

Summary
Crystal structures of substrate/product complexes of Methanobacterium thermoautotrophicum orotidine-5′-monophosphate decarboxylase, obtained at various steps in its catalysis of the unusual transformation of 6-CN-UMP into barbituric acid ribosyl monophosphate (BMP), show that the cyano substituent of the substrate, when bound to the active site, is first bent significantly from the plane of the pyrimidine ring and then replaced by an oxygen atom. Although the K72A and D70A/K72A mutants are either catalytically strongly impaired or even completely inactive they still display bending of the C6 substituent. Interestingly, high-resolution structures of the D70A and D75N mutants revealed a covalent bond between C6 of UMP and the Lys72 side chain after the –CN moiety’s release. The same covalent bond was observed when the native enzyme was incubated with 6-N3-UMP and 6-I-UMP; in contrast, the K72A mutant transformed 6-I-UMP to BMP. These results demonstrate that, given a suitable environment, native ODCase as well as several of its mutants are not restricted to the physiologically relevant decarboxylation; they are able to catalyze even nucleophilic substitution reactions but consistently maintain distortion on the C6 substituent as an important feature of catalysis.
Keywords: ODCase, orotidine monophosphate decarbxylase, OMPDC, covalent inhibition, time-resolved crystallography
Introduction
Orotidine-5′-monophosphate decarboxylase (ODCase) catalyzes the transformation of orotidine-5′-monophosphate (OMP) to uridine-5′-monophosphate (UMP) (Fig. 1a), the last step of de novo pyrimidine biosynthesis. ODCase is also known as one of the most proficient enzymes, accelerating the decarboxylation reaction by 17 orders of magnitude when compared to the spontaneous reaction in water at neutral pH.1 ODCase achieves this enormous catalytic power without employing cofactors, metal ions or delocalization effects.
Fig. 1. Structure and catalysis of MtODCase.

(a) Reaction catalyzed by ODCase. (b) Chemical structures of the inhibitors discussed in this report. (c) Ribbon diagram of the BMP complex of M. thermoautotrophicum ODCase (PDB ID: 1X1Z). Secondary structure elements are indicated in blue and green for one subunit and in red and yellow for the other. The ligands are shown as stick models indicating the location of the active sites. (d) Hydrogen-bonding and electrostatic network surrounding the inhibitor BMP in the active site. The numbers indicate the distances between the various atoms in Ångstrom. Figs. 1(c)–(d) were prepared with PyMol (http://pymol.sourceforge.net).
Thus far, X-ray crystal structures of ODCases from eleven organisms have been determined; they all assume a TIM-barrel fold and their active sites are structurally quite similar (Fig. 1c). The reaction center is located at the C-terminal side of the β-sheet barrel and at the interface of the catalytically active dimer.2 Crystal structures of active site mutants and of complexes of ODCases from several species with various inhibitors established the very restricted and rigid nature of their active sites. Bound ligands are buried deeply at the dimer interface. Usually, about 15 hydrogen bonds are formed between mononucleotide ligands and enzyme matrix; one counts about 20 bonds if those to tightly bound water molecules are included. Consistently, two thirds of these bonds involve the ribose monophosphate part of the ligands. Residues Val182 to Gly190 are disordered in the apo-form of the enzyme but assume a stable loop conformation when interacting with the 5′-phosphate of nucleotide ligands. 3
One of the most characteristic features of the active site is an extended network of hydrogen bonds and electrostatic interactions anchored by alternating lysine and aspartate residues. While Lys42, Asp70 and Lys72 are from one subunit, the fourth residue Asp75B* originates from the second subunit. Any substituent at C6 of the uracil ring directly faces this network, placed especially close to Asp70 and Lys72 (Fig. 1d), residues for which mutagenesis and kinetics studies of several orthologous enzymes consistently postulate a leading role in catalysis.4–8
As part of our efforts to investigate in further detail the reaction mechanism underlying this astonishing catalytic power and to supplement our mutation studies,9,10 we undertook the crystallographic analysis of the complexes of the three substrate analogues 6-CN-UMP, 6-N3-UMP and 6-I-UMP with M. thermoautotrophicum ODCase at resolutions between 1.85 Å and 1.53 Å. Interest in these compounds was also heightened by the recent finding that 6-CN-UMP is a slow substrate of MtODCase, being converted into BMP with a half-life of 5 h for the reaction in solution11,12 and by the fact that 6-N3-UMP and 6-I-UMP are irreversible inhibitors of Plasmodium falciparum ODCase.13
Initially, 6-CN-UMP inhibits MtODCase competitively with a Ki of 2.9 × 10−5 M; the product BMP is the most potent competitive ODCase inhibitor known (Ki = 8.8 × 10−12 for yeast ODCase).14 Again from the initial rates, one can estimate a competitive Ki of 1.9 × 10−7 M for 6-N3-UMP but then this compound modifies the enzyme covalently with a KI and kinact of 6.3 × 10−7 M and 61.2 h−1, respectively.13,15 To explain the mixture of non-covalent and covalent interactions combined with catalytic transformations experienced when MtODCase is incubated with substrate analogues poses an interesting challenge.
To provide a structural framework for the mechanistic studies, we analyzed the crystal structures of these compounds in complex with native MtODCase as well as several of its active site mutants. The slowness of the unexpected and mechanistically highly interesting conversion of 6-CN-UMP to BMP allowed us to investigate different time points along the reaction coordinate. Several structures of the complexes of 6-CN-UMP with electrostatic network mutants were also determined to further probe the contributions of individual residues to catalysis.
Results and Discussion
Crystallography
Crystals of the complexes of native MtODCase with 6-CN-UMP and 6-N3-UMP, as well as those of the enzyme’s D70A, K72A and D75N mutants with 6-CN-UMP all grew in space group P21 with approximate cell dimensions of a = 58 Å, b = 74 Å, c = 59 Å and β = 119° (for details, see Table 1). Crystals of the double mutant D70A/K72A complexed with 6-CN-UMP also adopted space group P21 but with a = 59.6 Å, b = 64.4 Å, c = 61.5 Å; β = 115.5°. Crystals of the K72A mutant complexed with 6-I-UMP belonged to space group C2221 (a = 57.9 Å, b = 103.3 Å, c = 73.5 Å).
Table 1.
