

Abstract
Telomere shortening is the mechanism underlying replicative aging in fibroblasts. A variety of reports now claim that inactivation of the p16INK4a/pRB pathway is required in addition to telomere maintenance for the immortalization of cells such as skin keratinocytes and breast epithelial cells. We here show that the premature growth arrest of these cell types can be explained by an inadequate culture environment. Providing mesenchymal/epithelial interactions by cultivating the telomerase-expressing cells on feeder layers avoids the growth arrest associated with increased p16INK4a. These results do not support a telomere-independent mechanism of replicative aging.
Keywords: Replicative aging, cellular senescence, telomeres, p16INK4a, aging, cancer
The ability to proliferate indefinitely in tissue culture is one of many differences between normal human cells and established tumor cell lines. For nearly forty years (Hayflick and Moorhead 1961), the contrast between the immortality of cancer cells and the senescence of normal cells has been controversial. The major source of skepticism rests on the knowledge that in vitro culture conditions represent a departure from the in vivo environment, and that the aneuploidy and altered growth controls accumulated by tumor cells might simply allow them to divide under conditions inadequate to support the long-term proliferation of normal cells. Although greatly diminished by the recent advances uncovering cellular mechanisms for counting cell divisions (see below), concerns that cellular senescence represents a tissue-culture artifact have continued (Rubin 1997, 1998).The best understood mechanism for replicative aging involves telomere shortening. Telomerase, the enzyme responsible for maintaining telomere length (Greider and Blackburn 1985), is down-regulated or turned off in most human tissues early in development (Wright et al. 1996). The inability of the normal replication machinery to copy all the way to the end of a linear chromosome, combined with potential end-processing events (Wellinger et al. 1996; Huffman et al. 2000), results in the progressive shortening of telomeres with each subsequent cell division (Harley et al. 1990). The eventual growth arrest of cultured cells is thought to occur when some of the telomeres become sufficiently short to compromise their interaction with specific telomere-binding proteins such as TRF2 (van Steensel et al. 1998). The resulting failure of these proteins to mask the end of the chromosome from being recognized as a double-strand break induces a DNA-damage response that in turn produces the final growth arrest (Harley 1991). The ability of cDNAs encoding the catalytic subunit of human telomerase (hTERT) to produce telomerase activity, maintain telomere length, and immortalize normal human skin fibroblasts (Bodnar et al. 1998; Vaziri and Benchimol 1998), retinal pigment epithelial cells (Bodnar et al. 1998) and endothelial cells (Yang et al. 1999) has greatly strengthened this model.
Replicative aging is thought to provide a barrier against the accumulation of mutations and the formation of cancer. If each mutation requires 20–40 cell divisions, limiting the total to less than 100 doublings would prevent most pre-malignant cells with a few mutations from accumulating the additional mutations needed to become malignant. The ability to induce a growth arrest as a consequence of counting cell divisions is thus the essential feature distinguishing the mortality of normal cells and the immortality of tumor cells. Confusion has arisen because specific markers that can distinguish the growth arrest occurring as the end-stage of replicative aging versus arrest from other causes have not been identified. Hundreds of differences in senescent versus young cells have been cataloged (Hayflick 1980; Shelton et al. 1999). However, these changes differ between cell types and laboratories. One marker initially proposed for senescent cells, the senescence-associated beta galactosidase (SA-β gal) (Dimri et al. 1995), might better be described as a stress-associated β gal because it is induced by a variety of DNA damaging agents (Robles and Adami 1998). If short telomeres lose the capacity to be hidden from the DNA damage recognition apparatus, the phenotype of the senescent cell should be nonspecific and overlap with other damage/stress responses that use similar signal transduction pathways. The use of growth arrest as a surrogate marker for the process of replicative aging risks misidentifying mechanisms that induce cell cycle checkpoint controls from mechanisms that are actually counting cell divisions.
