

Abstract
It is widely believed that dopamine (DA) mediates methamphetamine (METH)-induced toxicity to brain dopaminergic neurons, because drugs that interfere with DA neurotransmission decrease toxicity, whereas drugs that increase DA neurotransmission enhance toxicity. However, temperature effects of drugs that have been used to manipulate brain DA neurotransmission confound interpretation of the data. Here we show that the recently reported ability of l-dihydroxyphenylalanine to reverse the protective effect of alpha-methyl-para-tyrosine on METH-induced DA neurotoxicity is also confounded by drug effects on body temperature. Further, we show that mice genetically engineered to be deficient in brain DA develop METH neurotoxicity, as long as the thermic effects of METH are preserved. In addition, we demonstrate that mice genetically engineered to have unilateral brain DA deficits develop METH-induced dopaminergic deficits that are of comparable magnitude on both sides of the brain. Taken together, these findings demonstrate that DA is not essential for the development of METH-induced dopaminergic neurotoxicity and suggest that mechanisms independent of DA warrant more intense investigation.
Keywords: dopamine, dopamine-deficient mice, methamphetamine, neurotoxicity, temperature
The toxic potential of methamphetamine (METH) toward brain dopamine (DA) neurons is well established (Lew et al. 1997; Riddle et al. 2006; Cadet and Krasnova 2009). Despite considerable research, the precise mechanism(s) by which METH induces degeneration of DA axon terminals remain(s) to be identified. However, a substantial body of data has implicated endogenous brain DA as a mediator of METH neurotoxicity (see Gibb et al. 1994; Volz et al. 2007; Kuhn et al. 2008; for reviews). For example, drugs that deplete brain DA [e.g. alpha-methyl-para-tyrosine (AMPT)] afford neuroprotection (Gibb and Kogan 1979; Schmidt et al. 1985), and drugs that replenish DA stores [e.g. l-dihydroxyphenylalanine (l-DOPA)] reinstate METH neurotoxicity (Gibb and Kogan 1979; Thomas et al. 2008). These observations have led to that DA mediates METH neurotoxicity (Volz et al. 2007; Guillot et al. 2008; Thomas et al. 2008; Kita et al. 2009).
As evidence implicating DA in METH neurotoxicity has accrued, the importance of body temperature in the development and extent of METH neurotoxicity has become apparent (see Sharma et al. 2007; Bowyer et al. 2008; Krasnova and Cadet 2009). In particular, higher body temperatures enhance toxicity, whereas lower body temperatures generally afford neuroprotection (Bowyer et al. 1992; Ali et al. 1994; Albers and Sonsalla 1995; Cappon et al. 1997; Miller and O’Callaghan 2003). Notably, temperature effects may have direct bearing on studies demonstrating a protective effect of dopaminergic drugs, because most drugs that alter DA neurotransmission also cause alterations in core temperature in METH-treated animals.
We previously reported that near total depletion of brain DA by AMPT or reserpine did not protect animals from METH-induced DA neurotoxicity, as long as drug effects on body temperature were controlled (Yuan et al. 2001). Although that study cast doubt on the role of DA in METH neurotoxicity, the experiments involved administration of multiple drugs and complex drug administration paradigms. This left open the possibility that other factors may have been involved in the reinstatement of DA neurotoxicity in DA depleted animals.
The present study sought to more directly evaluate the role of DA in METH neurotoxicity by further taking into account drug effects on body temperature and by using mice genetically engineered to lack DA, on either one or both sides of the brain. DA-deficient (DD) mice are particularly useful for evaluating the role of DA in METH neurotoxicity because they make it possible to circumvent problems associated with complex drug regimens and because animals with unilateral brain DA deficits obviate the need for any DA replenishment. Using these approaches, we now present evidence demonstrating that endogenous DA does not play a role in METH-induced dopaminergic neurotoxicity.
Materials and methods
Drugs and chemicals
(+)Methamphetamine (METH) hydrochloride was obtained from the National Institute on Drug Abuse (Rockville, MD, USA). AMPT methyl ester, DA hydrochloride, dihydroxyphenylacetic acid (DOPAC), l-DOPA and benserazide were purchased from the Sigma Chemical Co. (St. Louis, MO, USA). Drug doses were calculated as the free base.
