

Abstract
Cells survive exposure to bacterial pore-forming toxins, such as streptolysin O (SLO), through mechanisms that remain unclear. Previous studies have suggested that these toxins are cleared by endocytosis. However, the experiments reported here failed to reveal any evidence for endocytosis of SLO, nor did they reveal any signs of damage to endosomal membranes predicted from such endocytosis. Instead, we illustrate that SLO induces a characteristic form of plasma membrane blebbing that allows cells to shed SLO by the process known as ectocytosis. Specifically, ‘deep-etch’ electron microscopy of cells exposed to SLO illustrates that the toxin is rapidly sequestered into domains in the plasmalemma greatly enriched in SLO pores, and these domains bleb outwards and bud from the cell surface into the medium. Such ectocytosis is even observed in cells that have been chemically fixed before exposure to SLO, suggesting that it is caused by a direct physical action of the toxin on the cell membrane, rather than by an active cellular reaction. We conclude, therefore, that ectocytosis is an important means for SLO clearance and hypothesize that this is a primary method by which cells defend themselves generally against pore-forming toxins.
Key words: Blebbing, Ectocytosis, Pore-forming toxin, Cytolysin
Introduction
Many strains of bacteria secrete pore-forming toxins (PFTs) that are important for virulence (Awad et al., 1995; Cossart et al., 1989; Hirst et al., 2004; Limbago et al., 2000; Portnoy et al., 1988). These PFTs generally bind to cholesterol in target-cell membranes and oligomerize into complexes of 35–50 subunits that form ~30-nm pores in their membranes (Rossjohn et al., 2007; Tweten, 2005). By doing so, they can severely permeabilize cells and cause lysis and death. However, cells can survive exposure to low sublytic doses of bacterial PFTs, although the mechanism(s) for how they do so remain largely unknown; all that is known for certain is that cell survival depends upon a Ca2+-dependent mechanism (Walev et al., 2001). Proposals for what this mechanism might be (reviewed by Aroian and van der Goot, 2007) have included: active pore-disassembly, pore-blockade through the formation of some type of cytoplasmic ‘clot’, and endocytosis of the pores. The current model for the inactivation of the archetypal bacterial PFT streptolysin O (SLO) is that endocytosis serves as the primary mechanism (Idone et al., 2008; Thiery et al., 2010).
Endocytosis has also been implicated in eliminating other PFTs from the plasmalemma, including Staphylococcus aureus α-toxin and Vibrio cholera cytolysin (Gutierrez et al., 2007; Husmann et al., 2009). However, if endocytosis were the primary survival mechanism that cells used to clear PFTs, cells would probably have to deal with the problem of potentially permeabilizing their endosomal membranes, which could expose them to toxic hydrolases from their lysosomal systems. Indeed, the PFT listeriolysin O does not act until after it is endocytosed, simply because its activity is pH dependent (Geoffroy et al., 1987). Likewise, other bacteria have evolved complex strategies to ensure endocytosis of their pathogenic products, such as that of the AB family of toxins, where one subunit triggers uptake and delivery of a second, toxic subunit. For example, the AB-toxin of Bacillus anthracis possesses an ‘A’ subunit (the protective antigen) that promotes tyrosine phosphorylation of its own receptor in order to provoke uptake of the ‘B’ subunit (the lethal factor) (Abrami et al., 2010). However, B. anthracis also expresses the PFT anthrolysin O (Shannon et al., 2003), leaving it unclear why an AB mechanism would be necessary if anthrolysin O itself were capable of triggering endocytosis.
These varieties of bacterial attack, in which endocytosis of the toxin is designed by nature to promote cellular damage, illustrate why there might be a serious problem with cells using endocytosis as a mechanism of toxin clearance. In an effort to reconcile these potential problems, some observers have proposed that internalized toxins are promptly destroyed by autophagy (Gutierrez et al., 2007) or rapidly sorted away from lysosomes, perhaps by entering multivesicular bodies (Husmann et al., 2009). However, no experimental evidence to support either of these alternatives has been uncovered to date.
An alternative possibility is that bacterial toxins are shed directly from the plasma membrane, a process termed ectocytosis (Eken et al., 2008; Gasser and Schifferli, 2005; Stein and Luzio, 1989). Indeed, there is already evidence that SLO-treated cells do shed SLO along with some cellular proteins (Babiychuk et al., 2009; Walev et al., 1995; Walev et al., 2000; Walev et al., 1996; Xie and Low, 1995). However, to date it has been thought that cells shed only monomeric SLO (Walker et al., 1995), and most attention has focused on the cellular proteins that are shed, including glycosylphosphatidylinositol (GPI)-anchored proteins, L-selectin, CD14 and interleukin (IL)-6 receptor (Babiychuk et al., 2009; Walev et al., 2000; Walev et al., 1996; Xie and Low, 1995). In fact, the conclusion that SLO was shed in monomeric form came exclusively from sucrose-gradient sedimentation studies, but no ultrastructural studies were performed to confirm this conclusion.
Here, we tested the hypothesis that SLO pores are eliminated from the cell through ectocytosis, rather than being internalized by endocytosis. To do this, we re-examined the mechanism of membrane repair in cells treated with SLO by ‘deep-etch’ electron microscopy (EM) and we designed experiments to test whether blocking endocytosis rendered cells more vulnerable to SLO. We show that cells exposed to SLO sequester the toxin into distinct blebs that bulge from the plasma membrane and are promptly shed from cells, thereby satisfying the definition of ectocytosis. Additionally, and surprisingly, such ectocytosis occurs readily in chemically fixed cells, suggesting that it is a physical process, probably involving physicochemical changes in the plasma membrane. When this process occurs in living cells, it mediates the shedding of SLO from the cell surface, most probably promoting cell survival and serving to prevent the entry of pathogenic molecules into the cell.
Results
Microscopic titration of SLO action
We first confirmed, as previously reported (Walev et al., 2001), that the absence of external Ca2+ heightens cell permeability and mortality following SLO treatment (supplementary material Fig. S1A). We used this Ca2+ dependence to experimentally uncouple SLO-induced membrane permeabilization from Ca2+-induced resealing, as previously described (Babiychuk et al., 2009; Walev et al., 2001). To create a gradient of cell damage, we first cultured cells on glass-bottomed dishes, washed them and removed all medium except for a thin film of Ca2+-free Ringer's solution. We then gently applied a small bolus of SLO to the center of the dish, which created a concentration gradient of SLO across the dish through diffusion (Fig. 1A–H). We documented the permeabilization by including the membrane-impermeant dye FM1-43, which remains in the plasmalemmae of undamaged cells but enters and stains the organelles of SLO-permeabilized cells. After 5 minutes of this exposure to SLO, we washed the dishes with complete medium containing normal levels of Ca2+ and incubated them for 10 minutes to allow the cell membranes to reseal, if they could. This also washed the FM1-43 out of the membranes of non-permeabilized cells, leaving this dye only in cells that had been permeabilized during the 5-minute exposure to SLO. Finally, we added propidium iodide (PI) to mark cells that failed to reseal.
Fig. 1.

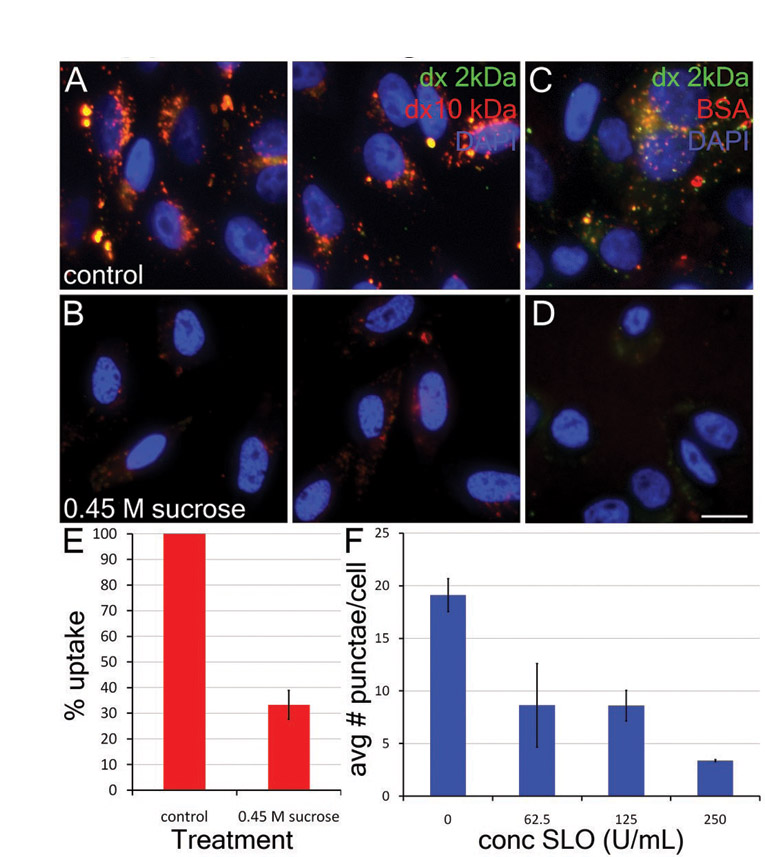
Endocytosis is not required for plasma membrane repair. (A–H) CHO cells in Ca2+-free Ringer's solution in the presence of FM1-43 (green, marks transiently permeabilized cells) were given a bolus of SLO to the center of the dish. SLO was allowed to diffuse across the dish for 5 minutes at 37°C. Cells were then given Ca2+-containing full medium (A–D) or full medium plus 0.45 M sucrose (E–H) for 10 minutes at 37°C, followed by PI to mark cells that failed to reseal. The panels show various regions of the dish, ranging from the edge, where no permeabilization occurred (A,E), through to regions where cells were permeabilized but resealed (B,C,F,G) and to the center of the dish, where cells were permanently permeabilized by SLO (D,H). (I–P) HeLa cells were pretreated with 2 mg/ml HRP for 4 minutes at 37°C, permeabilized for 3 minutes at 37°C with a bolus of SLO to the center of the coverslip in Ca2+-free Ringer's solution with HRP, incubated in full medium with HRP for 11 minutes at 37°C and either fixed immediately (I–L) or incubated for 1 hour in full medium without HRP at 37°C and fixed (M–P). At 1 hour, HRP internalization can be seen as punctate structures (arrowheads) in unpermeabilized (M) and transiently permeabilized cells (N,O), whereas cells that failed to reseal show residual HRP staining of the nuclear envelope and endoplasmic reticulum (L,P). Nuclear HRP is indicative of transient permeabilization. (Q–T) CHO cells were washed and challenged with SLO at 0 units per ml (Q), 31.25 units per ml (R), 62.5 units per ml (S) or 125 units per ml (T) in the presence of 2.5 mg/ml dx647 (red) for 4 minutes at 37°C. Then the cells were washed in PBS, fixed and stained with DAPI (blue). Micrographs show representative fields of cells from one of three independent experiments. Scale bars: 10 μm.
