

Abstract
Inhibition of platelet-derived growth factor-B (PDGF-B) has multiple effects on tumors, including loss of pericytes, regression of some vessels, normalization of other vessels, and reduction of interstitial pressure. PDGF-B inhibition also increases the efficacy of cancer therapeutics, but the role on tumor vessel efficiency and drug delivery is unclear. We sought to determine whether inhibition of PDGF-B signaling can increase delivery and efficacy of cyclophosphamide in Lewis lung carcinomas or RIP-Tag2 tumors. PDGF-B blockade in Lewis lung carcinoma tumors by the DNA aptamer AX102 for 14 days increased the number of perfused tumor vessels marked by lectin in the bloodstream by 50%. AX102 also increased the width of sleeves of viable tumor cells around blood vessels by 66%, increased tumor cell proliferation by 90%, and increased intratumoral delivery of Hoechst 33342 by 78%. A low dose of cyclophosphamide (20 mg/kg) reduced tumor cell proliferation by 31% when combined with AX102 but not when given alone. Synergy of cyclophosphamide and AX102 on tumor cell proliferation also was found in RIP-Tag2 tumors. Similarly, the PDGF receptor signaling inhibitor imatinib increased delivery of cyclophosphamide and reduced tumor burden in RIP-Tag2 mice, without evidence of tumor cell sensitization to chemotherapy. Together, these findings indicate that inhibition of PDGF-B signaling promotes the delivery and efficacy of chemotherapeutic agents by increasing the efficiency of tumor blood vessels.
Blood vessels in tumors are structurally and functionally abnormal and inefficient. Many tumor vessels are irregularly shaped; lack the normal hierarchical arrangement of arterioles, capillaries, and venules; and have endothelial cells that are disorganized and leaky.1,2 Pericytes, which contribute to the stability of normal blood vessels, do not have a normal association with endothelial cells of tumor vessels.1,2 These abnormalities of tumor vessels not only impair blood flow and delivery of nutrients and oxygen but also reduce the delivery of cancer therapeutics.2,3
Drugs that block vascular endothelial growth factor prevent angiogenesis, cause regression of some tumor vessels, and normalize tumor vessels that do not regress.4–7 The normalization of tumor vessels may improve the delivery of chemotherapy to tumors.2
Platelet-derived growth factor-B (PDGF-B) from endothelial cells and its receptor (PDGFR-β) on pericytes play important roles in pericyte recruitment to blood vessels.8-10 Pericytes contribute to blood vessel maturation, maintain normal endothelial cell function, promote vascular stability, and participate in the regulation of blood flow.11–15 In the absence of pericytes during development, blood vessels are leaky, have microaneurysms, continue to proliferate, and have impaired blood flow.15,16
Inhibition of PDGF-B signaling in tumors reduces pericyte coverage of tumor vessels, overall tumor vascularity, and interstitial pressure.6,17-19 Tumor vessels that are stripped of pericytes are preferentially pruned and leave behind tumor vessels that have a more normalized phenotype that can support more rapid tumor growth despite the reduced vascularity.6 PDGF-B inhibitors can increase the efficacy of low-dose metronomic chemotherapy,20 but it is unclear whether the increased efficacy is a consequence of normalization of tumor vessels, increased tumor vessel efficacy, and improved drug delivery after PDGF-B inhibition.6
The aim of the present study was to determine whether inhibition of PDGF-B can increase the efficiency of tumor blood vessels and improve the delivery and efficacy of the cytotoxic agent, cyclophosphamide (CTX), in tumors. To address this, we examined the effects of selective sequestration of PDGF-B by a DNA oligonucleotide aptamer (AX102, Archemix Corporation)6,17 or inhibition of PDGF receptor signaling by imatinib mesylate (Gleevec, Novartis)21 on Lewis lung carcinoma (LLC) and on islet cell tumors in RIP-Tag2 transgenic mice. Tumor vessel labeling by FITC–Lycopersicon esculentum lectin (FITC-LEA lectin), which was injected i.v. and circulated for 2 minutes, was used as an index of tumor vessel efficiency. We also measured the width of sleeves of viable perivascular tumor cells, abundance of proliferating tumor cells, and amount of Hoechst 33342 dye leakage around tumor vessels. Finally, we asked whether PDGF-B inhibition increased the antitumor effect of CTX in LLC and in RIP-Tag2 tumors. We found that inhibition of PDGF-B increased all indices of tumor vessel efficiency, improved the delivery of CTX, and augmented the reduction of tumor burden by CTX.
Materials and Methods
Animals and Treatment
LLC tumors were implanted in C57BL/6 mice at 8 to 10 weeks of age and allowed to grow for 5 to 7 days before treatment was started.6 RIP-Tag2 mice in a C57BL/6 background were treated beginning at 12 weeks of age. AX102 or vehicle (0.9% NaCl) was injected at a dose of 50 mg/kg i.p. once daily for 7, 14, 21, or 28 days. CTX (Sigma, St. Louis, MO) was given in a dose of approximately 20 mg/kg in the drinking water as previously described22 or 20 mg/kg or 105 mg/kg injected i.p. every other day for 7 days. In studies of AX102 and CTX administered together, mice were treated with vehicle or AX102 for 21 days and received an i.p. injection of vehicle or CTX at a dose of 20 or 105 mg/kg every other day during the final 7 days.
LLC-bearing mice used for functional studies of the tumor vasculature were treated with vehicle of AX102 for 14 days and then received an i.v. injection of one of several tracers. FITC-LEA lectin [1 mg/kg; molecular weight (MW), 100,000 Da; Vector Laboratories, Burlingame, CA] or Hoechst 33342 dye (20 mg/kg; MW, 616 Da; Invitrogen, Carlsbad, CA) was injected and allowed to circulate for 2 minutes before the perfusion of fixative. Dextran labeled with tetramethyl-rhodamine isothiocyanate (500 mg/kg; MW, 155,000 Da; Sigma), normal rat IgG (2.5 mg/kg; Jackson ImmunoResearch, West Grove, PA), or anti-fibrinogen antibody (2.5 mg/kg; Dako, Carpinteria, CA) was injected via tail vein 6 hours before the perfusion.23
Mice used in the studies of AX102 and CTX administered together received an injection of pimonidazole hydrochloride (Hypoxyprobe; 60 mg/kg i.p.; HPI, Inc., Burlington, MA) 1 hour before the perfusion. Hypoxic regions of tumor sections were detected using a FITC-conjugated mouse antibody that detects adducts of pimonidazole hydrochloride (1:100).
The effect of imatinib and CTX on tumor burden was examined in RIP-Tag2 mice treated for 28 days (8–17 mice per group). The mice were given food containing 50% sugar and provided water containing 5% sugar to offset the hypoglycemia associated with the functional insulinomas. Imatinib (University of California San Francisco Medical Center Pharmacy, San Francisco, CA) was administered by gavage at a total dose of 150 mg/kg/day, divided into a dose of 50 mg/kg in the morning and a dose of 100 mg/kg in the afternoon for 28 days. Mice received CTX at a dose of 105 mg/kg by i.p. injection on days 0, 2, and 4 followed by a 17-day rest period, and then on days 21, 23, and 25.22 After euthanasia, tumors larger than 1 mm2 were excised, measured, and the tumor volume was calculated from the following formula: V = a × b2 × Π/6, where a is the longest dimension and b is the shortest dimension. All experimental procedures were approved by and performed in accordance with the University of California, San Francisco, Institutional Animal Care and Use Committee.
Fixation and IHC
After the tissues were preserved by systemic vascular perfusion of fixative (1% paraformaldehyde in phosphate-buffered saline), the tumors were removed and frozen, sectioned with a cryostat (thickness, 60–80 μm), and stained by IHC with combinations of two or three primary antibodies.5,6 Endothelial cells were marked by hamster anti-CD31 (platelet endothelial cell adhesion molecule 1, clone 2H8, 1:500; Thermo Scientific, Hudson, NH) or rat anti-CD31 (platelet endothelial cell adhesion molecule 1, clone MEC 13.3, 1:500; Pharmingen, San Diego, CA), pericytes by Cy3-conjugated mouse anti–α-smooth muscle actin (α-SMA; 1:1000; Sigma), proliferating cells by rabbit anti–phosphohistone-H3 (PHH3; 1:1000; Millipore/Upstate Biotechnology, Billerica, MA), apoptotic cells by rabbit anti-activated caspase-3 (caspase-3; 1:1000; R&D Systems, Minneapolis, MN), and erythrocytes by rat anti–TER-119 (1:250; BD Biosciences, Bedford, MA).