Animals
For AMPT/L-DOPA studies, we used male albino Swiss-Webster mice (Taconic Farms, Germantown, NY, USA) that were 8 weeks of age and weighed 25–35 g at the beginning of the study. For studies involving genetically induced DA deficiency, we used DD mice generated by gene targeting, as previously described (Zhou and Palmiter 1995); these mice (Th−/−; DbhTh/+) carry two inactive Tyrosine hydroxylase (Th) alleles, one intact Dopamine β-hydroxylase allele (Dbh+), and one Dbh allele with a targeted insertion of the Th gene (DbhTh). DD mice were maintained on a mixed C57BL/6 X 129Sv genetic background and required daily treatment with l-DOPA (50 mg/kg; i.p.) for survival. The METH toxicity experiments were initiated 16 h after the last l-DOPA injection, when mice are > 99% depleted of striatal DA (Kim et al. 2000). For studies in animals genetically engineered to lack DA on only one side of the brain, we used dopamine-deficient floxed stop (DDfs) mice that had undergone unilateral viral rescue of dopamine signaling as described (Heusner et al. 2003; Hnasko et al. 2006; Darvas and Palmiter 2009) using CAV2-Cre virus (Kremer et al. 2000). This virus is retrogradely transported from the site of injection and reactivates the endogenous Th gene to all DA neurons that project to the injection site, thereby restoring dopamine synthesis and release exclusively in that brain region. The CAV2-Cre virus (0.5 μL at a titer of 6 × 1012 particles/mL) was injected into the striatum (0.9 mm anterior to bregma, ± 2.0 mm lateral to midline, 3.0 mm ventral from the skull surface) of anesthetized (isoflurane) 2- to 3-month-old male and female DDfs and control mice that have at least one functional Th gene (referred to as sham controls). Virally injected DD mice were removed from l-DOPA treatment 2 weeks after viral injection, and those mice that maintained body weight after 1 week without l-DOPA treatment were designated as virally rescued DDfs (vrDDfs) mice and allowed at least five more weeks of recovery before testing. All mice were housed individually in transparent plastic cages and maintained on a 12 : 12 light-dark cycle (light from 7 am to 7 pm) in a temperature-controlled room (22 ± 1°C), except when otherwise dictated by the experimental design. Food and water were provided ad libitum. All animal care and experimental manipulations were approved by the Institutional Animal Care and Use Committees at Johns Hopkins University and the University of Washington, and were in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. The facilities for housing and care of the animals are accredited by the American Association for the Assessment and Accreditation of Laboratory Animal Care.
Drug treatments
For AMPT/L-DOPA studies, METH was administered i.p at a dose of 7.5 mg/kg every 2 h for a total of four doses, AMPT was given at a dose of 100 mg/kg, was given i.p. 24, 16, 4 and 1 h before commencing METH treatment, and l-DOPA was given i.p at a dose of 50 mg/kg 1 h before and 3 h after starting METH treatment, always along with benserazide (25 mg/kg; i.p.) to block peripheral l-DOPA decarboxylation. Doses and schedules of drug administration for the AMPT/L-DOPA study were selected on the basis of previous studies in which METH was administered alone and in combination with other drugs (Yuan et al. 2001; Thomas et al. 2008). For studies involving DD mice, METH was given i.p. at a dose of 10 mg/kg, every 2 h for a total of four doses. The 10 mg/kg dose (instead of 7.5 mg/kg) of METH was selected because it has been used extensively in the past to produce DA lesions (Albers and Sonsalla 1995; see Cadet and Krasnova 2009) and because METH was not used in combination with other drugs in these studies.
Temperature measurements
Core (rectal) temperature was measured using a Bat-12 thermometer coupled to a RET-3 rectal probe (Physitemp, Inc., Clifton, NJ, USA). Temperature was measured at baseline and every 30 min after METH for up to 4–8 h. In experiments with DD mice that required artificial heating, heat lamps were used, as needed, to ‘yoke’ core temperature in control animals given METH. As before (Yuan et al. 2001), this was accomplished by carrying out drug treatments and temperature studies in control mice 1 day prior to carrying out identical studies in DD mice, under identical conditions.
Chemical and protein determinations
For DA and DOPAC determinations, striatal tissue was dissected 1 week after drug treatment and stored frozen in liquid nitrogen until assay by means of HPLC coupled with electrochemical detection, as described (Villemagne et al. 1998). DA transporter (DAT) levels were determined by binding of [3H]WIN-35,428, while VMAT was measured by binding of [3H]DTBZ as described (Villemagne et al. 1998). Tyrosine hydroxylase (TH) was detected in coronal 30-μm brain sections through the mid-brain by using rabbit anti-TH (1 : 2000; Chemicon, Temecula, CA, USA) and CY3-(TH) labeled IgG secondary antibodies (1 : 200; Jackson Immuno-Research, West Grove, PA, USA).
Glial fibrillary acidic protein immunohistochemistry
Mice were treated with METH (10 mg/kg i.p., every 2 h × 4; n = 3) or the vehicle (n = 3) and examined 3 days after treatment. Under deep pentobarbital anesthesia (40 mg/kg, i.p.), the animals underwent intracardiac perfusion with 10% formol saline. The brain was then removed from the skull and tissue was sectioned and processed for glial fibrillary acidic protein (GFAP) immunocytochemistry. Tissue blocks were placed in buffered 4% paraformaldehyde for 6 h and then in 10% dimethylsulfoxide in phosphate-buffered saline overnight. Blocks were frozen-sectioned (30 μm) using a sliding microtome and collected in cold phosphate-buffered saline. Free-floating sections were incubated at 4°C for 60 h with rabbit anti-GFAP diluted 1 : 40 in SuperBlock blocking buffer with 0.2% Triton X-100 and 1% normal goat serum. Bound immunoglobulins were visualized with the Vectastain ABC-peroxidase method and staining was enhanced with a standard osmiophilic reaction sequence.
