proteins containing a similar luminal motif, and whose gene deletion results in heart defects. Interestingly, the only substrate matching these criteria and exhibiting a paired basic aa motif was BMP10 (supplemental Table S1). All the other hits contained a single Arg at the P1 cleavage site. We chose to synthesize five 12–13-aa peptides (shown in bold in supplemental Table S1) and tested their cleavage in vitro by furin, PC5/6, PACE4, or PC7. This prospective screening revealed that only BMP10 was cleaved to any significant extent by the PCs (not shown), making it the focus of the present study.
Pro-BMP10 Processing in Adult Mouse Heart
Analysis of the BMP10 precursor (pro-BMP10) sequence (supplemental Fig. S1) strongly suggested that cleavage of the N-terminal prosegment at DSSARIRR313↓NAKG (mouse nomenclature) would release active BMP10. The prosegment contains two potential N-glycosylation sites at Asn67 and Asn131 and its size is predicted to be ∼50 kDa (supplemental Fig. S1). It was reported that, under non-reducing conditions, both purified human BMP10 precursor (pro-BMP10) and mature BMP10 migrate as a mixture of monomers and disulfide-linked homodimers (13).
Accordingly, we first expressed a recombinant human pro-BMP10 in COS-1 cells and analyzed the secreted products by SDS-PAGE under non-reducing conditions, followed by Western blotting using an antibody that only recognizes the native precursor and its mature BMP10 form (nonreducing conditions are recommended by the R&D manufacturer of the BMP10 antibody) (Fig. 1A, left panel). The data showed that endogenous enzymes in COS-1 cells are able to only partially process pro-BMP10 (present as monomer and dimer) into mature BMP10. However, co-expression of furin completely transformed both monomeric and dimeric forms of the precursor into the mature ∼17-kDa BMP10, which in our hands migrates as a monomer even under non-reducing, but denaturing conditions.
FIGURE 1.

In vivo processing of mouse pro-BMP10. A, right panel: Western blot of tissue extracts from RA and LA isolated from WT adult (3 months old) male mice using a mouse BMP10 Ab or anti-mouse HRP Ab (control). Left panel: ex vivo control, Western blot (WB) analysis of 20-h conditioned media from COS-1 cells transiently transfected with non-tagged pro-BMP10 alone or with furin using the same mouse BMP10 Ab. Proteins were resolved by 8% Tris-Tricine SDS-PAGE gels under non-reducing conditions. B, BMP10 mRNA levels were measured by QPCR in RA (dark gray bar) and LA from WT adult male mice. Mean ± S.D. are given and n = 5 mice per group. Expression of BMP10 mRNA is restricted to adult mouse RA. C, PACE4, furin, and PC5/6 mRNA levels were determined by QPCR in RA (dark gray bars) and LA (light gray bars) from WT adult male mice. Mean ± S.D. are given and n = 5 mice per group. No statistical difference was observed between the mRNA levels of the RA and LA.
Using the same antibody, Western blot analysis of left (LA) and right (RA) mouse heart atria extracts revealed the presence of mature ∼17-kDa BMP10 only in extracts of the RA (Fig. 1A, right panel). Because this antibody was raised in mouse, we could not identify with certainty the pro-BMP10 form, as it migrates close to non-reduced mouse IgGs recognized by the secondary anti-mouse HRP-labeled antibody (Fig. 1A, right panel). Nevertheless, the presence of the ∼17-kDa BMP10 only in heart RA extracts agrees with our QPCR analysis (Fig. 1B) and the previously reported in situ hybridization and QPCR analyses (11), all of which revealed the restricted expression of BMP10 mRNA in adult mouse RA.
In conclusion, mature BMP10 (∼17 kDa) is detected in mouse RA extracts and the cognate processing enzyme(s) of pro-BMP10 in the RA is/are yet to be defined. Thus, we first measured by QPCR the mRNA levels of the three plausible processing enzymes furin, PC5/6, and PACE4 in RA and LA of adult mice. Transcripts of the above three convertases were similarly expressed in both RA and LA (Fig. 1C), with furin and PACE4 as the major convertases found in this tissue.
In Vitro Processing of a Mouse BMP10 Peptide Encompassing Its PC-like Cleavage Site
To define the best processing enzyme(s) of pro-BMP10, we first undertook an in vitro kinetic analysis of the cleavage of a 12-mer peptide mimicking the processing site of wild type (WT) mouse pro-BMP10: DSSARIRR313↓NAKG. This peptide was digested for 2 h with equal activity units of furin, PACE4, PC5/6, or PC7 and the products were separated by RP-HPLC and identified by mass spectroscopy (Fig. 2A). The data showed that 86% of the 12-mer peptide was cleaved by furin at the predicted Arg313↓ site, as compared with 67, 68, and 4% by PACE4, PC5/6, and PC7, respectively (Fig. 2B and supplemental Table S2A). Kinetic analyses revealed that furin is ∼3-fold more efficient in processing the 12-mer peptide as compared with PACE4 and PC5/6 (Fig. 2C), with an apparent Km(app) of ∼35 μm and Vmax(app)/Km(app) of ∼1.9 h−1 (supplemental Table S2B). Finally, the replacement of the P1′ Asn314 by Asp314 (DSSARIRR313↓DAKG) reduced by more than 50% the cleavage kinetics by the three PC candidates, without drastically affecting their cleavage selectivity (supplemental Table S2A). Thus, in contrast to the P1′ Asn of pro-Gdf11 (5), the P1′ Asn of pro-BMP10 is not critical for PC selectivity. We conclude that in vitro furin is the best pro-BMP10 convertase, followed by PACE4 or PC5/6, whereas PC7 is an unlikely candidate.