Imaging and Image Analysis
Specimens were examined with a Zeiss Axiophot fluorescence microscope equipped with single-, dual-, and triple-fluorescence filters and a low-light, externally cooled, three-chip, charge-coupled device camera (480 × 640 pixel RGB color images, CoolCam; SciMeasure Analytical Systems, Atlanta, GA) and a Zeiss LSM 510 laser-scanning confocal microscope with argon, helium-neon, and UV lasers (1024 × 1024 pixel RGB color images).
The number of functional tumor vessels was estimated by measuring the proportion of vessels stained for CD31 (all vessels, red channel) that were labeled by FITC-LEA lectin (functional vessels, green channel) after i.v. injection. The extent of colocalization of the two fluorophores was measured in digital images of 80-μm sections of LLC tumors (10× objective, 1× Optovar) by using the Colocalization plug-in function of ImageJ. Colocalization was defined as pixels that had fluorescence intensities of the red and green channels equal to or greater than the threshold value, ranging from 30 to 45. The threshold was determined by measuring the fluorescence intensity (background fluorescence) of tumors in mice not injected with FITC-LEA lectin. Colocalization was expressed as the percentage of FITC-LEA lectin pixels that colocalized with CD31 pixels.
The area density of CD31, FITC-LEA lectin, and other immunofluorescence staining recorded in digital microscopic images were calculated as the proportion of pixels having a fluorescence intensity equal to or greater than an empirically determined threshold value of 30 to 50 with ImageJ software.24 The mean fluorescence intensity of FITC-LEA lectin was determined by calculating the sum of the pixels at each intensity, times its corresponding intensity, divided by the total number of pixels. Values <25 were considered background and excluded.
Pericyte coverage was estimated from tumor vessels stained for CD31 and for α-SMA–positive pericytes by counting the vessels that had α-SMA staining or not. The results are presented as the percentage of tumor blood vessels with α-SMA–positive pericyte coverage.
The thickness of sleeves of viable tumor cells around blood vessels, extending from the abluminal surface of CD31 staining to the nearest region of apoptotic cells marked by activated caspase-3 staining, was measured using Zeiss LSM software on confocal images of LLC tumors treated with vehicle or AX102 for 14 days.
The area density of proliferating tumor cells marked by PHH3 immunoreactivity was measured in viable, nonhypoxic (pimonidazole-negative) regions of LLC tumors of mice treated with vehicle or AX102 in combination with CTX. The area density of PHH3-positive cells was measured within regions of interest outlined with the freehand selection tool of ImageJ.
In Vitro Sensitivity to Antiproliferative Effect of 4-OH-CTX
βTC-3 cells, a cell line established from a RIP-Tag2 tumor,25 were seeded into 96-well plates at a density of 104 cells per well, allowed to attach, and incubated for 2 days with or without 3 μmol/L imatinib plus 0 to 100 μmol/L 4-OH-Cyclophosphamide (4-OH-CTX), the active metabolite of CTX. Cell viability then was measured by the CellTiter 96 AQueous Non-Radioactive Cell Proliferation assay based on cellular reduction of the tetrazolium product MTS [3-(4,5-dimethylthiazol-2-yl)−5-(3-carboxymethoxyphenyl)−2-(4-sulfophenyl)-2H-tetrazolium], and subsequent spectrophotometric measurement (Promega, Madison, WI).25
Measurement of 4-OH-CTX in RIP-Tag2 Tumors
The CTX metabolite 4-OH-CTX was measured26 in tumors of RIP-Tag2 mice pretreated with vehicle or imatinib for 5 days before i.p. injection of 105 mg/kg 4-OH-CTX 1 hour after the final dose of vehicle/imatinib. Groups of mice (n = 4 for each treatment and time point) were euthanized at 1 or 6 hours after the injection of CTX. Tumors were excised, snap frozen, and pulverized with a mortar and pestle. Tumors from all mice in each group were combined into one trapping tube to ensure derivatization of CTX in a manner compatible with gas chromatography–mass spectrometry measurement of 4-OH-CTX concentration. Gas chromatography–mass spectrometry was performed in duplicate samples from each treatment group.
Statistical Analysis
Values expressed as means ± SEM reflect 3 to 6 mice per experimental group unless noted otherwise. Significance of differences was determined using a two-sided, unpaired Student's t-test or analysis of variance followed by the Fisher PLSD post hoc test. Differences with P values <0.05 were considered statistically significant.
Results
Increased Tumor Vessel Efficiency after Inhibition of PDGF-B by AX102
Blood vessels in LLC tumors and RIP-Tag2 tumors are known to have a more normal appearance after inhibition of PDGF-B by AX102.6 To determine whether the normalized vessels were more efficient, we assessed the pericyte coverage and determined the following: i) the proportion of vessels that were labeled by FITC-LEA lectin injected into the bloodstream, ii) the width of the sleeve of perivascular tumor cells, iii) the abundance of proliferating tumor cells, and iv) the amount of leakage of Hoechst 33342 dye or macromolecular tracers.
Blood vessels accompanied by α-SMA–positive pericytes were scattered in untreated LLC tumors, but many vessels lacked pericytes that stained for α-SMA (Figure 1A, arrows). After AX102 daily for 14 days, tumor vascularity and abundance of α-SMA pericytes were decreased (Figure 1A and Sennino et al6), but the proportion of vessels having α-SMA pericytes was increased from a baseline value of 40% to 59% after AX102 (Figure 1A).
Figure 1.
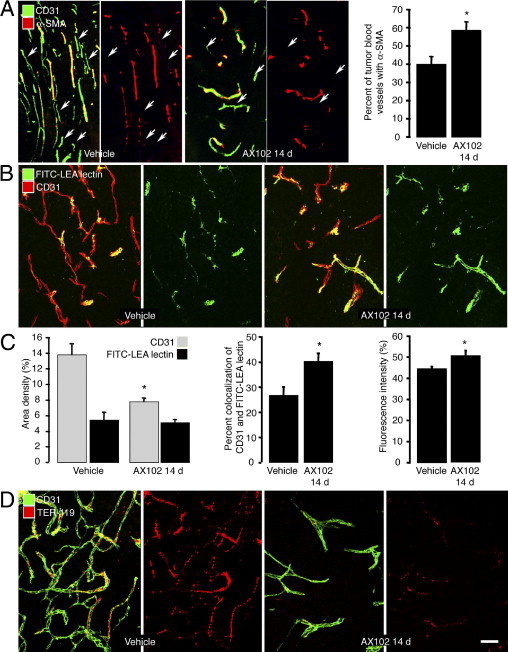
Greater pericyte coverage and patency of LLC tumor blood vessels after AX102. Confocal microscopic images of pericytes (α-SMA, red) on blood vessels (CD31, green) in LLC tumors after treatment with vehicle or AX102 for 14 days (A). Proportion of tumor vessels covered by α-SMA–positive pericytes (A, graph). Identification of functional blood vessels (CD31, red) in LLC tumors by green staining after i.v. injection of FITC-labeled LEA lectin revealed that many vessels in LLC tumors were not stained green under baseline conditions (B). By comparison, after AX102 for 14 days, tumor vessels were less numerous, but most had lectin staining (B). Measurements showed that, after treatment with AX102 for 14 days, tumor vascularity was reduced significantly, but the abundance of lectin-stained vessels was unchanged (C, left), the proportion of surviving vessels that had lectin staining was significantly greater (C, middle), and the fluorescence intensity of the lectin staining was stronger (C, right). Many blood vessels (CD31, green) in vehicle-treated LLC tumors contained erythrocytes (TER-119, red) despite vascular perfusion of fixative, but after AX102 for 14 days few tumor vessels had trapped erythrocytes (D). *P < 0.05 compared to vehicle. Scale bars: 150 μm (A); 50 μm (B and D).