Data analyses
Temperature results were analyzed by two-way analysis of variance (anova) for repeated measures with treatment as the between subjects factor and time as within subjects factor. When appropriate, group means at individual time points were compared by one-way anova, and post hoc comparisons were performed using Duncan’s Multiple Range Test. Striatal DA, DOPAC, DAT and VMAT data were analyzed by one-way anova, followed by Duncan’s Multiple Range post hoc comparisons. Correlations were explored using Pearson’s product moment correlation. Results were considered significant when p was less than 0.05, using a two-tailed test. Data analyses were performed using a statistical program for the social sciences (SPSS v.16, Chicago, IL, USA).
Results
In keeping with previous reports (Albers and Sonsalla 1995; Kuhn et al. 2008), METH injections produced increases in body temperature (Fig. 1). METH-induced increases in body temperature were attenuated by AMPT, particularly at the earlier time points (Fig. 1). l-DOPA increased body temperature in METH-treated animals pre-treated with AMPT (Fig. 1). AMPT alone and l-DOPA alone (or in combination with AMPT) tended to lower core temperature (not shown in figure for the sake of clarity).
Fig 1.
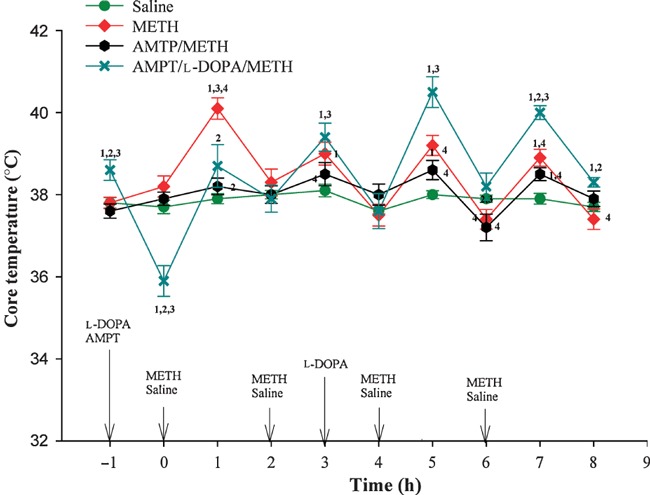
Effect of treatment with METH alone or in combination with AMPT and other drugs on core temperature in mice housed at an ambient temperature of 22 (± 1)°C. For doses and schedules of drug administration, please refer to Methods. Values represent the mean ± SEM.; n = 6–9 per group. 1Designates significant difference from saline control group, 2Designates significant difference from METH group, 3Designates significant difference from AMPT/METH group and 4Designates significant difference from AMPT/L-DOPA/METH group. Significant differences were determined by one-way anova followed by Duncan’s multiple range test. For the sake of clarity, control groups treated with only AMPT, l-DOPA or AMPT + l-DOPA are not shown in the figure.
Striatal DA and DOPAC levels measured 1 week after drug treatment in the various groups of mice described above showed that METH produced significant depletions of striatal DA and DOPAC, and that the effects of METH on DA and DOPAC levels were blocked by AMPT (Fig. 2). l-DOPA reversed the protective effects of AMPT on striatal DA and DOPAC concentrations (Fig. 2). METH also produced increases in GFAP staining (Fig. 3), suggesting that decreases in DA neuronal markers (DA, DOPAC and, as shown below, DAT and VMAT) are secondary to METH-induced dopaminergic neurotoxicity.
Fig 2.
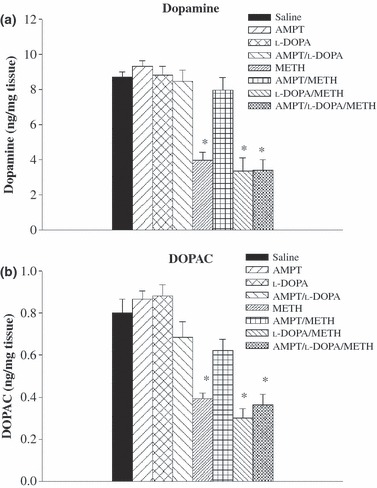
Effect of treatment with METH alone or in combination with other drug treatments on DA and DOPAC concentrations in the striata of mice whose core temperature changes after drug treatment are shown in Fig. 1. DA and DOPAC determinations were performed 1 week after drug treatment. For doses and schedules of drug administration, please see Methods. Values represent the mean ± SEM; n = 6–9 per group. *Designates significant difference from saline control group. Significant differences were determined by one-way anova followed by Duncan’s multiple range test.
Fig 3.
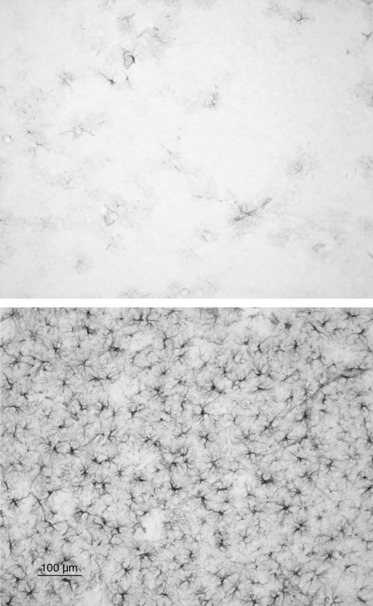
Astroglial activation, as revealed by increased in GFAP staining, in the striatum of DD mouse treated with METH (10 mg/kg, given i.p. every 2 h × 4) 3 days previously (lower panel) compared to control (upper panel).
