

Abstract
Eukaryotic cells have a powerful RNA decay machinery that plays an important and diverse role in regulating both the quantity and the quality of gene expression. Viral RNAs need to successfully navigate around this cellular machinery to initiate and maintain a highly productive infection. Recent work has shown that viruses have developed a variety of strategies to accomplish this, including inherent RNA shields, hijacking host RNA stability factors, incapacitating the host decay machinery and changing the entire landscape of RNA stability in cells using virally encoded nucleases. In addition to maintaining the stability of viral transcripts, these strategies can also contribute to the regulation and complexity of viral gene expression as well as to viral RNA evolution.
The cellular RNA decay machinery: a major player in determining the level and quality of gene expression
mRNAs in the cell are subjected to a coordinated, highly regulated attack by a rather complex decay machinery to both fine tune and, in many cases, control gene expression [1] (Figure 1 ). The major pathway of mRNA decay involves a two-step process. First, the poly(A) tail is shortened by one or more members of a set of deadenylase enzymes encoded by the cell, in particular Ccr4, Caf1, Pan2/3 or Parn [2]. The body of the mRNA is then subjected to exonucleolytic decay either in the 3′-to-5′ direction by the exosome [3] or in the 5′-to-3′ direction by the enzyme Xrn1 [4] subsequent to decapping by Dcp2 or related enzymes [5]. In addition, several mRNA quality control pathways involving RNA decay exist, including nonsense-mediated decay (NMD) and the related Staufen1-mediated decay (SMD) [6], no-go decay [7] and nonstop decay [8]. Transcripts can be targeted to RNA decay pathways by RNA-binding proteins 9, 10, 11 or via interactions with small RNAs in the RNAi system [12]. Select endoribonucleases, such as RNase L, can also be induced in cells in response to viral infections [13]. Thus, a veritable minefield of decay enzymes must be successfully navigated by viral transcripts when they are produced in the cell.
Figure 1.
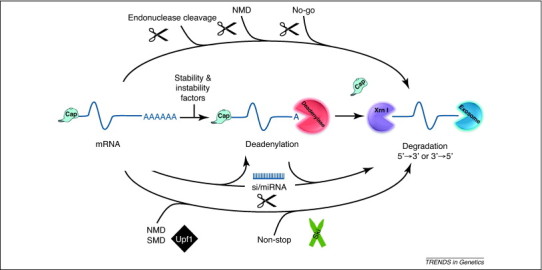
A multitude of ways to degrade an mRNA. The cellular mRNA decay machinery consists of multiple components and strategies to decay transcripts. The ultimate goal of all these strategies is to make the mRNA an effective substrate for exonucleolytic decay. This can be accomplished by a combination of at least four strategies that have been described to date. First, the central portion of the diagram illustrates the major pathway of mRNA decay. RNA turnover is generally initiated by the removal of the poly(A) tail (deadenylation) using a variety of deadenylase enzymes, in particular Ccr4 and Pan2/3. The deadenylated transcript is then degraded by highly processive exonucleases either in the 5′-to-3′ direction (Xrn1) following the removal of the 5′ cap by a decapping enzyme (e.g. Dcp2) or in the 3′-to-5′ direction by the exosome. As indicated in the figure, this process is naturally influenced by a variety of factors that promote transcript stability or instability. Second, mRNA decay can be targeted for direct endonucleolytic cleavage by specific protein factors and proceed to be degraded by exonucleases without deadenylation as a prerequisite (top arrow). These factors include endoribonucleases that naturally target specific mRNAs, the NMD pathway that can target the cleavage of mRNAs near the site of premature termination codons or a specific endonuclease that targets mRNAs that possess stalled ribosomes in the no-go decay pathway. Third, miRNA- and/or siRNA-mediated RNA decay can be initiated through a variety of mechanisms that feed transcripts into the exonucleolytic decay step. These include the direct endonucleolytic cleavage of the mRNA by Argonaut proteins or, in some cases, deadenylation. Finally, as depicted in the bottom arrow, a variety of quality control pathways exist in cells that target exonuclease-mediated RNA decay in the absence of deadenylation or endonucleolytic cleavage. These include NMD in some organisms/transcripts, SMD and the decay of mRNAs that lack a termination codon (nonstop decay). These pathways require specialized decay factors to mark the RNA for decay, including Upf1 (NMD and SMD) and the cytoplasmic Ski proteins for nonstop decay. For additional information, see [1].
The RNA decay machinery is not simply the garbage disposal in the cell but plays a vital role in integrating RNA levels in response to cellular needs and environmental cues. In several cases where it has been examined, regulated mRNA stability accounts for up to 50% of the regulation of gene expression in cells [14]. In addition, the cellular mRNA decay machinery plays a major role in maintaining the quality control of gene expression by rapidly removing unwanted (e.g. overexpressed, mutated, misfolded or not properly processed) RNAs. When a virus starts to flood the cell with its RNAs, surely viral transcripts fall into the ‘unwanted’ category of transcripts that the RNA decay machinery is designed to control. Thus, how viruses successfully avoid or interface with the cellular RNA decay machinery during infection is an important molecular aspect of host–pathogen interactions. Until recently, this was a largely unexplored question. Investigations over the past couple of years, however, have revealed numerous insights into the many diverse strategies viruses use to maintain and regulate the stability of their transcripts. These strategies, and their implications to viral replication and gene expression, are discussed below.
Why do viruses need to protect their RNAs during infection?
Because viral transcripts accumulate to such a high level during infection, it might be assumed that they simply overwhelm the cellular RNA decay machinery rather than find ways to stabilize individual transcripts. Several observations, however, suggest the high levels of viral transcripts might be a direct result of their intrinsic stability rather than reflect the indifference of viruses to the presence of the cellular RNA decay machinery. RNA decay enzymes are versatile and, in many cases, processive. We can find no evidence in the literature to suggest that strong promoters can saturate the capacity of mRNA decay processes. Furthermore, viruses must be susceptible to RNA decay because targeting nucleases to viral RNAs has been shown in several instances to be an effective approach to achieve a significant reduction in viral gene expression 15, 16. Thus, the consequences for failing to effectively protect viral transcripts from decay are low virus yields and an overall inefficient infection.