Labeling of tumor vessels by FITC-lectin was used as one indicator of blood vessel function in LLC tumors.5 Essentially all normal vessels were labeled by the lectin within minutes of i.v. injection, but fewer than half the vessels in untreated LLC tumors had lectin staining (Figure 1B). By comparison, after AX102 for 14 days, tumor vascularity was decreased but the abundance of lectin-stained vessels was unchanged and the proportion of lectin-stained vessels was greater (Figure 1, B and C). Measurements revealed that in AX102-treated tumors, overall vascularity was reduced 44%, but the abundance of lectin-stained vessels was reduced by only 6%, the proportion of tumor vessels stained by the lectin increased 51%, and the fluorescence intensity of the lectin was increased 13% (Figure 1C).
The presence of stagnant blood or intravascular coagulation, marked by the presence of erythrocytes (TER-119 immunoreactivity) in tumor vessels, was used as another indicator of vessel functionality at baseline and after AX102. Because erythrocytes were washed out of functionally normal blood vessels by vascular perfusion of fixative, any TER-119 staining indicated the presence of erythrocytes that could not be washed away. Many vessels in untreated LLC tumors had TER-119 staining, but few vessels had TER-119 staining after AX102 (Figure 1D).
The width of the sleeve of viable tumor cells around blood vessels, which is dependent on the availability of oxygen and nutrients,27 served as another functional indicator. Blood vessels in LLC tumors were surrounded by a sleeve of viable tumor cells that lacked activated caspase-3 immunoreactivity (Figure 2A). Beyond this region, staining for activated caspase-3 was abundant (Figure 2A). After treatment with AX102 for 14 days, perivascular sleeves of viable cells, from the edge of the blood vessel to the rim of apoptotic cells, were 49% wider (Figure 2A, graph).
Figure 2.
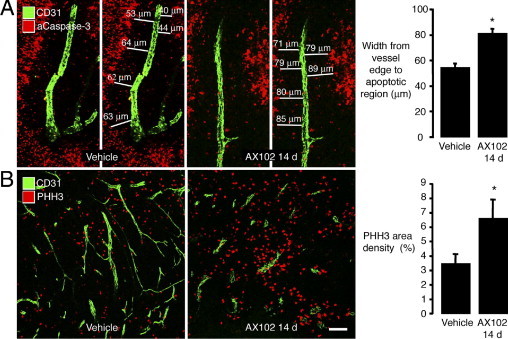
Greater perivascular cell proliferation in LLC tumors after AX102. Confocal microscopic images of blood vessels (CD31, green) and apoptotic cells (activated caspase-3, red) in LLC tumors with widths of sleeve of nonapoptotic tumor cells around tumor vessels measured from vessel to region of activated caspase staining (A). The sleeve of nonapoptotic tumor cells was significantly wider after AX102 for 14 days than after vehicle (A, graph). Fluorescent micrographs illustrating conspicuously more proliferating cells (PHH3, red) around blood vessels (CD31, green) in LLC tumors treated for 14 days with AX102 than with vehicle (B). The area density of PHH3 staining was significantly greater after AX102 (B, graph). *P < 0.05 compared to vehicle. Scale bars: 50 μm (A), 100 μm (B).
Another indicator of tumor viability was the presence of proliferating cells identified by PHH3 immunoreactivity, which was scattered in LLC tumors at baseline (Figure 2B). After AX102 for 14 days, cells with PHH3 immunoreactivity were almost double the abundance in the control (Figure 2B, graph).
Leakage of Hoechst 33342 dye, which binds to nuclear DNA, was used as a further indicator of vascular function in LLC tumors. After the dye was injected i.v. and circulated for 2 minutes, staining was more homogeneous in tumors treated with AX102 for 14 days than in those treated with vehicle (Figure 3A). Measurements showed that the area density of staining was 78% greater after AX102 (Figure 3A, graph).
Figure 3.
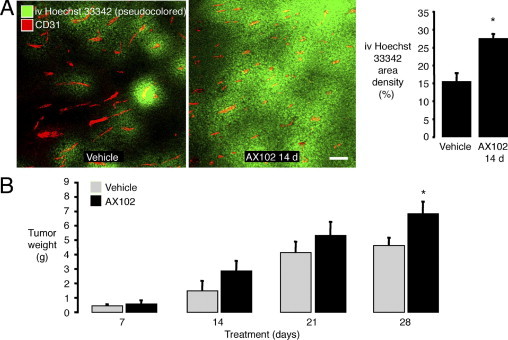
More widespread Hoechst 33342 dye leakage in LLC tumors after AX102. Fluorescence microscopic images of blood vessels (CD31, red) and Hoechst 33342 dye (pseudocolored green) in LLC tumors at 2 minutes after i.v. injection (A). Hoechst 33342 dye in LLC tumors was significantly more widespread after AX102 for 14 days than after vehicle (A, graph). Weight of LLC tumors was significantly greater after AX102 for 28 days than after vehicle (B). *P < 0.05 compared to vehicle. Scale bar = 100 μm.
To determine whether AX102 amplified the leakage of Hoechst 33342 by destabilizing tumor vessels as a result of PDGF-B blockade, we compared the amount and distribution of extravasation of three macromolecular tracers that have been used to assess leakage in tumors.23,28 Tetramethyl-rhodamine isothiocyanate-labeled dextran, normal rat IgG, or antifibrinogen antibody was injected i.v. into mice with LLC tumors, and after circulation for 6 hours the extravasated tracers were examined microscopically. All of the tracers were found to accumulate at the tumor–host interface, as reported previously for LLC tumors.23 Treatment with AX102 for 14 days had no noticeable effect on the amount or distribution of the three extravasated tracers (see Supplemental Figure S1 at http://ajp.amjpathol.org).
The overall consequence of PDGF-B blockade on vascular function in LLC tumors was tested by comparing tumor growth during 28 days of treatment with vehicle or AX102. Tumors treated with AX102 tended to be larger after 14 and 21 days of treatment and by 28 days were significantly larger than corresponding vehicle-treated controls (Figure 3B).
Dose-Dependent Effect of CTX on Vascularity and Cell Proliferation in LLC Tumors
The magnitude of the effect of CTX on cell proliferation in LLC tumors treated for 7 days varied with the dose and route of administration. Comparison of the effects of CTX in the drinking water, which gave a dose of approximately 20 mg/kg/day, and parenteral (i.p.) CTX at a dose of 20 or 105 mg/kg every other day revealed a reduction in tumor vascularity only with continuous oral dosing, but reductions in cell proliferation after all treatments, with the higher parenteral dose resulting in the largest reduction (Figure 4, A–C). The robust effect of 7-day parenteral CTX on cell proliferation without a reduction in tumor vascularity made it possible to separate the antitumor action of CTX from changes in the number of blood vessels.
Figure 4.
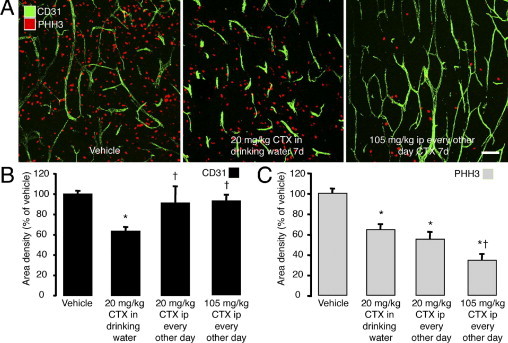
Dose-dependent effect of CTX on vascularity and cell proliferation in LLC tumors. Confocal microscopic images of blood vessels (CD31, green) and proliferating cells (PHH3, red) in LLC tumors treated with vehicle or CTX for 7 days (A). Tumors treated by low-dose CTX (20 mg/kg) given continuously in the drinking water for 7 days had fewer blood vessels and fewer proliferating cells than vehicle-treated controls (B and C). CTX given by i.p. injection every other day for 7 days at a low dose of CTX (20 mg/kg) or at the maximum tolerated dose (105 mg/kg) reduced cell proliferation but did not reduce tumor vascularity (B and C). P < 0.05 compared to *vehicle or †20 mg/kg CTX in drinking water. Scale bar = 100 μm.