There was a highly significant inverse relationship between body temperature and DA neurotoxicity in all METH-treated animals (i.e. animals treated with METH alone, METH plus AMPT, and METH plus AMPT plus l-DOPA), with animals achieving the highest core temperatures having the lowest DA levels 1 week after METH treatment, regardless of the drugs used in combination with METH (Fig. 4).
Fig 4.
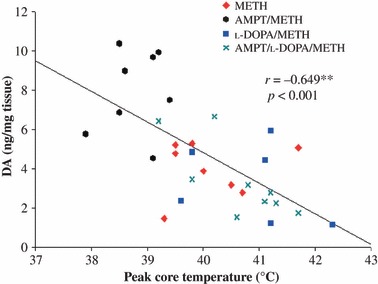
Significant inverse relationship between peak core temperature and striatal DA concentrations 1 week later in all METH-treated animals (i.e. METH alone, METH + AMPT and METH + AMPT + l-DOPA). Correlations were explored using Pearson’s product moment correlation. **p < 0.01.
To further examine the role of DA in METH neurotoxicity, we next evaluated the neurotoxic potential of METH in DD mice that were genetically engineered to have no DA by means of selective inactivation of the Th gene within DA neurons (Zhou and Palmiter 1995; Szczypka et al. 1999). Given the lack of DA (and, therefore, DOPAC) in these animals, DAT and VMAT served as surrogate DA nerve terminal markers in this experiment. As before, core temperature was monitored before, during and for a period after METH treatment, and DA nerve terminal markers were measured 1 week later. DD mice artificially warmed to develop comparable METH-induced increases in body temperature as those in control mice (Fig. 5) developed significant DAT and VMAT deficits (Fig. 6).
Fig 5.
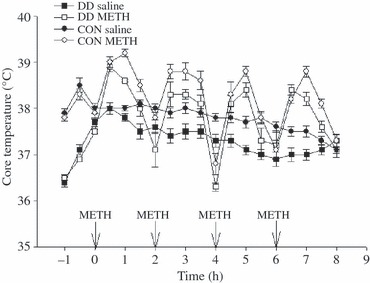
Body temperature responses of mice that received four i.p. injections of saline or METH (10 mg/kg) at 2-h intervals. Arrows indicate time of injection. DD mice were artificially warmed so that their temperature responses to METH paralleled those in WT mice. Values shown are the mean ± SEM; n = 5 per group.
Fig 6.
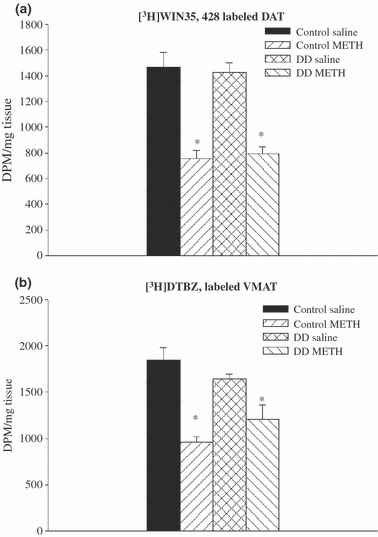
DAT and VMAT densities in the striata of DD and control mice treated 1 week previously with METH or saline. Core temperature responses in these animals are shown in Fig. 4. Values shown are the mean ± SEM; n = 5 per group. *Designates significant difference from control group. Significant differences were determined by one-way anova followed by Duncan’s multiple range test.
Although previous studies have shown that DA levels in DD mice are < 1% those in wild-type mice 16 h after treatment with l-DOPA (required for survival) (Szczypka et al. 1999; Kim et al. 2000), there was the remote possibility that a small amount of residual DA might have mediated the effect of METH on DA neuronal markers in DD mice. We, therefore, carried out an additional study in mice in which DA signaling had been restored to only one side of the striatum, DDfs mice (Hnasko et al. 2006; Darvas and Palmiter 2009). In contrast to DD mice with bilateral DA deficits, DDfs mice with unilateral DA restoration (Fig. 7) require no daily l-DOPA for survival (Hnasko et al. 2006; Darvas and Palmiter 2009). Further, these mice require no warming to develop METH-induced increases in body temperature comparable to those in controls (Fig. 7, panels A and B). As shown in Fig. 8(panels C and D), animals with DA restored to only the right striatum developed lasting METH-induced decreases in DAT density that were of comparable magnitude in both striata (right and left).
Fig 7.
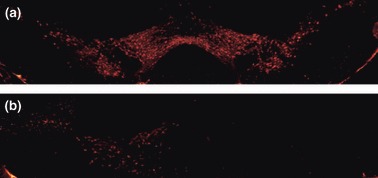
Tyrosine hydroxylase (TH) immunohistochemistry of coronal sections through midbrain of (a) control mouse (b) and a vrDD-DL mouse that had undergone unilateral DA rescue (see Methods). DD mice have no TH-positive cell bodies (Hnasko et al. 2006).
Fig 8.