Data and refinement statistics
| MtODCase | Wild-type | K72A | D70A | D75N | D70A/K72A | Wild-type | K72A |
|---|---|---|---|---|---|---|---|
| Ligand | 6-CN-UMP | 6-CN-UMP | 6-CN-UMP | 6-CN-UMP | 6-CN-UMP | 6-N3-UMP | 6-I-UMP |
| X-ray source | APS 14BM-C | APS 14ID-B | APS 14ID-B | In-house | APS 14BM-C | APS 14BM-D | APS 14ID-B |
| Data Statistics | |||||||
| Space group | P21 | P21 | P21 | P21 | P21 | P21 | C2221 |
| Cell constants a (Å) | 58.02 | 58.09 | 58.08 | 58.04 | 59.56 | 57.93 | 57.93 |
| b (Å) | 73.54 | 73.82 | 73.77 | 73.85 | 64.36 | 73.69 | 103.29 |
| c (Å) | 59.33 | 59.32 | 59.15 | 59.23 | 61.54 | 59.15 | 73.53 |
| β (°) | 119.3 | 119.3 | 119.4 | 119.3 | 115.52 | 119.3 | 90 |
| Resolution (Å) (Whole) | 100-1.57 | 100-1.53 | 100-1.85 | 100-1.67 | 55.6-1.56 | 100-1.66 | 200-1.58 |
| (Outmost) | (1.60-1.57) | (1.56-1.53) | (1.89-1.85) | (1.71-1.67) | (1.59-1.56) | (1.69-1.66) | (1.61-1.58) |
| No. Observed reflections | 229,504 | 224,906 | 133,818 | 366,648 | 178,717 | 193,043 | 223,943 |
| No. Unique reflections | 60,802 | 62,518 | 36,059 | 48,799 | 50,081 | 50,234 | 30,641 |
| Completeness† | 99.7% (99.8%) | 95.6% (76.6%) | 98.0% (92.4%) | 96.5% (93.5%) | 83.9% (30.2%) | 98.3% (97.0%) | 99.9% (100%) |
| Rmerge† | 6.0% (29.6%) | 5.5% (29.9%) | 6.2% (26.2%) | 5.1% (22.3%) | 8.3% (43.0%) | 6.0% (31.0%) | 5.7 % (30.1%) |
| I/σ(I)† | 22.1 (5.0) | 22.1(2.8) | 19.3 (3.8) | 37.3 (9.0) | 9.9 (1.4) | 22.6 (4.5) | 33.2 (7.0) |
|
| |||||||
| Refinement Statistics | |||||||
| Rcryst | 15.6% | 15.9% | 16.2% | 15.8% | 16.0% | 16.2% | 15.2% |
| Rfree | 18.2% | 18.7% | 19.1% | 18.5% | 18.7% | 18.7% | 17.1% |
| No. of Reflections used | 60,468 | 62,494 | 38,632 | 48,711 | 50,050 | 50,224 | 30,514 |
| Average B factors and No. of atoms‡ | |||||||
| Protein‡ | 14.1 (3,420) | 13.1 (3,416) | 15.9 (3,397) | 15.4 (3,484) | 18.2 (3,397) | 14.1 (3,392) | 12.2 (1,806) |
| Ligand‡ | 9.7 (48) | 9.7 (46) | 8.3 (42) | 9.1 (42) | 16.1 (46) | 6.9 (42) | 6.9(22) |
| Solvents‡ | 28.5 (311) | 26.4 (322) | 27.8 (263) | 30.3 (343) | 28.9 (199) | 27.6 (303) | 30.0 (201) |
| Ramachandran plot | |||||||
| Favored | 98.1% | 97.7% | 97.4% | 97.7% | 98.1% | 97.2% | 98.1% |
| Allowed | 100% | 100% | 100% | 100% | 100% | 100% | 100% |
| R.M.S. from ideal values | |||||||
| Lengths | 0.016 | 0.016 | 0.013 | 0.020 | 0.021 | 0.013 | 0.016 |
| Angles | 1.7 | 1.6 | 1.5 | 1.8 | 1.8 | 1.6 | 1.6 |
Values in parentheses are for the outmost shell
Values in parentheses are number of Atoms
Rcryst = Σ||Fobs| − |Fcalc||/Σ|Fobs|
Rfree is the same as R, but for a 5 % of all reflections that were never used in crystallographic refinement.
Structural and Biochemical Analyses
The overall molecular structures of all the complexes of native and mutant MtODCases with the inhibitors 6-CN-UMP, 6-N3-UMP and 6-I-UMP are almost identical, independent of the space group of their individual crystals (Fig. 1c).3,9–11,15 The asymmetric unit of the K72A:6-I-UMP crystal contains one ligand-bound subunit of the physiological MtODCase homodimer, whereas all other asymmetric units hold the dimer, with each subunit binding an inhibitor molecule. The RMSD distances between all Cα atoms, with the BMP complex of native MtODCase serving as reference, are 0.08 Å, 0.10 Å, 0.16 Å, 0.16 Å, 0.33 Å, 0.13 Å and 0.10 Å, for the complexes native:6-CN-UMP, K72A:6-CN-UMP, D70A:6-CN-UMP, D75N:6-CN-UMP, D70A/K72A:6-CN-UMP, native:6-N3-UMP and K72A:6-I-UMP, respectively. Individual subunits superimpose well with even lower RMSD values of between 0.03 Å and 0.17 Å for 213 to 215 Cα atoms, establishing overall structural integrity and conservation for the various MtODCase mutants investigated, independent of their ligation state.
Although the overall structure did not change, there were remarkable differences in the way the active sites of the various forms of the enzyme interacted with the inhibitors. Figure 2a shows 6-CN-UMP in the active site of the K72A mutant. The “omit” electron density map, calculated by excluding from phase calculation the atoms belonging to the pyrimidine ring and the surrounding residues, thereby avoiding model bias, clearly reflects the angle by which the cyano group is bent out of the plane of the pyrimidine ring. As seen computationally, the energy to deform the angle between the cyano moiety and the plane of the pyrimidine ring was significantly lower than that necessary to bend the angle C6-C7-N8 (Fig. 3); therefore, the latter was restrained to 180° during the refinement. Such an assignment is also supported by the fact that all cyano groups in the Cambridge Structural Database16 assume a linear geometry. With this parameter set, the best fit to the electron density was obtained when the cyano group was bent by approximately 50° out of the plane of the pyrimidine base and away from residue Lys72. A similar deformation has been reported for the carboxylate substituent of the substrate OMP when bound to the D70A/K72A mutant of MtODCase9 and to the D312N mutant of human ODCase; in the latter case, nucleotide binding also induced conformational changes in parts of the active site.17
Fig. 2. Close-ups of the active sites of native and mutant MtODCase complexes with 6-CN-UMP.