In addition to the simple notion that mRNA stability is inherently more preferable than is instability, there are at least four specific reasons why viral mRNAs might have evolved a specialized means to ensure their stability in the cell. First, several viral mRNAs lack fundamental RNA stability determinants. Flaviviruses, bunyaviruses and arenaviruses, for example, all encode mRNAs that lack a poly(A) tail. Thus, these viral transcripts could theoretically be recognized as deadenylated degradation intermediates and subjected to decapping/exonucleolytic decay. Second, the cell seems to have evolved several ways to distinguish self from nonself mRNAs. Recent work has demonstrated that differential 2′O cap methylation likely serves as a means of host cell restriction of viral mRNAs [17]. In addition, because it is thought that many cellular transcripts assemble key aspects of their mRNA–protein (mRNP) complexes in the nucleus [18], mRNAs encoded by cytoplasmic viruses probably contain significantly different complements of RNA-binding proteins from cellular mRNAs. Third, cells express proteins that can actively target viral transcripts for destruction. These include at least one host restriction factor, zinc finger antiviral protein, which specifically destabilizes viral mRNAs 19, 20, 21. Finally, the arrangement of the open reading frames in viral mRNAs can serve as a signal for transcript instability. The presence of consecutive open reading frames in retroviral transcripts and perhaps those of other viruses is a signal for NMD [22], and recent evidence indicates that signals for ribosomal frameshifting, which are commonly used in viruses to increase the coding capacity of the genome, are also an mRNA decay signal [23]. In summary, many viral transcripts are cast into the cytoplasm with several strikes already against them. It is perhaps therefore not surprising that viruses have developed specific strategies to stabilize their mRNAs.
A plethora of viral strategies to interfere with the host cell mRNA decay system
Viral strategies to evade regulation by the host RNA decay machinery seem to run the gamut of possibilities. Although this might make viral RNA stabilization a more challenging target for the development of antivirals, it provides a rich source of information on ways for molecules to interface with the cellular RNA decay machinery that should provide new insight into natural modes of ribonuclease targeting, action and regulation. Based on the published findings to date, we suggest four broad strategies for viral interference with host RNA decay: inherent viral defenses, the hijacking of cellular RNA stability factors, the viral-induced shutdown of aspects of the cellular RNA decay machinery and commandeering the RNA decay process itself by viral nucleases.
Inherent mechanisms for viral RNA stability
Inherent viral RNA defense mechanisms against the cellular RNA decay machinery can be divided into three fundamental types (Figure 2a). First is the membranous ‘lifestyle choice’ made by many viruses inside the cell. By surrounding viral replication centers with induced cellular phospholipid membranes, coronaviruses, for example, create a nuclease-resistant microenvironment for their replication [24]. Although this strategy might protect progeny RNA viral genomes from ribonucleases, it is important to keep in mind that viral mRNAs cannot be protected by this strategy if they are to be translated by cytoplasmic ribosomes. Thus, membranous compartments serve as excellent, nuclease-privileged sites of viral replication, but they cannot be relied upon to protect protein-encoding viral mRNAs.
Figure 2.
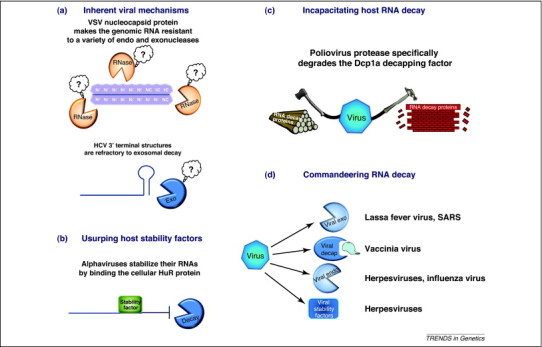
Four viral strategies for evading the host cell RNA decay machinery. Viruses have developed a variety of interesting approaches to stabilize their transcripts during infection. (a) Inherent resistance to decay. Viral RNA coated with nucleocapsid proteins, as observed with vesicular stomatitis virus, are resistant to nuclease degradation. Likewise, secondary structures at the termini of viral RNAs, for example the stem loop present at the precise 3′ terminus of HCV, are refractory to exonuclease degradation. (b) Usurping cellular stability factors. Viral RNAs may bind proteins that normally stabilize cellular transcripts, making the transcripts difficult for the cellular decay machinery to destroy. This has been demonstrated for Sindbis and other alphavirus transcripts that bind to the cellular HuR protein. (c) Viruses sometimes target selected enzymes and factors of the cellular RNA decay machinery for destruction. A clear example of this is the poliovirus-encoded protease that targets the cellular decapping factor Dcp1a for proteolytic cleavage. (d) Viruses can encode a variety of their own decay enzymes and regulatory factors that effectively change the entire landscape of RNA decay in the infected cell. As noted in the figure, these include exo- and endonucleases, decapping enzymes and factors that have a major impact on RNA decay such as the KSHV SOX protein.
Viruses might encode their own cis-acting RNA elements to protect individual transcripts from degradation by cellular enzymes. The clearest example of this is the U-rich expression and nuclear retention element at the 3′ end of the Kaposi's sarcoma-associated herpesvirus (KSHV) long noncoding polyadenylated nuclear RNA [25]. This element base pairs with the poly(A) tail of the transcript, forming an inhibitory structure for ribonucleases [26]. Another common RNA structure that provides protection from a class of RNA decay enzymes is the stable terminal stem loop found at the 3′ end of all viruses that lack a poly(A) tail [27]. These structures prevent exosome-associated exoribonuclease from gaining an effective grip on viral RNAs, thereby preserving the integrity of the 3′ end and blocking the 3′-to-5′ degradation of the transcript.