Typically, we observed that the cells at the edge of these dishes had not permeabilized at all (as indicated by their total lack of FM 1-43 or PI staining; Fig. 1A), whereas more proximally along the SLO gradient, some cells had permeabilized but resealed successfully (as shown by their retention of FM1-43 but exclusion of PI; Fig. 1B). By contrast, at the center of the dish, where SLO concentration was maximal, all cells had permeabilized and very few had been able to recover successfully (as shown by their minimal FM1-43 staining and positive PI staining; Fig. 1C,D). Thus, this method did indeed permit us to observe the full spectrum of SLO effects in each culture, even although SLO activity can vary uncontrollably between experiments (a well-known problem).
Endocytosis does not protect cells from SLO attack
Next, we used the above protocol to test whether endocytosis was necessary for membrane repair during or after SLO exposure. We reasoned that, if endocytosis was necessary for plasma membrane repair, then blocking this process during and after SLO exposure should prevent or retard the resealing and recovery of SLO-permeabilized cells. To block endocytosis abruptly, we placed cells in a hypertonic sucrose medium (Heuser and Anderson, 1989; Keyel et al., 2004). In Chinese-hamster ovary (CHO) cells, such hypertonic sucrose treatment reduced internalization of dextran and/or BSA to 30% of normal levels (supplementary material Fig. S2A–E). When we treated CHO cells with SLO and allowed them to recover in the presence of 0.45 M sucrose, we observed the same range of cell phenotypes as those observed in the absence of sucrose (Fig. 1E–H). Importantly, the same proportion of cells, and in the same distribution, were FM1-43-positive but PI-negative, indicating that they had been transiently permeabilized by the SLO but had managed to reseal by the time PI was added. These data show that membrane repair can occur normally under conditions that impair endocytosis and thus cast doubt on the notion that endocytosis is important for toxin clearance.
Viability of SLO-treated cells
Given the above evidence that CHO cells can recover from SLO-mediated permeabilization within 10 minutes, both with or without sucrose, it next became important to determine whether they remained viable at later time points. We tested this by performing 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) cell viability assays after SLO treatment (supplementary material Fig. S1B). Even at SLO exposures of up to ~65 units per ml, >90% of our CHO cells remained viable. Moreover, we determined by fluorescence-activated cell sorting (FACS) that, at such doses of SLO, many of the cells that remained viable had been permeabilized. We observed a substantial population of cells that were weakly PI-positive when PI was added before, but not after, SLO exposure (supplementary material Fig. S1C–F). This was in addition to the population of strongly PI-positive cells that always appeared after SLO treatment, regardless of when PI was added – indicating the population of cells permanently damaged by the SLO – and which correlated with the percentage of dead cells determined by the MTT assay. Taken together, these data indicate that the majority of cells that recover from a transient permeabilization by SLO remain viable at later time-points thereafter.
Levels of endocytosis in SLO-treated cells
If endocytosis were triggered in transiently permeabilized cells following SLO challenge, we would have expected to observe an increase in their uptake of extracellular tracers, such as horseradish peroxidase (HRP) or dextran, if we added such tracers along with the SLO. To examine this possibility, we incubated CHO cells with HRP during the initial SLO permeabilization in Ca2+-free Ringer's solution, continued their exposure to HRP during their first 10 minutes of recovery in complete medium and then either fixed the cells then or let them recover for a full hour before fixation (Fig. 1I–P).
At 10 minutes after SLO exposure in the presence of HRP, cells on the edge of the dishes were again unperturbed, and again served as controls for monitoring the background level of HRP endocytosis. By contrast, cells more central on the dishes were transiently permeabilized, given that they showed uptake of HRP into their cytosol rather than into their endosomes (this is like the uptake and retention of FM1-43 described above; Fig. 1I–K). Finally, cells in the very center of the dishes, exposed to the highest doses of SLO, were again irreparably lysed, as shown by the faint diaminobenzidine (DAB) staining of their nuclear envelopes but their inability to retain observable amounts of HRP in their cytoplasm (Fig. 1L).
In contrast to the above results, the phenotype of cells at 1 hour after recovery from SLO in the presence of HRP was quite different. Unperturbed cells at the periphery of each dish looked the same as at 10 minutes and contained the expected number of HRP-containing endosomes given their earlier 10-minute exposure to HRP (Fig. 1M, arrowheads). However, cells in the regions of the dishes that had shown uptake and retention of HRP into their cytoplasm at 10 minutes now showed clear and strong DAB staining of their nuclei, indicating that HRP was transferred from the cytoplasm to the nucleus (Fig. 1N,O).
This sequestration of HRP into the nucleus has never been reported before and deserves further analysis for its own sake. In addition, it served two purposes here. First, it demonstrated that the transiently permeabilized cells remained viable for the full hour following exposure. Permanently permeabilized cells exposed to HRP for prolonged periods never showed such nuclear retention; all they showed was a faint positive staining of their residual membranes (Fig. 1P). Second, the nuclear uptake allowed us to assess the level of endocytosis in these cells because it cleared the cytoplasm of HRP. We could therefore count the number of HRP-positive endosomes in cells that were transiently permeabilized by SLO and compared with this number in control cells at the edges of the dishes (Fig. 1M–P, arrowheads). We scored over 120 cells in each condition, counting all the HRP-positive vesicles observed within each cell, and found 9±4 endosomes in non-permeabilized cells near the edges of the dishes compared with 7±2 endosomes in transiently permeabilized cells more centrally. Thus, we found a small but significant decrease in endocytosis in transiently permeabilized cells compared with that in control cells (P=0.057, as determined by a Student's t-test). We observed no sign of any increase in endocytosis of HRP, not even in cells that were viable enough after SLO-exposure to move HRP from their cytoplasm to their nuclei. These results do not fit well with the notion that endocytosis is important for recovery from, or for resistance to, SLO (Idone et al., 2008).
To examine further the possibility that SLO exposure stimulates some sort of compensatory endocytosis, we repeated the protocols originally used to develop the idea (Idone et al., 2008). We incubated CHO cells simultaneously with a lysine-fixable 10-kDa dextran conjugated to Alexa-Fluor-647 and various concentrations of SLO for 4 minutes at 37°C and then examined these cells by fluorescence microscopy. We found that untreated control cells showed an abundance of fluorescent dextran-containing punctae, presumably representing endosomes (Fig. 1Q). By contrast, CHO cells treated with sublytic amounts of SLO exhibited a variable degree of permeabilization (as seen by variable retention of dextran within their cytoplasm; Fig. 1R,S), but displayed 50% fewer fluorescent dextran-containing punctae than did control cells (supplementary material Fig. S2F). Interestingly, at SLO doses that killed 40% of the cells (supplementary material Fig. S1F), cells generally appeared to be flooded with cytoplasmic dextran, but still displayed curious dextran-positive structures that did not look like bona fide endosomes because they were generally much larger and more tightly grouped about the nucleus than normal endosomes (Fig. 1T). These structures could have been interpreted as being endosomes in earlier studies. However, EM studies of cells damaged this severely by SLO in the presence of HRP suggested that these structures are inverted fragments of plasmalemma that passively trap extracellular materials as they form (data not shown).