(a)–(d) Fo−Fc omit electron density maps superimposed on the atomic model. The pyrimidine ring and residues Lys72, Asp70, Lys42 and Asp75B (or the corresponding mutant residues) are omitted for the map calculation. (a) K72A mutant. Map contoured at 2.5 σ. (b) Native MtODCase incubated with 6-CN-UMP for 1 day at RT and 1 day at 4 °C. (c) Native MtODCase incubated with 6-CN-UMP for 2 months at RT. D) D70A/K72A double mutant. Maps (b)–(d) are contoured at 3.0 σ. (e) View approximately perpendicular to (b). The Fo−Fc omit electron density map was calculated with the pyrimidine ring and the surrounding water molecules excluded from the phasing model. (f) Fo(2days)-Fo(2months) difference electron density map superimposed on the model of the native MtODCase 6-CN-UMP complex. The ligand molecule was excluded from the model used in calculating the phases for this map. Green (positive difference density) and red (negative difference density) meshes are contoured at +6.5 σ and −6.5 σ, respectively. (g)–(h) Stereo views of the side chain arrangements in the reaction center of MtODCase. Numbers colored in both blue and red indicate the distances between atoms in Ångstrom. (g) The K72A:6-CN-UMP complex. Lys72 of native MtODCase is superimposed for comparison and shown in transparent gray. (h) Native MtODCase:6-CN-UMP incubated for 1 day at RT and 1 day at 4 °C. The hydroxyl group of the product BMP is shown in transparent gray and red. (i) D70A/K72A:6-CN-UMP. Asp70 and Lys72 of native ODCase is superimposed for comparison and shown in transparent gray. (j) D70A/K72A:OMP (PDB ID: 1KM6). The D70A/K72A:6-CN-UMP complex was superimposed and 6-CN-UMP is shown in transparent grey for comparison. Water molecules bound in the D70A/K72A:6-CN-UMP complex are shown in transparent green. These panels were prepared with PyMol (http://pymol.sourceforge.net).
Fig. 3. Energy plot of -CN bending out of the plane of the uridine ring.
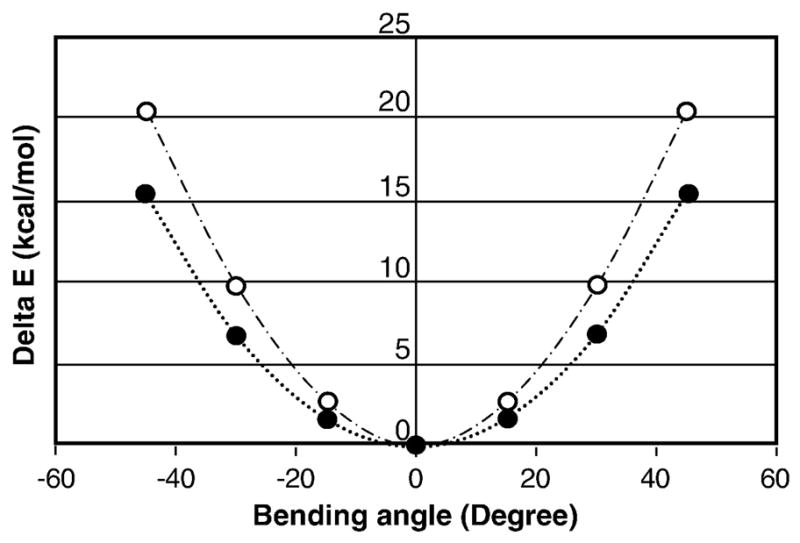
The energies for various geometries of 6-CN-UMP with C6–C7 bent with respect to the plane of the pyrimidine moiety were computed using Gaussian 98, using the basis set 6-31+G*, at the DFT/B3-LYP theory level in gas phase. The potential energy scans (PES) were conducted at 15° angle steps. Solid circles trace the energy for bending the angle (centre of the aromatic ring) – C6 – C7 and open circles trace the energy for the angle C6 – C7 – N8.
Two water molecules are bound near the cyano group. As the distance between the two molecules is only 1.8 Å (Figs. 2a and g), too close for simultaneous residency, they are assigned as a pair of water molecules HOH463(A) and HOH463(B) with alternate binding. Their respective occupancies are estimated at 0.6 (A) and 0.4 (B) based on the heights of the corresponding electron density peaks and appropriate B-factors of 13–16 Å2. HOH463(A) occupies a position very similar to that of Nζ of Lys72 in native MtODCase (in transparent gray in Fig. 2g); it is hydrogen-bonded to Asp70 and Asp75B. HOH463(B) is coordinated by the main chain carbonyl of Ala74B and by Asp75B. In native MtODCase, the alkyl side chain of Lys72 occupies the corresponding space, precluding water from binding at this location. Similarly located waters (HOH3015 and HOH3016) were found in the K72A:UMP complex (PDB ID: 1KM4). As the distance between these two waters is 2.6 Å they can be modeled as independent binders with full occupancy.
For the 6-CN-UMP complex of native MtODCase, flash-frozen two days after mixing, a more complex electron density is observed. Although density fitting a bent cyano moiety is seen (Fig. 2b, red arrow) it is weaker than the corresponding feature in the K72A complex and significant additional density, contiguous to the plane of the pyrimidine ring, is visible (Fig. 2b, green arrow). As MtODCase will convert 6-CN-UMP to BMP,11 a picomolar inhibitor of the enzyme,14 density representing the intermediate state was refined against a protein model with a 1:1 mixture of the two ligands in its active site. The B-factors for both O1 of BMP (hydroxyl at C6) and C7 of 6-CN-UMP refine to ~10 Å2, in the range of those of the aromatic ring atoms, indicating that the estimated occupancies of “substrate” and “product” are consistent with the results obtained in solution.11 In the best fit, the cyano group is bent by approximately 30° (angle between the segment of C6 and N8 and the pyrimidine plane), in the same direction (red arrow, Fig. 2b) but by slightly less than what had been observed in the K72A mutant structure (Figs. 2b vs. 2a).