Greater Efficacy of CTX on LLC Tumors When Given Together with AX102
To examine the interaction of CTX and PDGF-B inhibition in LLC tumors, we compared the effects of treatment with vehicle or CTX, at a dose of 20 or 105 mg/kg, given alone or in combination with AX102. The abundance of proliferating cells, marked by PHH3 immunoreactivity, was used as a readout. Proliferating cells were abundant in LLC tumors treated with vehicle and were even more numerous in the presence of AX102 (Figure 5A, left top and bottom, and 5B).
Figure 5.
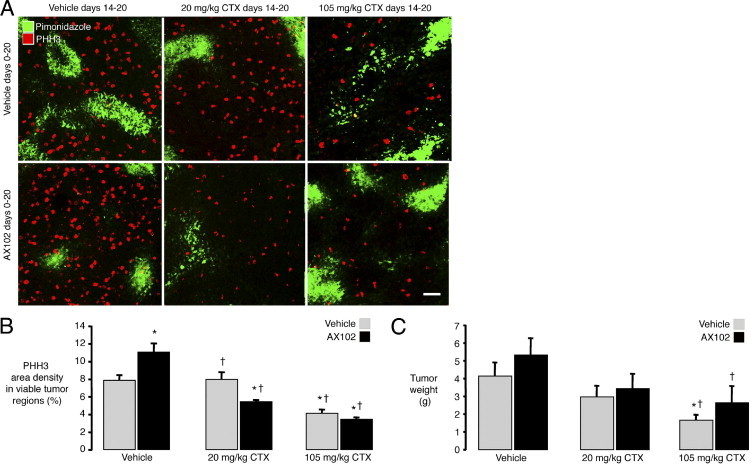
Greater efficacy of CTX on LLC tumors when given together with AX102. Confocal microscopic images of proliferating cells (PHH3, red) and hypoxic regions (pimonidazole, green) in LLC tumors treated with vehicle, 20 mg/kg CTX, or 105 mg/kg CTX on days 14 to 20 together with vehicle or AX102 on days 0 through 20 (A). Proliferating cells were more numerous in nonhypoxic regions of tumors than in hypoxic regions marked by pimonidazole (A). PHH3 staining was reduced significantly by treatment with 20 mg/kg CTX when accompanied by AX102 but not when accompanied by vehicle (A and B). PHH3 staining was reduced even more by a high dose of CTX (105 mg/kg), but the reduction was not augmented by the addition of AX102. Tumor weights were reduced by CTX in a dose-dependent manner (C). P < 0.05 compared to *vehicle + vehicle or †vehicle + AX102. Scale bar = 75 μm.
The lower dose of CTX (20 mg/kg i.p., every other day) had no effect on cell proliferation when given alone but caused a significant reduction in proliferation when combined with AX102 (Figure 5A, middle top and bottom, and 5B). By comparison, the higher dose of CTX (105 mg/kg i.p., every other day) caused similar reductions in proliferating tumor cells regardless of whether it was combined with vehicle (48% reduction) or AX102 (56% reduction) (Figure 5A, right top and bottom, and 5B).
When assessed at the end of the 21-day treatment schedule, the weight of LLC tumors was not significantly reduced by the lower dose of CTX (20 mg/kg), but tumor weight after the higher dose of CTX (105 mg/kg) was reduced by 67% when it was given with vehicle and by 43% when given with AX102 (Figure 5C).
Greater Efficacy of CTX on RIP-Tag2 Tumors When Given with AX102
To determine whether the effects of AX102 on CTX delivery were unique to LLC tumors, we performed similar experiments on spontaneous pancreatic islet tumors of RIP-Tag2 transgenic mice. Although treatment of RIP-Tag2 tumors with AX102 for 21 days without CTX caused a small (25%) but significant reduction in tumor vascularity (Figure 6A), it did not result in the increase in proliferating cells (PHH3 staining) as found in LLC tumors (Figure 6, B and C). This may be explained by the greater abundance of proliferating cells at baseline in RIP-Tag2 tumors (area density, 26%) than in LLC tumors (area density, 3%–8%). Importantly, however, in RIP-Tag2 tumors as found in LLC tumors, the lower dose of CTX (20 mg/kg) caused a significant (43%) reduction in proliferating cells when combined with AX102 but not when combined with vehicle (6% reduction). Also, similar to the findings in LLC tumors, the higher dose of CTX (105 mg/kg) caused relatively similar reductions in proliferating cells when combined with vehicle (54% reduction) or with AX102 (64% reduction) (Figure 6C). These findings are consistent with AX102 increasing the efficacy of CTX when given in submaximal doses but not at the maximal tolerated dose.
Figure 6.
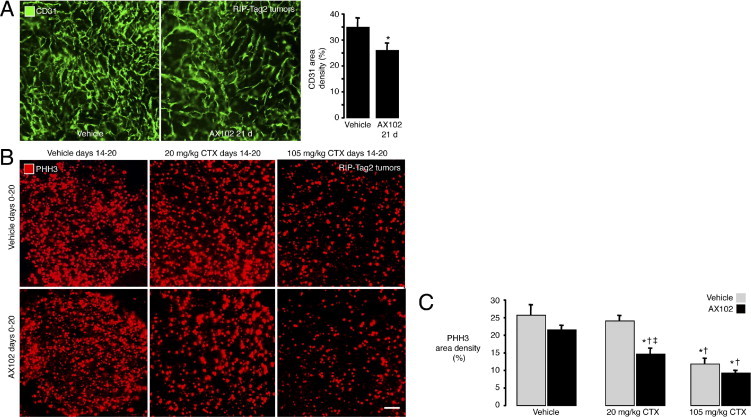
Greater efficacy of CTX on RIP-Tag2 tumors when given with AX102. Microscopic images showing small but significant reduction in vascularity (CD31, green) of RIP-Tag2 tumors after AX102 for 21 days (A), with confirmation by area density measurements (A, graph). Fluorescence microscopic images of RIP-Tag2 tumors compare the number of proliferating cells stained for PHH3 (red) after treatment with vehicle, 20 mg/kg CTX, or 105 mg/kg CTX on days 14 to 20, without or with AX102 on days 0 through 20 (B). Amount of PHH3 staining in RIP-Tag2 tumors was not reduced by 20 mg/kg CTX given with vehicle but was reduced significantly when given together with AX102 (C). PHH3 staining was reduced after the high dose of CTX (105 mg/kg) given alone and was not reduced further by concurrent AX102. *P < 0.05 compared to vehicle (A) or vehicle + vehicle (C). †P < 0.05 compared to vehicle + AX102. ‡P < 0.05 compared to 20 mg/kg CTX + vehicle. Scale bars: 80 μm (A), 90 μm (B).
Synergy of Imatinib and CTX in RIP-Tag2 Tumors in Vivo but Not in Vitro
As another strategy to test the effect of inhibition of PDGF-B signaling on the action of CTX on RIP-Tag2 tumors, we used the receptor tyrosine kinase inhibitor imatinib in place of AX102. Imatinib was chosen for these studies because it is known to increase the effect of CTX on RIP-Tag2 tumors20 and currently is used in the clinic. Tumor burden was the readout. Tumor burden naturally increased in RIP-Tag2 mice from 12 to 15 weeks of age (Figure 7A). Tumor growth was not slowed by imatinib by itself, but was stopped by a high dose of CTX (105 mg/kg, every other day) alone. However, CTX given together with imatinib not only stopped tumor growth but also reduced tumor burden to a level below that present at the onset of treatment (Figure 7A).
Figure 7.
