

Abstract
Gene expression is stochastic, and noise that arises from the stochastic nature of biochemical reactions propagates through active regulatory links. Thus, correlations in gene-expression noise can provide information about regulatory links. We present what to our knowledge is a new approach to measure and interpret such correlated fluctuations at the level of single microcolonies, which derive from single cells. We demonstrated this approach mathematically using stochastic modeling, and applied it to experimental time-lapse fluorescence microscopy data. Specifically, we investigated the relationships among LuxO, LuxR, and the small regulatory RNA qrr4 in the model quorum-sensing bacterium Vibrio harveyi. Our results show that LuxR positively regulates the qrr4 promoter. Under our conditions, we find that qrr regulation weakly depends on total LuxO levels and that LuxO autorepression is saturated. We also find evidence that the fluctuations in LuxO levels are dominated by intrinsic noise. We furthermore propose LuxO and LuxR interact at all autoinducer levels via an unknown mechanism. Of importance, our new method of evaluating correlations at the microcolony level is unaffected by partition noise at cell division. Moreover, the method is first-order accurate and requires less effort for data analysis than single-cell-based approaches. This new correlation approach can be applied to other systems to aid analysis of gene regulatory circuits.
Introduction
The biological processes that occur inside cells involve interactions among many proteins. In general, when one considers a regulatory interaction between two protein molecules, three questions arise: 1), What regulates what? 2), Is this regulation positive or negative? 3), How tight is the regulation? In some situations, knowledge about biochemical properties suffices to answer these questions. However, at certain times, even when the regulatory link physically exists, it may not be operational. In such cases, changes in the upstream concentration of a regulator do not affect the downstream processes. This may occur because the concentration of the upstream regulator lies beyond an effective range, or a critical cofactor is lacking (1,2). A number of techniques have been developed to address the above questions, such as the deletion and overexpression methods that are widely used in molecular biology (3). An alternate approach is to improve our quantitative understanding of how a gene regulation circuit functions as a whole (4), especially with all fully functional factors in the circuit.
Gene expression is inherently a stochastic process. As a result, cells develop nongenetic individuality even under uniform growth conditions, as has been confirmed by single-cell microscopy (5–7). Here we exploit the variability of protein levels and the temporal dynamics of this variability to address the three questions mentioned above. In steady-state growth, cellular protein levels are assumed to fluctuate around a mean value. With an active regulatory link, upstream protein level fluctuations can propagate downstream. For positive regulation, the downstream level fluctuates in the same direction as the upstream regulator, albeit with a time lag. For negative regulation, the downstream level fluctuates in the opposite direction to the upstream regulator, also with a time lag. This time lag exists because it takes time to dilute and degrade existing molecules while new molecules are synthesized. One can infer information about the activity level of the regulatory link and the time lag for fluctuation propagation by investigating certain temporal correlation functions of protein-level fluctuations.
Correlation techniques have previously been used to quantify and analyze fluctuations at the molecular level in biochemical reactions (8,9). The cross correlation reports the time lag between similar patterns in upstream and downstream levels, and thus answers the first question (What regulates what?). Most of the time, the sign of the correlation where the amplitude is largest answers the second question (Is regulation positive or negative?), because a positive (negative) correlation indicates positive (negative) regulation. The value of the cross correlation can be used to address the third question (How tight is the regulation?). Not all downstream level fluctuations can be correlated with the upstream level fluctuation. For tight (loose) regulation, the correlation between upstream and downstream is high (low), and downstream components respond strongly (weakly) to the upstream level changes. Autocorrelation also carries information. It reveals how long it takes for cells to eliminate the memory of previous protein levels. The width of the autocorrelation peak is essentially the timescale required for a cell with above- or below-average protein levels to relax to average levels. The process of memory elimination is strongly affected by active regulation, especially active autoregulation (10).
Fluctuations also arise during cell division when molecules are partitioned stochastically between two daughters. The resulting noise profiles are remarkably difficult to separate from gene expression noise (11). Here, we introduce a fluctuation-based approach that is applied at the level of the microcolony, which is a collection all cells derived from the same single cell. By taking averages over a microcolony, we can eliminate partition noise. Compared with correlations at the single-cell level, correlations at the microcolony level therefore provide more-relevant information about gene expression and active regulatory links.
In this work, we investigated the regulatory pathways in the bioluminescent marine bacterium Vibrio haryeyi using fluctuation correlation. V. harveyi communicates by synthesizing, releasing, and detecting the population-dependent accumulation of extracellular signal molecules called autoinducers (AIs) (12–15). This cell-to-cell communication process, which coordinates collective behaviors, is called quorum sensing (QS). At the heart of the V. harveyi QS circuit (Fig. 1 A) are five small quorum regulatory RNAs called Qrr1–5 (16,17). At low AI concentrations, phosphorylated LuxO (LuxO∼P) (3) activates transcription of the qrr1–5 genes. At high AI concentrations, LuxO is not phosphorylated, and transcription of the qrr genes ceases. The small RNAs Qrr1–5 block translation and destabilize several mRNA targets, including that encoding luxR (18). LuxR is the master transcriptional regulator that activates and represses >70 genes (12,19–21). The V. harveyi QS circuit produces highly uniform behavior in individual cells, suggesting that the QS circuit has been optimized to synchronize the response to AIs (22). The QS circuit apparently differs from regulatory loops designed to generate diversity among members of the population, e.g., in competence and sporulation in Bacillus subtilis (23,24).
Figure 1.