Lysosomal exocytosis is not provoked by SLO exposure
Our counts of HRP-positive endosomes in cells exposed to SLO could have been grossly misleading if exocytosis of endosomes and/or lysosomes occurred in recovering cells at the very same time as endocytosis, which is known to occur in some forms of membrane damage (Reddy et al., 2001). Conceivably, such exocytosis could have perfectly counterbalanced whatever increase in HRP endocytosis occurred, and left us with the mistaken impression that no change in endosome numbers had occurred. To rule out this possibility, we first preloaded the endosomal-lysosomal system of cells with HRP, by giving them one full hour of exposure to exogenous HRP, and then applied SLO. Importantly, this did not lead to any sign of decline or diminution in the number of HRP-positive endosomes in any of our cells, in any region of the dishes – damaged or undamaged (data not shown). Thus, we conclude that lysosomal exocytosis was not masking any increase in HRP endocytosis.
Localization of SLO to the surface of cells
If endocytosis was not actively involved in SLO clearance, then SLO would be expected to remain on the surface of cells. To test for this, we fixed SLO-treated CHO cells and immunostained them for SLO without detergent permeabilization (Fig. 2A–D). Antibodies against the Golgi protein giantin (also known as golgin subfamily B member 1) were included to delineate those cells permanently permeabilized by SLO (Fig. 2, asterisks). This showed that, at all doses, a substantial fraction of cells were SLO-positive but giantin-negative, indicating that these cells remained alive but retained SLO on their surface (Fig. 2B–D). Curiously, this labeling of SLO of giantin-negative cells was often punctate, as observed by light microscopy (Fig. 2, arrowheads), and staining intensity increased in a dose-dependent manner (Fig. 2E). Such punctae might have been taken to be endosomes in earlier studies (Idone et al., 2008); however, we would not expect endosomes to stain with antibodies against SLO in cells where the Golgi complex did not stain with antibodies against giantin, indicating that they had not been permeabilized. Rather, the observed SLO punctae are likely to be surface structures – possibly clumps of SLO pores on the cell surface.
Fig. 2.
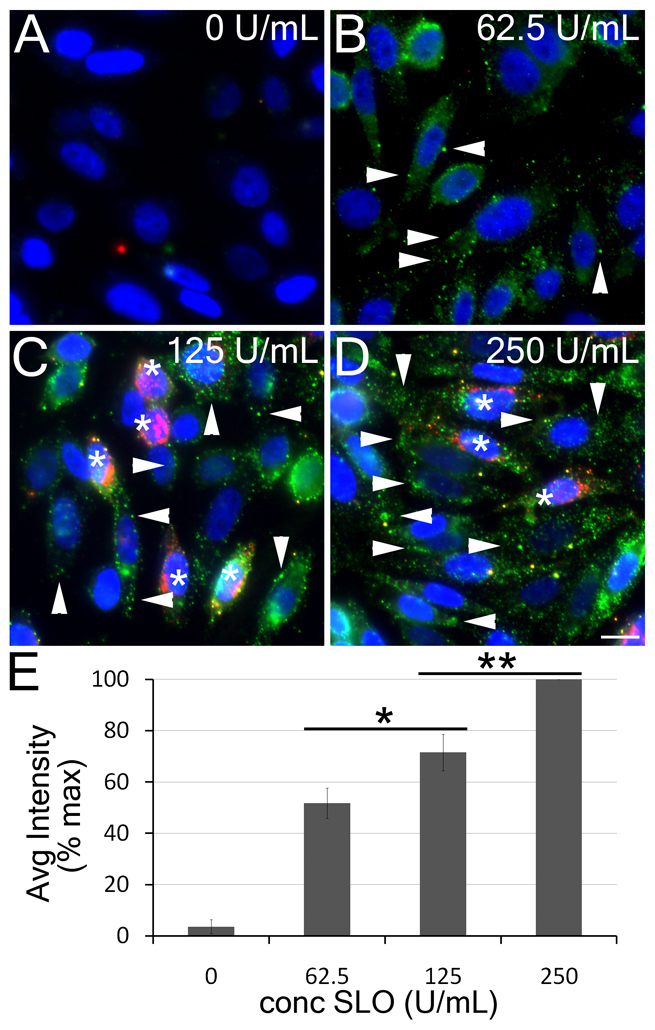
SLO remains on the cell surface. CHO cells were treated with 0 units per ml (A), 62.5 units per ml (B), 125 units per ml (C) or 250 units per ml (D) SLO and fixed. Cells were stained without chemical permeabilization with antibodies against SLO (green), giantin (red) and DAPI (blue). Giantin marks the Golgi and only stains those cells that lost plasma membrane integrity, indicated by asterisks, whereas arrowheads denote SLO punctae on non-permeabilized cells. (E) Mean (±s.e.m.) intensity of anti-SLO antibody staining on the cell surface of the non-permeabilized cells from the experiments described in A–D, normalized to the maximum intensity. Over 75 cells per treatment in three independent experiments were analyzed. Intensity changes were significant (*P=0.083; **P=0.029) between treatments. Micrographs show representative fields of cells from one of three independent experiments. Scale bar: 10 μm.
EM localization of SLO on the surface of cells
To gain further insight into this punctate distribution of SLO on the cell surface, we turned to deep-etch EM (DEEM), which resolves individual SLO pores, in order to determine their distribution on the cell surface. This required titrating the dose of SLO to a level where cells were not grossly permeabilized, which we determined by phase-contrast light microscopy before EM.
Cells treated with sublytic doses of SLO displayed by DEEM plasma membranes enriched in pores; but these pores were not distributed uniformly or randomly on the cell surface. Rather, they were clustered into discrete highly enriched domains; in fact they were so enriched that they typically appeared to exclude most other membrane proteins (Fig. 3A, black arrowheads). Quantification of the number of pores revealed an average of 24 pores per μm2 on blebs, whereas pores were undetectable on the plasma membrane (P=0.004, as determined by a Student's t-test). Importantly, we never observed these clusters of SLO pores on any indentations of the plasma membrane that might have been incipient endosomes, despite the fact that the openings of clathrin-coated pits and caveolae were always readily apparent in our deep-etch replicas, even when they were very shallowly indented (e.g. when they are presumably at a very early stage of formation; Fig. 3A, white arrowheads). Rather, all the clusters of SLO we observed occurred on eversions of the plasma membrane, on structures that could only be called ‘blebs’.
Fig. 3.
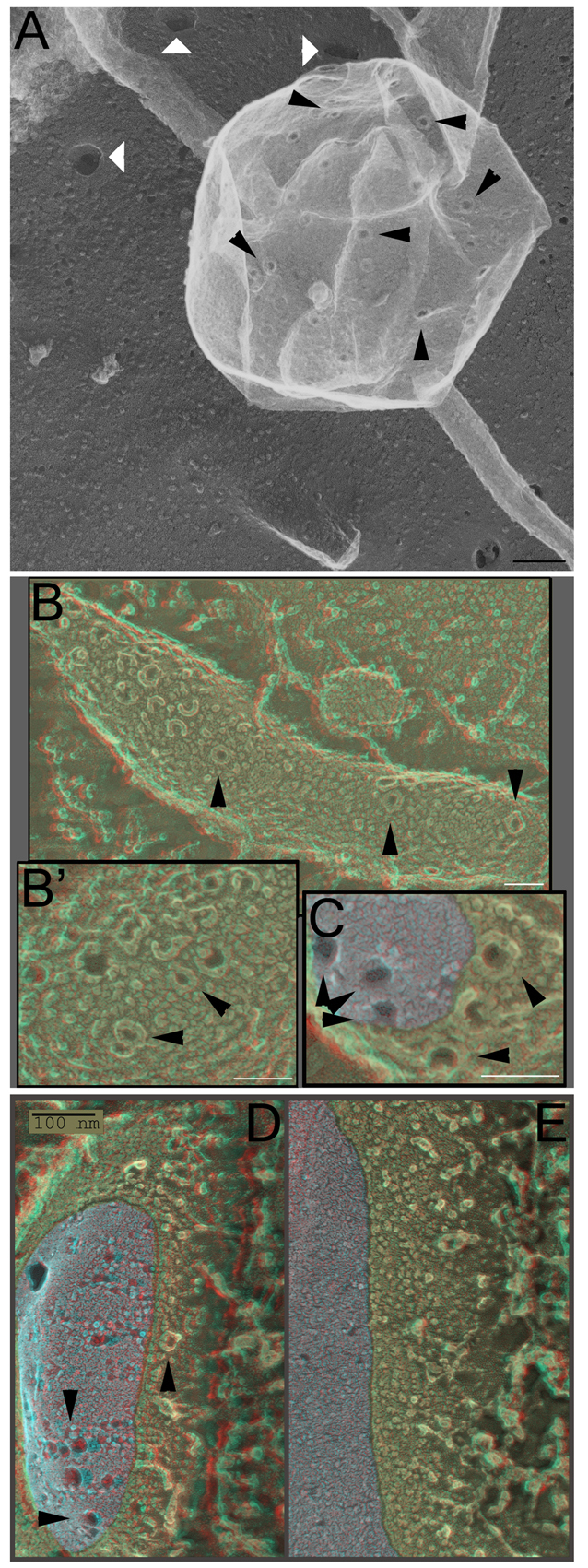
SLO provokes blebbing from living cells. (A) CHO cells were treated for 30 minutes with 500 units per ml SLO at 37°C, fixed and prepared for DEEM. Black arrowheads mark the ring-shaped SLO pores present on blebs. White arrowheads denote coated pits. (B–E) HeLa cells were treated with 125,000 units per ml SLO for 1 minute, fixed and either prepared for DEEM (B,B′) or freeze-fracture EM (C–E). Blebs (C,D) or plasma membrane (E) are shown. The fracture region is depicted in gray for clarity. Arrowheads mark the assembled ring-shaped SLO pores. Micrographs are representative of two independent experiments. Scale bars: 100 nm (A,D,E); 50 nm (B,B′,C).