A water molecule, HOH461, is found next to the O1 atom of BMP (Fig. 2e). A corresponding water molecule was identified in the complex of native enzyme with BMP (PDB codes: 1X1Z and 1LOR).10,11 No such water is detected in the complexes of native MtODCase with UMP and 6-azaUMP (PDB codes: 1LOQ and 1DVJ).10 The B-factor of this BMP-specific water molecule refines to 23 Å2 when its occupancy is estimated at 0.50, whereas the B-values of the corresponding waters in native MtODCase complexed with BMP are 16 Å2 and 19 Å2 (PDB codes: 1X1Z and 1LOR) when set to full occupancy.
Hydrolysis of 6-CN-UMP to BMP continued in the crystal even after 2 days of incubation. Figure 2c represents the same view of a map calculated from data collected after a two-month incubation (PDB ID: 1X1Z).11 The long wait ensured that the reaction had gone to completion. The cyano group has been replaced by a hydroxyl group, BMP is now easily identified in representative electron density. The map and molecular structure of the product complex are identical to those of native MtODCase co-crystallized with BMP, reported previously.10
The three crystal structures described above, the K72A mutant with 6-CN-UMP and the native enzyme incubated with 6-CN-UMP for two days and for two months, respectively, can be considered as a series of “time-resolved” snapshots (Figs. 2a–c). The K72A mutant, although catalytically impaired, is still capable of distorting the ligand upon binding, thereby trapping the warped conformation of 6-CN-UMP (Fig. 2a), mimicking the Michaelis complex or “0 time” snapshot of this slow reaction. A different scenario is observed with catalytically active, native ODCase. Figure 2b presents the arguably most interesting result, a snapshot of the partial transformation of 6-CN-UMP into BMP. Electron densities corresponding to the substrate and the product are superimposed. After two days, approximately half of the cyano group has been replaced by a hydroxyl group in the crystal (Fig. 2b). As slower reaction rates are not unusual in a crystalline environment, this result is consistent with the relatively long half-life (t1/2) of the reaction of ca. five hours in solution.11 Bending the C6-C7 bond out of the plane of the aromatic ring by 30° or more will help facilitate the attack of a water molecule at C6 instead of the generally more reactive C7 position. In the end, the action of native ODCase on 6-CN-UMP will produce BMP, the most potent inhibitor of ODCase (Fig. 2c).
The electron density map calculated with structure factors Fo (2 d) - Fo (2 months) illustrates nicely the change that occurs when the substrate 6-CN-UMP is slowly transformed into the product BMP (Fig. 2f). The decrease of the density level representing the cyano group and the concomitant increase at the position of the oxygen atom reflect the progress of the catalytic reaction.
The distortion observed in 6-CN-UMP could be caused either by electrostatic or steric repulsion between the ligand’s cyano group and the surrounding side chains of the active site as well as tightly bound water molecules, with the carboxyl group of Asp70, the amino group of Lys72 and the water molecule HOH463(A) as the prime candidates for such interactions. Among these three groups, Asp70 probably makes the strongest contribution to the bending of the linear substituent in both the K72A:6-CN-UMP and native enzyme:6-CN-UMP complexes. In the K72A mutant, the distance between the cyano group and Asp70 Oε2 is 2.9 Å (Fig. 2g). HOH463(A) is close to the ligand’s C6 and C7 atoms, with distances of 3.3 Å to 3.4 Å. In native ODCase, the corresponding numbers are 3.2 Å and 3.4 Å (Fig. 2h).
The double mutant D70A/K72A has completely lost its capability to transform OMP to UMP, as shown by the crystallization and structural analysis of the substrate complex.9 As the removal of the two side chains generates significant space that should easily accommodate the cyano substituent, one could expect the 6-CN-UMP complex to harbour the cyano moiety that extends from the aromatic ring in an unstrained coplanar configuration. However, the corresponding electron density clearly shows that the cyano segment is still bent out of the plane of the pyrimidine ring (Fig. 2d; red arrow). Three characteristic waters, HOH465, HOH467 and HOH469, can also be seen around the cyano group in the omit electron density map shown in Fig. 2d. Held by an extensive network of hydrogen bonds, they are located in density visible up to contour levels of 5.5 σ, 8.0 σ and 3.7 σ, respectively, indicating relatively tight binding. HOH465 resides halfway between the positions assumed by Nζ of Lys72 and the carboxylate of Asp70 in the native enzyme (Fig. 2i). It is anchored by hydrogen bonds to Oδ2 of Asp75B (2.8 Å) and Nζ of Lys42 (3.1 Å). Forming close contacts of 3.5 Å, 3.1 Å and 3.2 Å to C6, C7 and N8, respectively, this water could now contribute to the bending of the C6-CN bond. The other two waters, HOH467 and HOH469, with distances to the cyano substituent ranging from 3.5 to 4.0 Å, might add, although more weakly, to the distortion (Fig. 2i).
All 208 Cα atoms of one subunit of the D70A/K72A:6-CN-UMP complex superimpose well (RMSD = 0.34 Å) with their counterparts in the previously analyzed D70A/K72A:OMP complex (PDB ID: 1KM6).9 This similarity extends to the respective substituents. The carboxylate of OMP is bent in the same direction as the cyano group in 6-CN-UMP; its rotational component obviously absent in the latter (Fig. 2j). Of the three waters bound near the reaction center in the OMP complex, HOH3015 and HOH3017 are located almost in the same positions as HOH467 and HOH469 in the 6-CN-UMP complex, respectively. HOH3016 (OMP complex) bridges Oδ2 of Asp75B and Nζ of Lys42 as does HOH465 in the 6-CN-UMP complex; the actual positions of these two waters, however, are about 1.5 Å apart. HOH3016 is 3.5 – 4.0 Å away from the carboxylate group of OMP (Fig. 2j), whereas HOH465 approaches C7 of the cyano group to 3.1 Å (Fig. 2i).
The catalytic activity of the D91A mutant of yeast ODCase is reduced by at least 5 orders of magnitude.8 Co-crystallization of the equivalent D70A mutant of the archaeal enzyme with 6-CN-UMP, undertaken in an effort to better isolate the role of Lys72 in catalysis, produced another surprising result; a covalent bond between the ligand and the enzyme (Fig. 4a). The Fo−Fc omit map of electron density unequivocally shows a strong connection between Nζ of Lys72 and C6 of the pyrimidine ring. The cyano moiety of the original ligand has been completely replaced. Mass spectrometry confirmed the covalent modification (Figs. 4b–c). With the loss of the negative charge of Asp70, the side chain of Lys42 is anchored less strongly, assuming alternate conformations.