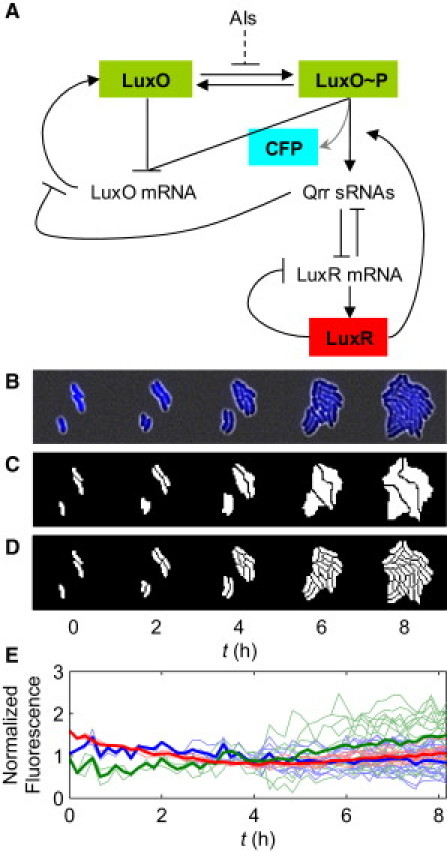
A portion of the QS circuit in V. harveyi is shown along with time-lapse images of gene expression within microcolonies. (A) A portion of the QS pathway in V. harveyi. The concentration of AIs regulates the phosphorylation of the LuxO protein. LuxO∼P activates production of the Qrr sRNAs, which repress LuxR protein production by binding and degrading its mRNA. Together with LuxO∼P, LuxR activates the production of Qrr sRNAs. LuxO represses its own transcription regardless of its phosphorylation status. Qrr sRNAs repress LuxO protein production via direct basepairing with luxO mRNA. Most lines denote regulation native to V. harveyi. For imaging purposes, LuxO is fused to YFP, and LuxR is fused to mCherry. CFP is produced together with Qrr4 (gray arrow). (B) Typical time-lapse images of V. harveyi cells. This series shows the cyan fluorescence channel (qrr4-cfp) overlaid with the white-light channel. (C) Image partitioned at the microcolony level. The three cells at the beginning of image acquisition grew into three microcolonies. (D) Image partitioned at the single-cell level. Pixels denoting cells or microcolonies are shown in white; background pixels are shown in black. (E) Typical time traces of fluorescence per unit area within a microcolony in different channels: qrr4-cfp (blue), YFP-LuxO (green), and mCherry-LuxR (red). The normalized fluorescence values are very close to unity over time. The thick curves are the microcolony averages, and the thin curves are from individual cells within the microcolony.
This study focuses on regulation among the luxO, luxR, and qrr genes. In our experiments, LuxO protein was fused to yellow fluorescent protein (YFP), and LuxR protein was fused to mCherry. The fusion proteins preserve the function of the native proteins. The strains also contain a transcriptional fusion of cfp fused to the qrr4 to report the promotor activity through the cyan fluorescence (Fig. 1 A). The dynamics of fluorescence and fluorescent-tagged proteins in individual cells were followed by time-lapse fluorescence microscopy (Fig. 1, B–D). We calculated the cross- and autocorrelations of different fluorescence fluctuation signals to analyze this regulatory network. We showed that in V. harveyi, LuxR tightly activates the qrr4 promoter at low AI levels, which agrees with previous findings (18). We observed a low correlation between the total YFP-LuxO level and the qrr4 promoter activity, which helps to elucidate how LuxO∼P regulates qrr4. We also found that although LuxO represses its own expression, there was no signature of this feedback in the measured autocorrelation under our experimental conditions. Fluctuation of LuxO concentration is apparently dominated by intrinsic noise. At 0, 50 nM, and 1 μM AI, a positive correlation was observed between early YFP-LuxO and subsequent mCherry-LuxR level fluctuations. We therefore propose that LuxO interacts with LuxR at all AI levels via a mechanism other than the currently known small-RNA-based regulation.
Materials and Methods
The strains used in this study, KT651 (wild-type (WT)) and KT653 (luxO-ar), both have three fluorescent protein fusions: YFP-LuxO replaces the native LuxO, mCherry-LuxR replaces the native LuxR, and qrr4-cfp is used to report the activity of the qrr4 promoter. In KT653, site-directed mutagenesis was performed to disrupt LuxO autorepression. Cells were initially grown in liquid culture and then continued to grow between a glass coverslip and an agarose pad on an inverted microscope. Images were acquired in multiple fluorescent channels as well as in transmitted light at 10-min intervals (Fig. 1, B–D). We analyzed the images to obtain the size and total fluorescence of individual cells and microcolonies at each time point. More details about the cell strains, microscopy, and image analysis are provided in the Supporting Material.
To calculate the fluctuation correlation, we define Xi(t) and Yi(t) to represent any two protein concentrations at time t, and use i to index the single microcolony or single cell. We also define xi(t) and yi(t) to be any of the concentration deviations from the expected values. The expected values and the concentration deviations (fluctuations) are expressed by
| (1) |
The correlation S and normalized correlation R between the fluctuations x and y are given by
| (2) |
| (3) |
The normalized correlation matrix Rx,y(t1, t2) defined in Eq. 3 plays a major role in our subsequent analysis. The time coordinates (t1, t2) serve as the indices specifying a matrix element of Rx,y(t1, t2). In practice, Sx,x(t1, t1) and Sy,y(t2, t2) in the denominator are not directly calculated but are extrapolated from neighboring elements to exclude imaging noise (Supporting Material).
Ideally, when the regulation process is time-invariant, the values of the Rx,y(t1, t2) elements depend only on the time difference t1 – t2. Hence, elements that lie on lines parallel to the diagonal have identical values (these lines are defined by t1 – t2 = τ, where τ is a constant). In a heat map of Rx,y(t1, t2) values, all of the contour lines should be parallel to the diagonal. The two-dimensional correlation heat map Rx,y(t1, t2) can then be reduced to a one-dimensional correlation curve:
| (4) |
However, when the correlation matrix is constructed from experimental data, deviations from this simple pattern indicate that the regulation parameters have changed over the course of the experiment. Our experiments were designed to minimize such changes. Data sets in which large regions of the correlation heat map display horizontal or vertical swaths with nominally uniform values might be caused by individual cells that failed to follow the population behavior. Such individual cells and their associated microcolonies were carefully excluded from the analysis.
Results
Correlation at the microcolony level
Analysis of fluorescence microscopy at the single-cell level usually requires integration over each whole cell volume. However, cell volume usually grows during the cell cycle and halves at cell division. As a result, even though its protein concentration or fluorescence per unit volume is at steady state, the fluctuation amplitude is not steady. Because the fluctuation amplitude is related to the total molecule number in the cell, the amplitude changes with cell volume. One approach is to limit the sampling volume to a constant value so it is not affected by cell growth. Here we introduce an alternative approach that still utilizes data from the whole cell volume.