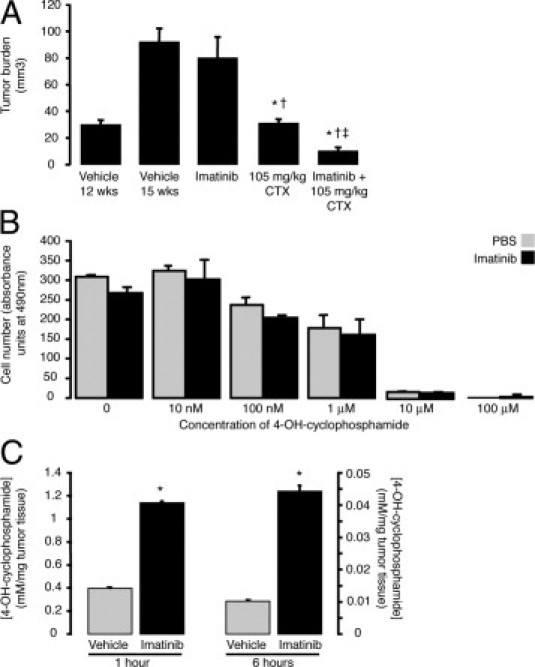
Synergy of imatinib and CTX in RIP-Tag2 tumors in vivo but not in vitro. Tumor growth in RIP-Tag2 mice treated from 12 weeks of age was unaffected by imatinib alone daily for 28 days, but was stopped by CTX alone and was reversed by CTX administered together with imatinib, indicative of synergistic actions of the two agents in vivo (A). However, imatinib did not increase the effect of a broad range of concentrations of 4-OH-CTX on tumor cell proliferation in vitro, consistent with an indirect effect of imatinib on tumor cells in vivo (B). Pretreatment with imatinib for 5 days led to significantly higher 4-OH-CTX concentrations in RIP-Tag2 tumors at both 1 (left) and 6 hours (right) after an injection of CTX (C). P < 0.05 compared to *vehicle, †imatinib alone, or ‡CTX alone.
To determine whether this reduction in tumor burden was a consequence of imatinib increasing the sensitivity of tumor cells to CTX, we performed in vitro studies with the βTC-3 cell line derived from RIP-Tag2 tumors and a standard MTS based cell viability assay (CellTiter 96; Promega). The addition of imatinib did not increase the effect of 4-OH-CTX on βTC-3 cell proliferation at any 4-OH-CTX concentration tested, from 10 nmol/L to 100 μmol/L (Figure 7B).
We therefore determined whether imatinib instead increased the delivery of CTX to tumors. Here, we compared the amount of CTX in tumors at 1 or 6 hours after a dose of 4-OH-CTX in RIP-Tag2 mice after pretreatment with vehicle or imatinib for 5 days. Gas chromatography–mass spectrometry measurements revealed that the concentration of 4-OH-CTX in tumors was significantly greater at both time points in imatinib-pretreated mice (Figure 7C).
Discussion
The purpose of this study was to determine whether inhibitors of PDGF-B signaling can increase the delivery and efficacy of chemotherapy by improving the efficiency of tumor blood vessels. Our experiments in preclinical tumor models provide multiple lines of evidence for this mechanism of synergy between PDGF-B inhibitors and cytotoxic agents. In particular, we found that selective PDGF-B blockade by AX102 reduced the vascularity of LLC tumors, but the surviving vessels had greater pericyte coverage and were functionally more efficient. Evidence of improved tumor vessel efficiency included fewer stagnated erythrocytes, stronger and more uniform staining by a circulating fluorescent lectin, more widespread leakage of Hoechst 33342 dye, wider sleeves of proliferating perivascular tumor cells, and faster tumor growth.
Administration of AX102 together with CTX at a low dose (20 mg/kg i.p., every other day) reduced cell proliferation in tumors, but this dose of CTX had little effect when given alone. This synergy of AX102 and CTX was found both in LLC and in RIP-Tag2 tumors. Similarly, administration of CTX with imatinib, which blocks PDGF-receptor signaling, not only stopped growth of RIP-Tag2 tumors but also reduced tumor burden to below the level present at the onset of treatment. By comparison, when given alone, CTX stopped growth but did not cause regression of tumors, and imatinib did not even slow tumor growth. The synergy was probably the result of increased CTX delivery to tumors, rather than sensitization of tumor cells to CTX, because imatinib did not increase the effect of CTX in vitro. Together, these results indicate that inhibition of PDGF-B signaling improved the delivery and antiproliferative effect of CTX.
Strategies to Improve Drug Delivery to Tumors
Many strategies are being explored to improve the delivery of cytotoxic agents to tumors. Liposomes, nanoparticles, bacteria-derived minicells, and antibody–drug conjugates are among the approaches.29–34 Strategies that promote vascular dilatation, constriction, leakiness, or normalization also are being tried to promote delivery of chemotherapeutics.20,35–42 Our studies extend previous work indicating that the delivery and efficacy of CTX can be improved by inhibition of PDGF-B signaling.20
Paradoxical Effects of AX102 on Tumor Growth
We used AX102 to block the actions of PDGF-B because of its selectivity, robust effects on tumor blood vessels, and seemingly paradoxical actions of reducing vascularity while stimulating growth of LLC tumors.6 Use of AX102 provided an opportunity to determine whether the apparent paradox could be explained by increased efficiency of tumor vessels that did not regress after PDGF-B blockade. As the highest-affinity ligand for PDGF-β, PDGF-B is essential for pericyte function in normal organs and in tumors.6,43–46 Balance of signaling PDGF-B and other growth factors is required for normal pericyte function and for blood vessel stability. This balance is lost in tumor microenvironments, where the stoichiometry of cytokines is profoundly disturbed, but is somehow recalibrated by inhibitors that result in vessel normalization.
Synergy of AX102 or Imatinib and Cyclophosphamide
We examined the effect of inhibition of PDGF-B signaling on tumor vessel efficiency and on the antiproliferative action of CTX in LLC and RIP-Tag2 tumors. The two models provided complementary data and made it possible to determine whether the reduction in tumor cell proliferation was owing to increased vascular delivery of CTX or to an additive effect of PDGF-B signaling inhibition and CTX on tumor cells. LLC tumors are known to be sensitive to PDGF-B inhibition.6 Pancreatic islet tumors in RIP-Tag2 mice form spontaneously, are highly vascular, have abundant pericytes, and lack large regions of necrosis,5,44,47,48 which can interfere with measurement of drug accumulation in tumors.
We found that CTX caused a greater reduction in tumor cell proliferation when given together with AX102 or imatinib. This synergy was accompanied by increased tumor delivery of CTX without sensitization of tumor cells to CTX. A likely basis of the synergy is improved efficiency of tumor vessels as a feature of vessel normalization after inhibition of PDGF-B signaling.6 Tumor vessels, which are usually leaky and have impaired blood flow, can become more normal during anti-angiogenic therapy.49 Vascular endothelial growth factor inhibitors are known to cause pruning of tumor vessels, which decreases the surface area for molecules in plasma to extravasate, but the normalization of the surviving vessels can compensate by providing more efficient delivery of nutrients, oxygen, and drugs.2
Inhibition of PDGF-B by AX102 can cause regression of approximately 80% of blood vessels in LLC tumors.6 Many vessels that survive have a more normal appearance. To determine whether the surviving tumor vessels are more efficient, we determined the proportion of functional tumor vessels, extent of Hoechst 33342 dye leakage into tumors, and amount of perivascular tumor cell viability and proliferation. As expected, after AX102, LLC tumors had fewer blood vessels,6 but most of those remaining were patent and functional, as reflected by fluorescent LEA lectin staining, Hoechst 33342 dye leakage, and thickness and proliferation rate of perivascular tumor cells.
Greater Importance of Vessel Function Than Number
The functional properties of tumor vessels are more important than the number of vessels that survive anti-angiogenic therapy. Inhibition of Delta-like ligand 4 promotes vessel sprouting and angiogenesis, but the new blood vessels are poorly perfused, and the tumors grow more slowly.50 The opposite occurred in LLC tumors after AX102, where fewer tumor vessels led to faster growing tumors (shown here and by Sennino et al6). These results are consistent with previous studies showing LLC tumors grow faster in mice that lack the PDGF-B retention motif.51
Transvascular transport of drugs into tumors is dependent on multiple factors, including drug concentration, blood flow, effective vascular surface area determined by the number and size of functional vessels, endothelial barrier function, and transmural driving force.35,52 The balance of intravascular and interstitial hydrostatic and colloid osmotic pressures determines the transmural driving force. Vessel normalization is thought to increase the driving force by decreasing intravascular resistance. Inhibition of PDGF-B also can decrease interstitial pressure in tumors through effects on stromal and perivascular cells.53
Extravasation of low-molecular-weight drugs depends largely on diffusion, but transport of macromolecules is governed more by convection.35,52 AX102 increased the leakage of low-molecular-weight Hoechst 33342 dye (MW, 616 Da) and the delivery of cyclophosphamide (MW, 279 Da), but did not noticeably change the leakage of macromolecular dextran (MW, 155,000 Da) or IgG (MW, 150,000 Da). These findings favor improved intratumoral blood flow over changes in endothelial barrier function or transmural driving force as a more important effect of AX102. Greater leakage of Hoechst 33342 argues that better vessel function outweighs any reduction in vascular surface area from vessel pruning. Improved vessel function, manifested by increased blood flow, reduced leakage, and reduced interstitial pressure, would all be expected to improve delivery and efficacy of chemotherapeutics.2
Improved blood flow also would influence the rate of cell proliferation in tumors through increased availability of oxygen and nutrients. The mitotic index in tumors decreases with increasing distance from functional blood vessels.54 Our findings of thicker perivascular sleeves of tumor cells and more cellular proliferation after treatment with AX102 are consistent with this mechanism.