FIGURE 2.

In vitro, furin is better than PC5/6 or PACE4 at processing the 12-mer mouse BMP10 peptide at the predicted R311IRR314↓ cleavage site, whereas PC7 does not cleave this peptide. A, typical RP-HPLC profile for in vitro digestion of the 12-mer mouse BMP10 peptide with soluble furin. The synthetic peptide (200 μm) was incubated for 2 h in vitro with 2 units of purified soluble furin, PC5/6, PACE4, or PC7, as described under “Experimental Procedures.” The products were separated by RP-HPLC on a Varian C18 column (5 μm, 100 Å, 4.6 × 250 mm). The cleavage site RIRR↓ was confirmed by MS/MS. The % cleavage was calculated as the ratio of the normalized peak areas (peak area/number of peptide bonds) of C-terminal fragment NAKG and the intact 12-mer peptide (at time 0). B, summary of the in vitro 2-h digestions of the 12-mer mBMP10 peptide with furin, PC5/6, PACE4, and PC7, respectively, based on RP-HPLC analyses. The results represent an average of two independent experiments. C, time dependent in vitro cleavage of the 12-mer mBMP10 peptide. The synthetic peptide was incubated with purified PCs in vitro for variable amounts of time. For each time point, the incubation mixture was subjected to RP-HPLC separation and the % cleavage was calculated and plotted as a function of time. Averages of two independent experiments are presented. Based on the linear range of the respective rate profiles (e.g. % cleavage at 20 min), the 12-mer mBMP10 peptide is a ∼3-fold better substrate for furin than for PC5/6 or PACE4.
Ex Vivo Processing of Human Pro-BMP10
Human pro-BMP10 carrying a protein C tag (ProtC) just after the signal peptide site (Fig. 3A), as for pro-Gdf11 (5), was expressed in COS-1 cells, known to lack PC5/6 expression (21, 24). Although WT (ProtC)-BMP10 is well processed, the single R316A, R313A, and double R316A/R313A mutants are not cleaved (Fig. 3B, media). This proves that the cleavage site of pro-BMP10 at RIRR316↓NA requires both P1 and P4 Arg, befitting the cleavage recognition motif RXRR↓ of basic aa-specific convertases (2). The cleaved prosegment did not accumulate in cells (Fig. 3B, WT lanes), suggesting that in COS-1 cells pro-BMP10 processing is a late cellular event occurring at the cell surface, or in the media. This is supported by Endo H and PNGase F digestions that show that whereas cellular pro-BMP10 is sensitive to both enzymes, suggesting an endoplasmic reticulum localization, the secreted pro-BMP10 and prosegment are only digested by PNGase F and are resistant to Endo H treatment (supplemental Fig. S2). Furthermore, the endogenous pro-BMP10 convertase is likely to be a member of the PC-family, as overexpression of potent secretory PC inhibitors composed of either the prosegment of furin (ppFurin) (20) or PACE4 (ppPACE4) (21, 25) completely abrogated pro-BMP10 maturation (Fig. 3B, media).
FIGURE 3.

Ex vivo validation of the R313IRR316↓ cleavage site by site-directed mutagenesis. A, schematic representation of the 424-aa human prepro-BMP10 and its derived forms, pro-BMP10, prosegment, and mature BMP10 (BMP10). Depicted are the signal peptide (SP), N-terminal ProtC tag, potential N-glycosylation sites (N67, N131), predicted PC-processing site (R313IRR316↓) and its mutants: P1 (R316A), P4 (R313A), and P1/P4 (R316A/R313A). B, cell lysates (left) and 20-h conditioned media (right) from COS-1 cells transiently expressing (ProtC)-BMP10 carrying either no mutation (WT; lane 1) or mutations R316A (lane 2), R313A (lane 3), and R316A/R313A (lane 4), or (ProtC)-BMP10 WT and either prepro-furin (ppFurin; lane 5) or prepro-PACE4 (ppPACE4; lane 6), or no protein (vector, lane 7) were analyzed by Western blotting using a rabbit ProtC-Ab. Processing of pro-BMP10 (pro) WT into its prosegment is detected only in the medium (right). The mutant forms of pro-BMP10 (R316A, R313A, and R316A/R313A) are no longer cleaved. In cell lysates only the uncleaved form (pro-BMP10) is detected (left). *, nonspecific band. The data are representative of at least two independent experiments. WB, Western blot.
To verify whether the high efficacy of endogenous furin to process pro-BMP10 is cell-type dependent, we expressed (ProtC)-BMP10 in different cells containing varying mixtures of the convertases, in the absence or presence of co-transfected ppFurin (Fig. 4). Thus, we tested HEK293 cells (expressing furin, PC5/6, and PACE4), COS-1 cells (lacking PC5/6) (24), CHO-K1 cells (lacking PACE4) (17), CHO-FD11 cells (lacking endogenous furin and PACE4) (17, 26), CHO-FD11 cells overexpressing furin in a stable fashion (lacking PACE4) (25), and LoVo cells (lacking furin and PC5/6) (18, 24). Western blot analysis of the proteins secreted in the medium show that all cells expressing ppFurin cannot process pro-BMP10. Furthermore, cells that are deficient in furin expression invariably exhibit lack of processing (see CHO-FD11 and LoVo cells). Cells that lack only PACE4 still can process pro-BMP10, albeit less efficiently (32%) than the same cells overexpressing furin (CHO-FD11/Fur or furin-transfected LoVo cells) (100%) (Fig. 4). In human LoVo cells, it seems that full-length furin (Furin) and soluble furin (sFurin) lacking the transmembrane and cytosolic tail (27), can both completely process pro-BMP10. This, however, is not always the case, as will be next demonstrated in CHO-FD11 cells (Fig. 5). We conclude that, among the processing enzymes examined, furin cleaves pro-BMP10 most efficiently ex vivo, as it did in vitro, and that this result does not seem to be cell-type dependent.