The sources of fluctuation in protein levels inside the cell may be complicated. The most fundamental source of fluctuation is the stochastic nature of molecule synthesis, degradation, and dilution. Given fixed synthesis, degradation, and dilution rates, the steady-state molecule-number fluctuation inside a fixed volume is Poissonian. Hence, the molecule-number standard deviation (SD) is the square root of the total molecule number in that volume. Now consider an exponentially growing volume with the concentration at steady state. The number SD will be proportional to the square root of the total volume. Therefore, for the measured concentration (total intensity divided by total volume), its fluctuation amplitude is proportional to . As a result, in the dynamics of the normalized fluctuation levels, the effective dilution rate is halved due to normalization (Eqs. S66–S68), whereas the synthesis and degradation rates are unchanged. Therefore, for an exponentially growing volume, as in a microcolony, the fixed-volume correlation results can still be used only with a change of the dilution rate. As an example, the autocorrelation of an unregulated stable protein is at the microcolony level, whereas if it is measured in a fixed volume, Rx,x(τ) = e–β|τ|, where β = 2β0 = log 2 generation–1 (for details, see Supporting Material).
Many proteins are regulated or affected by extrinsic noise. Their fluctuations are not Poissonian; however, even a complicated regulation process can be dissected into small steps. Given fixed rates, the fluctuation introduced in each step will be Poissonian and can be normalized as above. The final fluctuation is an integration of the Poissonian fluctuations of all of the intermediate steps. Thus, in many cases, even with complicated regulation, protein-level fluctuations can still be normalized and analyzed as in the fixed-volume case. Eq. 3 automatically yields the fluctuation correlation normalized to as .
Cross correlation between mCherry-LuxR and qrr4-cfp
We used mCherry-LuxR (red fluorescence) and qrr4-cfp (cyan fluorescence) to investigate regulation between LuxR and Qrr4. The cross correlation between red and cyan fluorescence fluctuations was compared only at 0 and 50 nM AI because at 1 μM AI, the expression of qrr4 is minimal and the cyan fluorescence is too weak to be resolved from the cell's autofluorescence background.
According to Eq. 3, with the assignment X → red fluorescence from mCherry-LuxR and Y → cyan fluorescence from qrr4-cfp, we computed the cross-correlation matrix RmCherry, CFP between fluctuations in the red and cyan fluorescence. Heat maps of the correlation matrixes are displayed at 0 nM (Fig. 2 A) and 50 nM AI (Fig. 2 B). In Fig. 2 A, the diagonal clearly separates two regions with strikingly different cross-correlation values. The high values in the region above the diagonal contrasts sharply with the low values below. The enhanced correlation in the former implies that the qrr4-cfp fluctuation positively correlates with mCherry-LuxR fluctuations with a time lag. The correlation curve shown in Fig. 2 C (blue solid curve) displays a broad peak centered near −1.4 generations. This agrees well with the typical time lag between two proteins in a regulatory link (Supporting Material).
Figure 2.
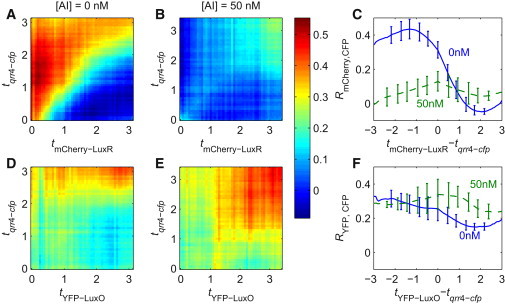
Fluctuation correlation between qrr4-cfp and concentrations of two upstream proteins (mCherry-LuxR and YFP-LuxO) in the WT strain. (A and B) Heat maps of the correlation matrix RmCherry, CFP between concentration fluctuations of mCherry-LuxR and qrr4-cfp, at 0 nM AI (A) and 50 nM (B). (A) The qrr4-cfp fluctuation correlates strongly with that of the earlier mCherry-LuxR, with a time lag of ∼1.5 generation. (B) At 50 nM AI, such an asymmetric correlation is essentially absent. (C) Correlation curves at 0 nM of AI (blue solid) and at 50 nM (green dashed). (D and E) Heat maps of the correlation matrix RYFP, CFP measuring correlations between fluctuations in qrr4-cfp and total YFP-LuxO concentrations. The relatively uniform values for both 0 nM AI (D) and 50 nM (E) implies a very weak correlation. (F) Correlation curves at 0 nM AI (blue solid) and 50 nM (green dashed). Each matrix was averaged over the matrixes produced from four independent movies.
By contrast, the correlation matrix at 50 nM AI is markedly lower in value over the entire heat map (Fig. 2 B). The corresponding correlation curve is roughly symmetric about tmCherry–LuxR – tqrr4–cfp = 0 (Fig. 2 C, green dashed curve). We attribute this weak, symmetric profile to extrinsic noise. Thus, at 0 nM AI, mCherry-LuxR tightly and positively regulates qrr4, whereas this regulation is significantly weakened at intermediate AI concentration (50 nM).
LuxR positive regulation of the qrr promoters was previously investigated in bulk assays. A co-activator, LuxO∼P, is required for such regulation (16). It was reported that LuxR directly activates the expression of qrr2, qrr3, and qrr4 genes by binding to their promoters (18,25). Similarly, our correlation results show that mCherry-LuxR positively regulates the qrr4 promoter at low AI concentrations, when YFP-LuxO∼P is abundant.
There is yet another regulatory link between LuxR and Qrr4: specifically, Qrr sRNAs destabilize luxR mRNA. If Qrr4 strongly represses luxR expression, the cross-correlation values might be negative for tmCherry–LuxR – tqrr4–cfp > 0. In principle, one would expect the amplitude of such negative cross correlation to be small, for the following reasons: When one fluorescent protein regulates another, such as mCherry-LuxR upregulating qrr4-cfp, the correlation time lag is the time required for relaxation of the upstream protein-level fluctuation and expression of the downstream protein. That is the time for protein to be degraded or diluted. When it comes to RNA interactions, assuming that the time periods required to synthesize fluorescent molecules from cfp mRNA and luxR-mCherry mRNA are similar, the time lag of the cyan-red correlation due to this regulation is the lifetime of the RNA molecules. The kinetics of Qrr-mRNA binding and degradation are much faster than the timescales for changes in protein levels (26,27). Therefore, the correlation due to this regulation should have a negative value and almost zero time lag. However, cells usually also have extrinsic noise that affects the synthesis of all proteins, resulting in a positive correlation between almost any two proteins. Generally, the correlation due to extrinsic noise has almost zero time lag. Because these two effects act in opposite directions, they may cancel, resulting in the apparent lack of correlation between qrr4-cfp and mCherry-LuxR for tmCherry–LuxR – tqrr4–cfp > 0. However, there appears to be a slightly negative lobe in Fig. 2 C (blue solid curve) that may be the result of this negative regulation.