Fig. 4. Complexes with covalent links between ligand and Lys72 of MtODCase.

(a) Fo−Fc omit electron density map around the ligand superimposed on the atomic model of the D70A:6-CN-UMP complex. Corresponding mass spectra of MtODCase (b) without any ligand (control) and (c) after incubation with 6-CN-UMP. (d) D75NB:6-CN-UMP complex. Corresponding mass spectra of MtODCase (e) without any ligand (control) and (f) after incubation with 6-CN-UMP. (g) Native MtODCase:6-N3-UMP complex. (h) without any ligand (control) and (i) after incubation. All maps are contoured at 2.5 σ. Figs. 4(a), (d), and (g) were prepared with PyMol (http://pymol.sourceforge.net).
The same covalent modification of the enzyme, again confirmed by mass spectrometry, was seen when the D75N mutant was exposed to 6-CN-UMP (Figs. 4d–f) but this time the side chain of Asn75 also adopted an alternate conformation (D75NB(B)), in addition to the one found in native enzyme (D75NB(A)). As judged by electron density peak height, approximately 85% of the residues are in this latter conformation, with 15 % residing in the new one (Fig. 4d). Confirming this assignment, the B-factors of all atoms in both orientations refine to a relatively narrow range of appropriate values (10–15 Å2).
6-CN-UMP is slowly converted to BMP by native ODCase.11 The D70A and D75N mutants of the enzyme, however, form a covalent bond to the side chain of Lys72, a neighboring active site residue. In the following, we attempt an explanation for the difference in behaviour between native and mutant proteins. The electrostatic network of Lys42-Asp70-Lys72-Asp75B (Fig. 1d) is conserved in all known ODCases 5,18; it is reasonable to assume that the arrangement keeps the Nζ atom of Lys72 protonated. Both mutations, D70A as well as D75NB, lead to a decrease of the pKa value of Lys72, increasing the probability that its Nζ carries a lone pair of electrons. As indicated by the alternate conformations of Lys42 in the D70A mutant and of Asn75B, breaking this electrostatic network significantly increases the mobility of the side chains that form it. The same result had been seen in the D70A:UMP complex structure, in which Lys72 also assumes two conformations.9 Both aspects, mobility and deprotonation, make it easier for Lys72 to undergo a nucleophilic attack on C6 of the pyrimidine ring, replacing the already strained cyano group (Fig. 6b). In contrast, in the native enzyme with its intact network of charges, Lys72 is always protonated rendering it an electrophile and leaving water as a substitute (Fig. 6c). As the active site in the native enzyme is rather crowded, water molecules will only rarely be able to approach the C6 atom closely enough to participate in the reaction, i.e. turnover will be very slow.11
Fig. 6. Cartoon of the proposed reaction schemes.

Distortions of the carboxyl, cyano, iodo and azido groups play an important role in the reaction catalyzed by ODCase. “P-Rib” abbreviates ribose-5′-monophosphate. (a) The reaction from OMP to UMP catalyzed by native ODCase. (b) Covalent bond formation between the mutant ODCase and 6-CN-UMP. (c) The conversion of 6-CN-UMP to BMP catalyzed by native ODCase. (d) Covalent bond formation between the native ODCase and 6-I-UMP or 6-N3-UMP. (e) The conversion of 6-I-UMP to BMP by K72A MtODCase. Distortion of the bond linking the pyrimidine ring to the C6-substituent is conserved in all these reactions, the physiologically relevant decarboxylation as well as the nucleophilic mechanisms.
The covalent bond between the ligand and Lys72 is also found when native MtODCase is incubated with 6-I-UMP,15 a reaction also observed in human ODCase17 and in the P. falciparum enzyme.13,15 For the latter protein, 6-N3-UMP was shown to result in the identical covalent modification.13 We now confirm that MtODCase, too, is capable of covalent bond formation between the pyrimidine ring and Lys72 when incubated with 6-N3-UMP (Figs. 4g–i).
The approximate pKa values for HI, HN3 and HCN are −10, 4.6 and 9.2, respectively, making I− a good and CN− a poor leaving group in this group of ligands. As discussed above, with Lys72 engaged in the tight Lys-Asp-Lys-Asp network of the ODCase active site with two carboxylates next to its side chain amino group, its pKa will shift to a higher value simultaneously reducing its ability to act as a nucleophile. This arrangement also results in severely reduced mobility for the Lys72 side chain. In the case of 6-I-UMP and 6-N3-UMP, despite these barriers, Lys72 undergoes a nucleophilic attack on the C6 atom (Fig. 6d). In the 6-CN-UMP complex, however, the side chain of Lys72—due to the poor leaving group status of the cyano moiety as well as the already mentioned restricted ability of the Lys72 side chain to function as a nucleophile—it is no longer able to release the cyano substituent (Fig. 6c).
The conversion of 6-I-UMP into BMP by the K72A mutant of MtODCase has been characterized both by crystallography and mass spectrometry (Fig. 5). Again due to the mutation to alanine, the lysine amino group is unavailable for a nucleophilic attack onto the C6 position of the pyrimidine; the accompanying increase in space allows for an easier access of water molecules which then are able to act as nucleophiles, generating the inhibitor BMP (Figs. 5 and 6e) as had been observed for the 6-CN-UMP complex of native MtODCase.
Fig. 5. BMP as product of exposure of 6-I-UMP to the K72A mutant of MtODCase.

(a) Fo−Fc electron density omit map (contoured at 3.5 σ) around the ligand superimposed on the atomic model of the K72A:6-I-UMP complex. Corresponding mass spectra of MtODCase (b) without any ligand (control) and (c) after incubation with 6-I-UMP. Fig. 5(a) was prepared with PyMol (http://pymol.sourceforge.net).
For the three substrate analogs described in this report, one could reasonably conclude that the type of catalytic reaction that is eventually taking place in the active site of ODCase depends on the potential availability of Lys72 as a nucleophile and on the degree of access water has to the reaction center. Dependent on the actual pKa of its side chain amino group and/or its mobility, this can involve a direct attack of Lys72 onto the C6 center, resulting in a covalent link between the ligand and the enzyme, or the activation of a water molecule to generate BMP.