FIGURE 4.

Pro-BMP10 is cleaved ex vivo by endogenous furin (CHO-K1 cells), stably expressed furin (CHO-FD11/Fur cells), or transiently expressed furin (LoVo cells). Western blot (WB) analyses of 20-h conditioned medium from cells (HEK293, COS-1, CHO-K1, CHO-FD11/Fur, and CHO-FD11 cells) transiently transfected with either (ProtC)-BMP10 and an empty vector, or (ProtC)-BMP10 and the prosegment of furin (ppFurin), and from cells (LoVo cells) transfected with either empty vector, (ProtC)-BMP10 and empty vector, (ProtC)-BMP10 and furin, or (ProtC)-BMP10 and sFurin. The % of pro-BMP10 cleavage into its prosegment, calculated as prosegment/(pro-BMP10 + prosegment) × 100, is indicated below each lane along with the average % cleavage and S.D. values of several (n) independent experiments. The deficiency in endogenous PC activity of each cell line is depicted as ΔPC. Note the lack of processing of pro-BMP10 overexpressed in cell lines deficient in endogenous furin activity (CHO-FD11 and LoVo cells).
FIGURE 5.

Ex vivo processing of human pro-BMP10 by overexpressed furin, PACE4, and PC5/6. Western blot (WB) analyses of 20-h conditioned media from CHO-FD11 cells transiently transfected with either empty vector (vector; lane 8), or with a vector expressing ProtC-tagged pro-BMP10 (lanes 1–7; (ProtC)-BMP10) and vectors expressing either no protein (vector), furin, PACE4, or PC5/6, or their truncated versions sFurin, PACE4-ΔC, or PC5/6-ΔC. Proteins were revealed by using a rabbit ProtC-Ab. The corresponding percentages of pro-BMP10 cleavage (%) calculated from the ratio of band intensities of prosegment/(pro-BMP10 + prosegment) are indicated. The average % of pro-BMP10 cleavage and corresponding S.D. values from 4 independent experiments (n = 4) are shown as a bar graph.
Processing of Human Pro-BMP10 by Furin, PACE4, and PC5/6 Occurs in Different Cellular Locations
The above data could not eliminate a possible contribution of PACE4 and PC5/6 in the processing of pro-BMP10. Hence, we tested the effect of co-expression of furin, PACE4, and PC5/6 with pro-BMP10 in cells where it is not processed, i.e. CHO-FD11 cells (Fig. 5). To determine whether localization to the cell surface is important, we also co-expressed pro-BMP10 with either sFurin, or mutant PACE4-ΔC and PC5/6-ΔC that lack the C-terminal Cys-rich domains and thus no longer interact with cell surface heparan sulfate proteoglycans (21, 25). Western blot analysis of the proteins in the media showed that furin, PACE4, and PC5/6 can process pro-BMP10 to release the prosegment to the extent of 100, 90, and 80%, respectively (Fig. 5). The truncated forms that do not bind the cell surface either have similar efficacies (PC5/6-ΔC; ∼90%), are less efficient (sFurin; ∼20–40%), or no longer cleave pro-BMP10 (PACE4-ΔC; 0%) (Fig. 5). These data suggest that in CHO-FD11 cells, membrane binding of furin and cell-surface association of PACE4 are needed for maximal activity on pro-BMP10 conversion, whereas cell surface association is dispensable for PC5/6 activity on this precursor.
Processing of a control protein pro-7B2 by furin-like convertases into 7B2 is known to occur in the trans Golgi network (TGN) (28). The pan-PC cell-permeable inhibitor decanoyl-RVKR-chloromethyl ketone (RVKR-cmk) (16, 29), and the cell-surface peptide inhibitor hexapeptide d-Arg (D6R) (30, 31) are good inhibitors of furin, PC5/6, and PACE4 activity on RTKR-MCA in vitro (not shown). However, only RVKR-cmk, and not D6R, was able to block the ex vivo pro-7B2 processing, as observed in the medium of COS-1 cells treated with these inhibitors (supplemental Fig. S3). Thus, we tested the efficacies of the above inhibitors on pro-BMP10 processing, by analyzing CHO-FD11 cells overexpressing the substrate and the convertases (Fig. 6A), and naive HEK293, or COS-1 cells overexpressing pro-BMP10 only (Fig. 6B). In CHO-FD11 cells, the presence of 25 μm RVKR-cmk inhibited the processing of pro-BMP10 by furin (∼60%) and sFurin (∼98%). In contrast, 10 μm D6R had no overt effect (Fig. 6A, left panel). This suggests that intracellular furin and sFurin are mostly responsible for the processing of pro-BMP10, and that in the media furin is not able to cleave this precursor.
FIGURE 6.