At the transition between low cell density (LCD) and high cell density (HCD), LuxO∼P and LuxR are simultaneously present. A previous study of V. cholerae showed that HapR (a homolog of V. harveyi's LuxR) dramatically accelerates the transition from HCD to LCD (26). In a recent study, the fluorescence from mCherry-LuxR revealed that the concentration of LuxR is significant in the LCD state (28) (Table 1). A strong positive correlation between early mCherry-LuxR levels and late qrr4-cfp levels at 0 nM AI indicates that LuxR is active and positively regulates the qrr4 gene at LCD. This finding suggests that in addition to accelerating the HCD-LCD transition, LuxR activation of the qrr expression also reduces LuxR concentration at LCD.
Table 1.
Steady-state fluorescence in V. harveyi at AI concentrations of 0 nM, 50 nM, and 1 μM
| 0 nM | 50 nM | 1 μM | ||
|---|---|---|---|---|
| qrr4-cfp | WT | 163 ± 61 | 92 ± 49∗ | 7.6 ± 22∗ |
| luxO-ar | 113 ± 40 | 135 ± 63∗ | 2.8 ± 21∗ | |
| YFP-LuxO | WT | 8.2 ± 5.3 | 17.6 ± 10.1 | 21.6 ± 9.2 |
| luxO-ar | 41.3 ± 15.5 | 69.5 ± 16.9 | 76.6 ± 18.6 | |
| mCherry-LuxR | WT | 101 ± 23 | 577 ± 97 | 670 ± 116 |
| luxO-ar | 90 ± 25 | 439 ± 89 | 634 ± 107 |
The fluorescence units are partition units per cell, which are calculated according to Teng et al. (28).
Partition unit can be a dimer or an oligomer if the protein dimerizes or oligomerizes; 260 cell division events were used for the estimation.
SD of cell autofluorescence is comparable to that of signal fluorescence, making the error bars large.
Because the qrr promoters are predominantly regulated by LuxO∼P, and not dramatically by LuxR (18), the weaker regulation between LuxR and qrr at intermediate AI concentrations (50 nM) may be a consequence of two possibilities: 1), LuxR saturates the binding site in the qrr4 promoter; or 2), the LuxO∼P concentration is too low for the LuxO∼P binding site to be occupied. At 50 nM AI, the significant level of cyan fluorescent protein (CFP; Table 1) suggests that LuxO∼P remains abundant. The CFP level becomes close to zero only in the limit of a very high AI concentration (1 μM), when almost all LuxO molecules are unphosphorylated. Our observation that the regulatory link between LuxR and the qrr4 promoter weakens considerably at 50 nM AI may therefore reflect saturation of the LuxR binding sites at the qrr4 promoter. A previous bioinformatics analysis showed that LuxR binding sites at the qrr promoters more closely resemble the LuxR consensus binding site than do LuxR binding sites at other known LuxR-regulated genes (25). Therefore, the LuxR binding sites at the qrr promoters are easier to saturate than other LuxR binding sites.
To summarize, our cross-correlation results show that at very low AI concentrations, LuxR proteins bind to the qrr4 promoter to activate the qrr4 gene expression, which agrees with previous findings. These results raise the possibility that the LuxR binding site at the qrr4 promoter is readily saturated by LuxR, and thus this regulatory link becomes less sensitive to LuxR concentration at intermediate or higher AI concentrations (50 nM).
Cross correlation between total YFP-LuxO and qrr4-cfp
We now consider cross correlation between the fluorescence signals from YFP-LuxO molecules and qrr4-cfp. Using Eq. 3 with the assignment X → YFP-LuxO and Y → qrr4-cfp, we obtained heat maps of the cross-correlation matrix RYFP, CFP at 0 and 50 nM AI (shown in Fig. 2, D and E, respectively). At 0 nM AI, there is a weak trend of matrix values being higher above the diagonal than below. At 50 nM AI, there is no discernible asymmetrical feature about the diagonal that would be suggestive of regulation. These heat maps suggest that the regulatory links between LuxO concentration and qrr4 expression are weak.
It is known that LuxO∼P activates the qrr genes, whereas unphosphorylated LuxO does not (3,16). LuxO molecules are phosphorylated through LuxU by AI receptors acting as kinases in the absence of AI ligands. We expect AI binding and unbinding and phosphate transfer to be rapid processes relative to protein turnover. We assume a fixed probability ρ that any particular LuxO molecule is phosphorylated. With a given total LuxO number NLuxO, the LuxO∼P number NLuxO∼P follows a binomial distribution with 〈NLuxO∼P〉 = ρNLuxO and . The SD over the mean is a measure of how NLuxO∼P tracks NLuxO, because a high value means a high noise level in NLuxO∼P and therefore loose tracking of NLuxO:
| (5) |
Either a low phosphorylation level or a low NLuxO number would result in NLuxO∼P not closely tracking NLuxO, which could explain the low correlation between YFP-LuxO and qrr4-cfp fluctuations. In the WT strain, the YFP-LuxO level is estimated to be as low as 8.3 partition units per cell (Table 1). However, because LuxO is likely to oligomerize (3), the true copy number per cell is unknown.
Alternatively, if unphosphorylated LuxO also binds to the qrr promoters but does not activate transcription, then only the ratio between LuxO∼P and LuxO concentrations controls the qrr transcription. This concentration ratio is independent of total LuxO levels, and thus the cross correlation between YFP-LuxO and qrr4-cfp is also expected to be low in this case.
In WT V. harveyi, luxO mRNA is one of the targets of Qrr regulation. Our chromosomal luxO-yfp fusion has the WT luxO promoter and ribosomal binding site, and is therefore subject to negative regulation by Qrrs (27). Such negative regulation is barely visible in the correlation matrix, likely for the same reason that the negative regulation between qrr4-cfp and mCherry-LuxR is not visible in the correlation matrix.
To summarize, although the qrr promoters are predominantly regulated by LuxO∼P, the total YFP-LuxO amount correlates poorly with qrr4-cfp expressed.