This is not to say, however, that such eversions or blebs looked like the standard reactive blebs that cells form when they are irritated or injured (Charras et al., 2008; Charras et al., 2006; Charras et al., 2005; Mitchison et al., 2008; Straight et al., 2003). They did not. The latter sorts of blebs are larger, more plump and their membranes bear a perfectly normal complement of proteins, as judged by the appearance of the small ‘bumps’ seen in replicas of the outside surface of cells (supplementary material Fig. S3A,B). By contrast, the SLO-enriched blebs were smaller, flatter, more pleomorphic and tended to exclude the normal plasma membrane ‘bumps’ and ‘blobs’ that are generally considered by freeze-etch practitioners to be the exofacial portions of intramembrane proteins.
‘Quick freezing’ of living cells reveals SLO pores in blebs
To confirm that SLO also concentrates into blebs in living cells, we next treated HeLa cells with SLO and immediately quick-froze them without any chemical fixation. In order to view the surfaces of these cells by EM, they had to be freeze-fractured and then deep-etched, rather than simply freeze-dried as in the experiments above (this is because the medium on top of living cells obscures them if they are directly freeze-dried). We could readily observe, on these freeze-fractured cells, similar-looking SLO structures to those described above, which were again concentrated into membrane blebs and eversions (Fig. 3B–D, arrowheads). By contrast, the number of SLO rings in the rest of the membrane of these cells appeared to be exceedingly low (Fig. 3E). Additionally, the freeze-fractured cells illustrated that many of the SLO rings formed large pores straight through the plasma membrane (Fig. 3C,D, gray indicates the fracture faces and arrowheads indicate pores). Taken together, these results suggest that the punctate distribution of SLO observed here, and previously by Idone and colleagues (Idone et al., 2008), probably represents SLO-induced blebbing rather than SLO endocytosis.
Shedding of SLO-enriched vesicles from treated cells
The above EM results are also entirely consistent with earlier biochemical evidence that SLO induces the shedding of small membrane vesicles from the plasma membrane (Xie and Low, 1995). Xie and Low concentrated on the release of GPI-linked proteins in SLO-treated cells, but they did not consider the possibility that SLO itself was released along with these other proteins (Xie and Low, 1995). Other studies have examined the direct shedding of material from the plasma membrane and named the process ectocytosis (Eken et al., 2008; Gasser and Schifferli, 2005; Stein and Luzio, 1989). The present finding of SLO on characteristic membrane blebs thus raised the possibility that SLO pores are shed through ectocytosis. To test for this directly, we collected the supernatants from cells exposed to sublytic doses of SLO and analyzed these supernatants, both biochemically and ultrastructurally. To ensure that we did not collect just cell debris from ruptured cells, we kept the dose of SLO sublytic, and we spun the supernatant at 1200 g for 5 minutes to sediment any large cell fragments or any whole cells that might have been released into the supernatant. The supernatant remaining after this low-speed spin was then concentrated by ultracentrifugation at 100,000 g for 40 minutes, resuspended in a small volume of buffer, adsorbed to glass coverslips and prepared for DEEM. We observed closed membranous vesicles that were often almost completely saturated with SLO pores (Fig. 4A). As a control in these experiments, we treated our cleared cell supernatants with Triton X-100 before high-speed centrifugation. This greatly reduced the amount of SLO in the pellet and left nothing at all to see by DEEM, confirming that the shed structures were indeed membranous in nature.
Fig. 4.
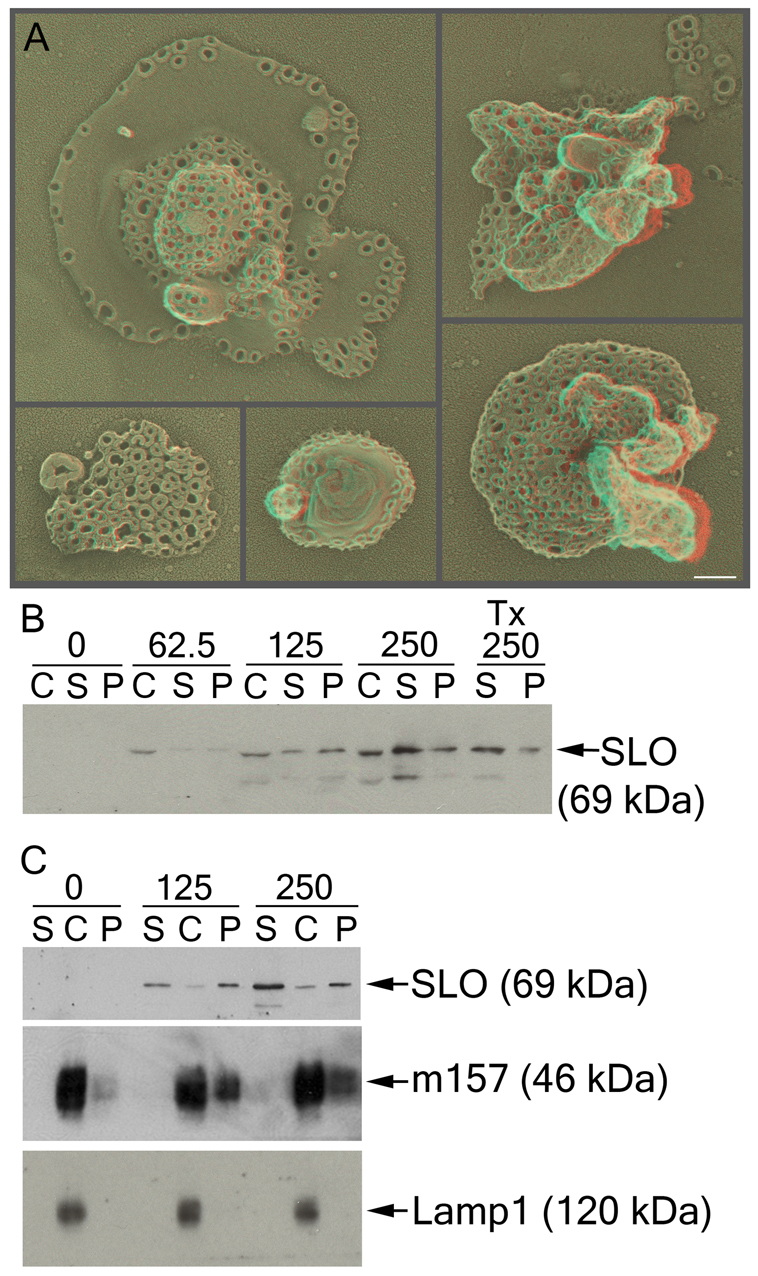
Cells shed SLO-rich microvesicles. (A) Platinum replicas of pellets formed after high-speed ultracentrifugation (high-speed pellet) from RMAS-m157 cells treated with 250 units per ml SLO were visualized by DEEM. (B,C) CHO (B) or RMAS-m157 (C) cells were incubated with the indicated concentration of SLO (in units per ml) for 5 minutes at 37°C and the cells sedimented. The pellets formed after low-speed cell centrifugation (cell pellets) were washed and solubilized in SDS sample buffer, whereas the supernatants were ultracentrifuged at 107,000 g for 40 minutes at 4°C to pellet any shed membranes. Triton X-100 was added to the supernatant marked Tx just before the high-speed spin. Cell pellets (C), high-speed supernatants (S) and high-speed pellets (P) were resolved by SDS-PAGE and probed with the HAB-003 anti-SLO monoclonal antibody, the 6H121 anti-m157 monoclonal antibody or the anti-Lamp1 rat antibody. SLO was detectable in the high-speed pellet along with the GPI-linked m157. Micrographs show representative cells or structures from one of three independent experiments, whereas blots are representative of ten experiments. Scale bar: 100 nm.
Parallel biochemical analyses of these putative ectosomes confirmed that they were indeed greatly enriched in SLO (Fig. 4B,C). Quantification of these immunoblots indicated that ~60% of the SLO was shed in the first 5 minutes. Along with SLO, ectosomes from RMAS cells stably transduced with the GPI-linked viral protein m157 (Tripathy et al., 2006) contained a rich complement of m157. This is consistent with the important previous study demonstrating SLO-induced release of GPI-anchored proteins (Xie and Low, 1995). By contrast, we did not detect the lysosomal protein Lamp1 in our ectosome fraction, suggesting that SLO-treated cells do not shed any of their lysosomal contents into the medium (Fig. 4C). Our ectosomes were heterogeneous in size, ranging from ~200 nm up to ~2 μm, which was similar in size and diversity to the blebs we observed protruding from the plasmalemma of whole cells (Fig. 3). This is considerably larger than the usual size of exosomes, the other major type of vesicle released from cells by exocytosis of multivesicular bodies (Raposo et al., 1996; Stoorvogel et al., 2002; Thery et al., 1999).