Ex vivo processing of pro-BMP10 by the PCs and their derivatives, and inhibition by D6R and RVKR-cmk. A, Western blot (WB) analysis of 20-h conditioned medium from CHO-FD11 cells co-transfected with (ProtC)-BMP10 and either an empty vector or furin, sFurin, PACE4, PC5/6, or PC5/6-ΔC. As indicated, the conditioned medium was collected after no treatment (−), or treatment (+) with either the cell permeable convertase inhibitor RVKR-cmk (25 μm) or the cell surface convertase inhibitor D6R (10 μm). For each condition, the average % of pro-BMP10 cleavage and corresponding S.D. values from three to four independent experiments are shown as a bar graph. B, Western blot analyses of 20-h conditioned medium from HEK293 cells (left) or COS-1 cells (right) transiently expressing (ProtC)-BMP10 or no protein (vector) and collected after no treatment (dimethyl sulfoxide, DMSO) or treatment with either the cell surface convertase inhibitor D6R (10 or 20 μm) or the cell permeable convertase inhibitor RVKR-cmk (25 or 50 μm). These data are representative of at least two independent experiments.
In contrast to furin, the processing of pro-BMP10 by either PACE4 or PC5/6 is almost completely (>95%) inhibited by D6R (Fig. 6A, right panel), suggesting that their activity on pro-BMP10 is primarily limited to the cell surface. In agreement, the secreted PACE4-ΔC that does not bind cell surface heparan sulfate proteoglycans (21, 25) also does not cleave pro-BMP10 (Fig. 5). Unexpectedly PC5/6-ΔC is only partially inhibited by D6R (∼30%; Fig. 6A, right panel), suggesting that this truncated form of PC5/6 can also process pro-BMP10 intracellularly.
To gauge the sensitivity of pro-BMP10 processing by endogenous convertases, we compared the effects of RVKR-cmk and D6R on HEK293 cells (Fig. 6B, left panel) and COS-1 cells (Fig. 6B, right panel). Analysis of the medium from both cell types revealed that RVKR-cmk completely abolished the processing of pro-BMP10. In contrast, D6R had either partial (HEK293 cells) or full (COS-1 cells) inhibitory effects on this processing. These findings suggested that, in HEK293 cells, pro-BMP10 processing is mostly achieved by endogenous intracellular proteases, most likely furin, with some contribution of cell surface/media enzymes. In contrast, in COS-1 cells, which are very rich in heparan sulfate proteoglycans (21) and express low levels of furin and PC5/6 (24), processing of pro-BMP10 mostly occurred at the cell surface/media, likely by endogenous PACE4.
We next studied the time-dependent processing of pro-BMP10 using a pulse-chase paradigm in HEK293 cells. Following a 15-min pulse with [35S]Met/Cys, the cells were washed and chased for 30, 60, and 120 min. Immunoprecipitation of BMP10 and SDS-PAGE (Fig. 7A) show that it took about 30 min of chase (P15C30) to detect mature BMP10 in the media. The lack of accumulation of BMP10 in the cells at all chase periods suggests that, once formed, mature BMP10 is rapidly secreted (Fig. 7A).
FIGURE 7.

Ex vivo, pro-BMP10 is processed in a post-medial Golgi compartment (likely TGN) and is rapidly secreted into the media. A, HEK293 cells transiently transfected with (ProtC)-BMP10 were pulse-labeled with [35S]Met/Cys for 15 min and chased for 0, 30, 60, and 120 min in the absence of the radiolabel. Cell lysates and medium samples were immunoprecipitated with a mouse BMP10 Ab and then resolved by SDS-PAGE (8% Tris-Tricine gels) followed by autoradiography (4 days). B, HEK293 cells transiently transfected with nontagged pro-BMP10 (BMP10) were pulse-labeled with [35S]Met/Cys for 2 h in the absence of any treatment, or in the presence of brefeldin A (BFA; 2.5 μg/ml), tunicamycin (Tun; 5 μg/ml), D6R (10 μm), RVKR-cmk (50 μm), or dimethyl sulfoxide (DMSO). In control experiments HEK293 cells were transiently transfected with (ProtC)-BMP10 or no protein (vector). Cell lysates and medium samples were immunoprecipitated (IP) with mouse BMP10 Ab and then resolved by SDS-PAGE (8% Tris-Tricine gels) followed by autoradiography (17 h for pro-BMP10 detection and 4 days for detection of mature BMP10).
Pro-BMP10 processing was also analyzed after a 2-h pulse with [35S]Met/Cys in the presence of specific compounds (Fig. 7B). Incubation of cells with brefeldin A, which causes the collapse of the cis/medial Golgi with the endoplasmic reticulum, prevented secretion and processing of pro-BMP10, in accordance with the activity of PCs starting at the TGN level (3, 32). Interestingly, incubation of cells with the N-glycosylation inhibitor tunicamycin resulted in much lower secretion and processing of pro-BMP10, emphasizing the importance of one or both potential N-glycosylation sites of the prosegment (Fig. 3A) in the folding and/or stability of pro-BMP10. Finally, whereas RVKR-cmk completely blocked the processing of pro-BMP10, the inhibitor D6R only blocked ∼60% of this cleavage in the media, without any effect in the cells (compare lanes 2 and 5 of Fig. 7B). Altogether, these data lead us to conclude that in HEK293 cells pro-BMP10 processing occurs both intracellularly in a post-medial Golgi compartment, likely the TGN, and at the cell surface.