Autocorrelation of YFP-LuxO
LuxO is known to repress its own expression (27). In general, a protein can repress its own expression to accelerate the process of damping large concentration fluctuations and returning the concentration to the basal value (29–31). A too-low steady-state protein level or lack of an essential cofactor can result in ineffective repression. At the other extreme, the steady-state protein level can be so high that it saturates autorepression, in which case the mean protein level is repressed compared with a strain lacking autorepression. For both of these cases, the fluctuation autocorrelation timescales are simply those required for dilution and degradation of the protein, as if there were no autorepression occurring.
A LuxO binding site is located upstream from the luxO gene. Point mutations can impair this binding site and eliminate autorepression (the luxO-ar strain) (27). Compared with the WT strain, the luxO-ar strain produces more LuxO protein at all AI concentrations. Specifically, in our strains, when LuxO is substituted with YFP-LuxO, higher YFP fluorescence is observed in the luxO-ar strain than in the WT strain (27) (Table 1). We found that LuxO autorepression was significant at all AI levels in our experiments, even in the absence of AI, when LuxO levels are at their lowest.
To investigate whether autorepression reduces the correlation time of fluctuations in the LuxO level, we produced autocorrelation heat maps for fluctuations in the YFP fluorescence in both WT and luxO-ar strains, as shown in Fig. 3, A and B, respectively. The autocorrelations are compared at the 1 μM AI concentration to minimize the influence from sRNAs (Fig. 3 C). In fact, the autocorrelation curves are very similar at all three AI concentrations (data not shown). The fluctuation timescale is inferred from the width of the autocorrelation curve. The correlation curve is most reliable in the central region (τ near 0) where the data density is highest. In this region, the two autocorrelation curves for the WT and the luxO-ar strain overlap well within the error ranges. This overlap indicates that LuxO autorepression is saturated, because active autorepression would shorten the correlation time of fluctuations.
Figure 3.
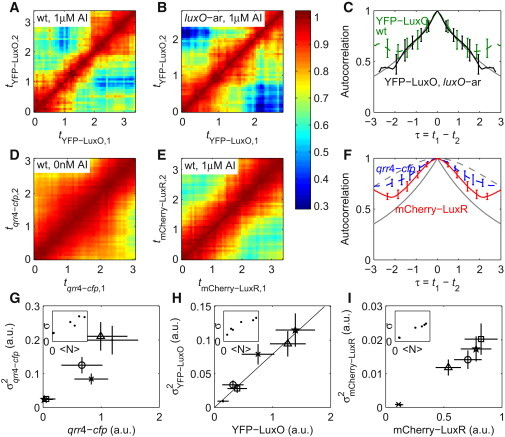
Autocorrelation of fluctuations for YFP-LuxO, qrr4-cfp, and mCherry-LuxR concentrations. (A, B, D, and E) Heat maps of concentration fluctuation autocorrelation matrixes for YFP-LuxO in the WT strain (A), YFP-LuxO in the luxO-ar strain (B), qrr4-cfp in the WT strain (D), and mCherry-LuxR in the WT strain (E). AI concentrations are indicated in the figure. (C) Autocorrelation curves inferred from A (green dashed) and B (black solid). The gray curve is the theoretical prediction of autocorrelation for a stable protein with noisy production (, where β0 = (log 2)/2, is half of the basic dilution rate). (F) Autocorrelation curves inferred from D (blue dashed) and E (red solid). The lower gray curve is the same as the gray curve in C. The upper dashed curve is the theoretical autocorrelation for proteins whose production rate depends on an upstream stable protein with noisy production, 10 (also Eq. S47). (G–I) Single-molecule fluorescence squared SD (σ2) versus mean (〈N〉) at the beginning of image acquisition for qrr4-cfp (G), YFP-LuxO (H), and mCherry-LuxR (I). σ2 is proportional to 〈N〉 if intrinsic noise dominates. Such a relation is observed for YFP-LuxO but not for qrr4-cfp or mCheery-LuxR, which are known to be regulated by multiple upstream factors. Symbols: WT 0 nM AI (dot), WT 50 nM AI (circle), WT 1 μM AI (square), luxO-ar 0 nM AI (cross), luxO-ar 50 nM AI (triangle), and luxO-ar 0 nM AI (pentagram). The insets show SD (σ) versus mean (〈N〉) on a linear scale.
The autocorrelation curves also overlap well with theoretical prediction for a stable protein without an upstream regulator , where β0 = (log 2)/2 generation –1 (Fig. 3 C, gray curve; also see Supporting Material), suggesting no active upstream protein regulators. On the contrary, it was previously reported that another protein LuxT represses the luxO promoter by binding between 117 and 149 bases upstream of the luxO initiation codon (32). Any upstream protein regulation noise or extrinsic noise should result in a broader autocorrelation peak, with tight regulation producing a wide peak with a round top.
In principle, if an upstream regulator or extrinsic noise coexists with autorepression, it is possible to obtain a similar autocorrelation peak as for an unregulated protein. If fluctuations in the copy number of YFP-LuxO proteins are dominated by intrinsic noise, one would expect the square of the fluorescence SD to scale linearly with the total fluorescence. Such scaling should not apply if there is a comparable amount of extrinsic noise. The SD squared σI2 versus total fluorescence intensity I per cell is plotted for qrr4-cfp, YFP-LuxO, and mCherry-LuxR in Fig. 3, G–I, at the single-cell level. A clear linear scaling is observed for YFP-LuxO, consistent with the case that intrinsic noise dominates. For the other two proteins, which are extrinsically regulated, simple linear scling does not apply. The linear fitting of σI2 versus I for YFP-LuxO has a near-zero y-intercept, suggesting that the extrinsic noise is almost zero for this protein. Because intrinsic noise appears to dominate the fluctuation of YFP-LuxO level, noise upstream of YFP-LuxO cannot propagate downstream to factors regulated by YFP-LuxO.
We found I2/σI2 for YFP-LuxO per cell at 0 nM, 50 nM, and 1 μM AIs to be 1.89, 4.04 and 4.97 for the WT strain; and 9.49, 16.0, and 17.6 for the luxO-ar strain, respectively. The total protein number N = (1 + b)I2/σI2, where the burst size b is the average number of protein produced per mRNA molecule (33) (Eq. S34). YFP-LuxO amount is estimated based on the partition at cell division (Table 1). If a partition unit is a YFP-LuxO monomer, we get b = 3.35. However, because LuxO is likely to oligomerize (3), an analysis based on partition noise would underestimate the real copy number and b would be larger.