Several reports have claimed that keratinocytes and other types of epithelial cells exhibit an additional mechanism of replicative aging involving the p16/pRB pathway that is independent of telomere length, and that inactivation of both mechanisms is required for cellular immortalization (Kiyono et al. 1998; Jarrard et al. 1999; Dickson et al. 2000; Farwell et al. 2000; Jones et al. 2000). If true, these observations would imply the existence of additional tumor suppressor mechanisms for counting cell divisions, and should stimulate research to identify the pathways involved and their relative contribution to physiological changes in vivo. However, the proliferative life span of only 15–20 doublings using chemically defined media in most of these reports was dramatically less than the ∼50 doublings described previously for the growth of keratinocytes on feeder layers (Green et al. 1977). Furthermore, 15–20 doublings appears grossly inadequate to explain the estimated number of keratinocyte doublings occurring in vivo (Rubin 1997). The longstanding concerns about the influence of the culture environment should be addressed before growth arrest can be legitimately described as reflecting replicative aging. Thus, we used three different model systems to examine whether the proposed p16/pRB pathway actually represented a telomere-independent second mechanism of cellular senescence or was a secondary consequence of particular culture conditions. We here report that human keratinocytes grown on appropriate feeder layers can be directly immortalized by hTERT maintenance of telomeres without any intervening inactivation of a p16/pRB pathway. Human mammary epithelial (HME) cells have been described as exhibiting a p16-mediated “mortality stage 0” mechanism of senescence (Foster et al. 1998). We find that the replicative capacity of HME cells can also be extended without inactivating p16 if these cells are cultured on feeder layers. Finally, culture of human fibroblasts under inadequate culture conditions (chemically defined medium in the presence of 0.25% serum) recapitulates the keratinocyte and HME scenarios. Such fibroblasts arrest after ∼25 doublings regardless of telomere length/telomerase, but can be rescued and immortalized following transfer to adequate culture media. Although one cannot theoretically exclude the existence of nontelomere based mechanisms for replicative aging, these results establish that at present there is no reliable evidence for the presence of a second mechanism of replicative aging in epithelial cells.
Results
Culture in the absence of feeder layers induces p16INK4a and prevents the immortalization of keratinocytes
We compared the proliferative life span of epidermal keratinocytes grown in chemically defined media versus on mitomycin-C treated 3T3 feeder layers. Growth of epidermal keratinocytes on feeder layers delayed the up-regulation of p16 (Fig. 1B) and extended the life span from 15 to 36 population doublings (PD) (Fig. 1A). Cells cultured on feeder layers were infected with hTERT just as they approached senescence, and dividing cells overgrew the culture and have been continuously dividing since then (current PD >160). Similar results were obtained with keratinocytes from a different donor (data not shown). The cells on feeder layers still expressed p16, and p16 levels increased upon transfer to defined media (Fig. 1B) or following UV irradiation (data not shown).
Figure 1.

Growth of keratinocytes on feeder-layers inhibits the premature growth arrest. (A) Human foreskin keratinocytes initially grown on mitomycin-C treated 3T3 feeder layers were maintained on feeder layers or transferred to chemically defined medium on plastic substrates at the first split. Cells grew for many more doublings on feeder layers. The insert shows a Western blot of p16INK4a levels for cells grown under the two conditions. (B) Keratinocytes growing on feeder layers were infected with hTERT just as growth slowed. Immortal cells overgrew the culture and have continued to divide. The Western blot shows that p16INK4a levels increased six doublings after the cells were shifted back into chemically defined medium on plastic. These cells stopped diving three doublings later.
Telomerase-expressing keratinocytes that were transferred to chemically defined medium on plastic substrates at PD 89 arrested nine doublings later, confirming that these conditions were inadequate for the long-term growth of keratinocytes. Several clones of dividing cells emerged after ∼6 weeks that could be continuously cultivated in the chemically defined media. These cells neither expressed p16 nor induced p16 following UV irradiation (data not shown). This demonstrates that the immortal keratinocytes grown on feeder layers still contained a functional p16 whose inactivation was required for continuous growth in defined medium on plastic substrates. The dependence of this premature growth arrest on the p16/pRB pathway was further confirmed by showing that the human papilloma virus protein E7 abrogated the arrest and permitted the continuous growth of hTERT-expressing keratinocytes in chemically defined medium on plastic substrates (Fig. 2; Kiyono et al. 1998).