Because membrane-bound furin is the most promising candidate pro-BMP10 convertase, it was necessary to define the importance of its transmembrane and/or cytosolic tail in this process. Although it is known that the cytosolic tail of furin regulates its cycling from the TGN to the cell surface (32), the role of the transmembrane domain is less known, except that it may regulate its trafficking preference to the conventional versus nonconventional secretory pathways (33). Thus, we compared the pro-BMP10 cleavage by WT furin and furin chimeras in which either the cytosolic tail (furinCT-PC7) or transmembrane/cytosolic tail (furinTMCT-PC7) were replaced by those of PC7 (33) (supplemental Fig. S4). The data in CHO-FD11 media did not reveal any difference between WT and either furin chimeras in their ability to process pro-BMP10, whereas soluble sFurin was much less efficient (supplemental Fig. S4B). Thus, pro-BMP10 processing requires membrane association of furin. The intracellular cleavage of pro-BMP10 by furin was further confirmed by the inability of D6R to inhibit the pro-BMP10 processing in CHO-FD11 overexpressing either furinCT-PC7 or furinTMCT-PC7 (not shown), although the latter chimera is mostly localized at the cell surface (34).
In Mouse Primary Hepatocytes and Right Atria, Furin Is the Physiological Convertase of Pro-BMP10
To probe the processing of pro-BMP10 under more physiological conditions, we analyzed it in mouse primary hepatocytes and in atria. Because the complete KO of mouse furin (4) and PC5/6 (5, 6) are embryonic lethal, we chose to study pro-BMP10 processing in mouse primary hepatocytes from WT and conditional KOs of either furin or PC5/6 in hepatocytes (22). The data show that the lack of furin results in an 81% reduction of pro-BMP10 processing (from 84 to 24%). The residual 24% cleavage is likely due to PC5/6, as its absence lowered the cleavage efficiency from 84% in WT hepatocytes to 63% in PC5/6 KO hepatocytes (Fig. 8). However, a possible contribution of PACE4 cannot be excluded. Nevertheless, it is clear that in the absence of endogenous furin, pro-BMP10 processing is largely compromised in mouse hepatocytes.
FIGURE 8.
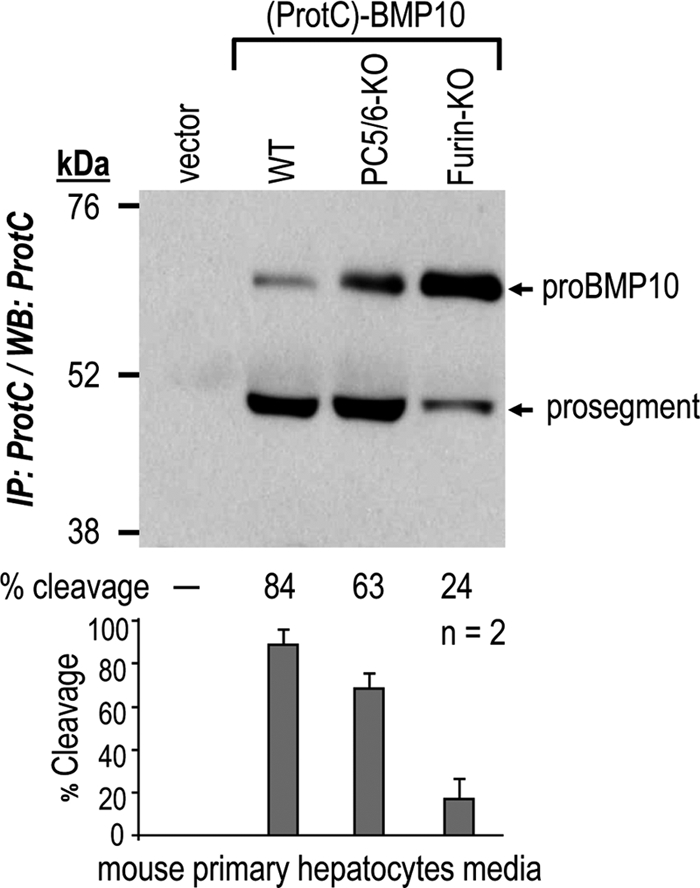
Furin is the major pro-BMP10 cleaving enzyme in hepatocytes. Primary hepatocytes isolated from WT mice and those lacking PC5/6 (PC5/6-KO) or furin (Fur-KO) in hepatocytes (22) were transiently transfected with plasmids expressing no protein (vector) or (ProtC)-BMP10. Processing of pro-BMP10 into its prosegment was analyzed in 48-h conditioned medium by immunoprecipitation (IP) with a rabbit ProtC-Ab followed by Western blotting (WB) using the same antibody. The percentages (%) of cleavages in this particular experiment are indicated, along with the average % cleavage and S.D. values of two independent experiments.
Because the main source of pro-BMP10 in adult mice is the RA of the heart (11), we needed to estimate the levels of BMP10 in this tissue. Because furin and PC5/6 KO result in embryonic lethal phenotypes, we could only compare the levels of BMP10 in adult atria from WT and PACE4 KO mice. Western blot analyses show that BMP10 levels are not significantly different between WT (5 mice) and PACE4 KO (7 mice) animals (supplemental Fig. S5). This suggests that the absence of PACE4 does not affect the processing of pro-BMP10 in the adult mouse RA.
DISCUSSION
We reported previously that Asn296 at the P1′ position in pro-Gdf11 (NTKRSRR295↓NLGLD) is critical for its selective cleavage by PC5/6 (5). In agreement, PC5/6 KO mice recapitulated all the phenotypes associated with Gdf11 deficiency. In addition, PC5/6 KO mice exhibited other specific phenotypes including heart defects, such as ventricular septal defects and transposition of great arteries, right-sided aortic arch, and ductus arteriosus (6). To identify other PC5/6 substrates, a bioinformatics search detected 9 secretory precursor proteins that exhibit a PC-like cleavage site with a P1′ Asn, and whose gene KO results in heart-related phenotypes (supplemental Table S1). In vitro testing of five selected candidates using synthetic peptides revealed that only pro-BMP10 is a plausible substrate for PC cleavage (Fig. 2 and supplemental Fig. S1). Kinetic analyses showed that furin is ∼3-fold better than PACE4 and PC5/6 to cleave a 12-mer peptide encompassing the predicted DSSARIRR313↓NAKG site (Fig. 2C). The associated Km(app) of 35 μm and Vmax(app)/Km(app) of 1.9 h−1 (supplemental Table S2B) compare very well with those published for good furin substrates (35). Unexpectedly, replacement of the P1′ Asn by Asp had no major impact on PC cleavage preference (supplemental Table S2A). We therefore concluded that the presence of an Asn at P1′ is not the only criterion governing PC5/6 selectivity.