Finally, some viruses encode proteins that will specifically protect viral RNAs from degradation during infection. A major example of this is the nucleocapsid protein of negative sense RNA viruses such as vesicular stomatitis virus that assembles on the viral genome and antigenomic RNAs cotranscriptionally and renders the RNA insensitive to RNases [28]. Another example of this strategy is the KSHV ORF57 protein, which recent evidence suggests binds to a specific viral element and protects intronless transcripts from degradation in the nucleus [29]. In summary, these inherent defense mechanisms seem to be used by numerous viruses as a general way of protecting noncoding RNAs or RNAs that are not destined to be translated from degradation by the cellular RNA decay machinery. This assists in the accumulation of viral templates for replication, transcription and packaging. Viral mRNAs, however, do not seem to employ this strategy for stabilization because it might be largely incompatible with efficient translation by cellular ribosomes.
Hijacking of host cell RNA stability factors
One attractive strategy for viruses to confound host cell RNA decay enzymes is for their mRNAs to borrow or usurp cellular factors whose natural function is to stabilize cellular transcripts (Figure 2b). The two most widely studied cellular RNA stability factors are HuR and poly(C)-binding protein 2 (PCBP2). The ubiquitously expressed HuR protein has been demonstrated to bind and stabilize well over 50 independent mRNAs in the cell by interacting with U-rich or AU-rich elements [30]. Alphaviruses such as Sindbis virus have been shown to contain high-affinity U-rich HuR protein-binding sites in their 3′ untranslated regions (UTRs) and to use HuR protein to stabilize viral mRNAs and promote a productive infection [31]. HuR has also been shown to interact with hepatitis C virus (HCV) [32] and human papillomavirus [33] transcripts and might serve a similar role in these viral infections. HuR protein also seems to contribute to foamy viral RNA export from the nucleus [34] as well as in human papillomavirus-induced cellular transformation [35]. PCBP2, also referred to as αCP or hnRNP E, plays a major role in stabilizing α-globin and probably other long-lived mRNAs in red blood cells 36, 37. A variety of picornaviruses (e.g. poliovirus, coxsackievirus and hepatitis A virus) bind PCBP2 and use protein to promote optimal viral translation and gene expression 38, 39.
In addition to these two well-studied regulators of RNA stability, additional cellular factors are usurped by certain viruses to maintain the integrity of their transcripts. Insulin-like growth factor mRNA-binding protein has recently been shown to interact with murine leukemia virus genomic RNAs and promote their stability and packaging [40]. Another retrovirus, Rous sarcoma virus, contains an RNA stability element that interacts with unknown cellular factors to allow viral mRNAs to avoid destruction by the NMD pathway and promote proper translation [41]. Many segmented RNA viruses remove the caps along with a small portion of the 5′ UTR from cellular transcripts and incorporate them into their own transcripts to promote the stability and translation of viral mRNAs [42]. Finally, viruses can also target cellular kinases that regulate RNA stability in the cell. For example, the double-stranded RNA (dsRNA)-dependent protein kinase PKR, which regulates the stability of interferon α/β mRNAs [43], is targeted by several viruses including adenovirus [44]. Thus, with a growing list of targeted cellular factors, many viruses seem to use this strategy to protect their mRNAs from the cellular RNA decay machinery. Furthermore, because our understanding of RNA stability factors is still in its infancy, the study of viral interactions with cellular RNA-binding proteins might also yield insights into novel roles for proteins in regulating RNA stability in the cell.
Shutting down host cell RNA decay enzymes
Instead of subtly trying to protect their own transcripts, viruses can adopt the strategy of simply disarming aspects of the cellular RNA decay machinery to render it ineffective (Figure 2c). Poliovirus, for example, has recently been shown to target Dcp1a – a major cofactor in decapping and the 5′-to-3′ mRNA decay pathway [45]. This results in a disruption of P bodies – cytoplasmic sites of accumulation of many RNA decay factors – and presumably disrupts the major aspects of the cellular RNA decay machinery [46]. Poliovirus proteases also target and degrade a variety of cellular proteins, including the cytoplasmic poly(A) binding protein PABPC1 [47], which is a cofactor that stimulates the Pan2/3 deadenylase. The targeting of other cellular RNA decay factors by picornaviruses remains to be elucidated, but it should be an interesting area for future research. Small miRNAs involved in the RNAi pathway can also target viral transcripts for translational repression and degradation [48]. Interestingly, small RNAs encoded by herpesvirus saimiri have recently been shown to bind and downregulate selected miRNAs, disarming this major entry point for RNAs into decay pathways [49]. Specialized ribonucleases can also be targeted by viruses for inactivation. Human cytomegalovirus infection causes the inhibition of RNase L [50]. In addition, many of the cystic leukoencephalopathy symptoms caused by congenital human cytomegalovirus infection are curiously similar to those caused by mutations in a cellular RNase T2 enzyme 51, 52. Finally, enzymes such as adenosine deaminase acting on RNA (ADAR) that can modify viral RNAs and cause transcript instability are targeted by an array of viruses [53]. Thus, simply directly attacking the source of the problem and destroying or inactivating RNA decay factors is also an effective approach to maintaining viral RNA stability.
Commandeering of the RNA decay process by viruses
Taking a cue from the sports cliché that sometimes the best defense is a good offense, another major viral strategy to interface with the cellular RNA decay machinery is to overwhelm it with additional nucleases that effectively reset the playing field (Figure 2d). This strategy is rather widespread among viruses, and several specific examples are outlined below.