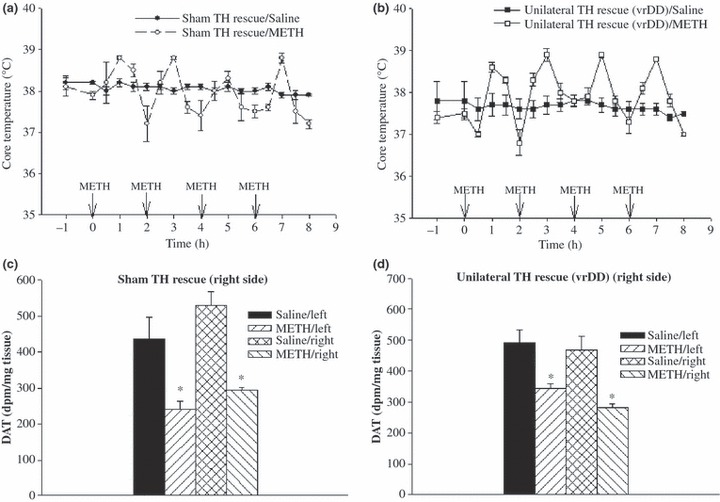
Core temperature responses (top panels) and striatal DAT densities (bottom panels) in unilateral rescue (vrDDL) mice and sham wild-type controls that received four i.p. injections of METH (10 mg/kg) or saline, at 2-h intervals. Core temperature was measured during the period of drug exposure; striatal DAT density was measured 1 week after drug or saline treatment. Given that rescue (or sham procedure) was unilateral and done on right side, right and left striata in each animal were analyzed separately. Note that METH elevates temperature in both sham and virally rescued mice and that the lesions (lasting loss of DAT) are the same on both sides – hence independent of DA. Values shown are the mean ± SEM; n = 4 per group. *Designates significant difference from ipsilateral control; significant differences were determined by Student’s t-test (p < 0.05).
Discussion
The importance of DA in METH-induced dopaminergic neurotoxicity has been largely inferred from pharmacological studies demonstrating a protective effect of the catecholamine synthesis inhibitor, AMPT, and a reversal of this effect by treatment with l-DOPA. The first such observation (Gibb and Kogan 1979) was made before the importance of temperature on METH-induced neurotoxicity had been discovered. Therefore, in these early studies in rats, temperature of animals was not measured. Further, DA markers in these studies were measured only 18 h after drug treatment, leaving open the possibility that the effects observed were related to pharmacological rather than toxic drug actions. More recently, Thomas et al. (2008) conducted a similar set of studies in mice, and these investigators also reached the conclusion that METH neurotoxicity was mediated by DA (newly synthesized pool). Of note, these recent studies also measured DA axonal markers only 2 days following drug treatment and, therefore, also may have been confounded by pharmacological drug effects. More importantly, as shown in the present study, l-DOPA not only has the potential to replenish DA stores depleted by AMPT but also to raise body temperature in METH-treated animals pre-treated with AMPT (Fig. 2). Thus, the effects of l-DOPA, like those of various other dopaminergic drugs on METH toxicity, are confounded by drug action on body temperature, making it impossible to exclude the possibility that effects of l-DOPA on body temperature, rather than on brain DA levels per se, underlie the ability of l-DOPA to reverse the neuroprotective effect of AMPT in METH-treated animals (Gibb and Kogan 1979; Thomas et al. 2008).
The important influence of temperature, as opposed to DA, in METH neurotoxicity, is readily apparent in an analysis of the relationship between peak temperature following METH and subsequent neurotoxic lesions in the four groups of METH-treated mice described here. In particular, peak temperature in all METH-treated mice (i.e. animals treated with METH alone, METH + AMPT. METH + l-DOPA and METH + AMPT + l-DOPA) is highly and significantly associated with extent of neurotoxicity (as measured by DA and DOPAC depletion 1 week later), both within and across treatment groups (Fig. 4). For example, animals that received METH + AMPT + l-DOPA and developed higher peak temperatures had greater DA deficits than those in the same group who developed lower peak temperatures. Further, lesion size in the METH + AMPT + l-DOPA group was similar to lesion size in the METH + AMPT group at any particular peak core temperature. These and previous observations (Bowyer et al. 1994; Albers and Sonsalla 1995; Yuan et al. 2001; Miller and O’Callaghan 2003) strongly suggest that it is the effects of AMPT, l-DOPA and other dopaminergic drugs on core temperature (rather than DA) that influence the expression of dopaminergic neurotoxicity in METH-treated animals. To our knowledge, there is no direct evidence that increased temperature per se can cause a lasting decrease of DA axon terminal markers such as DA or DAT, although other neural elements may be adversely affected by hyperthermia (see Kiyatkin 2010).
To further and more directly examine the importance of DA in METH neurotoxicity, we next evaluated the neurotoxic potential of METH in mice deficient in brain DA (DD mice). DD mice artificially warmed to develop comparable METH-induced increases in body temperature as METH-treated controls sustained DAT and VMAT decrements that were of comparable magnitude as those seen in wild-type controls (Fig. 6). This would not be expected if DA was essential for the expression of METH neurotoxicity. That is, if DA was needed for METH neurotoxicity, one would expect at least some decrement in METH-induced toxicity in DD mice, and this was not observed.
