

Abstract
Background
Cancer progenitor cells (CPC) have been postulated to promote treatment resistance and disease progression in prostate and other malignancies. We investigated whether the enzyme telomerase, which is active in cancer cells and in normal stem cells, plays an important role in CPC which can be exploited to neutralize these cells.
Methods
We used flow cytometry and assays of gene expression, clonogenicity and invasiveness to isolate and characterize a putative CPC subpopulation from freshly-resected human prostatectomy specimens. Telomerase activity was measured by qPCR-based Telomeric Repeat Amplification Protocol (TRAP). Telomerase interference was achieved by ectopic expression of a mutated telomerase RNA construct which reprograms telomerase to generate “toxic” uncapped telomeres. Treated cells were assayed for apoptosis, proliferation in culture, and xenograft tumor formation.
Results
CPC in prostate tumors expressed elevated levels of genes associated with a progenitor phenotype and were highly clonogenic and invasive. Significantly, CPC telomerase activity was 20 to 200-fold higher than in non-CPC from the same tumors, and CPC were exquisitely sensitive to telomerase interference which induced rapid apoptosis and growth inhibition. Similarly, induction of telomerase interference in highly-tumorigenic CPC isolated from a prostate cancer cell line abrogated their ability to form tumor xenografts.
Conclusions
Human prostate tumors contain a CPC subpopulation with markedly elevated telomerase activity which renders them acutely susceptible to telomerase interference. These findings offer the first tumor-derived and in vivo evidence that telomerase may constitute a CPC “Achilles heel” which may ultimately form the basis for more effective new CPC-targeting therapies.
Keywords: cancer progenitor cell, telomerase targeting, prostate cancer, cancer stem cell
Introduction
Cancer progenitor cells (CPC) are self-renewing, highly tumorigenic cancer cells recently identified in a broad spectrum of malignancies and implicated in tumor formation, therapy resistance and disease dissemination [1-2]. In prostate, as in other cancers, various strategies have been developed to enrich and isolate CPC from tumors, including flow cytometry for specific cell surface markers [3-10], isolation of a side population [11], and formation of cell spheres [5] or holoclones [12-13]. In addition, cancer cell lines, human xenograft tumors [5-8,10] and mouse models [14-15] have also been commonly used to characterize prostate CPC. The mostly robust prostate CPC markers to emerge from this collective work have been integrin α2β1, CD44 and CD133 [3-10]; these cell surface antigens enrich for a subpopulation of prostate cancer cells with an embryonic stem-like gene expression profile and with high clonogenic, metastatic, and tumorigenic potential relative to marker-negative cells [3-4,6-10,16-17].
In light of CPC’s high tumorigenicity and therapy resistance, it has been suggested that truly effective new cancer treatments should specifically aim to target this tumor subpopulation [1-2,18]; however, there are as yet no effective CPC-targeting treatments. Telomerase activity is a recognized hallmark of malignancy which has been explored extensively by our group and others for its therapeutic and biomarker potential [19-22]. Whereas benign, terminally differentiated tissues have extremely low levels of telomerase [23], malignant cells from a variety of cancers have significant telomerase expression and activity that correlate with malignant potential and high tumor initiating ability [24-26]. In addition to its role in cancer, telomerase recently has been found to play an equally important function in normal stem cell function, inducing stem cell activation in human and mouse epidermal, gastrointestinal, hematopoietic, neuronal, and reproductive niches [27]. Given its dual roles in carcinogenesis and stem cell activation, we speculated that perhaps telomerase activation plays an equally critical role in CPC and hence may constitute an attractive therapeutic strategy for targeting these cells.
To address this question, we investigated the relative telomerase activity and expression levels within cancer cell subpopulations isolated from freshly resected human prostate tumors and from prostate cancer cell lines. Specifically, we used FACS to isolate integrin α2β1highCD44+ cells, previously reported to possess a cancer progenitor-like phenotype [3-4,6-7,28-29]. When isolated from prostate tumors in our studies, these cells expressed higher levels of self-renewal genes and were more clongenic and invasive in vitro than α2β1highCD44− cells from the same tumors. Similarly, when isolated from the DU145 prostate cancer cell line, α2β1highCD44+ cells expressed CPC-like properties in vitro, as well as high tumor initiation in vivo compared to α2β1highCD44− cells. Remarkably, both in tumors and cell lines the putative CPC (α2β1highCD44+) possessed markedly elevated levels of telomerase expression and activity compared with bulk non-CPC (α2β1highCD44−). Therefore, we tested whether CPC were susceptible to telomerase interference, a therapeutic strategy that specifically exploits the presence of high telomerase activity [19,22,30-31]. Telomerase interference is a two-pronged approach consisting of: 1. telomerase RNA with a mutated template region (MT-hTer), and 2. siRNA against endogenous wild-type telomerase RNA. Ectopic co-expression of these two constructs from a single vector (MT-hTer/siRNA) effectively substitutes MT-hTer for wild-type hTer, thus reprogramming telomerase to encode incorrect telomeres. The altered “toxic” telomeres elicit a brisk DNA damage response and rapid apoptosis in cells with high levels of active telomerase. In the present study, MT-hTer/siRNA effectively reprogrammed the active telomerase of prostate CPC to induce rapid apoptosis and abrogate tumor initiation, thus underscoring the therapeutic potential of targeting CPC with telomerase interference.