Many DNA viruses encode nucleases or RNA decay factors that alter general RNA decay kinetics/mechanisms inside an infected cell. The KSHV SOX protein, interestingly, in addition to possessing RNase activity in vitro [54], causes the hyperadenylation of cellular mRNAs, the nuclear accumulation of the normally cytoplasmic PABPC1 and an increased rate of turnover of cellular mRNAs 55, 56. Thus, it is probable that the SOX protein must interface with the host machinery to achieve widespread mRNA turnover in cells. Other herpesviruses (e.g. herpes simplex virus) encode a powerful, nonselective ribonuclease as part of the viral tegument layer, which results in a massive degradation of cellular transcripts and helps generate the patterns of early- and late-stage gene expression by influencing the half-lives of viral transcripts and translation [57]. Epstein Barr virus, another member of the Herpesviridae, also encodes a Mn2+-dependent RNase in the BGLF5 protein that might contribute to host cell shutoff [58]. Finally, vaccinia virus encodes its own mRNA decapping enzymes (D9 and D10) 59, 60 that recognize the 5′ cap structure in a noncanonical fashion [61]. The large cytoplasmic poxviruses are the only DNA viruses shown to date to encode a decapping activity. Interestingly, in most cases, precisely how viral transcripts escape their own effectors of RNA degradation is not clear.
Perhaps somewhat surprisingly, RNA viruses also encode their own ribonucleases or factors that can stimulate RNA decay. Cap-stealing endonucleases [62] encoded by many segmented RNA viruses, for example, might selectively target cellular mRNAs for decay by 5′-to-3′ exonucleases and this dramatically changes the regulation of mRNA stability in infected cells. Segmented RNA viruses use the capped oligomers generated by endonuclease cleavage to initiate the transcription of their own mRNAs. The gag protein of the persistent dsRNA L-A virus of Saccharomyces cerevisiae possesses a potent decapping activity that exposes the 5′ end of cellular mRNAs to exonucleolytic degradation [63]. L-A virus transcripts are immune to this enzyme because they contain a simple diphosphate at their 5′ ends rather than a conventional cap [64]. The SARS coronavirus Nsp1 protein induces the degradation of cellular mRNAs by an apparently novel mechanism. Nsp1 protein associates with 40S ribosomes and induces a modification of some sort on the 5′ UTR region of mRNAs that leads to their degradation [65]. Finally, the nucleocapsid protein of Lassa fever virus, a member of the Arenaviridae, has recently been shown to possess 3′-to-5′ exonuclease activity 66, 67. This exonuclease is essential for the virus to interfere with aspects of the interferon and other innate immune responses. The only other RNA virus known to date to encode an exonuclease is the Nsp14 protein of the SARS coronaviruses [68]. This protein has a DEDDh motif similar to that of the Lassa fever virus nucleocapsid protein and thereby also probably attacks RNAs from the 3′ end using a catalytic mechanism involving two metal ions [69].
Not just a game of ‘keep away’ – additional benefits from interfacing with the cellular RNA decay machinery
Although stabilizing viral transcripts and/or destroying host cell mRNAs to enhance viral gene expression are obvious benefits of a successful interface with the cellular RNA decay machinery, could viruses reap additional benefits? Several recent pieces of evidence do indeed indicate that this could be the case. First, instead of using an internal promoter, one class of viruses uses the cellular RNA decay machinery to generate a small RNA from viral genomic transcripts (Figure 3a). All insect-borne flaviviruses use the cellular Xrn1 5′-to-3′ exonuclease to generate a small flavivirus RNA (sfRNA) from the 3′ UTR of the virus [70]. This small RNA is generated because of a set of pseudoknot structures capable of stalling the exonuclease 71, 72. The small sfRNA (or the generation thereof) has been determined to be important for viral replication and cytopathology in the case of a West Nile Virus infection [70]. Second, instead of doing everything they can to avoid the RNAi machinery, at least one virus has figured out a way to use it to benefit its replication (Figure 3b). HCV, another member of the Flaviviridae, uses the abundant liver miRNA-122 to increase the accumulation of its mRNA during infection. miRNA-122 binds to two sites in the 5′ UTR of HCV and forms a unique structure that seems to be required for efficient viral replication 73, 74, 75. Third, viruses might use nucleases to increase the rate of viral RNA evolution (Figure 3c). Mutations in the Nsp14 exonuclease of independent coronaviruses result in a 15–21-fold decrease in replication fidelity 76, 77. Thus, the presence of this exonuclease can clearly influence viral RNA mutation rates and the generation of quasispecies. Fourth, at least one negative sense RNA virus (viruses whose RNA genomes are not used as a message) has determined a way to use differential RNA stability to help regulate its gene expression post-transcriptionally (Figure 3d). Intron-containing Borna virus mRNAs contain 50–100 bp long instability elements in their introns that have been shown to influence levels of RNA accumulation [78]. This would provide a level of regulation of gene expression for this negative sense RNA virus in addition to the transcriptional gradient afforded by classical ‘stop/start’ transcription models [79]. Finally, several viruses or virus-like particles have determined how to use aspects of the RNA decay machinery in novel ways. Retrotransposon Ty1 Gag protein, for example, has been shown to colocalize with the decapping factors Dcp1 and Dcp2 as well as with the Xrn1 exonuclease and to use these 5′-to-3′ decay pathway factors and presumably P bodies that contain these factors to promote effective retrotransposition and RNA packaging [80]. The NMD factor Upf1 has been shown to be part of the HIV mRNP complex and to positively influence gene expression from viral transcripts [81]. Lastly, LSm proteins, Rck/p54 (DDX6) and PatL1 (several auxiliary factors involved in RNA decay and cytoplasmic mRNA fate 82, 83, 84) have been shown to play a role in the replication and gene expression of Brome mosaic virus [85], HCV 86, 87 and perhaps the stabilization of poxvirus mRNAs that contain a unique poly(A) ‘head’ [88]. Thus, many viruses seem to have found ways to turn these probable foes of a productive infection into beneficial friends.
Figure 3.