Figure 2.
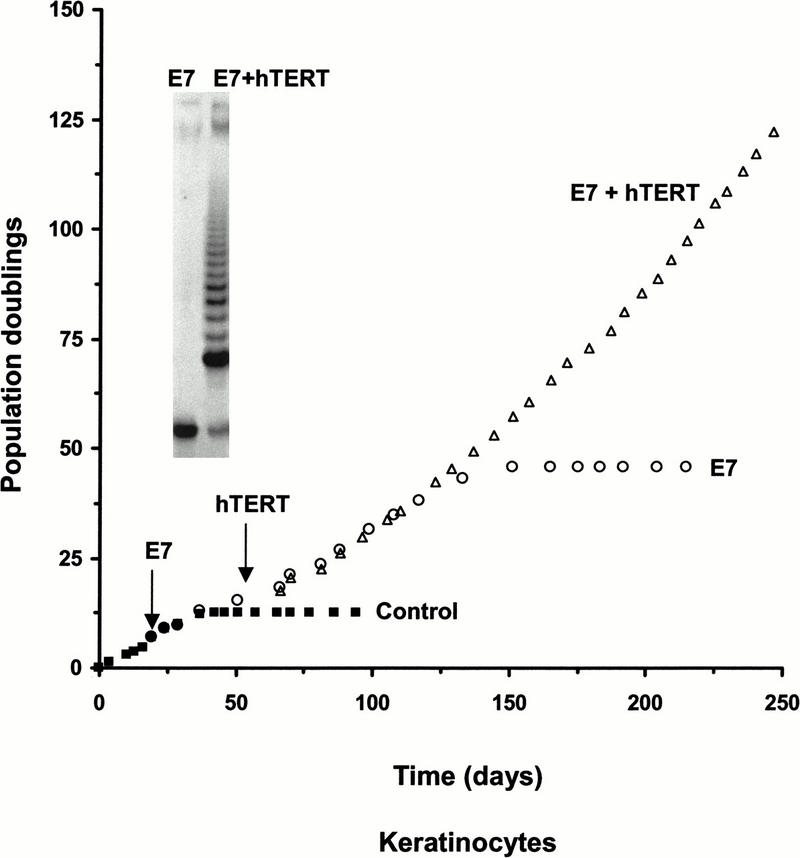
HPV E7 blocks the premature growth arrest. Keratinocytes growing in chemically defined medium on plastic substrates exhibited a greatly extended proliferative capacity following infection with E7-expressing retroviruses and exhibited indefinite growth following the addition of hTERT. The insert shows the lack of telomerase activity in E7-keratinocytes and its presence after infection with the hTERT-encoding retrovirus.
Human mammary epithelial cells cultured in the presence of feeder layers do not exhibit M0
When telomeres become sufficiently short, human fibroblasts arrest at mortality stage 1 (M1) (Wright and Shay 1992). Blocking the action of p53 and pRB with oncogenes allows an extended life span until terminal telomere shortening induces M2 (crisis). An additional stage in replicative aging has been reported for HME cells. After 10–15 doublings, HME cells undergo a p16-mediated growth arrest, described as mortality stage 0 (M0) (Foster et al. 1998). After a period of self-selection, cells that have methylated the INK4a locus emerge and divide until reaching telomere-based senescence. We tested whether the mesenchymal/epithelial microenvironment found to be effective with keratinocytes also influenced the M0 stage in HME cells. Cells grown in chemically defined medium on plastic substrates showed the previously described M0 with increased p16, growth arrest, and an eventual outgrowth of cells that did not express p16. The life span of HMEs was significantly extended and there was a dramatic delay in the accumulation of p16 (Fig. 3) when cells were cocultured on feeder layers. Thus, the phenomenon of M0 in HMEs depends on the growth of the cells in a chemically defined medium on plastic culture dishes and is not seen when cells are cultured on feeder layers.