Materials and Methods
Tissue collection, processing and cell culture
Prostate tumors freshly resected from prostate cancer patients at USC Norris Comprehensive Cancer Center were examined, inked, graded and staged by a pathologist in a de-identified, IRB-approved protocol. Cells were obtained as described previously [3-4] with minor modifications: Briefly, tissue was minced with scalpels and digested in DMEM/F12 (50:50 mix) media supplemented with 8.75 μg/ml liberase blendzymes 3 (Roche Applied Science, Mannheim, Germany) and 1μg/ml DNAse I (Invitrogen, Carlsbad, CA) overnight at 37 °C in a shaker incubator. Epithelial organoids were separated from the stromal fraction by unit gravity centrifugation and disaggregated into single cell suspension by incubation with trypsin/EDTA (Mediatech, Manassas, VA) for 5 min at 37°C. A portion of the cells were stained and analyzed by flow cytometry. The rest were plated on collagen coated plate (BD Pharmigen, San Diego, CA) and incubated at 37°C for 1 hr to enrich for an α2β1integrin+ cell population as described previously [3]. Non-adherent cells were verified to be α2β1integrin− CD44− (Supplementary Figure 1B). The adherent cells were collected by incubation for 5 min with accutase (Innovative Cell Technologies, San Diego, CA) for FACS. Tumor cells were maintained on collagen coated plate in CnT52 (PCT Prostate Epithelium Medium, Low BPE (Human), Millipore, CA) at 37°C, 5% CO2. To further molecularly validate the cancer identity of CPC, GSTP1 promoter methylation was confirmed in a subset of specimens and compared to adjacent benign tissue (Supplementary Table 1). Human prostate cancer cell line DU145 was cultured in RPMI 1640 with fetal bovine serum (FBS, 10%, Omega) at 37°C, 5% CO2.
Flow cytometry
Fluorescence-activated cell sorting (FACS) analysis was used for determination of marker expression by FACSAria (Becton Dickinson Immunocytometry Systems). The following antibodies were used: integrin α2 (PE-CD49b) and FITC-CD44 (BD PharMingen, San Diego, CA); CD133/1 (allophycocyanin (APC) labeled; Miltenyi Biotec, Inc., Auburn,CA). Single cell suspension was obtained from freshly resected and digested prostate tumor samples as described above and resuspended in cell staining buffer containing 2% FBS and 5mM EDTA. Cells were stained with the above antibodies on ice for 20 min. Data were analyzed by FACSDiva.
Colony-forming assay and cell migration assay
To test clonogenicity, sorted cells were seeded on collagen coated plates, and colonies were counted after 21 days. To test invasiveness, cell migration assays were performed using Matrigel-coated 24-well inserts (BD Pharmigen, San Diego, CA) per manufacturer’s instruction. Briefly, 104 cells were placed in the upper chamber with CnT52 medium while the lower chamber was filled with CnT52 containing 10 ng/ml SDF-1 (R&D systems, Minneapolis, MN). Non-migrated cells in the upper chamber were removed following fixation and staining of cells in the lower chamber. Migrated cells were analyzed using a Zeiss Imager.Z1 microscope with Axiovision software at 20X magnification.
RNA extraction, reverse transcription, and PCR
Total RNA was extracted by RNAqueous-micro Kit (Applied Biosytems Inc, Foster City, CA). First strand cDNA was synthesized using the RETROscript reverse transcription kit (Applied Biosytems Inc, Foster City, CA). PCR primers were described previously [7,32] and in Supplementary Table 2. GAPDH was used as internal control.
Quantitative PCR – Telomeric Repeat Amplification Protocol (qPCR-TRAP) and telomere length assay
Telomerase activity from cell extracts was analyzed using real-time PCR based telomeric repeat amplification protocol (TRAP) as described [33] and in Supplementary Table 3. Briefly, cells were lysed in TRAPeze® 1X CHAPS Lysis Buffer (Millipore, Temecula, CA). Cell lysate from 1000 cells was added for each reaction for each sample. The iQ5 optical system software version 2.0 was used to analyze the results. To determine telomere lengths, genomic DNA was extracted using Qiagen DNeasy Blood & Tissue Kit (Qiagen), and relative telomere lengths were analyzed in triplicate by real time PCR (Biorad MyIQ) as described previously using T and S primers [22].
Virus production and infection
Lentivirus was generated and cells were infected as previously described [19,30]. Briefly, 12 μg of lentiviral vector, along with 3 μg of pMD.G and 9 μg of pCMVdR8.91 plasmids were cotransfected into 293T cells by using the calcium phosphate co-precipitation method. Virus-containing medium was harvested 48 and 72 hr after transfection and filtered through 0.45 μm filter. Cells were seeded at 105 cells/well in 6-well plate overnight before adding lentiviruses packaged from various constructs supplemented with 8 μg/ml polybrene. After overnight infection, medium was changed and GFP signal was confirmed at 48 hours post infection to ensure >90% transduction (>90% of cells GFP+).
Cell growth curve and apoptosis assay
Sorted cells were seeded at 105 cells/well in 6 well plate and infected with control or MT-hTer/siRNA lentivirus. Cell proliferation was measured by cell counting using a hemocytometer at subsequent time points after infection. Apoptosis was analyzed at day 4 post infection with MT-hTer/siRNA by TUNEL assays performed following the protocol described in In Situ Cell Death Detection Kit, Fluorescein (Roche Applied Science, Indianapolis, IN) and analyzed on BD LSR-II.
Subcutaneous tumor xenografts
All experiments were approved and performed following the rules of the Institutional Animal Care and Use Committees at University of Southern California. 6-8 week old, male SCID mice were purchased from NIH. DU145 cells were infected overnight with control or MT-hTer/siRNA lentiviruses and cultured at 37°C, 5% CO2 for 1 day after changing media. For each mouse, 5000 cells were resuspended in media, mixed with 50 μl ice-cold matrigel (BD biosciences, San Jose, CA) and placed on ice until inoculation. 1 ml insulin syringe was used for subcutaneous inoculation onto the flank of each mouse (5 mice per treatment group). The growth of tumors was observed and recorded as tumor volume by caliper measurement. Ninety days after inoculation, mice were sacrificed, and tumors were resected and weighed.
Statistical analysis
Performed in collaboration with USC/Norris Biostatistics Core. All experiments were conducted in triplicate with error bars representing standard deviation around the mean. Student’s t-test was used to determine statistical significance when comparing mean values at one point in time (e.g. gene expression, % apoptosis, cell growth inhibition, relative telomerase activity in DU145 cells, tumor weights). Two-sided Wilcoxon matched-pairs signed rank test was performed by using Graphpad Prism5.0 software to compare statistical significance of telomerase activity and telomerase (hTERT) mRNA level for patients’ sample as well as tumor growth from DU145 cells over time.