Dosage Regimen of Cyclophosphamide
To explore possible synergy of AX102 and CTX, we injected CTX i.p. every other day because this regimen is standard for studying CTX at its maximum tolerated dose.55 This regimen also made it possible to examine the effect of CTX on tumor cell proliferation without an appreciable decrease in tumor vascularity. Concurrent administration of AX102 increased the efficacy of CTX when given at the lower dose of 20 mg/kg. However, AX102 did not have this synergistic effect at the maximum tolerated dose, 105 mg/kg, the dose at which CTX reached the tumor in a sufficient amount to suppress cell proliferation despite abnormalities in the tumor vasculature. However, the higher dose also resulted in greater systemic drug exposure. The combination of AX102 with a lower dose of CTX provides the promise of therapeutic efficacy with fewer side effects.
Other studies have illustrated the influence of CTX dose on mechanism of action and outcome. In the presence of low-dose, metronomic CTX, PDGF-B inhibition can amplify the reduction in tumor growth by sensitizing tumor vessel endothelial cells to the cytotoxic agent.20 Further studies are needed to determine whether the reduction in tumor vascularity found with continuous, low-dose CTX in drinking water would offset the improvement in CTX delivery after inhibition of PDGF-B signaling.
Variability in Tumor Response to PDGF-B Inhibition
AX102 increased the efficacy of low-dose CTX in reducing cell proliferation in both LLC and RIP-Tag2 tumors. Imatinib increased the efficacy of CTX at reducing tumor burden in RIP-Tag2 mice, when both agents were given over a period of 28 days and the maximal tolerated dose of CTX was used. Despite consistent synergy of the drug combinations, some effects of AX102 used as a single agent were tumor-specific. AX102 increased cell proliferation and growth of LLC tumors, as has been reported after PDGF inhibition in other tumor models,17,21,51 but this did not occur in RIP-Tag2 tumors. Such tumor-related differences could provide an opportunity to identify biomarkers that predict tumor response to these inhibitors. Experiments showing the beneficial effects of imatinib administered with CTX illustrate that continuous administration of both drugs is likely to be more efficacious than pretreatment with a PDGF signaling inhibitor before beginning the cytotoxic agent.
Conclusions
In conclusion, the results of these experiments show that treatment with inhibitors of PDGF-B signaling can make tumor vessels more efficient through normalization of the vessel wall. Normalized blood vessels can support a larger number of proliferating tumor cells and increase the growth of some tumors. However, the improved function of tumor vessels and corresponding changes in the tumor interstitium also can provide more efficient delivery of small-molecule cytotoxic agents. By promoting these changes, selective PDGF-B inhibitors can improve the antitumor effect of chemotherapeutics, even when overall tumor vascularity is reduced, with the potential of better efficacy at a lower dose and reduced side effects.
Acknowledgments
We thank Karen Olson (Archemix Corp., Cambridge, MA) for providing AX102 and invaluable advice and helpful discussions, Ryan Naylor (University of California San Francisco, San Francisco, CA) for critical review of the manuscript, and Susan Ludeman and Ivan Spasojevic (Duke University, Durham, NC) for measurement of 4-OH-CTX in tumors.
Footnotes
This research was supported in part by National Institutes of Health grants CA82923 from the National Cancer Institute; grants HL24136, HL59157, and HL96511 from the National Heart, Lung, and Blood Institute; a grant from Archemix Corp.; funding from The AngelWorks Foundation (D.M.M.); and a postdoctoral fellowship from the Tobacco-Related Disease Research Program (#14FT-0152 to B.L.F.).
None of the authors disclosed any relevant financial relationships.
Supplemental material for this article can be found at http://ajp.amjpathol.org or at doi: 10.1016/j.ajpath.2011.02.019.
Current address of D.H., Swiss Institute for Experimental Cancer Research (ISREC), Ecole Polytechnique Fédérale de Lausanne, Switzerland; of B.L.F., Lilly Research Laboratories, Eli Lilly and Company, Indianapolis, Indiana.
Supplementary data
Leakage of macromolecular tracers in LLC tumors. Extravasation of red fluorescent dextran (MW, 155,000 Da) (A), normal rat IgG (B), or antifibrinogen antibody (C) at the border of LLC tumors perfused with fixative 6 hours after injection of the macromolecular tracer. Tumor vessels marked by CD31 (green). The distribution of extravasated tracer was similar in tumors regardless of whether they were treated with vehicle or AX102 for 14 days.
References
- 1.Baluk P., Hashizume H., McDonald D.M. Cellular abnormalities of blood vessels as targets in cancer. Curr Opin Genet Dev. 2005;15:102–111. doi: 10.1016/j.gde.2004.12.005. [DOI] [PubMed] [Google Scholar]
- 2.Jain R.K. Normalization of tumor vasculature: an emerging concept in antiangiogenic therapy. Science. 2005;307:58–62. doi: 10.1126/science.1104819. [DOI] [PubMed] [Google Scholar]
- 3.Jain R.K. Molecular regulation of vessel maturation. Nat Med. 2003;9:685–693. doi: 10.1038/nm0603-685. [DOI] [PubMed] [Google Scholar]
- 4.Falcon B.L., Hashizume H., Koumoutsakos P., Chou J., Bready J.V., Coxon A., Oliner J.D., McDonald D.M. Contrasting actions of selective inhibitors of angiopoietin-1 and angiopoietin-2 on the normalization of tumor blood vessels. Am J Pathol. 2009;175:2159–2170. doi: 10.2353/ajpath.2009.090391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Inai T., Mancuso M., Hashizume H., Baffert F., Haskell A., Baluk P., Hu-Lowe D.D., Shalinsky D.R., Thurston G., Yancopoulos G.D., McDonald D.M. Inhibition of vascular endothelial growth factor (VEGF) signaling in cancer causes loss of endothelial fenestrations, regression of tumor vessels, and appearance of basement membrane ghosts. Am J Pathol. 2004;165:35–52. doi: 10.1016/S0002-9440(10)63273-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sennino B., Falcon B.L., McCauley D., Le T., McCauley T., Kurz J.C., Haskell A., Epstein D.M., McDonald D.M. Sequential loss of tumor vessel pericytes and endothelial cells after inhibition of platelet-derived growth factor B by selective aptamer AX102. Cancer Res. 2007;67:7358–7367. doi: 10.1158/0008-5472.CAN-07-0293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Winkler F., Kozin S.V., Tong R.T., Chae S.S., Booth M.F., Garkavtsev I., Xu L., Hicklin D.J., Fukumura D., di Tomaso E., Munn L.L., Jain R.K. Kinetics of vascular normalization by VEGFR2 blockade governs brain tumor response to radiation: role of oxygenation, angiopoietin-1, and matrix metalloproteinases. Cancer Cell. 2004;6:553–563. doi: 10.1016/j.ccr.2004.10.011. [DOI] [PubMed] [Google Scholar]
- 8.Armulik A., Abramsson A., Betsholtz C. Endothelial/pericyte interactions. Circ Res. 2005;97:512–523. doi: 10.1161/01.RES.0000182903.16652.d7. [DOI] [PubMed] [Google Scholar]