The above findings with a synthetic peptide are corroborated by ex vivo data with full-length pro-BMP10. In COS-1 cells, a cell line lacking PC5/6, pro-BMP10 is efficiently processed by endogenous PCs (Figs. 1A, 3B, and 4). The lack of processing of pro-BMP10 overexpressed in a furin- and PACE4-deficient cell line, CHO-FD11, and in furin-deficient LoVo cells, was restored by stable (CHO-FD11/Fur cells) or transient expression of furin (Fig. 4). Furthermore, based on the inhibition of pro-BMP10 processing by D6R, a cell surface PC inhibitor, it seems that the best convertase furin mostly acts intracellularly, likely in the TGN where it is largely concentrated (32, 36), whereas PACE4 and PC5/6 seem to cleave this substrate almost exclusively at the cell surface (Figs. 5 and 6).
To identify the implicated PC(s) in vivo, pro-BMP10 processing was analyzed in a more physiologically relevant environment. In both primary hepatocytes (Fig. 8) and RA (Fig. 1A and supplemental Fig. S5) furin is the major convertase responsible for the production of mature BMP10. Although PC5/6 is responsible for ∼20% of pro-BMP cleavage in primary hepatocytes (Fig. 8), its contribution to pro-BMP10 cleavage in RA can be excluded, as its mRNA is barely detectable in this tissue (Fig. 1C). The higher mRNA levels of PACE4 in the RA made it a putative PC for pro-BMP10 cleavage in vivo. However, mature BMP10 levels in heart RA did not differ between WT and viable PACE4 KO mice, ruling out a key in vivo contribution (supplemental Fig. S5).
Collectively, the sum of the above in vitro, ex vivo, and in vivo data strongly supports a major contribution of furin in the physiological processing of pro-BMP10. In agreement, both furin (4) and BMP10 (11) mRNAs are first expressed in the heart at E8.5–9.0, a critical time in cardiogenesis coinciding with a shift from cardiac patterning and looping to cardiac growth and chamber formation (11). The zygotic knock-out of furin has very severe embryonic abnormalities due to effects on patterning, including cardia bifida and failure of looping (4). The zygotic knock-out of Bmp10 exhibits a failure of myocardial growth, and abnormal endocardial cushion development in outflow tract and atrioventricular canal (11). Thus, the abnormalities in the complete KO of Bmp10 and furin are related, i.e. failure of atrioventricular canal development and myocardial growth, and both knock-out mice die at E10.5. Finally, our data exclude a significant in vivo PC5/6 and PACE4 contribution in the processing of heart pro-BMP10, which is in agreement with the observations that complete PC5/6 (5, 6) and PACE4 (7) KO mice survive until birth or beyond, respectively.
However, it is likely that BMP10 is not the only substrate that is affected by the absence of furin in heart embryos. Furin is likely implicated in the specific processing of multiple TGF-β like proteins (37) important for heart looping, such as lefty-1 and lefty-2 (4, 8) as well as other bone morphogenetic proteins such as BMP2 (38) and BMP4 (39). Interestingly, whereas BMP4 has been proven to have a second processing site in its prosegment, the cleavage of which enhances the half-life of active BMP4 (40), no second furin-like cleavage site has been found in either BMP10 (this work) or Gdf11 (5).
Screening databases for potential furin-like substrates that exhibit a motif RXRR↓N similar to that of BMP10 revealed two new candidates, namely the type II transmembrane growth factor ectodysplasin-A (RVRR156↓NK) (41), and the extracellular matrix collagen type V-α1 (collagen type V-α1; RTRR1585↓NI) (42), a minor component of collagen I fibrils. In E10.5 mice, collagen type V-α1 expression was found in the heart, and dorsal aorta wall, and collagen type V-α1 KO mice revealed that this protein is important in the development of functional connective tissues (43).
In many cases, members of the PC family might substitute or supplement furin activity, suggesting the existence of some functional redundancy between PCs (24, 44). In only a few cases was furin shown to be absolutely essential for the processing of a given substrate in vivo. These include the activation of the V-ATPase subunit regulating intragranular acidification in pancreatic β-cells (45), the regulation of TGF-β1 production (37) in immune T cells (46), and the inactivation of PCSK9 in hepatocytes (22). The present work provides evidence for yet another in vivo furin-specific substrate, namely BMP10 in the RA. In the future, inducible tissue-specific KO mice of furin in heart would certainly be of value to potentially identify other essential furin substrates implicated in key cardiac functions.
Supplementary Material
Acknowledgments
We thank Denis Faubert and Sylvain Tessier for technical assistance with mass spectrometry analyses. We are grateful to Brigitte Mary for efficacious editorial assistance.
This work was supported by Canadian Institutes of Health Research Grant MOP-44363, a Strauss Foundation grant, Canada Chair Grant 216684, British Heart Foundation Chair Award CH/09/003 (to S. Bhattacharya) and Project Grant Award PG/08/045/25069 (to S. B.), and Wellcome Trust Core Grant Award 075491/Z/04.