Results
Prostate tumor cells expressing integrin α2β1highCD44+ have a cancer progenitor cell (CPC) phenotype
Freshly resected human prostate tumors (Table 1 and Supplementary Figure 1) were disaggregated, digested, and dissociated into single cell suspensions as described previously [3-4] and in Materials and Methods. Cells were analyzed by FACS for expression of integrin α2β1 (CD49b), CD44 and CD133, surface markers widely reported in prostate cancer to enrich for CPC properties such as elevated expression of self-renewal genes, high in vitro clonogenicity and invasiveness, and increased tumor initiation after selection from xenografted human cell lines [3-4,6-7,29]. In all tumor samples, we found α2β1highCD44+ cells, which constituted 0.7% to 9.2% of all cancer cells (Table 1 and Supplementary Figure 1). In contrast, minimal CD133 expression was observed (Supplementary Figure 1), consistent with previous reports [28]. In all experiments, transient collagen adherence [3] was used to enrich for integrin α2β1+ cells, followed by FACS to further select for an α2β1highCD44+ subpopulation; purity (>98%) was confirmed by re-analysis with FACS (Supplementary Figure 1).
Table 1.
Clinical and histological features of prostatectomy specimens
| Tumor | Pathologic TNM Stage | Gleason Score | PSA | % CPC |
|---|---|---|---|---|
| 1 | pT2bN0MX | 3+4 | 6.76 | 7.0 |
| 2 | pT4N1MX | 5+5 | 3.71 | 0.7 |
| 3 | pT3bN1MX | 4+3 | 11.1 | 5.3 |
| 4 | pT3bN0MX | 4+5 | 9.32 | 3.5 |
| 5 | pT3aN0MX | 4+4 | 0.26 | 5.2 |
| 6 | pT3aN0MX | 3+3 | 8.67 | 6.4 |
| 7 | pT3bN0MX | 3+5 | 9.44 | 9.2 |
| 8 | pT2cN0MX | 3+5 | 5.14 | 5.2 |
| 9 | pT2bN0MX | 3+4 | 10.2 | 0.8 |
| 10 | pT3aN0MX | 4+3 | 19.7 | 6.6 |
| 11 | pT3bN0MX | 3+4 | 7.72 | 3.9 |
| 12 | pT4N1MX | 4+5 | 7.29 | 4.3 |
| 13 | pT2aN0MX | 4+4 | 6.77 | 2.8 |
To confirm the previously-reported CPC properties of the tumor-derived FACS-sorted cells, we conducted phenotypic analyses which revealed that – compared with the non-CPC majority of tumor cells (α2β1highCD44−) – the α2β1highCD44+ subpopulation expressed significantly higher mRNA levels of genes associated with self-renewal, proliferation and invasiveness, such as beta-catenin, NANOG, Oct3/4, SMO and Bmi1; conversely, mRNA levels of genes associated with a differentiated prostate phenotype, such as androgen receptor (AR) and prostate specific antigen (PSA), were significantly lower (Figure 1A). Functionally, α2β1highCD44+ subpopulations from all 8 tested tumors proliferated in culture, with three of these generating discrete colonies at an average of 44 colonies per 1000 cells seeded after 21 days in culture; in contrast, α2β1highCD44− cells from all 8 tumors tested did not proliferate in culture and produced no colonies (Figure 1B). Moreover, α2β1highCD44+ cells from 4 of 8 tumors migrated across the membrane in a matrigel invasiveness assay at an average of 39 migrated cells per 5 high powered fields after 24 hours (interestingly, the same subset of tumors yielded α2β1highCD44+ cells which were both clonogenic and invasive); in contrast, none of the α2β1highCD44− cells from all 8 tumors tested were invasive (Figure 1C).
Figure 1. CPC phenotypes of cell subpopulations isolated from human prostate tumors.
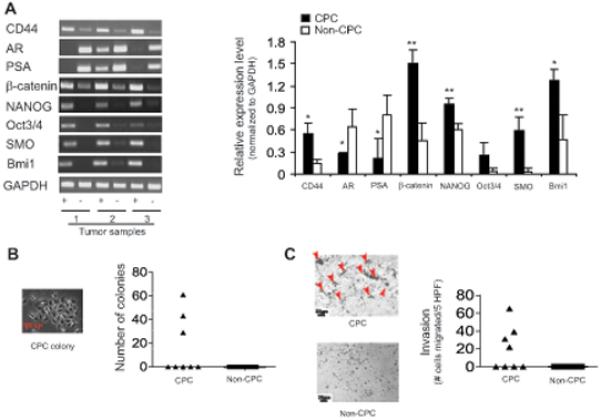
(A) Left: Gene expression (by RT-PCR) of cell subpopulations from 3 individual tumors (“+” = CPC, and “−” = non-CPC). Right: Semi-quantitative densitometric analysis of gel at left (mean values from 3 tumors). (B) Relative colony formation and (C) matrigel invasion of cell subpopulations from 8 individual tumors with sample micrographs (arrows on matrigel micrograph indicate cells that migrated across the membrane).
CPC have high telomerase expression and activity that can be targeted with telomerase interference
We measured relative telomerase activity from the putative tumor-derived CPC subpopulations (α2β1highCD44+) and non-CPC subpopulations (α2β1+CD44−) using qPCR-TRAP. Notably, we found that CPC had significantly higher (approximately 20-fold to 200-fold) telomerase activity than non-CPC in 6 of 6 tumors tested (Figure 2A, p=0.03). CPC also had significantly higher mRNA levels of hTERT, the chief determinant of telomerase activity (Figure 2B, p=0.008), whereas their mean bulk telomere lengths were not significantly different from those of non-CPC (Figure 2C).
Figure 2. Telomerase and telomere characterization of CPC and non-CPC cell subpopulations isolated from human prostate tumors.