To summarize, we conclude from the autocorrelation curves that LuxO saturates the binding site in the promoter that represses its own expression. Our results also suggest that the noise in the LuxO level is dominated by intrinsic noise.
LuxO fluctuations correlate positively with subsequent LuxR fluctuations
Broadened autocorrelation curves reflect regulation by slowly varying upstream components, such as an upstream regulatory protein. The heat maps of the autocorrelation of qrr4-cfp and mCherry-LuxR are shown in Fig. 3, D and E, respectively. The profiles of the autocorrelation curves for both qrr4-cfp and mCherry-LuxR (Fig. 3 F) differ considerably from those expected for stable proteins undergoing noisy production (Fig. 3 F, lower gray curve). This may result from the propagation of noise from upstream components or from extrinsic noise. In the case of qrr4-cfp, the qrr4 promoter is regulated by several upstream long-lived proteins (LuxR as shown in Fig. 2, and σ54). Therefore, the greater width of the autocorrelation curve of qrr4-cfp relative to unregulated proteins is expected. (The autocorrelation curve of qrr4-cfp fluctuations will differ from that of Qrr4 fluctuations, if measurable, because the sRNA is subject to rapid degradations.) Likewise, LuxR may have an active upstream regulator or LuxR may be affected by extrinsic noise.
The cross-correlation matrixes between YFP-LuxO and mCherry-LuxR show that YFP-LuxO fluctuations positively correlate with time-lagged mCherry-LuxR fluctuations at all three AI concentrations investigated (Fig. 4). The asymmetric structure of the cross-correlation matrix is significant enough to show that noise propagates positively from YFP-LuxO to mCherry-LuxR levels.
Figure 4.
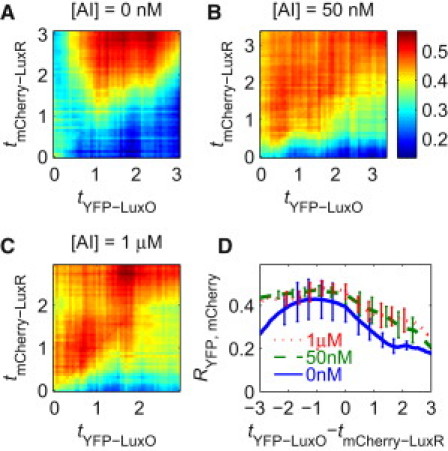
Heat maps of the cross-correlation matrix RYFP, mCherry measuring cross correlations between fluctuations in YFP-LuxO and mCherry-LuxR concentrations. (A–C) Heat maps of RYFP, mCherry at three AI concentrations: 0 nM (A), 50 nM (B), and 1 μM (C). The correlation curves are plotted in D. In panels A–C, the higher values above the diagonal correspond to the finding that YFP-LuxO actively and positively regulates mCherry-LuxR. Regulation does not depend on LuxO phosphorylation status, because it is observed at all three AI concentrations. Each matrix was averaged over the matrixes produced from four independent movies.
It is true that the presence of a correlation with a time lag can, in principle, occur without causal regulation in complex gene networks. Certain network architectures can produce qualitatively similar cross-correlation functions. An example would be A regulates both B and C, and B regulates D. In this case, the correlation between C and D is similar to the correlation between B and D, because part of B and D's fluctuation comes from their common regulator, A (9). Our data suggest that YFP-LuxO level fluctuation is mostly due to intrinsic noise. In this case, YFP-LuxO fluctuations do not reflect an upstream noise source, which disfavors the above scenario for noncausal correlation. A cross correlation similar to that observed for LuxO and LuxR can also occur if YFP-LuxO regulates some other factor that regulates mCherry-LuxR in this system. In this case, the time lag between YFP-LuxO and mCherry-LuxR fluctuation is expected to be longer than direct regulation. The theoretical peak position is −1.4 generations for direct regulation. Adding another layer of intermediate regulatory protein would move the correlation peak to between −1.4 and −2.0 generations (see Supporting Material). Protein degradation and feedback can also affect the peak position.
In the QS circuit, the main regulator of LuxR is the Qrr sRNAs. The lifetime of sRNA molecules can be enhanced upon binding to the sRNA chaperone Hfq (16), which protects the sRNA molecules from degradation (34). In this case, slow fluctuations of Qrr levels could also lead to a broad autocorrelation peak for mCherry-LuxR. In the QS circuit as it is currently known, LuxO and LuxR interact exclusively through the Qrr sRNAs. As a result, no interaction between LuxO and LuxR is expected at high AI levels when very few Qrr sRNAs are produced (Table 1).
Although the correlation analysis suggests that LuxO upregulates LuxR at all AI concentrations, the mCherry-LuxR level is not higher in the luxO-ar strain, where there are more YFP-LuxO proteins per cell. Because LuxR and LuxO also interact through sRNAs, if LuxO upregulates LuxR via an sRNA independent pathway, one would expect LuxR levels to be higher without sRNAs, e.g., at saturating AI concentrations. A previous bulk experiment that compared HCD cultures for the WT strain, ΔluxO-Cmr strain (no functional LuxO), and luxO D47A strain (LuxO locked in unphosphorylated state) showed no difference in relative light units (3), which are indicators of the LuxR level. It is therefore very puzzling that LuxR fluctuation correlates with LuxO with a time lag, and this finding suggests that there may be additional links within the regulatory circuit that are currently unknown.
Discussion
We studied protein concentration fluctuations in live V. harveyi cells. Time-lapse microscopy was used to monitor fluorescent fusion proteins and a fluorescent reporter. Single cells were traced as they grew into microcolonies. Protein-level fluctuations were also measured at the microcolony level, which treats the microcolony as if it were a single large cell. Autocorrelation and cross-correlation matrixes were calculated between different protein concentration fluctuations at the microcolony level.
The microcolony method of calculating fluctuation correlations is accurate when protein-level fluctuations can be modeled as linear perturbations around a steady state, because the microcolony is a linear combination of all descendent cells. Moreover, the microcolony method can improve the accuracy of single-cell-level analysis. Usually, the volume of a single cell doubles every cell cycle, which for Poissonian fluctuations implies that the concentration fluctuation amplitude decreases by a factor of . In previous studies, the protein concentration fluctuation amplitude at the single-cell level was assumed to be constant over time. This approximation was regarded as adequate when fluctuation data were obtained from a large number of unsynchronized cells (9) or when relative protein concentration ranks instead of the true protein concentrations were used for correlation calculations (10). We were able to significantly improve on this approximation by calculating correlations of microcolony-level fluctuations. Our theoretical predictions agree well with Monte Carlo simulations (see Supporting Material).