- 9.Betsholtz C., Lindblom P., Gerhardt H. Role of pericytes in vascular morphogenesis. EXS. 2005;94:115–125. doi: 10.1007/3-7643-7311-3_8. [DOI] [PubMed] [Google Scholar]
- 10.Enge M., Bjarnegard M., Gerhardt H., Gustafsson E., Kalen M., Asker N., Hammes H.P., Shani M., Fassler R., Betsholtz C. Endothelium-specific platelet-derived growth factor-B ablation mimics diabetic retinopathy. EMBO J. 2002;21:4307–4316. doi: 10.1093/emboj/cdf418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Allt G., Lawrenson J.G. Pericytes: cell biology and pathology. Cells Tissues Organs. 2001;169:1–11. doi: 10.1159/000047855. [DOI] [PubMed] [Google Scholar]
- 12.Hellstrom M., Kalen M., Lindahl P., Abramsson A., Betsholtz C. Role of PDGF-B and PDGFR-beta in recruitment of vascular smooth muscle cells and pericytes during embryonic blood vessel formation in the mouse. Development. 1999;126:3047–3055. doi: 10.1242/dev.126.14.3047. [DOI] [PubMed] [Google Scholar]
- 13.Hirschi K.K., D'Amore P.A. Pericytes in the microvasculature. Cardiovasc Res. 1996;32:687–698. [PubMed] [Google Scholar]
- 14.Lindahl P., Hellstrom M., Kalen M., Betsholtz C. Endothelial-perivascular cell signaling in vascular development: lessons from knockout mice. Curr Opin Lipidol. 1998;9:407–411. doi: 10.1097/00041433-199810000-00004. [DOI] [PubMed] [Google Scholar]
- 15.von Tell D., Armulik A., Betsholtz C. Pericytes and vascular stability. Exp Cell Res. 2006;312:623–629. doi: 10.1016/j.yexcr.2005.10.019. [DOI] [PubMed] [Google Scholar]
- 16.Hellstrom M., Gerhardt H., Kalen M., Li X., Eriksson U., Wolburg H., Betsholtz C. Lack of pericytes leads to endothelial hyperplasia and abnormal vascular morphogenesis. J Cell Biol. 2001;153:543–553. doi: 10.1083/jcb.153.3.543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lu C., Shahzad M.M., Moreno-Smith M., Lin Y.G., Jennings N.B., Allen J.K., Landen C.N., Mangala L.S., Armaiz-Pena G.N., Schmandt R., Nick A.M., Stone R.L., Jaffe R.B., Coleman R.L., Sood A.K. Targeting pericytes with a PDGF-B aptamer in human ovarian carcinoma models. Cancer Biol Ther. 2010;9:176–182. doi: 10.4161/cbt.9.3.10635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pietras K., Rubin K., Sjoblom T., Buchdunger E., Sjoquist M., Heldin C.H., Ostman A. Inhibition of PDGF receptor signaling in tumor stroma enhances antitumor effect of chemotherapy. Cancer Res. 2002;62:5476–5484. [PubMed] [Google Scholar]
- 19.Song S., Ewald A.J., Stallcup W., Werb Z., Bergers G. PDGFRbeta+ perivascular progenitor cells in tumours regulate pericyte differentiation and vascular survival. Nat Cell Biol. 2005;7:870–879. doi: 10.1038/ncb1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pietras K., Hanahan D. A multitargeted, metronomic, and maximum-tolerated dose “chemo-switch” regimen is antiangiogenic, producing objective responses and survival benefit in a mouse model of cancer. J Clin Oncol. 2005;23:939–952. doi: 10.1200/JCO.2005.07.093. [DOI] [PubMed] [Google Scholar]
- 21.Pietras K., Pahler J., Bergers G., Hanahan D. Functions of paracrine PDGF signaling in the proangiogenic tumor stroma revealed by pharmacological targeting. PLoS Med. 2008;5:e19. doi: 10.1371/journal.pmed.0050019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Man S., Bocci G., Francia G., Green S.K., Jothy S., Hanahan D., Bohlen P., Hicklin D.J., Bergers G., Kerbel R.S. Antitumor effects in mice of low-dose (metronomic) cyclophosphamide administered continuously through the drinking water. Cancer Res. 2002;62:2731–2735. [PubMed] [Google Scholar]
- 23.Nakahara T., Norberg S.M., Shalinsky D.R., Hu-Lowe D.D., McDonald D.M. Effect of inhibition of vascular endothelial growth factor signaling on distribution of extravasated antibodies in tumors. Cancer Res. 2006;66:1434–1445. doi: 10.1158/0008-5472.CAN-05-0923. [DOI] [PubMed] [Google Scholar]
- 24.Abramoff M.D., Magelhaes P.J., Ram S.J. Image processing with ImageJ. Biophotonics Int. 2004;11:36–42. [Google Scholar]
- 25.Efrat S., Linde S., Kofod H., Spector D., Delannoy M., Grant S., Hanahan D., Baekkeskov S. Beta-cell lines derived from transgenic mice expressing a hybrid insulin gene-oncogene. Proc Natl Acad Sci U S A. 1988;85:9037–9041. doi: 10.1073/pnas.85.23.9037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ichikawa T., Petros W.P., Ludeman S.M., Fangmeier J., Hochberg F.H., Colvin O.M., Chiocca E.A. Intraneoplastic polymer-based delivery of cyclophosphamide for intratumoral bioconversion by a replicating oncolytic viral vector. Cancer Res. 2001;61:864–868. [PubMed] [Google Scholar]
- 27.Hirst D.G., Hirst V.K., Joiner B., Prise V., Shaffi K.M. Changes in tumour morphology with alterations in oxygen availability: further evidence for oxygen as a limiting substrate. Br J Cancer. 1991;64:54–58. doi: 10.1038/bjc.1991.238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tozer G.M., Akerman S., Cross N.A., Barber P.R., Bjorndahl M.A., Greco O., Harris S., Hill S.A., Honess D.J., Ireson C.R., Pettyjohn K.L., Prise V.E., Reyes-Aldasoro C.C., Ruhrberg C., Shima D.T., Kanthou C. Blood vessel maturation and response to vascular-disrupting therapy in single vascular endothelial growth factor-A isoform-producing tumors. Cancer Res. 2008;68:2301–2311. doi: 10.1158/0008-5472.CAN-07-2011. [DOI] [PubMed] [Google Scholar]
- 29.MacDiarmid J.A., Mugridge N.B., Weiss J.C., Phillips L., Burn A.L., Paulin R.P., Haasdyk J.E., Dickson K.A., Brahmbhatt V.N., Pattison S.T., James A.C., AL Bakri G., Straw R.C., Stillman B., Graham R.M., Brahmbhatt H. Bacterially derived 400 nm particles for encapsulation and cancer cell targeting of chemotherapeutics. Cancer Cell. 2007;11:431–445. doi: 10.1016/j.ccr.2007.03.012. [DOI] [PubMed] [Google Scholar]
- 30.Baker J.H., Lam J., Kyle A.H., Sy J., Oliver T., Co S.J., Dragowska W.H., Ramsay E., Anantha M., Ruth T.J., Adam M.J., Yung A., Kozlowski P., Minchinton A.I., Ng S.S., Bally M.B., Yapp D.T. Irinophore C, a novel nanoformulation of irinotecan, alters tumor vascular function and enhances the distribution of 5-fluorouracil and doxorubicin. Clin Cancer Res. 2008;14:7260–7271. doi: 10.1158/1078-0432.CCR-08-0736. [DOI] [PubMed] [Google Scholar]
- 31.Chari R.V. Targeted cancer therapy: conferring specificity to cytotoxic drugs. Acc Chem Res. 2008;41:98–107. doi: 10.1021/ar700108g. [DOI] [PubMed] [Google Scholar]
- 32.Li S., Wang A., Jiang W., Guan Z. Pharmacokinetic characteristics and anticancer effects of 5-fluorouracil loaded nanoparticles. BMC Cancer. 2008;8:103. doi: 10.1186/1471-2407-8-103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Malam Y., Loizidou M., Seifalian A.M. Liposomes and nanoparticles: nanosized vehicles for drug delivery in cancer. Trends Pharmacol Sci. 2009;30:592–599. doi: 10.1016/j.tips.2009.08.004. [DOI] [PubMed] [Google Scholar]