Because DD mice can have a small amount of residual DA (< 1% of control), there was the remote possibility that the small amount of remaining DA may have mediated the toxic effect of METH in DD animals. To exclude this possibility, an additional study was carried out using mice genetically engineered to lack DA on only one side of the brain (Hnasko et al. 2006; Darvas and Palmiter 2009). Unlike animals with bilateral DA deficits, animals with unilateral DA deficits do not require daily administration of l-DOPA for survival and develop normal temperature elevations after METH, thereby eliminating potential concerns regarding the effects of these factors (l-DOPA, warming) on experimental results. Further, because each animal serves as its own control, issues of inter-animal variability are eliminated. In animals with unilateral brain DA deficits, METH produced lasting bilateral DAT loss that was of comparable magnitude on both sides of the brain, indicating that expression of METH neurotoxicity was not influenced by the presence or absence of DA. Given this finding, it is extremely unlikely that DA is a mediator of METH-induced dopaminergic neurotoxicity.
Potential mechanisms of METH toxicity that are independent of endogenous DA warrant attention. These include: (i) a direct toxic effect of METH (or metabolite) on mitochondrial function leading to energy compromise within DA terminals (Thrash et al. 2009); (ii) ionic disturbance within the DA terminals resulting from prolonged interaction between METH and the DAT (Callahan et al. 2001); (iii) glutamate-mediated production of free radicals and oxidative stress (Yamamoto and Zhu 1998); (iv) mitochondrial K+ ATP channel opening or other molecular events that lead to activation of toxic cellular processes (see Goñi-Allo et al. 2008; Cadet and Krasnova 2009); and/or (v) activation of cell death cascades that result in neural injury of the type produced by METH which, notably, can include nerve cell bodies that do not contain DA (see Stirling and Stys 2010). At present, it is not clear which, if any, of these mechanisms is responsible for destruction of DA neuron terminals in METH-treated animals (see Riddle et al. 2006; Krasnova and Cadet 2009). However, as detailed elsewhere (McCann and Ricaurte 2004), whatever mechanism is at work must account for the marked influence of body temperature, the key role of the DAT and the sparing of noradrenergic neurons invariably observed in animals that sustain METH-induced DA neural injury.
It is important to acknowledge potential limitations of the present study. First, like previous pharmacological studies, the AMPT/L-DOPA studies described herein involved multiple drugs and complex drug administration paradigms that complicate study interpretation. Second, it is possible that temperature curves in artificially warmed DD animals with bilateral DA deficits did not perfectly model those in control animals, or that the small amount of residual DA in DD animals (< 1%) played a role. However, if DA was essential for METH toxicity, some attenuation of METH toxicity would be expected in DD animals, and this was not observed. Further, DD animals with unilateral DA deficits did not receive l-DOPA or require warming and these animals had similar METH-induced lesions on both sides of the brain, another finding that would not be expected if DA was the mediator of METH neurotoxicity. Finally, there is the outside possibility that DAT and VMAT decreases in METH-treated DD animals are not due primarily to DA terminal destruction but rather reflect METH-induced decreases in the synthesis of these proteins related to actions of METH on the cell nucleus (Asanuma et al. 2000). At odds with this possibility is the fact that METH-induced deficits in DA axon terminal markers are associated with signs of glial activation, a finding that is more in keeping with a toxic action of METH on DA axon terminals.
In conclusion, the present findings strongly indicate that endogenous DA is not essential for the development of METH-induced dopaminergic neurotoxicity. Additional research will be needed to identify the series of cellular and subcellular processes that mediate METH-induced DA dopaminergic neurotoxicity. In light of the present findings, neurodegenerative mechanisms independent of endogenous brain DA warrant increased attention.
Acknowledgments
We thank Valerie Z. Wall for maintaining the DD mouse colony. We thank Dr. Miguel Chillon (Vector Production Unit of Centre de Biotecnologia Animal i Teràpia Gènica at Universitat Autonoma Barcelona) for preparing the CAV2-Cre virus. This work was supported by PHS Grants DA05938 and DA01796401 (Ricaurte).