Figure 3.
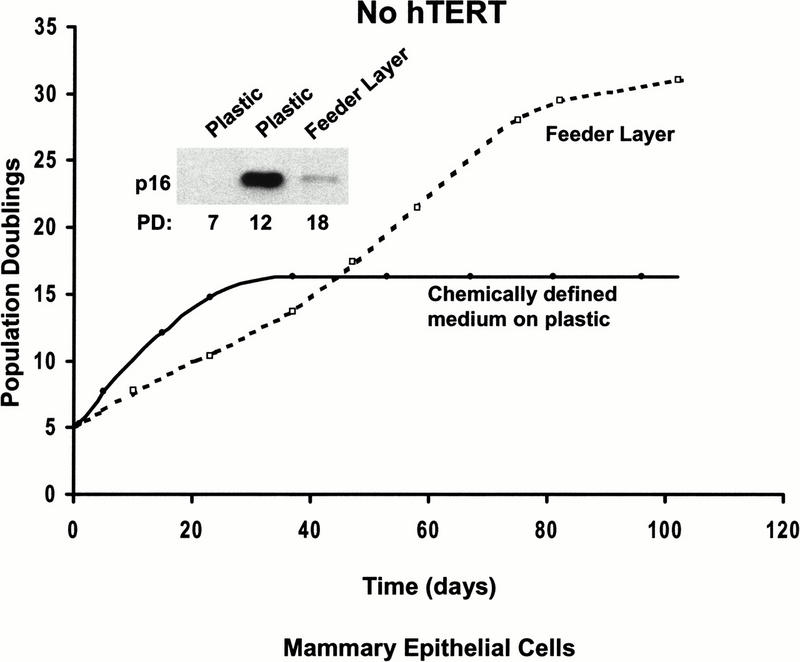
Human mammary epithelial cells do not exhibit M0 when grown on feeder layers. The premature growth arrest of HME grown in a chemically defined medium was prevented by cultivating the cells on mitomycin-C treated NIH 3T3 feeder layers. Growth on feeder layers also greatly delayed the increase of p16 INK4a, as seen in the western blot insert.
Premature growth arrest of dermal fibroblasts in defined media
Dermal fibroblasts represent a well-characterized system in which telomerase is sufficient to immortalize cells from many different normal donors and from patients with a variety of genetic syndromes (Bodnar et al. 1998; Vaziri and Benchimol 1998; Ouellette et al. 2000). These immortal cells contain normal cell-cycle checkpoint activities (Jiang et al. 1999; Morales et al. 1999) and do not exhibit a putative p16-mediated second mechanism of replicative aging. We tested whether we could create a premature growth arrest associated with increased p16 levels by cultivating skin fibroblasts under less than optimal conditions (Fig. 4A). These cells grow to approximately PD 90–95 when grown in 10% serum. If the skin fibroblasts were transferred to a defined medium designed for the growth of human fibroblasts supplemented with 0.25% serum at PD 29, they only divided an additional 30 times before growth arresting at a total PD of 59. If the cells were transferred back to medium with 10% serum at PD 58, they were able to recover and divide an additional 26 times. These results demonstrate that although the chemically defined medium appeared adequate, it was clearly incapable of supporting true long-term growth of these cells. Western blots showed that p16 became elevated when the fibroblasts growth arrested in chemically defined medium, but not when they were at much higher population doubling levels in 10% serum (Fig. 4A).
Figure 4.