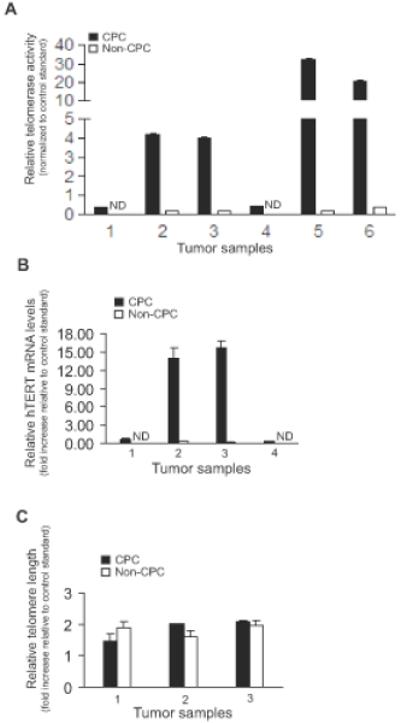
(A) Telomerase activity of cell subpopulations from 6 individual tumors by qPCR-TRAP, normalized to a standard control telomerase activity from DU145 cancer cells (p=0.03). (B) Relative hTERT mRNA levels of cell subpopulations in 4 individual tumors (p=0.008). (C) Telomere lengths of cell subpopulations in 3 individual tumors, respectively. In 2A-B, “ND” = not detectable. In all panels, 1000 cells were used for each experiment, and assays were conducted in triplicate and compared using a two-sided Wilcoxon matched-pairs signed rank test of statistical significance.
Having observed the markedly elevated telomerase expression and activity of tumor-derived CPC, we reasoned that this subpopulation may be particularly susceptible to telomerase interference, ectopic introduction of telomerase RNA with a mutated template region (MT-hTer/siRNA) that reprograms the telomerase enzyme to add incorrect telomeres, resulting in telomeric uncapping, DNA damage, and apoptosis [19,22,30]. We infected the tumor-derived CPC with lentivirus expressing either MT-hTer/siRNA or vector control. Two days after infection, MT-hTer expression by qPCR was ~6-fold compared to vector control (Figure 3A), and a modified qPCR-TRAP assay to detect mutant telomeric repeats showed a 5-fold increase in mutant telomerase activity (Figure 3B); together, these data confirmed successful MT-hTer/siRNA expression and reprogramming of telomerase activity in the CPC subpopulation. Importantly, telomerase interference resulted in 80% apoptosis by day 4 of infection and ~95% cell growth inhibition by day 6 (Figure 3C-D). Hence, the high telomerase expression and activity of tumor-derived CPC rendered them acutely susceptible to telomerase interference.
Figure 3. Induction of telomerase interference (reprogramming of telomerase) in tumor-derived CPC.
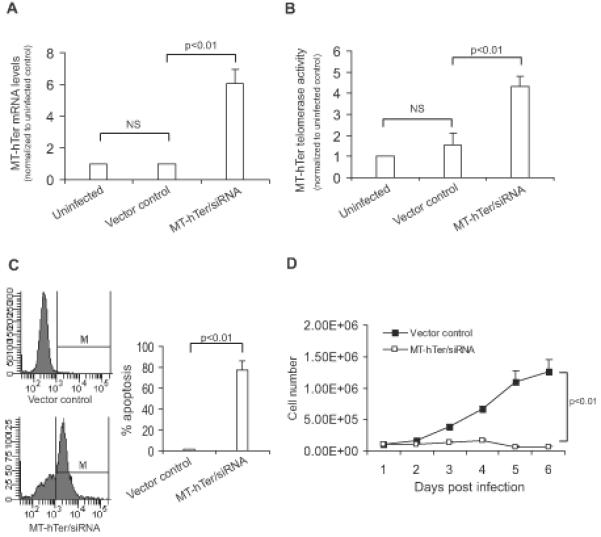
(A) MT-hTer expression by qPCR and (B) MT-telomerase activity levels by MT-specific qPCR-TRAP 48 hours after infection with lentivirus expressing MT-hTer/siRNA. (C) Percent apoptosis by TUNEL assay at day 4 after treatment with MT-hTer/siRNA or vector control. (D) Proliferation after treatment with MT-hTer/siRNA or vector control. Data in A-D reflects biological triplicate experiments conducted using CPC cells isolated from 1 patient tumor. All experiments were also repeated in triplicate using 2 additional patient tumors with similar results (data not shown).
CPC derived from a prostate cancer cell line have high tumorigenicity that is abrogated by telomerase interference
Having observed the marked efficacy of telomerase interference in vitro, next we tested whether this approach could also inhibit tumor formation in vivo. Currently there is no robust model for direct implantation and growth of human tumor-derived prostate cancer cells; therefore, we investigated tumor initiation in vivo using α2β1highCD44+ cells isolated from the DU145 prostate cancer cell line, a strategy previously shown to enrich for CPC properties [6-7,29]. Consistent with previously published cell line data [6-7,29] and similar to our primary tumor results, the DU145-derived putative CPC subpopulation (α2β1highCD44+) had significantly higher self-renewal gene expression (Figure 4A) and was significantly more clonogenic and invasive (Figure 4B) than the non-CPC (α2β1highCD44−) subpopulation. Further mirroring our findings in primary tumors, telomerase expression and activity of DU145-derived CPC were double those of non-CPC (Figure 4C), and telomere lengths were not significantly different (Figure 4D). Next, we tested the in vitro effects of telomerase interference (MT-hTer/siRNA) in the CPC fraction relative to the non-CPC fraction of DU145 cells, a direct comparison which had not been possible with primary tumor derived cells, because primary tumor-derived non-CPC did not propagate in culture. Notably, ectopic expression of MT-hTer/siRNA in DU145-derived CPC caused a significant 3-fold inhibition of proliferation relative to vector control by day 6 after lentiviral infection (Figure 5A). In contrast, proliferation of DU145-derived non-CPC was not significantly inhibited by telomerase interference, possibly because the lower telomerase expression and activity of these cells provided less substrate to be reprogrammed by MT-hTer/siRNA (Figure 5A).