The steadily increasing number of publications reporting covalent interactions of ODCases with various substrate analogs have led to a renewed interest in mechanistic proposals that include covalent intermediates. One has to consider, however, that all known covalent links are to ring atom C6 and generate “dead end” complexes. Wittmann et al.17 suggested that the 6-CN-UMP to BMP transformation could also involve a covalent interaction between an active site lysine residue and atom C6. They postulated that the slow reaction rate could allow for “neighboring group participation of the lysine Nζ atom” with formation of a reactive imine/iminium ion followed by hydrolysis. The even slower reaction of 6-N3-UMP with ODCase13, which nevertheless results in the lysine-nucleotide bond, would seem to contradict such an interpretation. The results, however, are fully consistent with an increased nucleophilic potential of the central lysine residue in the active site of ODCase enzymes. While the electron pair shared between atom C6 and substituents like cyano or azido groups will stay with the leaving group, the opposite is true when CO2 is released from OMP. In this case, a nucleophilic attack on atom C6 does not work. However, an elegant mechanism can be proposed that includes a Michael addition of an active site lysine residue at atom C5 instead.17,19 Although model compounds undergo rapidly reversible Michael addition at atom C519 there is no experimental evidence so far that the equivalent reaction takes place at the active site of ODCases. Further refinement of nucleotide analogs, generating molecules that will stop the catalytic reaction at intermediate points of the mechanistic pathway, combined with low activity mutants might shed more light on ODCase’s still enigmatic behavior.
Crystal structures have confirmed that the active sites of ODCase enzymes from eleven sources are all very similar,20 closely restricting the space they provide for the binding of ligands.21 A medium resolution crystallographic analysis of P. falciparum ODCase places OMP very close to the side chain of the central aspartate of the Lys-Asp-Lys-Asp network18 and even the best-binding inhibitor, BMP, fits very tightly into the reaction center6,7,10; e.g. in MtODCase, Oδ2 of Asp70 and O1 of BMP are only 3.1 Å apart (Fig. 1d).11 BMP has been suggested as a good mimic of the transition state of the decarboxylation reaction.18 It seems to represent the closest favorable approach between enzyme and ligand, fitting perfectly into the active site. Any substrate or substrate analog with a C6 substituent larger than an oxygen atom should experience distortion, the larger the C6-substituent the stronger the forces. Figure 2 shows how such stress affects 6-CN-UMP, which displays a clear bending of its C6-CN bond angle. Such distortions have also been described for the physiological substrate in its complex with the D312N mutant of human ODCase17 and, to a lesser degree, the D70A/K72A double mutant of MtODCase.9
ODCase, as a catalytic protein that accelerates the electrophilic substitution of the carboxylate group of OMP by a proton, has evolved into one of the most proficient enzymes known.22 It seems that one crucial aspect of its performance is the distortion of the bond that links the substituent to C6 of the pyrimidine ring (Fig. 6). When confronted with substrate analogues, however, this highly specialized catalyst can also increase the reaction rates of chemical transformations that involve nucleophilic substitutions, albeit by several orders of magnitude less than it achieves in the physiologically relevant reaction. Although the chemical mechanisms are quite different, the distortion of the bond to be cleaved, which will weaken the conjugation and increase the reactivity of the C6 center of the pyrimidine, is common to both the natural decarboxylation and the novel nucleophilic pathways. The nature of the product then is determined by the properties of the respective substrate. By donating a proton to the transient carbanion, the central lysine residue in the charge-network found in all ODCase active sites, Lys72 in MtODCase, is clearly crucial for the efficient catalysis of the OMP to UMP conversion. It also plays the decisive role in determining the direction taken in the transformation of other UMP C6-derivatives by an enzyme that displays a remarkable promiscuity not only in accepting a variety of substrate molecules but also in the chemical mechanisms employed in their transformation.
Materials and Methods
6-CN-UMP, 6-N3-UMP and 6-I-UMP were synthesized as published.11,12,15 Native orotidine 5′-monophosphate decarboxylase from Methanobacterium thermoautotrophicum (MtODCase) and its mutants were expressed and purified as described using Ni-NTA and gel-filtration chromatography.9,23 All proteins were dialyzed against crystallization buffer composed of 20 mM HEPES-NaOH pH 7.5, 150 mM NaCl and 5mM DTT. At RT, MtODCase (10 mg/mL) in crystallization buffer and incubated with 6-CN-UMP, 6-N3-UMP or 6-I-UMP (10 mM final concentrations) was mixed with equal amounts of various precipitant solutions for crystallization using the hanging-drop vapor diffusion method. Initially, clusters of crystals grew when 1.1–1.36 M sodium citrate and 5 % (v/v) dioxane at pH 6–8.5 were used as precipitant. Several cycles of microseeding were necessary to obtain single, diffraction-quality crystals. Crystals were dipped in a cryo-protectant buffer composed of 1.2 M sodium citrate, 15% glycerol and 0.1 M MES-Na at pH 6.5 before being flash-frozen in a nitrogen stream at 95 – 100 K.
To trap an intermediate time point of the reaction of 6-CN-UMP with native MtODCase, a special crystallization effort was required because the ligand was completely converted into BMP within a few days. Hanging drops were generated immediately after mixing the enzyme and ligand solutions. After ~10 hours, they were micro-seeded, followed by incubation for ~24 hours at 4 oC and another ~14 hours at room temperature. Thus in total, 6-CN-UMP was exposed to enzyme activity for one day at RT and another day at 4 oC before the ongoing reaction was stopped by flash-freezing the crystals.
Diffraction data sets were collected either at beamlines 14BM-C and 14ID-B at the Advanced Photon Source, Argonne, USA or on a Cu Kα laboratory X-ray source (Rigaku FR-C rotating anode, Xenocs multi-layer focusing optics, MAR345 image plate detector). Data were integrated, scaled, and truncated using DENZO, SCALEPACK or HKL2000,24 and TRUNCATE.25 The datasets collected from crystals of the D70A/K72A:6-CN-UMP and K72A:6-I-UMP complexes were phased with the help of the molecular replacement program MOLREP using native MtODCase as the search model. All other data sets could be phased directly from the molecular model of the BMP complex of MtODCase (PDB-ID 1X1Z). Model-building and refinement were done with the help of the program packages O,26 COOT,27 CNS28 and REFMAC29. The parameter file for uridine-5′-monophosphate was the one stored at the Hic-UP server (http://xray.bmc.uu.se/hicup). No restraints were applied to the characteristic bonds between Lys72 and UMP in the three covalent complexes (D70A:6-CN-UMP, D75N:6-CN-UMP, wild-type:6-N3-UMP). The parameters for BMP were generated with the help of the program SKETCHER using default settings (http://www.ccp4.ac.uk/dist/ccp4i/help/modules/sketcher.html). The 6-CN-UMP parameter file was created based on that of UMP with the bond distances of C6-C7 (1.47 Å) and C7-N8 (1.16 Å), and the bond angle of C6-C7-N8 (180°) restrained with standard energy terms. Other parameters, e.g. the angles N1-C6-C7 and C5-C6-C7, dihedral angles and planes around the -CN moiety, were not restrained during the refinement of the 6-CN-UMP structure. All refined structures were validated using MolProbity30 at the RCSB protein data bank validation server (http://deposit.pdb.org/validate/). Statistics of data collection and refinement are summarized in Table 1.