Glossary
- AMPT
alpha-methyl-para-tyrosine
- DA
dopamine
- DAT
DA transporter
- DD
DA-deficient
- DDfs
DA-deficient floxed stop
- DOPAC
dihydroxyphenylacetic acid
- GFAP
glial fibrillary acidic protein
- l-DOPA
l-dihydroxyphenylalanine
- METH
methamphetamine
- TH
tyrosine hydroxylase
- VMAT
vesicular monoamine transporter
References
- Albers DS, Sonsalla PK. Methamphetamine-induced hyperthermia and dopaminergic neurotoxicity in mice: pharmacological profile of protective and nonprotective gents. J. Pharmacol. Exp. Ther. 1995;275:1104–1114. [PubMed] [Google Scholar]
- Ali SF, Newport GD, Holson R, Slikker W, Jr, Bowyer JF. Low environmental temperatures or pharmacologic agents that produce hypothermia decrease methamphetamine neurotoxicity in mice. Brain Res. 1994;658:33–38. doi: 10.1016/s0006-8993(09)90007-5. [DOI] [PubMed] [Google Scholar]
- Asanuma M, Hayashi T, Ordonèz S, Ogawa N, Cadet JL. Direct interactions of methamphetamine with the nucleus. Brain Res. Mol. Brain Res. 2000;80:237–243. doi: 10.1016/s0169-328x(00)00128-5. [DOI] [PubMed] [Google Scholar]
- Bowyer JF, Tank AW, Newport GD, Slikker W, Jr, Ali SF, Holson RR. The influence of environmental temperature on the transient effects of methamphetamine on dopamine levels and dopamine release in rat striatum. J. Pharmacol. Exp. Ther. 1992;260:817–824. [PubMed] [Google Scholar]
- Bowyer JF, Davies DL, Schmued L, Broening HW, Newport GD, Slikker W, Jr, Holson RR. Further studies of the role of hyperthermia in methamphetamine neurotoxicity. J. Pharmacol. Exp. Ther. 1994;268:1571–1580. [PubMed] [Google Scholar]
- Bowyer JF, Thomas M, Schmued LC, Ali SF. Brain region-specific neurodegenerative profiles showing the relative importance of amphetamine dose, hyperthermia, seizures, and the blood-brain barrier. Ann. N Y Acad. Sci. 2008;1139:127–139. doi: 10.1196/annals.1432.005. [DOI] [PubMed] [Google Scholar]
- Cadet JL, Krasnova IN. Molecular bases of methamphetamine-induced neurodegeneration. Int. Rev. Neurobiol. 2009;88:101–119. doi: 10.1016/S0074-7742(09)88005-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callahan BT, Cord BJ, Yuan J, McCann UD, Ricaurte GA. Inhibitors of Na(+)/H(+)and Na(+)/Ca(2+) exchange potentiate methamphetamine-induced dopamine neurotoxicity: possible role of ionic dysregulation in methamphetamine neurotoxicity. J. Neurochem. 2001;77:1348–1362. doi: 10.1046/j.1471-4159.2001.00341.x. [DOI] [PubMed] [Google Scholar]
- Cappon GD, Morford L, Vorhees CV. Ontogeny of methamphetamine-induced neurotoxicity and associated hyperthermic response. Dev. Brain. Res. 1997;103:155–162. doi: 10.1016/s0165-3806(97)81791-9. [DOI] [PubMed] [Google Scholar]
- Darvas M, Palmiter RD. Restriction of dopamine signaling to the dorsolateral striatum is sufficient for many cognitive behaviors. Proc. Natl Acad. Sci. USA. 2009;106:14664–14669. doi: 10.1073/pnas.0907299106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibb JW, Kogan FJ. Influence of dopamine synthesis on methamphetamine-induced changes in striatal and adrenal tyrosine hydroxylase activity. Naunyn Schmiedebergs Arch. Pharmacol. 1979;310:185–187. doi: 10.1007/BF00500283. [DOI] [PubMed] [Google Scholar]
- Gibb JW, Hanson GR, Johnson M. Neurochemical mechanisms of toxicity. In: Cho AK, Segal DS, editors. Amphetamine and Its Analogs. New York: Academic Press; 1994. pp. 269–295. [Google Scholar]
- Goñi-Allo B, Puerta E, Ramos M, Lasheras B, Jordán J, Aguirre N. Minoxidil prevents 3,4-methylenedioxymethamphetamine-induced serotonin depletions: role of mitochondrial ATP-sensitive potassium channels, Akt and ERK. J. Neurochem. 2008;104:914–925. doi: 10.1111/j.1471-4159.2007.05042.x. [DOI] [PubMed] [Google Scholar]
- Guillot TS, Shepherd KR, Richardson JR, Wang MZ, Li Y, Emson PC, Miller GW. Reduced vesicular storage of dopamine exacerbates methamphetamine-induced neurodegeneration and astrogliosis. J. Neurochem. 2008;106:2205–2217. doi: 10.1111/j.1471-4159.2008.05568.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heusner CL, Hnasko TS, Szczypka MS, Liu Y, During MJ, Palmiter RD. Viral restoration of dopamine to the nucleus accumbens is sufficient to induce locomotor response to amphetamine. Brain Res. 2003;980:266–274. doi: 10.1016/s0006-8993(03)02986-x. [DOI] [PubMed] [Google Scholar]
- Hnasko TS, Perez FA, Scouras AD, Stoll EA, Gale SD, Luquet S, Phillips PE, Kremer EJ, Palmiter RD. Cre recombinase-mediated restoration of nigrostriatal dopamine in dopamine-deficient mice reverses hypophagia and bradykinesia. Proc. Natl Acad. Sci. USA. 2006;103:8858–8863. doi: 10.1073/pnas.0603081103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim DS, Palmiter RD, Cummins A, Gerfen CR. Reversal of supersensitive striatal dopamine D1 receptor signaling and extracellular signal-regulated kinase activity in dopamine-deficient mice. Neuroscience. 2000;137:1381–1388. doi: 10.1016/j.neuroscience.2005.10.054. [DOI] [PubMed] [Google Scholar]