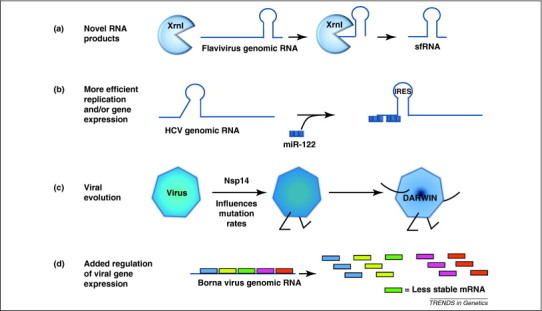
Novel uses of RNA decay factors by viruses. In addition to simply stabilizing or destabilizing viral RNAs, the cellular mRNA decay machinery and associated factors are also used by selected viruses to perform a variety of interesting roles in gene expression and replication. (a) Flaviviruses such as West Nile Virus use the cellular exoribonuclease Xrn1 to generate decay intermediates that result in novel subgenomic sfRNA transcripts, which have been shown to influence cytopathology. (b) HCV binds miRNA-122, which is abundant in liver cells, to generate/stabilize the structural elements at the 5′ end of its genomic RNA that are required for efficient viral gene expression/replication. (c) The Nsp14 exonuclease encoded by coronaviruses has been demonstrated to play a role in viral evolution and the generation of quasispecies diversity. (d) Selected transcripts encoded by Borna viruses contain instability elements that reduce the level of expression of specific mRNAs in a post-transcriptional fashion. This affords an additional level of control of gene expression for this RNA virus.
Concluding remarks
It has become clear in recent years that viruses have determined a variety of ways to successfully interact with the cellular RNA decay machinery to stabilize viral transcripts and promote productive infections. Given that viral interplay with the RNA decay machinery is still a rather understudied area, numerous fundamental questions remain to be addressed. Does every virus require a strategy to successfully interface with the RNA decay machinery? Although one might assume that the simple answer to this question is ‘of course’, this has not been formally demonstrated for many viruses. If indeed this is a requirement, disrupting the virus–RNA decay machinery interface in a manner to favor viral RNA decay represents a novel and attractive avenue for the development of a new class of antivirals that might have a broad spectrum of efficacy. The concept of using the cellular RNA decay machinery as an additional way of controlling viral gene expression is another attractive area for investigation, particularly because cells rely so heavy on RNA stability for regulation. Along these lines, do other negative sense RNA viruses in addition to the intron-containing Borna virus use the cellular RNA decay machinery to provide an unsuspected post-transcriptional level of regulation of gene expression? If so, models for gene expression for negative sense RNA viruses might need to be significantly expanded beyond the classical ‘stop/start’ transcription mechanism. Another area of investigation that might prove fruitful is the hunt for additional examples of using cellular RNA decay enzymes to generate novel viral noncoding transcripts that could expand the effective gene expression capacity of viral RNA genomes. The best current example of this is the flavivirus sfRNA that is generated by Xrn1. Because most members of the Flaviviridae utilize the cellular Xrn1 decay enzyme to generate a small RNA, why do pestiviruses and hepaciviruses fail to do so as well? Could miRNA-122 interactions at the 5′ ends of HCV RNAs be performing a similar function to block Xrn1 activity on viral genomes rather than relying on related structures in the 3′ UTR as with other flaviviruses? In summary, investigating the interplay of viral RNAs with the cellular decay machinery should yield plenty of interesting surprises, new concepts and broaden our molecular understanding of virus–host interactions for years to come.
Acknowledgments
Our apologies to colleagues whose work has not been cited because of space constraints. We wish to thank members of the Wilusz lab for critical comments. Virology and RNA stability research in the Wilusz lab is supported by grants from the National Institutes of Health (R01AI063434, R01GM072481 and U54 AI-065357).
References
- 1.Garneau N.L. The highways and byways of mRNA decay. Nat. Rev. Mol. Cell Biol. 2007;8:113–126. doi: 10.1038/nrm2104. [DOI] [PubMed] [Google Scholar]
- 2.Goldstrohm A.C., Wickens M. Multifunctional deadenylase complexes diversify mRNA control. Nat. Rev. Mol. Cell Biol. 2008;9:337–344. doi: 10.1038/nrm2370. [DOI] [PubMed] [Google Scholar]
- 3.Houseley J. RNA-quality control by the exosome. Nat. Rev. Mol. Cell Biol. 2006;9:529–539. doi: 10.1038/nrm1964. [DOI] [PubMed] [Google Scholar]
- 4.Chang J.H. Structural and biochemical studies of the 5’→3′ exoribonuclease Xrn1. Nat. Struct. Mol. Biol. 2011;18:270–276. doi: 10.1038/nsmb.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Song M.G. Multiple mRNA decapping enzymes in mammalian cells. Mol. Cell. 2010;40:423–432. doi: 10.1016/j.molcel.2010.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Maquat L.E., Gong C. Gene expression networks: competing mRNA decay pathways in mammalian cells. Biochem. Soc. Trans. 2009;37:1287–1292. doi: 10.1042/BST0371287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.van den Elzen A.M. Dissection of Dom34-Hbs1 reveals independent functions in two RNA quality control pathways. Nat. Struct. Mol. Biol. 2010;17:1446–1452. doi: 10.1038/nsmb.1963. [DOI] [PubMed] [Google Scholar]
- 8.Schaeffer D., van Hoof A. Different nuclease requirements for exosome-mediated degradation of normal and nonstop mRNAs. Proc. Natl. Acad. Sci. U.S.A. 2011;108:2366–2371. doi: 10.1073/pnas.1013180108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Clement S.L. Phosphorylation of tristetraprolin by MK2 impairs AU-rich element mRNA decay by preventing deadenylase recruitment. Mol. Cell. Biol. 2011;31:256–266. doi: 10.1128/MCB.00717-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zucconi B.E. Alternatively expressed domains of AU-rich element RNA-binding protein 1 (AUF1) regulate RNA-binding affinity, RNA-induced protein oligomerization, and the local conformation of bound RNA ligands. J. Biol. Chem. 2010;285:39127–39139. doi: 10.1074/jbc.M110.180182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gherzi R. The role of KSRP in mRNA decay and microRNA precursor maturation. Wiley Interdisciplinary Rev.: RNA. 2010;1:230–239. doi: 10.1002/wrna.2. [DOI] [PubMed] [Google Scholar]