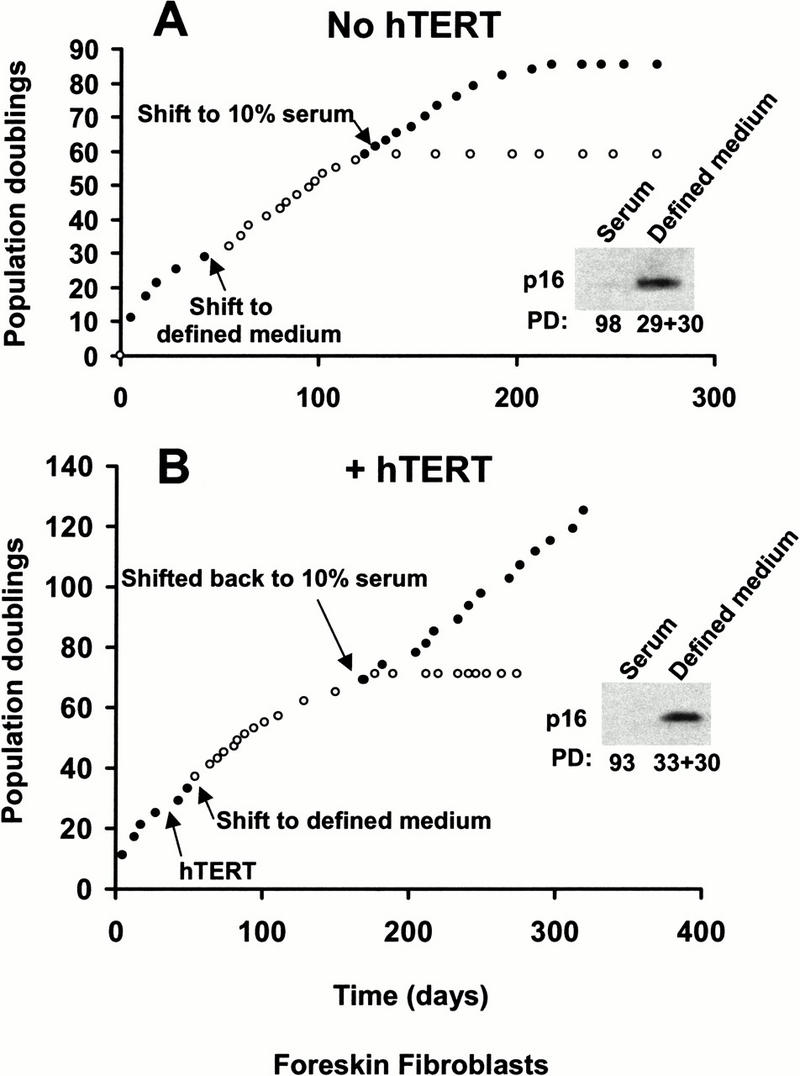
Defined medium induces a premature growth arrest of foreskin fibroblasts. (A) Human foreskin fibroblasts grown in 10% serum were transferred to chemically defined medium containing the minimum concentration of serum compatible with continued growth (0.25%) at population doubling 29. As growth slowed 30 doublings later, sister dishes were transferred back to medium containing 10% serum. Elevated p16INK4a levels were found in cells prematurely arrested by growth in defined medium but were not seen in foreskin fibroblasts grown in medium containing 10% serum. (B) Sister cultures to those shown in A were infected with hTERT, selected for drug resistance, and then transferred to defined medium as above at population doubling 33. As growth slowed 30 doublings later, sister dishes were transferred back to medium containing 10% serum. Telomerase-expressing fibroblasts growth arrested after 30 doublings in defined medium, but were immortal in medium containing 10% serum. Elevated p16INK4a levels were found in hTERT-expressing cells prematurely arrested by growth in defined medium but were not seen in the immortal cells grown in medium containing 10% serum.
Skin fibroblasts infected with hTERT at PD 28 were transferred to chemically defined medium after being selected for drug resistance. Similar to the controls, these telomerase-expressing fibroblasts growth-arrested after 30 doublings with increased levels of p16. Growth resumed when these cells were transferred back to 10% serum and continued beyond 130 doublings (Fig. 4B). Neither telomerase activity nor telomere-length maintenance was altered in the hTERT-infected cells when grown in the different media (data not shown). Thus, although the growth of foreskin fibroblasts in chemically defined medium containing 0.25% serum continued for many weeks and 30 doublings, it ultimately produced a telomere-independent growth arrest associated with an elevated p16.