The energies for various geometries of 6-CN-UMP with C6-C7 bent with respect to the plane of the pyrimidine moiety were computed using Gaussian 98, using the basis set 6-31+G* at the DFT/B3-LYP theory level in the gas phase. The potential energy scans (PES) were conducted at 15° angle steps (Fig. 3).
Mass spectral analyses (TOF ESI+) were performed at the Mass Spectrometry Facility, Advanced Protein Technology Center at the Hospital for Sick Children, Toronto, or at the Mass Spectrometry-AIMS Laboratory, Department of Chemistry, University of Toronto, Canada. The D70A mutant of MtODCase (12.3 mg/mL) mixed with 6-CN-UMP (12.3 mM, final concentration) and the K72A mutant (51 mg/mL) mixed with 6-I-UMP (40 mM) were both incubated for up to one week. At various time points, samples were drawn and subjected to mass spectral analysis. The D75NB mutant (11.7 mg/mL) was incubated with 15 mM 6-CN-UMP for 1 day, whereas native MtODCase (61 mg/mL) mixed with 6-N3-UMP (61 mM) was submitted after 16 hours incubation; all samples were analyzed within 24 h of submission.
Acknowledgments
We are grateful to the staff at the BioCARS beamlines of the Advanced Photon Source for their help with data collection and to Wing M. Lau and Yan Liu for their contributions to sample preparation and data analysis. MF expresses his special thanks to Wanda Gillon for her excellent help with the initial protein production and purification and to Kunio Miki for kindly granting access to a variety of experimental resources. This work was supported in part by a Japanese Grant-in-Aid for Young Scientists (16-7650, 17770087 and 20770081) and by the Uehara Memorial Foundation to MF. MF was also a recipient of an Overseas Fellowship and a Research Fellowship for Young Scientists from the Japanese Society for the Promotion of Science. EFP thanks the Canadian Institutes for Health Research (MOP-62704) and the Canada Research Chairs Program for support. LPK acknowledges support from the Canadian Institutes for Health Research (PPP-200606), and the Premier’s Research Excellence Award. Use of the Advanced Photon Source at Argonne National Laboratory was supported by the U. S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. DE-AC02-06CH11357. Use of the BioCARS Sector 14 was supported by the National Institutes of Health, National Center for Research Resources (RR007707).
Abbreviations
- ODCase
orotidine 5′-monophosphate decarboxylase
- OMP
orotidine 5′-monophosphate
- UMP
uridine 5′-monophosphate
- 6-CN-UMP
6-cyano uridine 5′-monophosphate
- BMP
barbituric acid ribosyl 5′-monophosphate
- 6-N3-UMP
6-azido uridine 5′-monophosphate
- 6-I-UMP
6-iodo uridine 5′-monophosphate
- RMSD
root mean square deviation
- RT
room temperature
Footnotes
Accession numbers
The atomic coordinates and structure factors have been deposited in the PDB (Protein Data Bank). The accession codes are 2ZZ1, 2ZZ2, 2ZZ3, 2ZZ4, 2ZZ5, 2ZZ6, and 2ZZ7 for native:6-CN-UMP, K72A:6-CN-UMP, D70A:6-CN-UMP, D75N:6-CN-UMP, K72A/D70A:6-CN-UMP, native:6-N3-UMP, and K72A:6-I-UMP, respectively.
If not stated otherwise, residue numbers refer to the enzyme from Methanobacterium thermoautotrophicum. The superscript B, as in Asp75B, indicates that the residue is contributed by the second subunit.
References
- 1.Radzicka A, Wolfenden R. A proficient enzyme. Science. 1995;267:90–3. doi: 10.1126/science.7809611. [DOI] [PubMed] [Google Scholar]
- 2.Wierenga RK. The TIM-barrel fold: a versatile framework for efficient enzymes. FEBS Lett. 2001;492:193–8. doi: 10.1016/s0014-5793(01)02236-0. [DOI] [PubMed] [Google Scholar]
- 3.Wu N, Mo Y, Gao J, Pai EF. Electrostatic stress in catalysis: structure and mechanism of the enzyme orotidine monophosphate decarboxylase. Proc Natl Acad Sci U S A. 2000;97:2017–22. doi: 10.1073/pnas.050417797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Appleby TC, Kinsland C, Begley TP, Ealick SE. The crystal structure and mechanism of orotidine 5′-monophosphate decarboxylase. Proc Natl Acad Sci U S A. 2000;97:2005–10. doi: 10.1073/pnas.259441296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Harris P, Navarro Poulsen JC, Jensen KF, Larsen S. Structural basis for the catalytic mechanism of a proficient enzyme: orotidine 5′-monophosphate decarboxylase. Biochemistry. 2000;39:4217–24. doi: 10.1021/bi992952r. [DOI] [PubMed] [Google Scholar]
- 6.Harris P, Poulsen JC, Jensen KF, Larsen S. Substrate binding induces domain movements in orotidine 5′-monophosphate decarboxylase. J Mol Biol. 2002;318:1019–29. doi: 10.1016/S0022-2836(02)00200-0. [DOI] [PubMed] [Google Scholar]
- 7.Miller BG, Hassell AM, Wolfenden R, Milburn MV, Short SA. Anatomy of a proficient enzyme: the structure of orotidine 5′-monophosphate decarboxylase in the presence and absence of a potential transition state analog. Proc Natl Acad Sci U S A. 2000;97:2011–6. doi: 10.1073/pnas.030409797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Miller BG, Snider MJ, Wolfenden R, Short SA. Dissecting a charged network at the active site of orotidine-5′-phosphate decarboxylase. J Biol Chem. 2001;276:15174–6. doi: 10.1074/jbc.M011429200. [DOI] [PubMed] [Google Scholar]