- 12.Ding S.W. RNA-based antiviral immunity. Nat. Rev. Immunol. 2010;10:632–644. doi: 10.1038/nri2824. [DOI] [PubMed] [Google Scholar]
- 13.Tomecki R., Dziembowski A. Novel endoribonucleases as central players in various pathways of eukaryotic RNA metabolism. RNA. 2010;16:1692–1724. doi: 10.1261/rna.2237610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cheadle C. Control of gene expression during T cell activation: alternate regulation of mRNA transcription and mRNA stability. BMC Genomics. 2005;6:75. doi: 10.1186/1471-2164-6-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bai Y. Salmonella-mediated delivery of RNase P-based ribozymes for inhibition of viral gene expression and replication in human cells. Proc. Natl. Acad. Sci. U.S.A. 2010;107:7269–7274. doi: 10.1073/pnas.0912813107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chou C.F. Tethering KSRP, a decay-promoting AU-rich element-binding protein, to mRNAs elicits mRNA decay. Mol. Cell. Biol. 2006;26:3695–3706. doi: 10.1128/MCB.26.10.3695-3706.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Daffis S. 2’-O methylation of the viral mRNA cap evades host restriction by IFIT family members. Nature. 2010;2468:452–456. doi: 10.1038/nature09489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Trcek T., Singer R.H. The cytoplasmic fate of an mRNP is determined cotranscriptionally: exception or rule? Genes Dev. 2010;24:1827–1831. doi: 10.1101/gad.1972810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Law L.M. Identification of a dominant negative inhibitor of human zinc finger antiviral protein reveals a functional endogenous pool and critical homotypic interactions. J. Virol. 2010;84:4504–4512. doi: 10.1128/JVI.02018-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang N. Viral induction of the zinc finger antiviral protein is IRF3-dependent but NF-kappaB-independent. J. Biol. Chem. 2010;285:6080–6090. doi: 10.1074/jbc.M109.054486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gao G. Inhibition of retroviral RNA production by ZAP, a CCCH-type zinc finger protein. Science. 2002;297:1703–1706. doi: 10.1126/science.1074276. [DOI] [PubMed] [Google Scholar]
- 22.Lee E.G. A premature termination codon mutation at the C terminus of foamy virus Gag downregulates the levels of spliced pol mRNA. J. Virol. 2008;82:1656–1664. doi: 10.1128/JVI.00990-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Belew A.T. Endogenous ribosomal frameshift signals operate as mRNA destabilizing elements through at least two molecular pathways in yeast. Nucleic Acids Res. 2011;39:2799–2808. doi: 10.1093/nar/gkq1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.van Hemert M.J. SARS-coronavirus replication/transcription complexes are membrane-protected and need a host factor for activity in vitro. PLoS Pathog. 2008;4:e1000054. doi: 10.1371/journal.ppat.1000054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Conrad N.K. Mutational analysis of a viral RNA element that counteracts rapid RNA decay by interaction with the polyadenylate tail. Proc. Natl. Acad. Sci. U.S.A. 2007;104:10412–10417. doi: 10.1073/pnas.0704187104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mitton-Fry R.M. Poly(A) tail recognition by a viral RNA element through assembly of a triple helix. Science. 2010;330:1244–1247. doi: 10.1126/science.1195858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ford L.P., Wilusz J. 3′-Terminal RNA structures and poly(U) tracts inhibit initiation by a 3′-->5′ exonuclease in vitro. Nucleic Acids Res. 1999;27:1159–1167. doi: 10.1093/nar/27.4.1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sarkar A. Structural and functional properties of the vesicular stomatitis virus nucleoprotein-RNA complex as revealed by proteolytic digestion. Virology. 2010;401:61–69. doi: 10.1016/j.virol.2010.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sahin B.B. Kaposi's sarcoma-associated herpesvirus ORF57 protein binds and protects a nuclear noncoding RNA from cellular RNA decay pathways. PLoS Pathog. 2010;6:e1000799. doi: 10.1371/journal.ppat.1000799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Abdelmohsen K., Gorospe M. Postranscritpional regulation of cancer traits by HuR. Wiley Interdisciplinary Rev.: RNA. 2010;1:214–229. doi: 10.1002/wrna.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sokoloski K.J. Sindbis virus usurps the cellular HuR protein to stabilize its transcripts and promote productive infections in mammalian and mosquito cells. Cell Host Microbe. 2010;8:196–207. doi: 10.1016/j.chom.2010.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Korf M. Inhibition of hepatitis C virus translation and subgenomic replication by siRNAs directed against highly conserved HCV sequence and cellular HCV cofactors. J. Hepatol. 2005;43:225–234. doi: 10.1016/j.jhep.2005.02.046. [DOI] [PubMed] [Google Scholar]
- 33.Cumming S.A. The RNA stability regulator HuR regulates L1 protein expression in vivo in differentiating cervical epithelial cells. Virology. 2009;383:142–149. doi: 10.1016/j.virol.2008.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bodem J. Foamy virus nuclear RNA export is distinct from that of other retroviruses. J. Virol. 2011;85:2333–2341. doi: 10.1128/JVI.01518-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kuroshima T. Viral-mediated stabilization of AU-rich element containing mRNA contributes to cell transformation. Oncogene. 2011 doi: 10.1038/onc.2011.14. [DOI] [PubMed] [Google Scholar]