The microcolony method eliminates some noise sources. Because it averages over the offspring cells, differences between sister cells are averaged out. Such differences are mainly the partition noise at cell division. This partition noise at the single-cell level is passed on to the descendant cells. In autocorrelation curves obtained at the single-cell level, the peaks appear wider due to partition noise. This can be seen, for example, in the YFP-LuxO autocorrelation (Fig. S2 e). There are fewer copies of YFP-LuxO in the WT strain than in the luxO-ar strain, and thus there is a higher fraction of partition noise in the former. At the single-cell level, the autocorrelation curve is wider for the WT stain than for the luxO-ar strain. In contrast, both curves have the same width at the microcolony level (Fig. 3 C). In this sense, correlation at the microcolony level provides more-relevant information about gene expression and protein regulation, with less disturbance from cell division and molecule partitioning.
Measuring protein-level fluctuation at the microcolony level can also reduce the complexity of image processing. Fluorescence microscopy image resolution is limited by optical resolution and camera pixel size. In practice, accurately partitioning each image from the microcolony into single-cell areas is the most error-prone and time-consuming part of the data analysis. Because most regulatory links have a timescale of ∼1 generation, to accurately study a regulatory link, one must follow the cells for n ≥ 3 generations. With the same set of time-lapse images, an ∼2n – 1 greater effort is required to partition the images into the single-cell level compared with the microcolony level. Moreover, in a microscopy image, the fluorescent intensity reading of a single cell is affected by the neighboring cells. Therefore, even if the image can be accurately divided into regions of single cells, the fluorescence reading may not truly reflect the protein level in each cell. The microcolonies usually have smaller ratios of border length to area than do single cells, so they are less affected by neighboring microcolonies compared with single cells. It was recently reported that a microfluidic device enables multiple cell lineages to be tracked in parallel (35). Such a device could reduce the image-processing complexity to a minimum for microcolony fluctuation correlation analysis.
Furthermore, if we can accurately follow the fluorescence of individual microcolonies, we will no longer have to perform microscopy when using correlation to study regulatory links in cell circuits. Single cells can be distributed to multi-well plates, and the total fluorescence can be accurately followed in each well to generate correlation matrixes. With current wavelength demixing technology, we can use multiple fluorescent tags in the same cells to study the interaction between multiple regulators.
We applied the improved correlation method to analyze V. harveyi strains that were engineered to harbor YFP-LuxO, mCherry-LuxR, and qrr4-cfp. Our results suggest that not all regulatory interactions are active all times, and not all genes are equally affected by extrinsic noise. We have demonstrated that fluctuation correlation can lead to a better understanding of a regulatory circuit as a whole. Fluctuation correlation is increasingly being used as a tool to study genetic regulation and noise (36); however, the process of calculating fluctuation correlation at the single-cell level can be cumbersome and time-consuming. Our approach for studying fluctuation correlation at the microcolony level can simplify this process and facilitate its use as a tool for investigating regulatory circuits.
Acknowledgments
We thank Tao Long, Pankaj Mehta, Shu-wen Teng, and Sine L. Svenningsen of Princeton University for insightful discussions. Y.W. thanks Hongye Sun and Joe Beechem at Life Technologies for encouragement.
This work was funded by the Howard Hughes Medical Institute, National Institutes of Health (grant 5R01GM065859), and National Science Foundation (grant MEB-0343821), and was partially supported by the Defense Advanced Research Projects Agency (grant HR0011-05-1-0055). Y.W. and N.P.O. received support from Princeton University.
Footnotes
Yufang Wang's present address is Life Technologies, Foster City, CA.
Supporting Material
References
- 1.Toledo F., Wahl G.M. Regulating the p53 pathway: in vitro hypotheses, in vivo veritas. Nat. Rev. Cancer. 2006;6:909–923. doi: 10.1038/nrc2012. [DOI] [PubMed] [Google Scholar]
- 2.Piggot P.J., Hilbert D.W. Sporulation of Bacillus subtilis. Curr. Opin. Microbiol. 2004;7:579–586. doi: 10.1016/j.mib.2004.10.001. [DOI] [PubMed] [Google Scholar]
- 3.Freeman J.A., Bassler B.L. A genetic analysis of the function of LuxO, a two-component response regulator involved in quorum sensing in Vibrio harveyi. Mol. Microbiol. 1999;31:665–677. doi: 10.1046/j.1365-2958.1999.01208.x. [DOI] [PubMed] [Google Scholar]
- 4.Süel G.M., Garcia-Ojalvo J., Elowitz M.B. An excitable gene regulatory circuit induces transient cellular differentiation. Nature. 2006;440:545–550. doi: 10.1038/nature04588. [DOI] [PubMed] [Google Scholar]
- 5.Spudich J.L., Koshland D.E., Jr. Non-genetic individuality: chance in the single cell. Nature. 1976;262:467–471. doi: 10.1038/262467a0. [DOI] [PubMed] [Google Scholar]
- 6.Elowitz M.B., Levine A.J., Swain P.S. Stochastic gene expression in a single cell. Science. 2002;297:1183–1186. doi: 10.1126/science.1070919. [DOI] [PubMed] [Google Scholar]
- 7.Rosenfeld N., Young J.W., Elowitz M.B. Gene regulation at the single-cell level. Science. 2005;307:1962–1965. doi: 10.1126/science.1106914. [DOI] [PubMed] [Google Scholar]
- 8.Arkin A., Shen P., Ross J. A test case of correlation metric construction of a reaction pathway from measurements. Science. 1997;277:1275–1278. [Google Scholar]
- 9.Dunlop M.J., Cox R.S., 3rd, Elowitz M.B. Regulatory activity revealed by dynamic correlations in gene expression noise. Nat. Genet. 2008;40:1493–1498. doi: 10.1038/ng.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sigal A., Milo R., Alon U. Variability and memory of protein levels in human cells. Nature. 2006;444:643–646. doi: 10.1038/nature05316. [DOI] [PubMed] [Google Scholar]