SLO localizes to blebs on chemically fixed cells
Having found that the SLO-enriched structures on the surfaces of cells (and the structures shed from SLO-treated cells) were relatively depleted of other membranous proteins, we next needed to consider the possibility that SLO caused some sort of phase-separation in the plasma membrane and that this was somehow coupled to the membrane eversion and blebbing observed. To test for this, we took advantage of the observation that SLO can bind to, and polymerize into rings, on aldehyde-fixed cells (Duncan and Schlegel, 1975). Indeed, we found a great abundance of SLO-enriched blebs on cells that had been prefixed either in formaldehyde, glutaraldehyde, or both (Fig. 5). Importantly, we never observed any such abundance of blebs in control cells that were fixed comparably but not treated with SLO (supplementary material Fig. S3C,D). These experiments also helped us distinguish between ectosomes and exosomes; the latter could not possibly be released from fixed cells because their discharge requires processing in multivesicular bodies before their exocytic release from within these bodies, both of which are living processes that do not continue after chemical fixation (Raposo et al., 1996; Stoorvogel et al., 2002; Thery et al., 1999).
Fig. 5.

SLO localizes to and induces blebs in fixed cells. HeLa cells were fixed, then treated with either 12,500 units per ml (A) or 125,000 units per ml SLO (B–D), fixed and prepared for DEEM. (A) Pore segregation into blebs is observed at low magnification (insets show higher magnification images contrasting the plasma membrane and bleb). Arrowheads indicate SLO pores. (B,C) A high-magnification three-dimensional anaglyph view of plasma membrane (B) or a two-dimensional view of bleb membrane (C) reveals an overabundance of pores on the bleb membrane. (D) SLO pores are observed in smooth phase-separated regions of the plasma membrane (colored gray for contrast). Micrographs are representative of four independent experiments. Scale bars: 500 nm (A); 100 nm (B–D).
Prefixation of cells before exposure to SLO also permitted the use of a higher concentration of SLO than was possible for living cells; thus, we could study pore formation itself in greater detail. When we increased SLO concentrations far beyond lytic levels, we continued to observe preferential segregation of SLO into plasmalemmal blebs, with up to 320 pores per μm2 on some blebs, although the general plasma membrane also began to display an average of 2 pores per μm2 (Fig. 5A–C). We could also observe a phase separation in small portions of the plasma membrane. These portions of the plasma membrane were characterized by smooth membrane, again lacking observable intramembrane particles, and these regions supported SLO polymerization (pore formation) (Fig. 5D).
Ca2+ is required for the shedding, but not the formation, of SLO-enriched blebs
Given the importance of Ca2+ for both membrane repair and the formation of large blebs (Babiychuk et al., 2009; Charras et al., 2008; Walev et al., 2001), we next sought to determine whether Ca2+ plays any role in the generation of the blebs observed here or the localization of SLO to these blebs using the prefixed cell system. To do this, we prefixed cell cultures in the presence of Mg2+ and 3 mM EGTA (in place of the usual 2 mM Ca2+ in our fixatives), and then exposed the cells to SLO without changing their cation environment. SLO pores and SLO-enriched blebs were just as abundant on cells fixed and treated in Mg2+ and EGTA as in those fixed in the presence of Ca2+, with a global average of 7.4 pores per μm2 in the presence of Ca2+ and 6.7 pores per μm2 in Mg2+ and EGTA (Fig. 6A,B). This demonstrated that Ca2+ is not necessary for the insertion of SLO into membranes, polymerization into pores in the membrane or for the segregation of SLO into plasmalemmal blebs. Thus, SLO-induced bleb formation is independent of Ca2+.
Fig. 6.

Ca2+-dependent SLO shedding. (A,B) Three-dimensional DEEM anaglyphs illustrate CHO cells that were prefixed, treated with 4,000 units per ml SLO for 4 minutes at 37°C, and fixed, all in the absence of Ca2+. Black arrowheads denote SLO rings present in blebs. White arrowheads mark clathrin-coated pits. SLO pores are pseudo-colored gray in the higher magnification view of the boxed area (shown in B) for clarity. (C–F) 3T3 cells labeled with Cellmask Orange (red), in the presence of Sytox Green (green), were untreated (C) or treated with 250 units per ml SLO in the presence (D,E) or absence (F) of Ca2+. The lower panels show a higher magnification view of the boxed area in the upper panels. Arrowheads in C–D denote blebs separating from the plasma membrane, whereas arrowheads in F denote filopodia. The time following SLO addition is displayed in minutes:seconds. Micrographs are representative of three independent experiments. Scale bars: 100 nm (A); 200 nm (B); 10 μm (C–F); 5 μm (C–F, lower panels).
We next performed three-dimensional confocal imaging on 3T3 cell cultures labeled with the membrane dye Cellmask Orange to determine whether the shedding of SLO-enriched blebs in living cells was Ca2+ dependent. Control cells labeled with Cellmask Orange excluded Sytox Green, showed normal cellular activity and did not undergo blebbing, indicating that the dye was nontoxic (Fig. 6C; supplementary material Movie 1). Next, 3T3 cultures were treated with moderate doses of SLO in the presence of a normal concentration of Ca2+, and blebbing and release of vesicles was observed (Fig. 6D; supplementary material Movie 2). Interestingly, we also observed blebs that did not release from cells, yet these blebs accumulated Sytox Green (Fig. 6E; supplementary material Movie 3), suggesting that blebs can lose communication with the cytoplasm even before they are shed. This has been previously described for the PFT perforin (Keefe et al., 2005), suggesting that this is a common mechanism among PFTs. Under Ca2+-free conditions, however, we did not observe any ectocytosis (Fig. 6F; supplementary material Movie 4). Instead, we observed the formation, and wiggling about, of long filopodial structures (Fig. 6F, arrowhead). This suggests that Ca2+ is required for the final scission event.
To determine whether Ca2+ plays a role in the final step of ectocytosis, we collected the 107,000 g pellet from clarified supernatants of cells that had been prefixed and then treated with SLO in the presence of either Ca2+ or Mg2+ and EGTA and analyzed them for SLO content. We found a dramatic increase in the amount of SLO recovered from cells that had been treated in the presence of Ca2+ (supplementary material Fig. S4). Because SLO partitions into blebs independently of Ca2+ (Fig. 6), the results provide a measure of the amount of vesicles recovered. Taken together, we conclude that Ca2+ is needed for the scission event of ectocytosis.
Discussion
Here, we provide evidence that a prototypical PFT, streptolysin O, provokes a cell-survival mechanism that involves shedding of the toxin, not internalization and degradation. Specifically, we show that sublytic SLO doses, which create pores in the plasma membrane, provoke a cellular reaction in which the pores rapidly become segregated into plasmalemmal blebs, and the blebs become rapidly shed into the surrounding medium, in the process currently termed ectocytosis.
Contrary to earlier reports, we find no evidence for sequestration of SLO pores into membrane invaginations or into incipient endosomes, and we observe no increase in endocytosis in SLO-treated cells (Idone et al., 2008; Thiery et al., 2010). Furthermore, we find that blocking endocytosis does not compromise the ability of cells to survive SLO. From these results, we conclude that endocytosis is not likely to be the primary mechanism of SLO clearance, but that ectocytosis probably is.
While this manuscript was under review, Babiychuk et al. described a Ca2+-dependent cell blebbing that is important for membrane repair, although they did not link this blebbing to sequestration and removal of SLO pores (Babiychuk et al., 2010). However, the active blebbing that occurs in a Ca2+-dependent fashion is the abrupt protrusion of big (micron-sized) cytoplasmic protrusions depleted of cortical cytoskeleton and filled with readily mobile cytoplasmic components (most notably ribosomes) (Zeligs and Wollman, 1977). This blebbing also occurs during mitosis (zeiotic blebbing), and upon exposure to oxidizing and reducing agents, as well as occuring generally when the cortical cytoskeleton of the cell is constitutively weakened, such as in filamin-deficient melanoma cells (Charras et al., 2008; Charras et al., 2006; Charras et al., 2005; Cunningham et al., 1992; Mitchison et al., 2008; Straight et al., 2003). Such ‘active’ blebs display a characteristic behavior; they not only protrude extremely abruptly, like herniations of the plasmalemma generated by internal turgor pressure (which they probably are), but also retract actively after a minute or so, coincident with (and consequent to) the reconstruction of an actomyosin-rich cortex immediately under their membranes (Mitchison et al., 2008).