The on-line version of this article (available at http://www.jbc.org) contains supplemental Tables S1 and S2 and Figs. S1–S5.
D. Constam, personal communications.
- aa
- amino acid
- BMP10
- bone morphogenetic protein 10
- D6R
- hexa d-arginine
- Endo H
- endonuclease H
- KO
- knockout
- LA
- left atria
- PCs
- proprotein convertases
- PC5/6 and PC7
- proprotein convertases 5/6 and 7
- PNGase F
- N-glycosidase F
- RA
- right atria
- TGN
- trans Golgi network
- MCA
- 7-amido-4-methylcoumarin
- QPCR
- quantitative PCR
- Gdf11
- growth differentiating factor 11
- Ab
- antibody
- Tricine
- N-[2-hydroxy-1,1-bis(hydroxymethyl)ethyl]glycine
- sFurin
- soluble furin
- cmk
- chloromethyl ketone
- E
- embryonic day
- ProtC
- Protein C tag.
REFERENCES
- 1. Srivastava D., Olson E. N. (2000) Nature 407, 221–226 [DOI] [PubMed] [Google Scholar]
- 2. Seidah N. G., Chrétien M. (1999) Brain Res. 848, 45–62 [DOI] [PubMed] [Google Scholar]
- 3. Seidah N. G., Mayer G., Zaid A., Rousselet E., Nassoury N., Poirier S., Essalmani R., Prat A. (2008) Int. J. Biochem. Cell. Biol. 40, 1111–1125 [DOI] [PubMed] [Google Scholar]
- 4. Roebroek A. J., Umans L., Pauli I. G., Robertson E. J., van Leuven F., Van de Ven W. J., Constam D. B. (1998) Development 125, 4863–4876 [DOI] [PubMed] [Google Scholar]
- 5. Essalmani R., Zaid A., Marcinkiewicz J., Chamberland A., Pasquato A., Seidah N. G., Prat A. (2008) Proc. Natl. Acad. Sci. U.S.A. 105, 5750–5755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Szumska D., Pieles G., Essalmani R., Bilski M., Mesnard D., Kaur K., Franklyn A., El Omari K., Jefferis J., Bentham J., Taylor J. M., Schneider J. E., Arnold S. J., Johnson P., Tymowska-Lalanne Z., Stammers D., Clarke K., Neubauer S., Morris A., Brown S. D., Shaw-Smith C., Cama A., Capra V., Ragoussis J., Constam D., Seidah N. G., Prat A., Bhattacharya S. (2008) Genes Dev. 22, 1465–1477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Constam D. B., Robertson E. J. (2000) Genes Dev. 14, 1146–1155 [PMC free article] [PubMed] [Google Scholar]
- 8. Constam D. B., Robertson E. J. (2000) Development 127, 245–254 [DOI] [PubMed] [Google Scholar]
- 9. Villeneuve P., Feliciangeli S., Croissandeau G., Seidah N. G., Mbikay M., Kitabgi P., Beaudet A. (2002) J. Neurochem. 82, 783–793 [DOI] [PubMed] [Google Scholar]
- 10. Neuhaus H., Rosen V., Thies R. S. (1999) Mech. Dev. 80, 181–184 [DOI] [PubMed] [Google Scholar]
- 11. Chen H., Shi S., Acosta L., Li W., Lu J., Bao S., Chen Z., Yang Z., Schneider M. D., Chien K. R., Conway S. J., Yoder M. C., Haneline L. S., Franco D., Shou W. (2004) Development 131, 2219–2231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chen H., Yong W., Ren S., Shen W., He Y., Cox K. A., Zhu W., Li W., Soonpaa M., Payne R. M., Franco D., Field L. J., Rosen V., Wang Y., Shou W. (2006) J. Biol. Chem. 281, 27481–27491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Mazerbourg S., Sangkuhl K., Luo C. W., Sudo S., Klein C., Hsueh A. J. (2005) J. Biol. Chem. 280, 32122–32132 [DOI] [PubMed] [Google Scholar]
- 14. Bessa P. C., Cerqueira M. T., Rada T., Gomes M. E., Neves N. M., Nobre A., Reis R. L., Casal M. (2009) Protein Expr. Purif. 63, 89–94 [DOI] [PubMed] [Google Scholar]
- 15. Fugère M., Limperis P. C., Beaulieu-Audy V., Gagnon F., Lavigne P., Klarskov K., Leduc R., Day R. (2002) J. Biol. Chem. 277, 7648–7656 [DOI] [PubMed] [Google Scholar]
- 16. Ozden S., Lucas-Hourani M., Ceccaldi P. E., Basak A., Valentine M., Benjannet S., Hamelin J., Jacob Y., Mamchaoui K., Mouly V., Desprès P., Gessain A., Butler-Browne G., Chrétien M., Tangy F., Vidalain P. O., Seidah N. G. (2008) J. Biol. Chem. 283, 21899–21908 [DOI] [PubMed] [Google Scholar]
- 17. Gordon V. M., Rehemtulla A., Leppla S. H. (1997) Infect. Immun. 65, 3370–3375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Takahashi S., Kasai K., Hatsuzawa K., Kitamura N., Misumi Y., Ikehara Y., Murakami K., Nakayama K. (1993) Biochem. Biophys. Res. Commun. 195, 1019–1026 [DOI] [PubMed] [Google Scholar]
- 19. Takahashi S., Nakagawa T., Kasai K., Banno T., Duguay S. J., Van de Ven W. J., Murakami K., Nakayama K. (1995) J. Biol. Chem. 270, 26565–26569 [DOI] [PubMed] [Google Scholar]
- 20. Zhong M., Munzer J. S., Basak A., Benjannet S., Mowla S. J., Decroly E., Chrétien M., Seidah N. G. (1999) J. Biol. Chem. 274, 33913–33920 [DOI] [PubMed] [Google Scholar]