Figure 4. CPC phenotypes of cell subpopulations isolated from DU145 human prostate cancer cell line.
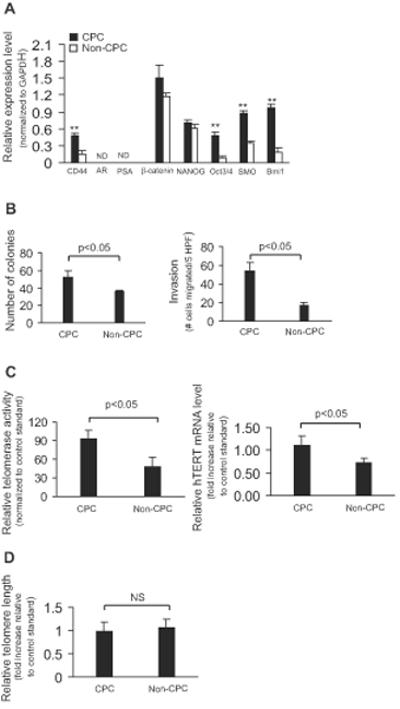
(A) Relative gene expression and (B) Colony formation (left), and invasiveness (right) of CPC and non-CPC subpopulations. (C) Relative telomerase activity (left) and hTERT mRNA levels (right), and bulk telomere lengths (D) of CPC and non-CPC subpopulations normalized to control standard (LNCaP cell line).
Figure 5. Effect of telomerase interference on in vivo tumor formation by DU145-derived CPC.
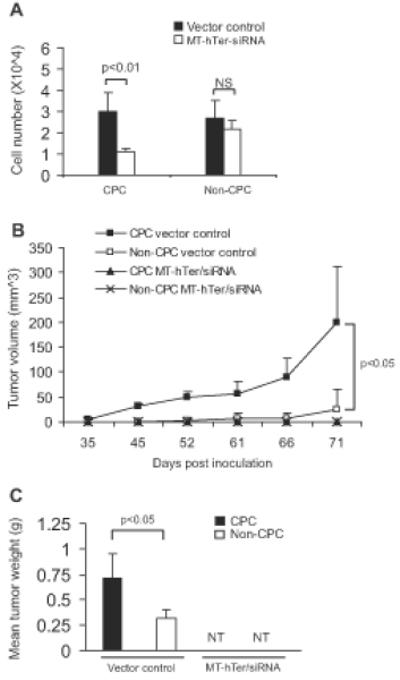
(A) Relative growth inhibition of CPC and non-CPC subpopulations 6 days after treatment with MT-hTer/siRNA. (B) CPC were more tumorigenic than non-CPC, and tumor formation by CPC was abrogated by telomerase interference. (C) Mean tumor weights at excision on day 90 post inoculation (NT = no tumors).
We investigated the in vivo impact of telomerase interference on the tumor initiating ability of CPC and non-CPC subpopulations derived from the DU145 cell line. MT-hTer/siRNA or vector control were ectopically overexpressed by lentiviral infection in CPC and non-CPC, and 5000 cells were inoculated subcutaneously into SCID mice (4 groups, 5 mice per group). In the vector control groups, CPC generated measurable tumors in 5 of 5 mice by day 45 with mean wet weight of 0.72 g at excision on day 90, whereas non-CPC generated measurable tumors in only 3 of 5 mice by day 61 with mean wet weight of only 0.32 g at excision on day 90 (i.e. fewer and smaller tumors with greater lag time), thus confirming the greater baseline tumorigenicity of CPC relative to non-CPC (Figure 5B, C). Notably, both subpopulations formed no tumors over 90 days of follow-up when ectopically expressing MT-hTer/siRNA, suggesting that telomerase interference effectively abrogated tumor initiation in vivo (Figure 5B, C).
Discussion
Telomerase activity is considered a nearly universal characteristic of cancer cells, an assumption that may exist because early surveys of telomerase activity were conducted indiscriminantly from lysates of entire cancer populations [25], and because the oncogenic role of telomerase was demonstrated by ectopically introducing the enzyme into unselected cell populations [34-35]. Contrary to this model of homogeneous telomerase activation, studies of normal tissue stem cell compartments have demonstrated a unique role for telomerase in stem cell activation [36-37], raising the possibility that perhaps telomerase plays a parallel unique role in so-called cancer progenitor (CPC) or tumor initiating cells. In support of a unique telomerase role in CPC, ectopic overexpression of telomerase in cancer cell lines has indeed been shown to enhance tumor initiation, perhaps reflecting a potentiation of the CPC phenotype [24,26]. Hence, we sought to determine whether telomerase activity in prostate tumors and cell lines is not uniformly distributed as previously assumed, but rather is focused predominantly in the CPC subpopulation where it can be used to neutralize these cells.
A variety of experimental systems have been described previously for the study of prostate CPC, resulting in a wide array of observations. For instance, a luminal epithelial stem cell was shown to be a cell of origin for prostate cancer using a mouse model in one study [38], while in another report only basal cells from primary benign human prostate tissue could initiate prostate cancer in immunodeficient mice [39]. Moreover, direct inoculation of primary prostate tumor subpopulations into mice as proof of a CPC phenotype has not been achieved as is done routinely in several other tumor types [40-44]. Given this background of multiple models and reports, we elected to conduct our study using primary human prostate tumors enriched for α2β1highCD44+, the most robust and consistently cited markers for a CPC phenotype in the prostate cancer literature [3-4,6-7,10,29]. Functionally, CD44, the primary receptor for hyaluronic acid (HA), plays critical roles in cancer cell adhesion, migration and drug resistance, consistent with a CPC phenotype [45]. This choice of markers was further reaffirmed by a recent report wherein integrin α6highCD44+ cell spheres generated in vitro from human prostate tumor tissues were successfully implanted (along with rat urogenital sinus mesenchyme) into a new strain of highly immunocompromised NOD-SCID/IL2rγNull mice, further substantiating the stem-like phenotype of this population [28].