- 36.Ji X. The 3′ untranslated region complex involved in stabilization of human alpha-globin mRNA assembles in the nucleus and serves an independent role as a splice enhancer. Mol. Cell. Biol. 2007;27:3290–3302. doi: 10.1128/MCB.02289-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Waggoner S.A. Depletion of the poly(C)-binding proteins alphaCP1 and alphaCP2 from K562 cells leads to p53-independent induction of cyclin-dependent kinase inhibitor (CDKN1A) and G1 arrest. J. Biol. Chem. 2009;284:9039–9049. doi: 10.1074/jbc.M806986200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sean P. Altered interactions between stem-loop IV within the 5’ noncoding region of coxsackievirus RNA and poly(rC) binding protein 2: effects on IRES-mediated translation and viral infectivity. Virology. 2009;389:45–58. doi: 10.1016/j.virol.2009.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Spear A. Protein-RNA tethering: the role of poly(C) binding protein 2 in poliovirus RNA replication. Virology. 2008;374:280–291. doi: 10.1016/j.virol.2007.12.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mai Y., Gao G. Expression of IMP1 enhances production of murine leukemia virus vector by facilitating viral genomic RNA packaging. PLoS ONE. 2010;5:e15881. doi: 10.1371/journal.pone.0015881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Withers J.B., Beemon K.L. Structural features in the Rous sarcoma virus RNA stability element are necessary for sensing the correct termination codon. Retrovirology. 2010;7:65. doi: 10.1186/1742-4690-7-65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Reguera J. Bunyaviridae RNA polymerases (L-protein) have an N-terminal, influenza-like endonuclease domain, essential for viral cap-dependent transcription. PLoS Pathog. 2010;6:e1001101. doi: 10.1371/journal.ppat.1001101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schulz O. Protein kinase R contributes to immunity against specific viruses by regulating interferon mRNA integrity. Cell Host Microbe. 2010;7:354–361. doi: 10.1016/j.chom.2010.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Launer-Felty K. Magnesium-dependent interaction of PKR with adenovirus VAI. J. Mol. Biol. 2010;402:638–644. doi: 10.1016/j.jmb.2010.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Borja M.S. Dcp1 links coactivators of mRNA decapping to Dcp2 by proline recognition. RNA. 2011;17:278–290. doi: 10.1261/rna.2382011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dougherty J.D. Poliovirus-mediated disruption of cytoplasmic processing bodies. J. Virol. 2011;85:64–75. doi: 10.1128/JVI.01657-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bonderoff J.M. Cleavage of poly(A)-binding protein by poliovirus 3C proteinase inhibits viral internal ribosome entry site-mediated translation. J. Virol. 2008;82:9389–9399. doi: 10.1128/JVI.00006-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Santhakumar D. Combined agonist-antagonist genome-wide functional screening identifies broadly active antiviral microRNAs. Proc. Natl. Acad. Sci. U.S.A. 2010;2107:13830–13835. doi: 10.1073/pnas.1008861107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cazalla D. Down-regulation of a host microRNA by a Herpesvirus saimiri noncoding RNA. Science. 2010;328:1563–1566. doi: 10.1126/science.1187197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Child S.J. Evasion of cellular antiviral responses by human cytomegalovirus TRS1 and IRS1. J. Virol. 2004;78:197–205. doi: 10.1128/JVI.78.1.197-205.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Luhtala N., Parker R. T2 Family ribonucleases: ancient enzymes with diverse roles. Trends Biochem. Sci. 2010;35:253–259. doi: 10.1016/j.tibs.2010.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Henneke M. RNASET2-deficient cystic leukoencephalopathy resembles congenital cytomegalovirus brain infection. Nat. Genet. 2009;41:773–775. doi: 10.1038/ng.398. [DOI] [PubMed] [Google Scholar]
- 53.George C.X. Adenosine deaminases acting on RNA, RNA editing, and interferon action. J. Interferon Cytokine Res. 2011;31:99–117. doi: 10.1089/jir.2010.0097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bagnéris C. Crystal structure of a KSHV-SOX-DNA complex: insights into the molecular mechanisms underlying DNase activity and host shutoff. Nucleic Acids Res. 2011 doi: 10.1093/nar/gkr111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lee Y.J., Glaunsinger B.A. Aberrant herpesvirus-induced polyadenylation correlates with cellular messenger RNA destruction. PLoS Biol. 2009;7:e1000107. doi: 10.1371/journal.pbio.1000107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kumar G.R., Glaunsinger B.A. Nuclear import of cytoplasmic poly(A) binding protein restricts gene expression via hyperadenylation and nuclear retention of mRNA. Mol. Cell. Biol. 2010;30:4996–5008. doi: 10.1128/MCB.00600-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Saffran H.A. Evidence for translational regulation by the herpes simplex virus virion host shutoff protein. J. Virol. 2010;84:6041–6049. doi: 10.1128/JVI.01819-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Buisson M. A bridge crosses the active-site canyon of the Epstein-Barr virus nuclease with DNase and RNase activities. J. Mol. Biol. 2009;391:717–728. doi: 10.1016/j.jmb.2009.06.034. [DOI] [PubMed] [Google Scholar]
- 59.Parrish S. Vaccinia virus D10 protein has mRNA decapping activity, providing a mechanism for control of host and viral gene expression. Proc. Natl. Acad. Sci. U.S.A. 2007;104:2139–2144. doi: 10.1073/pnas.0611685104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Parrish S., Moss B. Characterization of a second vaccinia virus mRNA-decapping enzyme conserved in poxviruses. J. Virol. 2007;81:12973–12978. doi: 10.1128/JVI.01668-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Soulière M.F. Insights into the molecular determinants involved in cap recognition by the vaccinia virus D10 decapping enzyme. Nucleic Acids Res. 2010;38:7599–7610. doi: 10.1093/nar/gkq628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Morin B. The N-terminal domain of the arenavirus L protein is an RNA endonuclease essential in mRNA transcription. PLoS Pathog. 2010;6:e1001038. doi: 10.1371/journal.ppat.1001038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tang J. The structural basis of recognition and removal of cellular mRNA 7-methyl G ‘caps’ by a viral capsid protein: a unique viral response to host defense. J. Mol. Recognit. 2005;18:158–168. doi: 10.1002/jmr.724. [DOI] [PubMed] [Google Scholar]