- 11.Huh D., Paulsson J. Non-genetic heterogeneity from stochastic partitioning at cell division. Nat. Genet. 2011;43:95–100. doi: 10.1038/ng.729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Henke J.M., Bassler B.L. Quorum sensing regulates type III secretion in Vibrio harveyi and Vibrio parahaemolyticus. J. Bacteriol. 2004;186:3794–3805. doi: 10.1128/JB.186.12.3794-3805.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.González J.E., Marketon M.M. Quorum sensing in nitrogen-fixing rhizobia. Microbiol. Mol. Biol. Rev. 2003;67:574–592. doi: 10.1128/MMBR.67.4.574-592.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cao J.G., Meighen E.A. Purification and structural identification of an autoinducer for the luminescence system of Vibrio harveyi. J. Biol. Chem. 1989;264:21670–21676. [PubMed] [Google Scholar]
- 15.Higgins D.A., Pomianek M.E., Bassler B.L. The major Vibrio cholerae autoinducer and its role in virulence factor production. Nature. 2007;450:883–886. doi: 10.1038/nature06284. [DOI] [PubMed] [Google Scholar]
- 16.Lenz D.H., Mok K.C., Bassler B.L. The small RNA chaperone Hfq and multiple small RNAs control quorum sensing in Vibrio harveyi and Vibrio cholerae. Cell. 2004;118:69–82. doi: 10.1016/j.cell.2004.06.009. [DOI] [PubMed] [Google Scholar]
- 17.Tu K.C., Bassler B.L. Multiple small RNAs act additively to integrate sensory information and control quorum sensing in Vibrio harveyi. Genes Dev. 2007;21:221–233. doi: 10.1101/gad.1502407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tu K.C., Waters C.M., Bassler B.L. A small-RNA-mediated negative feedback loop controls quorum-sensing dynamics in Vibrio harveyi. Mol. Microbiol. 2008;70:896–907. doi: 10.1111/j.1365-2958.2008.06452.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Waters C.M., Bassler B.L. Quorum sensing: cell-to-cell communication in bacteria. Annu. Rev. Cell Dev. Biol. 2005;21:319–346. doi: 10.1146/annurev.cellbio.21.012704.131001. [DOI] [PubMed] [Google Scholar]
- 20.Fuqua C., Winans S.C., Greenberg E.P. Census and consensus in bacterial ecosystems: the LuxR-LuxI family of quorum-sensing transcriptional regulators. Annu. Rev. Microbiol. 1996;50:727–751. doi: 10.1146/annurev.micro.50.1.727. [DOI] [PubMed] [Google Scholar]
- 21.Hammer B.K., Bassler B.L. Quorum sensing controls biofilm formation in Vibrio cholerae. Mol. Microbiol. 2003;50:101–104. doi: 10.1046/j.1365-2958.2003.03688.x. [DOI] [PubMed] [Google Scholar]
- 22.Long T., Tu K.C., Wingreen N.S. Quantifying the integration of quorum-sensing signals with single-cell resolution. PLoS Biol. 2009;7:e68. doi: 10.1371/journal.pbio.1000068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Losick R., Desplan C. Stochasticity and cell fate. Science. 2008;320:65–68. doi: 10.1126/science.1147888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Maamar H., Raj A., Dubnau D. Noise in gene expression determines cell fate in Bacillus subtilis. Science. 2007;317:526–529. doi: 10.1126/science.1140818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pompeani A.J., Irgon J.J., Bassler B.L. The Vibrio harveyi master quorum-sensing regulator, LuxR, a TetR-type protein is both an activator and a repressor: DNA recognition and binding specificity at target promoters. Mol. Microbiol. 2008;70:76–88. doi: 10.1111/j.1365-2958.2008.06389.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Svenningsen S.L., Waters C.M., Bassler B.L. A negative feedback loop involving small RNAs accelerates Vibrio cholerae's transition out of quorum-sensing mode. Genes Dev. 2008;22:226–238. doi: 10.1101/gad.1629908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tu K.C., Long T., Bassler B.L. Negative feedback loops involving small regulatory RNAs precisely control the Vibrio harveyi quorum-sensing response. Mol. Cell. 2010;37:567–579. doi: 10.1016/j.molcel.2010.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Teng S.W., Wang Y., Ong N.P. Measurement of the copy number of the master quorum-sensing regulator of a bacterial cell. Biophys. J. 2010;98:2024–2031. doi: 10.1016/j.bpj.2010.01.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Becskei A., Serrano L. Engineering stability in gene networks by autoregulation. Nature. 2000;405:590–593. doi: 10.1038/35014651. [DOI] [PubMed] [Google Scholar]
- 30.Rosenfeld N., Elowitz M.B., Alon U. Negative autoregulation speeds the response times of transcription networks. J. Mol. Biol. 2002;323:785–793. doi: 10.1016/s0022-2836(02)00994-4. [DOI] [PubMed] [Google Scholar]
- 31.Nevozhay D., Adams R.M., Balázsi G. Negative autoregulation linearizes the dose-response and suppresses the heterogeneity of gene expression. Proc. Natl. Acad. Sci. USA. 2009;106:5123–5128. doi: 10.1073/pnas.0809901106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lin Y.H., Miyamoto C., Meighen E.A. Cloning and functional studies of a luxO regulator LuxT from Vibrio harveyi. Biochim. Biophys. Acta. 2000;1494:226–235. doi: 10.1016/s0167-4781(00)00236-0. [DOI] [PubMed] [Google Scholar]
- 33.Thattai M., van Oudenaarden A. Intrinsic noise in gene regulatory networks. Proc. Natl. Acad. Sci. USA. 2001;98:8614–8619. doi: 10.1073/pnas.151588598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Moll I., Afonyushkin T., Bläsi U. Coincident Hfq binding and RNase E cleavage sites on mRNA and small regulatory RNAs. RNA. 2003;9:1308–1314. doi: 10.1261/rna.5850703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rowat A.C., Bird J.C., Weitz D.A. Tracking lineages of single cells in lines using a microfluidic device. Proc. Natl. Acad. Sci. USA. 2009;106:18149–18154. doi: 10.1073/pnas.0903163106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cox R.S., 3rd, Dunlop M.J., Elowitz M.B. A synthetic three-color scaffold for monitoring genetic regulation and noise. J. Biol. Eng. 2010;4:10. doi: 10.1186/1754-1611-4-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.