In our present study, prostate tumor-derived α2β1highCD44+ cells expressed elevated levels of genes associated with self-renewal and also were more clonogenic and invasive than bulk unselected cells. Similarly, DU145-derived α2β1highCD44+ cells also had CPC-like in vitro properties as well as increased tumorigenicity in vivo relative to α2β1highCD44− cells. Another study recently examined prostate cancer cell lines for CPC-like cells and observed this subpopulation to have elevated telomerase activity which was inhibited with an anti-telomerase oligonucleotide [13]. Our present studies sought to further advance this line of investigation in several ways: Comparing telomerase activity between CPC and non-CPC subpopulations from freshly resected human tumors, testing the impact of telomerase reprogramming (via MT-hTer/siRNA) on these tumor-derived subpopulations, and investigating the effects of telomerase interference on tumor initiation in vivo using a similar cell line-derived CPC subpopulation.
Strikingly, we found that a subpopulation of primary tumor cells with CPC properties had markedly higher telomerase activity and hTERT expression than non-CPC from the same prostate tumors. Moreover, the non-CPC fractions, which comprised the vast majority (>90%) of tumor cells, did not propagate in vitro, a finding that further highlighted the biologic dichotomy between the two subpopulations: The large non-CPC subpopulation had very low telomerase activity and was unable to proliferate in culture, whereas the small CPC subpopulation had very high telomerase expression and activity, did proliferate in vitro, and underwent rapid apoptosis with exposure to telomerase interference, a strategy which specifically reprograms and exploits telomerase to generate toxic uncapped telomeres. This dichotomy in telomerase phenotype was further borne out in the DU145 experiments, where telomerase interference significantly inhibited the proliferation of CPC but exerted minimal effects on non-CPC. One possible explanation for this differential effect was that CPC had much more telomerase available to be reprogrammed by MT-hTer/siRNA, although contribution from other biological differences between CPC and non-CPC cannot be ruled out. Lastly, in the in vivo experiments, non-CPC derived from DU145 were unable to form tumors, whereas DU145-derived CPC had brisk tumor formation that was efficiently abrogated by telomerase interference with MT-hTer/siRNA.
Collectively, our experiments demonstrate that telomerase expression and activity are not a uniform phenotype common to all cancer cells as generally assumed, but rather are concentrated in a subpopulation of cells with CPC-like properties in prostate tumors and cell lines. Their highly active telomerase phenotype rendered prostate CPC exceedingly susceptible to telomerase interference, which induced apoptosis, inhibited proliferation, and abrogated tumor formation. Moreover, the potent effects of MT-hTer/siRNA on DU145-derived CPC (telomerase-high) versus its minimal effects on DU145-derived non-CPC (telomerase-low) suggests that telomerase interference may have a degree of therapeutic selectivity for CPC vis-à-vis their high telomerase. Therefore, targeting telomerase in this manner may constitute a promising new therapeutic strategy for neutralizing CPC in prostate and other cancers, ultimately leading to more effective control of tumor recurrence and progression.
Supplementary Material
Acknowledgments
We are grateful to Drs. Daniel Weisenberger and Peter Laird (USC Epigenome Center) for their generous help with the DNA methylation experiments. We thank Drs. Pradip Roy-Burman and Parkash Gill for their critical reading of this manuscript.
Contract grand acknowledgement: NIH/NCI K08 CA126983-01, Tower Cancer Research Foundation, Wright Foundation.
References
- 1.Gupta PB, Chaffer CL, Weinberg RA. Cancer stem cells: mirage or reality? Nat Med. 2009;15(9):1010–1012. doi: 10.1038/nm0909-1010. [DOI] [PubMed] [Google Scholar]
- 2.Visvader JE, Lindeman GJ. Cancer stem cells in solid tumours: accumulating evidence and unresolved questions. Nat Rev Cancer. 2008;8(10):755–768. doi: 10.1038/nrc2499. [DOI] [PubMed] [Google Scholar]
- 3.Collins AT, Habib FK, Maitland NJ, Neal DE. Identification and isolation of human prostate epithelial stem cells based on alpha(2)beta(1)-integrin expression. J Cell Sci. 2001;114(Pt 21):3865–3872. doi: 10.1242/jcs.114.21.3865. [DOI] [PubMed] [Google Scholar]
- 4.Collins AT, Berry PA, Hyde C, Stower MJ, Maitland NJ. Prospective identification of tumorigenic prostate cancer stem cells. Cancer Res. 2005;65(23):10946–10951. doi: 10.1158/0008-5472.CAN-05-2018. [DOI] [PubMed] [Google Scholar]
- 5.Dubrovska A, Kim S, Salamone RJ, Walker JR, Maira SM, Garcia-Echeverria C, Schultz PG, Reddy VA. The role of PTEN/Akt/PI3K signaling in the maintenance and viability of prostate cancer stem-like cell populations. Proc Natl Acad Sci U S A. 2009;106(1):268–273. doi: 10.1073/pnas.0810956106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hurt EM, Kawasaki BT, Klarmann GJ, Thomas SB, Farrar WL. CD44+ CD24(−) prostate cells are early cancer progenitor/stem cells that provide a model for patients with poor prognosis. Br J Cancer. 2008;98(4):756–765. doi: 10.1038/sj.bjc.6604242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Patrawala L, Calhoun T, Schneider-Broussard R, Li H, Bhatia B, Tang S, Reilly JG, Chandra D, Zhou J, Claypool K, Coghlan L, Tang DG. Highly purified CD44+ prostate cancer cells from xenograft human tumors are enriched in tumorigenic and metastatic progenitor cells. Oncogene. 2006;25(12):1696–1708. doi: 10.1038/sj.onc.1209327. [DOI] [PubMed] [Google Scholar]