The blebs we describe here are different from these ‘reactive blebs’ in several ways: they are generally considerably smaller; they do not contain normal cytoplasmic components but appear to be isolated from the cytoplasm almost from the moment of their formation; they do not bear the normal complement of membrane proteins that occur elsewhere in the plasmalemma (‘reactive blebs’ do contain these proteins) but appear to be considerably depleted of cytoplasmic proteins and often lack them entirely (membrane proteins being largely replaced here with SLO pores); and bleb formation can occur independently of Ca2+, although shedding is Ca2+ dependent. How the formation and scission of nascent ectosomes coordinates with internal membrane fusion events (McNeil and Kirchhausen, 2005; McNeil et al., 2000) remains to be determined.
These ectosomes might induce host inflammatory effects, although any immunomodulatory functions remain to be determined. It is possible that ectosomes might aid in the generation of anti-toxin antibodies. Anti-SLO antibodies are known to form during group-A streptococcal infections and are important in the diagnosis of scarlet fever, rheumatic fever and post-infectious glomerulonephritis (Cunningham, 2000). Ectocytosis might also be the mechanism through which cellular proteins, such as alkaline phosphatase, the IL-6 receptor and CD14 (Walev et al., 1996; Xie and Low, 1995), are shed in response to SLO.
The evidence we provide here that chemically fixed cells also undergo ectocytosis when exposed to SLO argues that the mechanism could be a physicochemical response of the plasmalemma to perturbations of its lipids, which are not fixed by aldehydes. Because the lipid composition of cells can be modified both genetically and post-translationally (Bishop and Bell, 1988; Schujman and de Mendoza, 2005), it is highly probable that cells will vary in their ability to respond to toxins in this manner. Various cell types do show differential sensitivity to PFTs (Keefe et al., 2005), which could be the result of different propensities to engage in ectocytosis. Additionally, cell survival after exposure to the PFT aerolysin involves the caspase-1-dependent upregulation of sterol regulatory protein and other lipid-modifying enzymes (Gurcel et al., 2006). Thus, ectocytosis might be an important mechanism for cell survival.
Ectocytosis could also protect cells from the deleterious effects of internalized bacterial PFTs. Bacterial toxins that reach the secretory granules of T cells kill the cells through permeabilization of secretory granules containing granzymes (Carrero et al., 2008). Even toxins that act at the surface, such as anthrolysin O, can be active at acidic pH (Wei et al., 2005), underscoring the importance for cell survival of preventing these toxins from having access to the endosomal-lysosomal system. Thus, we describe a repair response that is beneficial to the cell because it permits cell survival, eliminates the bacterial toxin and prevents the toxin from further manipulating the cell or ‘sneaking into’ the endosomal-lysosomal system of the cell.
Materials and Methods
Plasmids and antibodies
The pBAD-gIII plasmid encoding His-tagged SLO was a gift from Michael Caparon (Washington University in St Louis, St Louis, MO) (Magassa et al., 2010). The pmx-m157 plasmid, encoding m157, was as previously described (Smith et al., 2002). The anti-giantin polyclonal antibody was from Covance, CD19 conjugated to allophycocyanin (APC), CD86-APC, the anti-Lamp1 rat antibody specific to murine Lamp1 and the anti-Lamp1 monoclonal antibody H4A3 specific for human Lamp1 were from BD Biosciences, the anti-SLO monoclonal antibody HAB-003 was from Abcam (Cambridge, MA) and anti-m157 monoclonal antibody 6H121 was as previously described (Tripathy et al., 2006). Fluorescently labeled secondary antibodies were from Invitrogen and HRP-conjugated antibodies were from Jackson ImmunoResearch.
Cell culture
HeLa cells were cultured in DMEM supplemented with 10% fetal calf serum (FCS) and 23 L-glutamine, CHO cells were cultured in similarly supplemented Ham's F12 medium and YAC-1 cells in similarly supplemented RPMI. 3T3 cells were cultured in HeLa medium supplemented with sodium pyruvate and non-essential amino acids. RMAS-m157 cells were generated by retroviral transduction of pmx-m157, as previously described (Smith et al., 2002), and cultured in RPMI with 10% FCS, 23 L-glutamine and 4 μg/ml puromycin.
Reagents
All reagents were from Sigma unless otherwise specified. SLO was either purified from Streptococcus pyogenes (Sigma) or induced in pBAD-gIII SLO-transformed Escherichia coli strain BL21 (Agilent Technologies) and purified on nickel-NTA agarose beads (Qiagen). SLO from Sigma was reconstituted in water and 10 mM DTT and snap-frozen on dry ice in aliquots. We observed a ~50% reduction in SLO activity following each round of freeze-thaw, and rapid inactivation at 37°C, and hence reported activity, rather than protein concentration, for SLO. Both proteins were used at a similar hemolytic activity, and this hemolytic activity corresponded to the same cytolytic activity in both sources of SLO. Any endotoxin present in the toxin preparations did not affect cell killing (data not shown). The SLO sensitivity of MyD88−/− cells was comparable to that of wild-type cells (data not shown), ruling out any role of SLO binding to toll-like receptor 4 (Park et al., 2004) in our system.
Purification of SLO
SLO was induced in exponentially growing cells with 0.2% arabinose for 3 hours at 25°C. Cells were collected, centrifuged, frozen at −80°C and purified as described previously (Mishra et al., 2005). Purity was assessed by SDS-PAGE, protein concentration by BCA assay (Thermofisher Scientific) and hemolytic activity by erythrocyte lysis. One unit was the amount of SLO causing 50% lysis of a 2% suspension of human erythrocytes in 10 mM HEPES pH 7.4, 2 mM CaCl2, 0.3% BSA in PBS after 30 minutes at 37°C, as determined by the A405 of the supernatant, and matched the hemolytic activity reported by Sigma. Following one freeze-thaw, commercially available SLO typically had an activity >15,700 units per ml (517,200 units per mg of protein), whereas His-tag-purified SLO had an activity of 650,000 units per ml (260,000 units per mg of protein).
FACS assay
Cells were harvested, centrifuged and resuspended in R/B medium (RPMI, 2 mM CaCl2 and 0.5% BSA), with 20 μg/ml PI, at 2×106 cells per ml. Cells were incubated with SLO for 5 minutes at 37°C. PI incorporation was immediately assessed by FACS using a FACScan (BD Biosciences). For some experiments, the incubation was extended to 30 minutes, PI was not added until after incubation with SLO, immediately before FACS, or cells were challenged with SLO in the presence of 2 mM EGTA. Cell viability was assessed using the Vybrant MTT cell proliferation assay (Invitrogen).
Electron microscopy
CHO or HeLa cells were grown on coverslips, treated with SLO at 37°C in Ringer's solution (155 mM NaCl, 3 mM KCl, 2 mM CaCl2, 1 mM MgCl2, 3 mM NaH2PO4, 5 mM HEPES pH 7.2, and 10 mM glucose) for 4 or 30 minutes, washed and fixed in 2% glutaraldehyde in 100 mM NaCl, 30 mM HEPES pH 7.2, 2 mM CaCl2 and 150 mM sucrose, (NaHCa-Suc) for 1 hour. To measure SLO binding in fixed cells, CHO or HeLa cells grown on coverslips were fixed in 2% glutaraldehyde in NaHCa-Suc for 8 minutes at room temperature, washed in Ringer's solution, incubated in the indicated concentration of SLO at 37°C in Ringer's solution for 1 or 4 minutes and fixed again in 2% glutaraldehyde in NaHCa-Suc. To eliminate Ca2+, Mg2+ replaced the Ca2+ present in all the buffers used and buffers were supplemented with EGTA. Platinum replicas were prepared as described previously (Heuser, 2000), examined in a 100CX microscope (JEOL, Peabody, MA) and imaged with an AMT digital camera (Danvers, MA).
Fluorescence microscopy
CHO cells were treated with SLO at the indicated concentrations for 5 minutes at 37°C, fixed in 2% paraformaldehyde, probed with anti-SLO and anti-giantin antibodies followed by goat Alexa-Fluor-488-conjugated anti-mouse-IgG and goat Alexa-Fluor-568-conjugated anti-rabbit-IgG, DAPI stained, mounted in gelvatol and imaged using a 1.40 NA, 633 Plan-Apochromat objective on an Eclipse 80i microscope (Nikon, Melville, NY) with a Coolsnap HQ2 camera (Photometrics, Tucson, AZ). Image acquisition, analysis and quantification were performed using Metamorph software (MDS Analytical Technologies, Downington, PA). Student's t-tests were used to assess significance.
Endocytosis assays
CHO cells were grown on Delta T dishes (Bioptechs, Butler, PA), washed, mounted onto a 510 confocal microscope stage running LSM510 acquisition software and equipped with a 403 1.40 NA Plan-Apochromat objective and AxioCam HR camera (Carl Zeiss, Thornwood, NY) and labeled with 1.1 μM FM1-43 in Ca2+-free Ringer's solution at 37°C. A bolus of SLO was added to the center of the dish, and the cells were incubated for 5 minutes to allow permeabilization. Permeabilization was quenched by addition of full medium (25 mM HEPES, pH 7.4) with or without 0.45 M sucrose for 10 minutes. PI was added to give a 20 μg/ml final concentration and cells were imaged immediately without fixation. Bulk-phase endocytosis was measured by incubating CHO cells in 0.5 mg/ml 2 kDa dextran coupled to Alexa Fluor 488 (Invitrogen) and either 0.5 mg/ml 10-kDa lysine-fixable dextran coupled to Alexa Fluor 647 (dx647) (Invitrogen) or BSA coupled to Alexa Fluor 633 (Invitrogen) for 40 minutes at 37°C, and cells were fixed, stained with DAPI and imaged. Rapid endocytosis was measured by incubating CHO cells in 2.5 mg/ml dx647 with SLO in R/B medium for 4 minutes at 37°C, followed by fixation and DAPI staining.