- Kita T, Miyazaki I, Asanuma M, Takeshima M, Wagner GC. Dopamine-induced behavioral changes and oxidative stress in methamphetamine-induced neurotoxicity. Int. Rev. Neurobiol. 2009;88:43–64. doi: 10.1016/S0074-7742(09)88003-3. [DOI] [PubMed] [Google Scholar]
- Kiyatkin EA. Brain temperature homeostasis: physiological fluctuations and pathological shifts. Front. Biosci. 2010;1:73–92. doi: 10.2741/3608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krasnova IN, Cadet JL. Methamphetamine toxicity and messengers of death. Brain Res. Rev. 2009;60:379–407. doi: 10.1016/j.brainresrev.2009.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kremer EJ, Boutin S, Chillon M, Danos O. Canine adenovirus vectors: an alternative for adenovirus-mediated gene transfer. J. Virol. 2000;74:505–512. doi: 10.1128/jvi.74.1.505-512.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn DM, Francescutti-Verbeem DM, Thomas DM. Dopamine disposition in the presynaptic process regulates the severity of methamphetamine-induced neurotoxicity. Ann. N Y Acad. Sci. 2008;1139:118–126. doi: 10.1196/annals.1432.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lew R, Malberg JE, Ricaurte GA, Seiden LS. Evidence for and mechanism of action of neurotoxicity of amphetamine related compounds. In: Kostrzewa RM, editor. Highly Selective Neurotoxins: Basic and Clinical Applications. Totowa: Humana Press Inc; 1997. pp. 235–268. [Google Scholar]
- McCann UD, Ricaurte GA. Amphetamine neurotoxicity: accomplishments and remaining challenges. Neurosci. Biobehav. Rev. 2004;27:821–826. doi: 10.1016/j.neubiorev.2003.11.003. [DOI] [PubMed] [Google Scholar]
- Miller DB, O’Callaghan JP. Elevated environmental temperature and methamphetamine neurotoxicity. Environ. Res. 2003;92:48–53. doi: 10.1016/s0013-9351(02)00051-8. [DOI] [PubMed] [Google Scholar]
- Riddle EL, Fleckenstein AE, Hanson GR. Mechanisms of methamphetamine-induced dopaminergic neurotoxicity. AAPS J. 2006;8:E413–E418. doi: 10.1007/BF02854914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt CJ, Ritter JK, Sonsalla PK, Hanson GR, Gibb JW. Role of dopamine in the neurotoxic effects of methamphetamine. J. Pharmacol. Exp. Ther. 1985;233:539–544. [PubMed] [Google Scholar]
- Sharma HS, Sjöquist PO, Ali SF. Drugs of abuse-induced hyperthermia, blood-brain barrier dysfunction and neurotoxicity: neuroprotective effects of a new antioxidant compound H-290/51. Curr. Pharm. Des. 2007;13:1903–1923. doi: 10.2174/138161207780858375. [DOI] [PubMed] [Google Scholar]
- Stirling DP, Stys PK. Mechanisms of axonal injury: internodal nanocomplexes and calcium deregulation. Trends Mol. Med. 2010;16:160–170. doi: 10.1016/j.molmed.2010.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szczypka MS, Rainey MA, Kim DS, Alaynick WA, Marck BT, Matsumoto AM, Palmiter RD. Feeding behavior in dopamine-deficient mice. Proc. Natl Acad. Sci. USA. 1999;96:12138–12143. doi: 10.1073/pnas.96.21.12138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas DM, Francescutti-Verbeem DM, Kuhn DM. The newly synthesized pool of dopamine determines the severity of methamphetamine-induced neurotoxicity. J. Neurochem. 2008;105:605–616. doi: 10.1111/j.1471-4159.2007.05155.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thrash B, Karuppagounder SS, Uthayathas S, Suppiramaniam V, Dhanasekaran M. Neurotoxic effects of methamphetamine. Neurochem. Res. 2009;35:171–179. doi: 10.1007/s11064-009-0042-5. [DOI] [PubMed] [Google Scholar]
- Villemagne V, Yuan J, Wong DF, et al. Brain dopamine neurotoxicity in baboons treated with doses of methamphetamine comparable to those recreationally abused by humans: evidence from [11C]WIN-35,428 positron emission tomography studies and direct in vitro determinations. J. Neurosci. 1998;18:419–427. doi: 10.1523/JNEUROSCI.18-01-00419.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volz TJ, Fleckenstein AE, Hanson GR. Methamphetamine-induced alterations in monoamine transport: implications for neurotoxicity, neuroprotection and treatment. Addiction. 2007;102(Suppl. 1):44–48. doi: 10.1111/j.1360-0443.2007.01771.x. [DOI] [PubMed] [Google Scholar]
- Yamamoto BK, Zhu W. The effects of methamphetamine on the production of free radicals and oxidative stress. J. Pharmacol. Exp. Ther. 1998;287:107–114. [PubMed] [Google Scholar]
- Yuan J, Callahan BT, McCann UD, Ricaurte GA. Evidence against an essential role of endogenous brain dopamine in methamphetamine-induced dopaminergic neurotoxicity. J. Neurochem. 2001;77:1338–1347. doi: 10.1046/j.1471-4159.2001.00339.x. [DOI] [PubMed] [Google Scholar]
- Zhou QY, Palmiter RD. Dopamine-deficient mice are severely hypoactive, adipsic, and aphagic. Cell. 1995;83:1197–1209. doi: 10.1016/0092-8674(95)90145-0. [DOI] [PubMed] [Google Scholar]