- 21. Mayer G., Hamelin J., Asselin M. C., Pasquato A., Marcinkiewicz E., Tang M., Tabibzadeh S., Seidah N. G. (2008) J. Biol. Chem. 283, 2373–2384 [DOI] [PubMed] [Google Scholar]
- 22. Essalmani R., Susan-Resiga D., Chamberland A., Abifadel M., Creemers J. W., Boileau C., Seidah N. G., Prat A. (2011) J. Biol. Chem. 286, 4257–4263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Dubuc G., Chamberland A., Wassef H., Davignon J., Seidah N. G., Bernier L., Prat A. (2004) Arterioscler. Thromb. Vasc. Biol. 24, 1454–1459 [DOI] [PubMed] [Google Scholar]
- 24. Seidah N. G., Chrétien M., Day R. (1994) Biochimie 76, 197–209 [DOI] [PubMed] [Google Scholar]
- 25. Nour N., Mayer G., Mort J. S., Salvas A., Mbikay M., Morrison C. J., Overall C. M., Seidah N. G. (2005) Mol. Biol. Cell. 16, 5215–5226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Gordon V. M., Klimpel K. R., Arora N., Henderson M. A., Leppla S. H. (1995) Infect. Immun. 63, 82–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Decroly E., Wouters S., Di Bello C., Lazure C., Ruysschaert J. M., Seidah N. G. (1996) J. Biol. Chem. 271, 30442–30450 [DOI] [PubMed] [Google Scholar]
- 28. Paquet L., Bergeron F., Boudreault A., Seidah N. G., Chrétien M., Mbikay M., Lazure C. (1994) J. Biol. Chem. 269, 19279–19285 [PubMed] [Google Scholar]
- 29. Hallenberger S., Bosch V., Angliker H., Shaw E., Klenk H. D., Garten W. (1992) Nature 360, 358–361 [DOI] [PubMed] [Google Scholar]
- 30. Cameron A., Appel J., Houghten R. A., Lindberg I. (2000) J. Biol. Chem. 275, 36741–36749 [DOI] [PubMed] [Google Scholar]
- 31. Wender P. A., Mitchell D. J., Pattabiraman K., Pelkey E. T., Steinman L., Rothbard J. B. (2000) Proc. Natl. Acad. Sci. U.S.A. 97, 13003–13008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Thomas G. (2002) Nat. Rev. Mol. Cell. Biol. 3, 753–766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Rousselet E., Benjannet S., Hamelin J., Canuel M., Seidah N. G. (2011) J. Biol. Chem. 286, 2728–2738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Rousselet E., Benjannet S., Marcinkiewicz E., Asselin M. C., Lazure C., Seidah N. G. (2011) J. Biol. Chem. 286, 9185–9195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lazure C., Gauthier D., Jean F., Boudreault A., Seidah N. G., Bennett H. P., Hendy G. N. (1998) J. Biol. Chem. 273, 8572–8580 [DOI] [PubMed] [Google Scholar]
- 36. Molloy S. S., Thomas L., VanSlyke J. K., Stenberg P. E., Thomas G. (1994) EMBO J. 13, 18–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Dubois C. M., Blanchette F., Laprise M. H., Leduc R., Grondin F., Seidah N. G. (2001) Am. J. Pathol. 158, 305–316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zhang H., Bradley A. (1996) Development 122, 2977–2986 [DOI] [PubMed] [Google Scholar]
- 39. Cui Y., Jean F., Thomas G., Christian J. L. (1998) EMBO J. 17, 4735–4743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Goldman D. C., Hackenmiller R., Nakayama T., Sopory S., Wong C., Kulessa H., Christian J. L. (2006) Development 133, 1933–1942 [DOI] [PubMed] [Google Scholar]
- 41. Chen Y., Molloy S. S., Thomas L., Gambee J., Bächinger H. P., Ferguson B., Zonana J., Thomas G., Morris N. P. (2001) Proc. Natl. Acad. Sci. U.S.A. 98, 7218–7223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Unsöld C., Pappano W. N., Imamura Y., Steiglitz B. M., Greenspan D. S. (2002) J. Biol. Chem. 277, 5596–5602 [DOI] [PubMed] [Google Scholar]
- 43. Roulet M., Ruggiero F., Karsenty G., LeGuellec D. (2007) Cell. Tissue Res. 327, 323–332 [DOI] [PubMed] [Google Scholar]
- 44. Roebroek A. J., Taylor N. A., Louagie E., Pauli I., Smeijers L., Snellinx A., Lauwers A., Van de Ven W. J., Hartmann D., Creemers J. W. (2004) J. Biol. Chem. 279, 53442–53450 [DOI] [PubMed] [Google Scholar]
- 45. Louagie E., Taylor N. A., Flamez D., Roebroek A. J., Bright N. A., Meulemans S., Quintens R., Herrera P. L., Schuit F., Van de Ven W. J., Creemers J. W. (2008) Proc. Natl. Acad. Sci. U.S.A. 105, 12319–12324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Pesu M., Watford W. T., Wei L., Xu L., Fuss I., Strober W., Andersson J., Shevach E. M., Quezado M., Bouladoux N., Roebroek A., Belkaid Y., Creemers J., O'Shea J. J. (2008) Nature 455, 246–250 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.