- 9.Wu N, Gillon W, Pai EF. Mapping the active site-ligand interactions of orotidine 5′-monophosphate decarboxylase by crystallography. Biochemistry. 2002;41:4002–11. doi: 10.1021/bi015758p. [DOI] [PubMed] [Google Scholar]
- 10.Wu N, Pai EF. Crystal structures of inhibitor complexes reveal an alternate binding mode in orotidine-5′-monophosphate decarboxylase. J Biol Chem. 2002;277:28080–7. doi: 10.1074/jbc.M202362200. [DOI] [PubMed] [Google Scholar]
- 11.Fujihashi M, Bello AM, Poduch E, Wei L, Annedi SC, Pai EF, Kotra LP. An unprecedented twist to ODCase catalytic activity. J Am Chem Soc. 2005;127:15048–50. doi: 10.1021/ja054865u. [DOI] [PubMed] [Google Scholar]
- 12.Poduch E, Bello AM, Tang S, Fujihashi M, Pai EF, Kotra LP. Design of inhibitors of orotidine monophosphate decarboxylase using bioisosteric replacement and determination of inhibition kinetics. J Med Chem. 2006;49:4937–45. doi: 10.1021/jm060202r. [DOI] [PubMed] [Google Scholar]
- 13.Bello AM, Poduch E, Liu Y, Wei L, Crandall I, Wang X, Dyanand C, Kain KC, Pai EF, Kotra LP. Structure-Activity Relationships of C6-Uridine Derivatives Targeting Plasmodia Orotidine Monophosphate Decarboxylase. J Med Chem. 2008;51:439–448. doi: 10.1021/jm7010673. [DOI] [PubMed] [Google Scholar]
- 14.Levine HL, Brody RS, Westheimer FH. Inhibition of orotidine-5′-phosphate decarboxylase by 1-(5′-phospho-beta-d-ribofuranosyl)barbituric acid, 6-azauridine 5′-phosphate, and uridine 5′-phosphate. Biochemistry. 1980;19:4993–9. doi: 10.1021/bi00563a010. [DOI] [PubMed] [Google Scholar]
- 15.Bello AM, Poduch E, Fujihashi M, Amani M, Li Y, Crandall I, Hui R, Lee PI, Kain KC, Pai EF, Kotra LP. A potent, covalent inhibitor of orotidine 5′-monophosphate decarboxylase with antimalarial activity. J Med Chem. 2007;50:915–21. doi: 10.1021/jm060827p. [DOI] [PubMed] [Google Scholar]
- 16.Allen FH. The Cambridge Structural Database: a quarter of a million crystal structures and rising. Acta Crystallogr B. 2002;58:380–8. doi: 10.1107/s0108768102003890. [DOI] [PubMed] [Google Scholar]
- 17.Wittmann JG, Heinrich D, Gasow K, Frey A, Diederichsen U, Rudolph MG. Structures of the human orotidine-5′-monophosphate decarboxylase support a covalent mechanism and provide a framework for drug design. Structure. 2008;16:82–92. doi: 10.1016/j.str.2007.10.020. [DOI] [PubMed] [Google Scholar]
- 18.Tokuoka K, Kusakari Y, Krungkrai SR, Matsumura H, Kai Y, Krungkrai J, Horii T, Inoue T. Structural basis for the decarboxylation of orotidine 5′-monophosphate (OMP) by Plasmodium falciparum OMP decarboxylase. J Biochem. 2008;143:69–78. doi: 10.1093/jb/mvm193. [DOI] [PubMed] [Google Scholar]
- 19.Silverman RB, Groziak MP. Model chemistry for a covalent mechanism of action of orotidine 5′-phosphate decarboxylase. J Am Chem Soc. 1982;104:6434–6439. [Google Scholar]
- 20.Langley DB, Shojaei M, Chan C, Lok HC, Mackay JP, Traut TW, Guss JM, Christopherson RI. Structure and inhibition of orotidine 5′-monophosphate decarboxylase from Plasmodium falciparum. Biochemistry. 2008;47:3842–54. doi: 10.1021/bi702390k. [DOI] [PubMed] [Google Scholar]
- 21.Wu N, Pai EF. Crystallographic Studies of Native and Mutant Orotidine 5′-Phosphate Decarboxylases. Top Curr Chem. 2004;238:23–42. [Google Scholar]
- 22.Miller BG, Wolfenden R. Catalytic proficiency: the unusual case of OMP decarboxylase. Annu Rev Biochem. 2002;71:847–85. doi: 10.1146/annurev.biochem.71.110601.135446. [DOI] [PubMed] [Google Scholar]
- 23.Wu N, Christendat D, Dharamsi A, Pai EF. Purification, crystallization and preliminary X-ray study of orotidine 5′-monophosphate decarboxylase. Acta Crystallogr D Biol Crystallogr. 2000;56:912–4. doi: 10.1107/s090744490000576x. [DOI] [PubMed] [Google Scholar]
- 24.Otwinowski Z, Minor W. Processing of X-ray Diffraction Data Collected in Oscillation Mode. In: Carter CW Jr, Sweet RM, editors. Macromolecular Crystallography, Part A. Vol. 276. Academic Press; 1997. pp. 307–326. [DOI] [PubMed] [Google Scholar]
- 25.Collaborative Computional Project N. The CCP4 suite: programs for protein crystallography. Acta Crystallogr D Biol Crystallogr. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 26.Jones TA, Zou JY, Cowan SW. Improved Methods for Building Protein Models in Electron Density Maps and the Location of Errors in these Models. Acta Crystallogr A. 1991;47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 27.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–32. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 28.Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr D Biol Crystallogr. 1998;54:905–21. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 29.Murshudov GN, Vagin AA, Lebedev A, Wilson KS, Dodson EJ. Efficient anisotropic refinement of macromolecular structures using FFT. Acta Crystallogr D Biol Crystallogr. 1999;55:247–55. doi: 10.1107/S090744499801405X. [DOI] [PubMed] [Google Scholar]
- 30.Lovell SC, Davis IW, Arendall WB, 3rd, de Bakker PI, Word JM, Prisant MG, Richardson JS, Richardson DC. Structure validation by Calpha geometry: phi,psi and Cbeta deviation. Proteins. 2003;50:437–50. doi: 10.1002/prot.10286. [DOI] [PubMed] [Google Scholar]