- 34.Chen X., Wang X., Wang Y., Yang L., Hu J., Xiao W., Fu A., Cai L., Li X., Ye X., Liu Y., Wu W., Shao X., Mao Y., Wei Y., Chen L. Improved tumor-targeting drug delivery and therapeutic efficacy by cationic liposome modified with truncated bFGF peptide. J Control Release. 2010;145:17–25. doi: 10.1016/j.jconrel.2010.03.007. [DOI] [PubMed] [Google Scholar]
- 35.Netti P.A., Hamberg L.M., Babich J.W., Kierstead D., Graham W., Hunter G.J., Wolf G.L., Fischman A., Boucher Y., Jain R.K. Enhancement of fluid filtration across tumor vessels: implication for delivery of macromolecules. Proc Natl Acad Sci U S A. 1999;96:3137–3142. doi: 10.1073/pnas.96.6.3137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dickson P.V., Hagedorn N.L., Hamner J.B., Fraga C.H., Ng C.Y., Stewart C.F., Davidoff A.M. Interferon beta-mediated vessel stabilization improves delivery and efficacy of systemically administered topotecan in a murine neuroblastoma model. J Pediatr Surg. 2007;42:160–165. doi: 10.1016/j.jpedsurg.2006.09.052. [DOI] [PubMed] [Google Scholar]
- 37.Dickson P.V., Hamner J.B., Sims T.L., Fraga C.H., Ng C.Y., Rajasekeran S., Hagedorn N.L., McCarville M.B., Stewart C.F., Davidoff A.M. Bevacizumab-induced transient remodeling of the vasculature in neuroblastoma xenografts results in improved delivery and efficacy of systemically administered chemotherapy. Clin Cancer Res. 2007;13:3942–3950. doi: 10.1158/1078-0432.CCR-07-0278. [DOI] [PubMed] [Google Scholar]
- 38.Dickson P.V., Hamner J.B., Streck C.J., Ng C.Y., McCarville M.B., Calabrese C., Gilbertson R.J., Stewart C.F., Wilson C.M., Gaber M.W., Pfeffer L.M., Skapek S.X., Nathwani A.C., Davidoff A.M. Continuous delivery of IFN-beta promotes sustained maturation of intratumoral vasculature. Mol Cancer Res. 2007;5:531–542. doi: 10.1158/1541-7786.MCR-06-0259. [DOI] [PubMed] [Google Scholar]
- 39.Sonveaux P. Provascular strategy: targeting functional adaptations of mature blood vessels in tumors to selectively influence the tumor vascular reactivity and improve cancer treatment. Radiother Oncol. 2008;86:300–313. doi: 10.1016/j.radonc.2008.01.024. [DOI] [PubMed] [Google Scholar]
- 40.Campbell N.E., Greenaway J., Henkin J., Moorehead R.A., Petrik J. The thrombospondin-1 mimetic ABT-510 increases the uptake and effectiveness of cisplatin and paclitaxel in a mouse model of epithelial ovarian cancer. Neoplasia. 2010;12:275–283. doi: 10.1593/neo.91880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Huang G., Chen L. Recombinant human endostatin improves anti-tumor efficacy of paclitaxel by normalizing tumor vasculature in Lewis lung carcinoma. J Cancer Res Clin Oncol. 2010;136:1201–1211. doi: 10.1007/s00432-010-0770-6. [DOI] [PubMed] [Google Scholar]
- 42.Sounni N.E., Dehne K., van Kempen L., Egeblad M., Affara N.I., Cuevas I., Wiesen J., Junankar S., Korets L., Lee J., Shen J., Morrison C.J., Overall C.M., Krane S.M., Werb Z., Boudreau N., Coussens L.M. Stromal regulation of vessel stability by MMP14 and TGFbeta. Dis Model Mech. 2010;3:317–332. doi: 10.1242/dmm.003863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Abramsson A., Lindblom P., Betsholtz C. Endothelial and nonendothelial sources of PDGF-B regulate pericyte recruitment and influence vascular pattern formation in tumors. J Clin Invest. 2003;112:1142–1151. doi: 10.1172/JCI18549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bergers G., Song S., Meyer-Morse N., Bergsland E., Hanahan D. Benefits of targeting both pericytes and endothelial cells in the tumor vasculature with kinase inhibitors. J Clin Invest. 2003;111:1287–1295. doi: 10.1172/JCI17929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Andrae J., Gallini R., Betsholtz C. Role of platelet-derived growth factors in physiology and medicine. Genes Dev. 2008;22:1276–1312. doi: 10.1101/gad.1653708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gaengel K., Genove G., Armulik A., Betsholtz C. Endothelial-mural cell signaling in vascular development and angiogenesis. Arterioscler Thromb Vasc Biol. 2009;29:630–638. doi: 10.1161/ATVBAHA.107.161521. [DOI] [PubMed] [Google Scholar]
- 47.Sennino B., Kuhnert F., Tabruyn S.P., Mancuso M.R., Hu-Lowe D.D., Kuo C.J., McDonald D.M. Cellular source and amount of vascular endothelial growth factor and platelet-derived growth factor in tumors determine response to angiogenesis inhibitors. Cancer Res. 2009;69:4527–4536. doi: 10.1158/0008-5472.CAN-08-3779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Paez-Ribes M., Allen E., Hudock J., Takeda T., Okuyama H., Vinals F., Inoue M., Bergers G., Hanahan D., Casanovas O. Antiangiogenic therapy elicits malignant progression of tumors to increased local invasion and distant metastasis. Cancer Cell. 2009;15:220–231. doi: 10.1016/j.ccr.2009.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fukumura D., Jain R.K. Tumor microvasculature and microenvironment: targets for anti-angiogenesis and normalization. Microvasc Res. 2007;74:72–84. doi: 10.1016/j.mvr.2007.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Noguera-Troise I., Daly C., Papadopoulos N.J., Coetzee S., Boland P., Gale N.W., Lin H.C., Yancopoulos G.D., Thurston G. Blockade of Dll4 inhibits tumour growth by promoting non-productive angiogenesis. Nature. 2006;444:1032–1037. doi: 10.1038/nature05355. [DOI] [PubMed] [Google Scholar]
- 51.Nisancioglu M.H., Betsholtz C., Genove G. The absence of pericytes does not increase the sensitivity of tumor vasculature to vascular endothelial growth factor-A blockade. Cancer Res. 2010;70:5109–5115. doi: 10.1158/0008-5472.CAN-09-4245. [DOI] [PubMed] [Google Scholar]
- 52.Jain R.K. Delivery of molecular and cellular medicine to solid tumors. Adv Drug Deliv Rev. 2001;46:149–168. doi: 10.1016/s0169-409x(00)00131-9. [DOI] [PubMed] [Google Scholar]
- 53.Pietras K., Ostman A., Sjoquist M., Buchdunger E., Reed R.K., Heldin C.H., Rubin K. Inhibition of platelet-derived growth factor receptors reduces interstitial hypertension and increases transcapillary transport in tumors. Cancer Res. 2001;61:2929–2934. [PubMed] [Google Scholar]
- 54.Tannock I.F. The relation between cell proliferation and the vascular system in a transplanted mouse mammary tumour. Br J Cancer. 1968;22:258–273. doi: 10.1038/bjc.1968.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Emmenegger U., Man S., Shaked Y., Francia G., Wong J.W., Hicklin D.J., Kerbel R.S. A comparative analysis of low-dose metronomic cyclophosphamide reveals absent or low-grade toxicity on tissues highly sensitive to the toxic effects of maximum tolerated dose regimens. Cancer Res. 2004;64:3994–4000. doi: 10.1158/0008-5472.CAN-04-0580. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Leakage of macromolecular tracers in LLC tumors. Extravasation of red fluorescent dextran (MW, 155,000 Da) (A), normal rat IgG (B), or antifibrinogen antibody (C) at the border of LLC tumors perfused with fixative 6 hours after injection of the macromolecular tracer. Tumor vessels marked by CD31 (green). The distribution of extravasated tracer was similar in tumors regardless of whether they were treated with vehicle or AX102 for 14 days.