Live imaging of blebbing
3T3 cells were labeled with Cellmask Orange (Invitrogen) for 5 minutes at 37°C on collagen-coated MatTek glass-bottomed dishes. Cells were imaged at 37°C in RPMI with Sytox Green (Invitrogen), 0.5% BSA, 25 mM HEPES pH 7.4 and either 2 mM CaCl2 or 2 mM EGTA, after treatment with a sublytic dose of SLO using a Yokagawa head (Tokyo, Japan), on a Nikon 200E microscope as previously described (Gao et al., 2010).
HRP assay
CHO cells were pretreated with 2 mg/ml HRP in Ringer's solution and exposed to a gradient of SLO in Ca2+-containing or Ca2+-free Ringer's solution, still in the presence of 2 mg/ml HRP, for 3 minutes at 37°C. Repair was initiated by washing out SLO and adding full medium, with 2 mg/ml HRP, for a further 11 minutes at 37°C. Cells were washed and either fixed in 2% glutaraldehyde or incubated for 1 hour in full medium at 37°C before fixation. HRP was visualized using standard techniques (Graham and Karnovsky, 1966). Cells were imaged and prepared for thin-section EM as previously described (Heuser, 2005). For HRP preloading experiments, cells were incubated in full medium supplemented with 5 mg/ml HRP for 2 hours before the SLO challenge.
Ultracentrifugation
Cells were harvested, spun, washed in RPMI with 2 mM CaCl2 and resuspended in RPMI with 2 mM CaCl2 with SLO for 5 minutes at 37°C and centrifuged at 1000 g for 5 minutes. Cell pellets were washed in PBS and solubilized at 95°C in SDS-sample buffer; supernatants were spun at 107,000 g (30,000 r.p.m., Sw50.1 Ti) for 40 minutes at 4°C to pellet all membranes. The supernatant was saved, whereas the pellet was washed, spun at 107,000 g for 40 minutes at 4°C and resuspended in either Ringer's solution, for EM, or in 95°C SDS-sample buffer, for SDS-PAGE. For prefixed cells, cells were grown to confluency in 10-cm dishes, washed in PBS, fixed for 8 minutes in 2% formaldehyde with 0.01% glutaraldehyde in Ringer's solution, washed in PBS, and incubated with 1000 units per ml SLO in Ringer's solution for 4 minutes at 37°C. Supernatants, together with one wash were collected, clarified by centrifugation at 300 g for 5 minutes at 4°C and ultracentrifuged at 103,000 g for 40 minutes at 4°C. Ca2+-free preparations used MgCl2 instead of CaCl2 and included 3 mM EGTA. The pellet was resuspended and either dissolved in SDS-sample buffer or spinoculated onto glass coverslips for DEEM.
Supplementary Material
Acknowledgments
We thank members of the Yokoyama laboratory for technical assistance and helpful discussion and the Heuser laboratory for transcribing dictations. This work was supported by the Howard Hughes Medical Institute and grants from the National Institutes of Health. The authors declare that they have no competing financial interest. P.K., W.Y. and J.H. devised the research approach. P.K., L.L., R.R., R.D.S., S.W. and J.H. performed experiments. R.R. and J.H. performed electron microscopy. P.K., R.R., J.H. and W.Y. analyzed data. P.K., W.Y. and J.H. wrote the manuscript. Deposited in PMC for release after 6 months.
Footnotes
Supplementary material available online at http://jcs.biologists.org/cgi/content/full/124/14/2414/DC1
References
- Abrami L., Kunz B., van der Goot F. G. (2010). Anthrax toxin triggers the activation of src-like kinases to mediate its own uptake. Proc. Natl. Acad. Sci. USA 107, 1420-1424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aroian R., van der Goot F. G. (2007). Pore-forming toxins and cellular non-immune defenses (CNIDs). Curr. Opin. Microbiol. 10, 57-61 [DOI] [PubMed] [Google Scholar]
- Awad M. M., Bryant A. E., Stevens D. L., Rood J. I. (1995). Virulence studies on chromosomal alpha-toxin and theta-toxin mutants constructed by allelic exchange provide genetic evidence for the essential role of alpha-toxin in Clostridium perfringens-mediated gas gangrene. Mol. Microbiol. 15, 191-202 [DOI] [PubMed] [Google Scholar]
- Babiychuk E. B., Monastyrskaya K., Potez S., Draeger A. (2009). Intracellular Ca(2+) operates a switch between repair and lysis of streptolysin O-perforated cells. Cell Death Differ. 16, 1126-1134 [DOI] [PubMed] [Google Scholar]
- Babiychuk E. B., Monastyrskaya K., Potez S., Draeger A. (2010). Blebbing confers resistance against cell lysis. Cell Death Differ. 18, 80-89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bishop W. R., Bell R. M. (1988). Assembly of phospholipids into cellular membranes: biosynthesis, transmembrane movement and intracellular translocation. Annu. Rev. Cell Biol. 4, 579-610 [DOI] [PubMed] [Google Scholar]
- Carrero J. A., Vivanco-Cid H., Unanue E. R. (2008). Granzymes drive a rapid listeriolysin O-induced T cell apoptosis. J. Immunol. 181, 1365-1374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charras G. T., Yarrow J. C., Horton M. A., Mahadevan L., Mitchison T. J. (2005). Non-equilibration of hydrostatic pressure in blebbing cells. Nature 435, 365-369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charras G. T., Hu C. K., Coughlin M., Mitchison T. J. (2006). Reassembly of contractile actin cortex in cell blebs. J. Cell Biol. 175, 477-490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charras G. T., Coughlin M., Mitchison T. J., Mahadevan L. (2008). Life and times of a cellular bleb. Biophys. J. 94, 1836-1853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cossart P., Vicente M. F., Mengaud J., Baquero F., Perez-Diaz J. C., Berche P. (1989). Listeriolysin O is essential for virulence of Listeria monocytogenes: direct evidence obtained by gene complementation. Infect. Immun. 57, 3629-3636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham C. C., Gorlin J. B., Kwiatkowski D. J., Hartwig J. H., Janmey P. A., Byers H. R., Stossel T. P. (1992). Actin-binding protein requirement for cortical stability and efficient locomotion. Science 255, 325-327 [DOI] [PubMed] [Google Scholar]
- Cunningham M. W. (2000). Pathogenesis of group A streptococcal infections. Clin. Microbiol. Rev. 13, 470-511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan J. L., Schlegel R. (1975). Effect of streptolysin O on erythrocyte membranes, liposomes, and lipid dispersions. A protein-cholesterol interaction. J. Cell Biol. 67, 160-174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eken C., Gasser O., Zenhaeusern G., Oehri I., Hess C., Schifferli J. A. (2008). Polymorphonuclear neutrophil-derived ectosomes interfere with the maturation of monocyte-derived dendritic cells. J. Immunol. 180, 817-824 [DOI] [PubMed] [Google Scholar]
- Gao W., Kang J. H., Liao Y., Ding W. X., Gambotto A. A., Watkins S. C., Liu Y. J., Stolz D. B., Yin X. M. (2010). Biochemical isolation and characterization of the tubulovesicular LC3-positive autophagosomal compartment. J. Biol. Chem. 285, 1371-1383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasser O., Schifferli J. A. (2005). Microparticles released by human neutrophils adhere to erythrocytes in the presence of complement. Exp. Cell Res. 307, 381-387 [DOI] [PubMed] [Google Scholar]
- Geoffroy C., Gaillard J. L., Alouf J. E., Berche P. (1987). Purification, characterization, and toxicity of the sulfhydryl-activated hemolysin listeriolysin O from Listeria monocytogenes. Infect. Immun. 55, 1641-1646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham R. C., Jr, Karnovsky M. J. (1966). The early stages of absorption of injected horseradish peroxidase in the proximal tubules of mouse kidney: ultrastructural cytochemistry by a new technique. J. Histochem. Cytochem. 14, 291-302 [DOI] [PubMed] [Google Scholar]
- Gurcel L., Abrami L., Girardin S., Tschopp J., van der Goot F. G. (2006). Caspase-1 activation of lipid metabolic pathways in response to bacterial pore-forming toxins promotes cell survival. Cell 126, 1135-1145 [DOI] [PubMed] [Google Scholar]
- Gutierrez M. G., Saka H. A., Chinen I., Zoppino F. C., Yoshimori T., Bocco J. L., Colombo M. I. (2007). Protective role of autophagy against Vibrio cholerae cytolysin, a pore-forming toxin from V. cholerae. Proc. Natl. Acad. Sci. USA 104, 1829-1834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heuser J. (2000). The production of ‘cell cortices’ for light and electron microscopy. Traffic 1, 545-552 [DOI] [PubMed] [Google Scholar]