- 8.Miki J, Furusato B, Li H, Gu Y, Takahashi H, Egawa S, Sesterhenn IA, McLeod DG, Srivastava S, Rhim JS. Identification of putative stem cell markers, CD133 and CXCR4, in hTERT-immortalized primary nonmalignant and malignant tumor-derived human prostate epithelial cell lines and in prostate cancer specimens. Cancer Res. 2007;67(7):3153–3161. doi: 10.1158/0008-5472.CAN-06-4429. [DOI] [PubMed] [Google Scholar]
- 9.Vander Griend DJ, Karthaus WL, Dalrymple S, Meeker A, DeMarzo AM, Isaacs JT. The role of CD133 in normal human prostate stem cells and malignant cancer-initiating cells. Cancer Res. 2008;68(23):9703–9711. doi: 10.1158/0008-5472.CAN-08-3084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Klarmann GJ, Hurt EM, Mathews LA, Zhang X, Duhagon MA, Mistree T, Thomas SB, Farrar WL. Invasive prostate cancer cells are tumor initiating cells that have a stem cell-like genomic signature. Clin Exp Metastasis. 2009;26(5):433–446. doi: 10.1007/s10585-009-9242-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brown MD, Gilmore PE, Hart CA, Samuel JD, Ramani VA, George NJ, Clarke NW. Characterization of benign and malignant prostate epithelial Hoechst 33342 side populations. Prostate. 2007;67(13):1384–1396. doi: 10.1002/pros.20620. [DOI] [PubMed] [Google Scholar]
- 12.Li H, Chen X, Calhoun-Davis T, Claypool K, Tang DG. PC3 human prostate carcinoma cell holoclones contain self-renewing tumor-initiating cells. Cancer Res. 2008;68(6):1820–1825. doi: 10.1158/0008-5472.CAN-07-5878. [DOI] [PubMed] [Google Scholar]
- 13.Marian CO, Wright WE, Shay JW. The effects of telomerase inhibition on prostate tumor-initiating cells. Int J Cancer. 2010;127(2):321–331. doi: 10.1002/ijc.25043. [DOI] [PubMed] [Google Scholar]
- 14.Liao CP, Adisetiyo H, Liang M, Roy-Burman P. Cancer-associated fibroblasts enhance the gland-forming capability of prostate cancer stem cells. Cancer Res. 2010;70(18):7294–7303. doi: 10.1158/0008-5472.CAN-09-3982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mulholland DJ, Xin L, Morim A, Lawson D, Witte O, Wu H. Lin-Sca-1+CD49fhigh stem/progenitors are tumor-initiating cells in the Pten-null prostate cancer model. Cancer Res. 2009;69(22):8555–8562. doi: 10.1158/0008-5472.CAN-08-4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cunha GR, Sekkingstad M, Meloy BA. Heterospecific induction of prostatic development in tissue recombinants prepared with mouse, rat, rabbit and human tissues. Differentiation. 1983;24(2):174–180. doi: 10.1111/j.1432-0436.1983.tb01317.x. [DOI] [PubMed] [Google Scholar]
- 17.Guzman-Ramirez N, Voller M, Wetterwald A, Germann M, Cross NA, Rentsch CA, Schalken J, Thalmann GN, Cecchini MG. In vitro propagation and characterization of neoplastic stem/progenitor-like cells from human prostate cancer tissue. Prostate. 2009;69(15):1683–1693. doi: 10.1002/pros.21018. [DOI] [PubMed] [Google Scholar]
- 18.Maitland NJ, Collins AT. Prostate cancer stem cells: a new target for therapy. J Clin Oncol. 2008;26(17):2862–2870. doi: 10.1200/JCO.2007.15.1472. [DOI] [PubMed] [Google Scholar]
- 19.Goldkorn A, Blackburn EH. Assembly of mutant-template telomerase RNA into catalytically active telomerase ribonucleoprotein that can act on telomeres is required for apoptosis and cell cycle arrest in human cancer cells. Cancer Res. 2006;66(11):5763–5771. doi: 10.1158/0008-5472.CAN-05-3782. [DOI] [PubMed] [Google Scholar]
- 20.Shay JW, Wright WE. Telomerase therapeutics for cancer: challenges and new directions. Nat Rev Drug Discov. 2006;5(7):577–584. doi: 10.1038/nrd2081. [DOI] [PubMed] [Google Scholar]
- 21.Xu T, Lu B, Tai YC, Goldkorn A. A Cancer Detection Platform which Measures Telomerase Activity from Live Circulating Tumor Cells Captured on a Microfilter. Cancer Res. 2010 doi: 10.1158/0008-5472.CAN-10-0686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xu T, Xu Y, Liao CP, Lau R, Goldkorn A. Reprogramming murine telomerase rapidly inhibits the growth of mouse cancer cells in vitro and in vivo. Mol Cancer Ther. 2010;9(2):438–449. doi: 10.1158/1535-7163.MCT-09-0682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wright WE, Piatyszek MA, Rainey WE, Byrd W, Shay JW. Telomerase activity in human germline and embryonic tissues and cells. Dev Genet. 1996;18(2):173–179. doi: 10.1002/(SICI)1520-6408(1996)18:2<173::AID-DVG10>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 24.Gu G, Yuan J, Wills M, Kasper S. Prostate cancer cells with stem cell characteristics reconstitute the original human tumor in vivo. Cancer Res. 2007;67(10):4807–4815. doi: 10.1158/0008-5472.CAN-06-4608. [DOI] [PubMed] [Google Scholar]
- 25.Shay JW, Bacchetti S. A survey of telomerase activity in human cancer. Eur J Cancer. 1997;33(5):787–791. doi: 10.1016/S0959-8049(97)00062-2. [DOI] [PubMed] [Google Scholar]