- 64.Fujimura T., Esteban R. Yeast double-stranded RNA virus L-A deliberately synthesizes RNA transcripts with 5’-diphosphate. J. Biol. Chem. 2010;285:22911–22918. doi: 10.1074/jbc.M110.138982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kamitani W. A two-pronged strategy to suppress host protein synthesis by SARS coronavirus Nsp1 protein. Nat. Struct. Mol. Biol. 2009;16:1134–1140. doi: 10.1038/nsmb.1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hastie K.M. Structure of the Lassa virus nucleoprotein reveals a dsRNA-specific 3’ to 5’ exonuclease activity essential for immune suppression. Proc. Natl. Acad. Sci. U.S.A. 2011;108:2396–2401. doi: 10.1073/pnas.1016404108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Qi L. Cap binding and immune evasion revealed by Lassa nucleoprotein structure. Nature. 2010;468:779–783. doi: 10.1038/nature09605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Minskaia E. Discovery of an RNA virus 3’->5’ exoribonuclease that is critically involved in coronavirus RNA synthesis. Proc. Natl. Acad. Sci. U.S.A. 2006;103:5108–5113. doi: 10.1073/pnas.0508200103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Steitz T.A., Steitz J.A. A general two-metal-ion mechanism for catalytic RNA. Proc. Natl. Acad. Sci. U.S.A. 1993;90:6498–6502. doi: 10.1073/pnas.90.14.6498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Pijlman G.P. A highly structured, nuclease-resistant, noncoding RNA produced by flaviviruses is required for pathogenicity. Cell Host Microbe. 2008;4:579–591. doi: 10.1016/j.chom.2008.10.007. [DOI] [PubMed] [Google Scholar]
- 71.Funk A. RNA structures required for production of subgenomic flavivirus RNA. J. Virol. 2010;84:11407–11417. doi: 10.1128/JVI.01159-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Silva P.A. An RNA pseudoknot is required for production of yellow fever virus subgenomic RNA by the host nuclease XRN1. J. Virol. 2010;284:11395–11406. doi: 10.1128/JVI.01047-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Jopling C.L. Position-dependent function for a tandem microRNA miR-122-binding site located in the hepatitis C virus RNA genome. Cell Host Microbe. 2008;4:77–85. doi: 10.1016/j.chom.2008.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lanford R.E. Therapeutic silencing of microRNA-122 in primates with chronic hepatitis C virus infection. Science. 2010;327:198–201. doi: 10.1126/science.1178178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Machlin E.S. Masking the 5’ terminal nucleotides of the hepatitis C virus genome by an unconventional microRNA-target RNA complex. Proc. Natl. Acad. Sci. U.S.A. 2011;108:3193–3198. doi: 10.1073/pnas.1012464108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Eckerle L.D. Infidelity of SARS-CoV Nsp14-exonuclease mutant virus replication is revealed by complete genome sequencing. PLoS Pathog. 2010;6:e1000896. doi: 10.1371/journal.ppat.1000896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Eckerle L.D. High fidelity of murine hepatitis virus replication is decreased in nsp14 exoribonuclease mutants. J. Virol. 2007;81:12135–12144. doi: 10.1128/JVI.01296-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Siemetzki U. Identification of RNA instability elements in Borna disease virus. Virus Res. 2009;144:27–34. doi: 10.1016/j.virusres.2009.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Whelan S.P. Transcription and replication of nonsegmented negative-strand RNA viruses. Curr. Top. Microbiol. Immunol. 2004;283:61–119. doi: 10.1007/978-3-662-06099-5_3. [DOI] [PubMed] [Google Scholar]
- 80.Dutko J.A. 5′ to 3′ mRNA decay factors colocalize with Ty1 gag and human APOBEC3G and promote Ty1 retrotransposition. J. Virol. 2010;84:5052–5066. doi: 10.1128/JVI.02477-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ajamian L. Unexpected roles for UPF1 in HIV-1 RNA metabolism and translation. RNA. 2008;14:914–927. doi: 10.1261/rna.829208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wilusz C.J., Wilusz J. Eukaryotic Lsm proteins: lessons from bacteria. Nat. Struct. Mol. Biol. 2005;12:1031–1036. doi: 10.1038/nsmb1037. [DOI] [PubMed] [Google Scholar]
- 83.Kedersha N., Anderson P. Mammalian stress granules and processing bodies. Methods Enzymol. 2007;431:61–81. doi: 10.1016/S0076-6879(07)31005-7. [DOI] [PubMed] [Google Scholar]
- 84.Totaro A. The human Pat1b protein: a novel mRNA deadenylation factor identified by a new immunoprecipitation technique. Nucleic Acids Res. 2011;39:635–647. doi: 10.1093/nar/gkq797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Galão R.P. LSm1-7 complexes bind to specific sites in viral RNA genomes and regulate their translation and replication. RNA. 2010;16:817–827. doi: 10.1261/rna.1712910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Scheller N. Translation and replication of hepatitis C virus genomic RNA depends on ancient cellular proteins that control mRNA fates. Proc. Natl. Acad. Sci. U.S.A. 2009;106:13517–13522. doi: 10.1073/pnas.0906413106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Jangra R.K. DDX6 (Rck/p54) is required for efficient hepatitis C virus replication but not for internal ribosome entry site-directed translation. J. Virol. 2010;84:6810–6824. doi: 10.1128/JVI.00397-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Bergman N. Lsm proteins bind and stabilize RNAs containing 5’ poly(A) tracts. Nat. Struct. Mol. Biol. 2007;14:824–831. doi: 10.1038/nsmb1287. [DOI] [PubMed] [Google Scholar]