- Heuser J. (2005). Deep-etch EM reveals that the early poxvirus envelope is a single membrane bilayer stabilized by a geodetic “honeycomb” surface coat. J. Cell Biol. 169, 269-283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heuser J. E., Anderson R. G. (1989). Hypertonic media inhibit receptor-mediated endocytosis by blocking clathrin-coated pit formation. J. Cell Biol. 108, 389-400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirst R. A., Kadioglu A., O'Callaghan C., Andrew P. W. (2004). The role of pneumolysin in pneumococcal pneumonia and meningitis. Clin. Exp. Immunol. 138, 195-201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Husmann M., Beckmann E., Boller K., Kloft N., Tenzer S., Bobkiewicz W., Neukirch C., Bayley H., Bhakdi S. (2009). Elimination of a bacterial pore-forming toxin by sequential endocytosis and exocytosis. FEBS Lett. 583, 337-344 [DOI] [PubMed] [Google Scholar]
- Idone V., Tam C., Goss J. W., Toomre D., Pypaert M., Andrews N. W. (2008). Repair of injured plasma membrane by rapid Ca2+-dependent endocytosis. J. Cell Biol. 180, 905-914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keefe D., Shi L., Feske S., Massol R., Navarro F., Kirchhausen T., Lieberman J. (2005). Perforin triggers a plasma membrane-repair response that facilitates CTL induction of apoptosis. Immunity 23, 249-262 [DOI] [PubMed] [Google Scholar]
- Keyel P. A., Watkins S. C., Traub L. M. (2004). Endocytic adaptor molecules reveal an endosomal population of clathrin by total internal reflection fluorescence microscopy. J. Biol. Chem. 279, 13190-13204 [DOI] [PubMed] [Google Scholar]
- Limbago B., Penumalli V., Weinrick B., Scott J. R. (2000). Role of streptolysin O in a mouse model of invasive group A streptococcal disease. Infect. Immun. 68, 6384-6390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magassa N., Chandrasekaran S., Caparon M. G. (2010). Streptococcus pyogenes cytolysin-mediated translocation does not require pore formation by streptolysin O. EMBO Rep. 11, 400-405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNeil P. L., Kirchhausen T. (2005). An emergency response team for membrane repair. Nat. Rev. Mol. Cell Biol. 6, 499-505 [DOI] [PubMed] [Google Scholar]
- McNeil P. L., Vogel S. S., Miyake K., Terasaki M. (2000). Patching plasma membrane disruptions with cytoplasmic membrane. J. Cell Sci. 113, 1891-1902 [DOI] [PubMed] [Google Scholar]
- Mishra S. K., Keyel P. A., Edeling M. A., Dupin A. L., Owen D. J., Traub L. M. (2005). Functional dissection of an AP-2 beta2 appendage-binding sequence within the autosomal recessive hypercholesterolemia protein. J. Biol. Chem. 280, 19270-19280 [DOI] [PubMed] [Google Scholar]
- Mitchison T. J., Charras G. T., Mahadevan L. (2008). Implications of a poroelastic cytoplasm for the dynamics of animal cell shape. Semin. Cell Dev. Biol. 19, 215-223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J. M., Ng V. H., Maeda S., Rest R. F., Karin M. (2004). Anthrolysin O and other gram-positive cytolysins are toll-like receptor 4 agonists. J. Exp. Med. 200, 1647-1655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Portnoy D. A., Jacks P. S., Hinrichs D. J. (1988). Role of hemolysin for the intracellular growth of Listeria monocytogenes. J. Exp. Med. 167, 1459-1471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raposo G., Nijman H. W., Stoorvogel W., Liejendekker R., Harding C. V., Melief C. J., Geuze H. J. (1996). B lymphocytes secrete antigen-presenting vesicles. J. Exp. Med. 183, 1161-1172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy A., Caler E. V., Andrews N. W. (2001). Plasma membrane repair is mediated by Ca(2+)-regulated exocytosis of lysosomes. Cell 106, 157-169 [DOI] [PubMed] [Google Scholar]
- Rossjohn J., Polekhina G., Feil S. C., Morton C. J., Tweten R. K., Parker M. W. (2007). Structures of perfringolysin O suggest a pathway for activation of cholesterol-dependent cytolysins. J. Mol. Biol. 367, 1227-1236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schujman G. E., de Mendoza D. (2005). Transcriptional control of membrane lipid synthesis in bacteria. Curr. Opin. Microbiol. 8, 149-153 [DOI] [PubMed] [Google Scholar]
- Shannon J. G., Ross C. L., Koehler T. M., Rest R. F. (2003). Characterization of anthrolysin O, the Bacillus anthracis cholesterol-dependent cytolysin. Infect. Immun. 71, 3183-3189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith H. R., Heusel J. W., Mehta I. K., Kim S., Dorner B. G., Naidenko O. V., Iizuka K., Furukawa H., Beckman D. L., Pingel J. T., et al. (2002). Recognition of a virus-encoded ligand by a natural killer cell activation receptor. Proc. Natl. Acad. Sci. USA 99, 8826-8831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein J. M., Luzio J. P. (1989). Membrane sorting during vesicle shedding from neutrophils during sublytic complement attack. Biochem. Soc. Trans. 17, 1082-1083 [DOI] [PubMed] [Google Scholar]
- Stoorvogel W., Kleijmeer M. J., Geuze H. J., Raposo G. (2002). The biogenesis and functions of exosomes. Traffic 3, 321-330 [DOI] [PubMed] [Google Scholar]
- Straight A. F., Cheung A., Limouze J., Chen I., Westwood N. J., Sellers J. R., Mitchison T. J. (2003). Dissecting temporal and spatial control of cytokinesis with a myosin II Inhibitor. Science 299, 1743-1747 [DOI] [PubMed] [Google Scholar]
- Thery C., Regnault A., Garin J., Wolfers J., Zitvogel L., Ricciardi-Castagnoli P., Raposo G., Amigorena S. (1999). Molecular characterization of dendritic cell-derived exosomes. Selective accumulation of the heat shock protein hsc73. J. Cell Biol. 147, 599-610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiery J., Keefe D., Saffarian S., Martinvalet D., Walch M., Boucrot E., Kirchhausen T., Lieberman J. (2010). Perforin activates clathrin- and dynamin-dependent endocytosis, which is required for plasma membrane repair and delivery of granzyme B for granzyme-mediated apoptosis. Blood 115, 1582-1593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tripathy S. K., Smith H. R., Holroyd E. A., Pingel J. T., Yokoyama W. M. (2006). Expression of m157, a murine cytomegalovirus-encoded putative major histocompatibility class I (MHC-I)-like protein, is independent of viral regulation of host MHC-I. J. Virol. 80, 545-550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tweten R. K. (2005). Cholesterol-dependent cytolysins, a family of versatile pore-forming toxins. Infect. Immun. 73, 6199-6209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walev I., Palmer M., Valeva A., Weller U., Bhakdi S. (1995). Binding, oligomerization, and pore formation by streptolysin O in erythrocytes and fibroblast membranes: detection of nonlytic polymers. Infect. Immun. 63, 1188-1194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walev I., Vollmer P., Palmer M., Bhakdi S., Rose-John S. (1996). Pore-forming toxins trigger shedding of receptors for interleukin 6 and lipopolysaccharide. Proc. Natl. Acad. Sci. USA 93, 7882-7887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walev I., Tappe D., Gulbins E., Bhakdi S. (2000). Streptolysin O-permeabilized granulocytes shed L-selectin concomitantly with ceramide generation via neutral sphingomyelinase. J. Leukoc. Biol. 68, 865-872 [PubMed] [Google Scholar]
- Walev I., Bhakdi S. C., Hofmann F., Djonder N., Valeva A., Aktories K., Bhakdi S. (2001). Delivery of proteins into living cells by reversible membrane permeabilization with streptolysin-O. Proc. Natl. Acad. Sci. USA 98, 3185-3190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker B., Braha O., Cheley S., Bayley H. (1995). An intermediate in the assembly of a pore-forming protein trapped with a genetically-engineered switch. Chem. Biol. 2, 99-105 [DOI] [PubMed] [Google Scholar]
- Wei Z., Schnupf P., Poussin M. A., Zenewicz L. A., Shen H., Goldfine H. (2005). Characterization of Listeria monocytogenes expressing anthrolysin O and phosphatidylinositol-specific phospholipase C from Bacillus anthracis. Infect. Immun. 73, 6639-6646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie M., Low M. G. (1995). Streptolysin-O induces release of glycosylphosphatidylinositol-anchored alkaline phosphatase from ROS cells by vesiculation independently of phospholipase action. Biochem. J. 305, 529-537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeligs J. D., Wollman S. H. (1977). Ultrastructure of blebbing and phagocytosis of blebs by hyperplastic thyroid epithelial cells in vivo. J. Cell Biol. 72, 584-594 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}