- 26.Stewart SA, Hahn WC, O’Connor BF, Banner EN, Lundberg AS, Modha P, Mizuno H, Brooks MW, Fleming M, Zimonjic DB, Popescu NC, Weinberg RA. Telomerase contributes to tumorigenesis by a telomere length-independent mechanism. Proc Natl Acad Sci U S A. 2002;99(20):12606–12611. doi: 10.1073/pnas.182407599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mimeault M, Batra SK. Recent insights into the molecular mechanisms involved in aging and the malignant transformation of adult stem/progenitor cells and their therapeutic implications. Ageing Res Rev. 2009;8(2):94–112. doi: 10.1016/j.arr.2008.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Garraway IP, Sun W, Tran CP, Perner S, Zhang B, Goldstein AS, Hahm SA, Haider M, Head CS, Reiter RE, Rubin MA, Witte ON. Human prostate sphere-forming cells represent a subset of basal epithelial cells capable of glandular regeneration in vivo. Prostate. 2010;70(5):491–501. doi: 10.1002/pros.21083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Patrawala L, Calhoun-Davis T, Schneider-Broussard R, Tang DG. Hierarchical organization of prostate cancer cells in xenograft tumors: the CD44+alpha2beta1+ cell population is enriched in tumor-initiating cells. Cancer Res. 2007;67(14):6796–6805. doi: 10.1158/0008-5472.CAN-07-0490. [DOI] [PubMed] [Google Scholar]
- 30.Li S, Rosenberg JE, Donjacour AA, Botchkina IL, Hom YK, Cunha GR, Blackburn EH. Rapid inhibition of cancer cell growth induced by lentiviral delivery and expression of mutant-template telomerase RNA and anti-telomerase short-interfering RNA. Cancer Res. 2004;64(14):4833–4840. doi: 10.1158/0008-5472.CAN-04-0953. [DOI] [PubMed] [Google Scholar]
- 31.Marie-Egyptienne DT, Brault ME, Nimmo GA, Londono-Vallejo JA, Autexier C. Growth defects in mouse telomerase RNA-deficient cells expressing a template-mutated mouse telomerase RNA. Cancer Lett. 2009;275(2):266–276. doi: 10.1016/j.canlet.2008.10.027. [DOI] [PubMed] [Google Scholar]
- 32.Wagner W, Ansorge A, Wirkner U, Eckstein V, Schwager C, Blake J, Miesala K, Selig J, Saffrich R, Ansorge W, Ho AD. Molecular evidence for stem cell function of the slow-dividing fraction among human hematopoietic progenitor cells by genome-wide analysis. Blood. 2004;104(3):675–686. doi: 10.1182/blood-2003-10-3423. [DOI] [PubMed] [Google Scholar]
- 33.Herbert BS, Hochreiter AE, Wright WE, Shay JW. Nonradioactive detection of telomerase activity using the telomeric repeat amplification protocol. Nat Protoc. 2006;1(3):1583–1590. doi: 10.1038/nprot.2006.239. [DOI] [PubMed] [Google Scholar]
- 34.Bodnar AG, Ouellette M, Frolkis M, Holt SE, Chiu CP, Morin GB, Harley CB, Shay JW, Lichtsteiner S, Wright WE. Extension of life-span by introduction of telomerase into normal human cells. Science. 1998;279(5349):349–352. doi: 10.1126/science.279.5349.349. [DOI] [PubMed] [Google Scholar]
- 35.Hahn WC, Counter CM, Lundberg AS, Beijersbergen RL, Brooks MW, Weinberg RA. Creation of human tumour cells with defined genetic elements. Nature. 1999;400(6743):464–468. doi: 10.1038/22780. [DOI] [PubMed] [Google Scholar]
- 36.Lee HW, Blasco MA, Gottlieb GJ, Horner JW, 2nd, Greider CW, DePinho RA. Essential role of mouse telomerase in highly proliferative organs. Nature. 1998;392(6676):569–574. doi: 10.1038/33345. [DOI] [PubMed] [Google Scholar]
- 37.Sarin KY, Cheung P, Gilison D, Lee E, Tennen RI, Wang E, Artandi MK, Oro AE, Artandi SE. Conditional telomerase induction causes proliferation of hair follicle stem cells. Nature. 2005;436(7053):1048–1052. doi: 10.1038/nature03836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang X, Kruithof-de Julio M, Economides KD, Walker D, Yu H, Halili MV, Hu YP, Price SM, Abate-Shen C, Shen MM. A luminal epithelial stem cell that is a cell of origin for prostate cancer. Nature. 2009;461(7263):495–500. doi: 10.1038/nature08361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Goldstein AS, Huang J, Guo C, Garraway IP, Witte ON. Identification of a cell of origin for human prostate cancer. Science. 2010;329(5991):568–571. doi: 10.1126/science.1189992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci U S A. 2003;100(7):3983–3988. doi: 10.1073/pnas.0530291100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.O’Brien CA, Kreso A, Dick JE. Cancer stem cells in solid tumors: an overview. Semin Radiat Oncol. 2009;19(2):71–77. doi: 10.1016/j.semradonc.2008.11.001. [DOI] [PubMed] [Google Scholar]
- 42.O’Brien CA, Pollett A, Gallinger S, Dick JE. A human colon cancer cell capable of initiating tumour growth in immunodeficient mice. Nature. 2007;445(7123):106–110. doi: 10.1038/nature05372. [DOI] [PubMed] [Google Scholar]
- 43.Quintana E, Shackleton M, Sabel MS, Fullen DR, Johnson TM, Morrison SJ. Efficient tumour formation by single human melanoma cells. Nature. 2008;456(7222):593–598. doi: 10.1038/nature07567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Singh SK, Hawkins C, Clarke ID, Squire JA, Bayani J, Hide T, Henkelman RM, Cusimano MD, Dirks PB. Identification of human brain tumour initiating cells. Nature. 2004;432(7015):396–401. doi: 10.1038/nature03128. [DOI] [PubMed] [Google Scholar]
- 45.Hao JL, Cozzi PJ, Khatri A, Power CA, Li Y. CD147/EMMPRIN and CD44 are potential therapeutic targets for metastatic prostate cancer. Curr Cancer Drug Targets. 2010;10(3):287–306. doi: 10.2174/156800910791190193. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.