Discussion
The apparent failure of telomerase to immortalize a variety of epithelial cell types has led many authors to propose that there may be other mechanisms of cellular senescence used as tumor suppressors (Kiyono et al. 1998; Jarrard et al. 1999; Dickson et al. 2000; Farwell et al. 2000; Jones et al. 2000). The implication of these proposals is that there are telomere-independent mechanisms for counting cell divisions during replicative aging. We have reexamined the evidence for these claims and the results demonstrate that the premature growth arrest observed with two different epithelial cell types is a consequence of the particular tissue culture conditions used, and can be prevented by growing the cells on appropriate feeder layers. Keratinocytes immortalized with telomerase on feeder layers still exhibit a growth arrest with elevated p16 after being transferred into chemically defined media on plastic culture dishes. Rare cells that eventually escape this checkpoint and resume growth no longer express p16, consistent with the previous demonstrations that inactivation of the INK4a locus is required for extended growth of these cells in chemically defined media on plastic substrates. However, because these cells were already immortal when grown on feeder layers, we believe it is inappropriate to describe this change as required for immortalization. Instead, p16 inactivation under these conditions should be characterized as an event necessary for proliferation in an inadequate culture environment. We thus conclude that the published evidence does not support the presence of telomere-independent mechanisms of replicative aging.
Chemically defined media for the growth of specialized cell types were developed for a variety of needs. One major advantage is the ability to create a culture environment that inhibits the overgrowth of fibroblasts while still permitting the proliferation of the desired cell type. It is not surprising that eliminating factors needed by fibroblasts might contribute to conditions that supported the short-term but not the long-term proliferation of target cells. These media were also developed to prevent terminal differentiation while maintaining the potential to differentiate. Our results demonstrate that growth for 15–25 doublings is not sufficient for defining a medium as being adequate. Skin fibroblasts expressing hTERT grown in a chemically defined medium supplemented with 0.25% fetal bovine serum were able to divide for 20–30 doublings, but then up-regulated p16 and growth arrested by telomere-independent mechanisms. Because these same fibroblasts were immortal when grown under conventional conditions, the premature growth arrest must have resulted from an inadequate environment rather than from an alternative counting mechanism.
HME cells have been described as having an M0 mechanism of cellular senescence that prevents proliferation beyond 10–15 doublings, and from which cells escape by methylating p16 (Foster et al. 1998). Given the large proliferative demands placed on these cells during multiple pregnancies, it appeared improbable that such a limited proliferative capacity would be present in the intact organism. The present results demonstrate that M0 represents a premature growth arrest due to inadequate culture conditions that can be avoided by culturing mammary epithelial cells on feeder layers.
Much of the confusion about replicative aging results from the nonspecific phenotype of the final nonproliferative state. The best evidence to date supports a model in which sufficiently short telomeres produce a DNA damage signal that induces growth arrest using the same basic checkpoint machinery that mediates many other phenomena. Factors like p53 and p16 may mediate the growth arrest induced by both replicative aging and DNA damaging agents like blastocidin and cis-platinum (Robles and Adami 1998). However, we do not believe it is biologically meaningful to describe the growth arrest induced by blastocidin as being cellular senescence, as there is no element of counting cell divisions involved. The same argument applies to the premature cellular senescence induced by Ras (Serrano et al. 1997). The Ras-induced growth arrest plays the same anti-hyperproliferative role as the apoptosis induced by Myc overexpression, and we believe it is inappropriate to describe its tumor suppressor function as being related to replicative aging.
The confusion between replicative aging and inadequate culture conditions illustrated above for human epithelial cells is similar to what is found for mouse embryo fibroblasts (Sherr and DePinho 2000; Wright and Shay 2000). The growth arrest that occurs after 10–20 doublings is telomere-independent, because it occurs after the same number of doublings in cells from mice lacking the RNA component of telomerase (Blasco et al. 1997) whose telomeres are shorter than those in wild-type mice. The observation that the transcriptional activity of p53 is induced 10- to 40-fold within the first three passages after mouse embryo fibroblasts are put in culture (Mendrysa and Perry 2000) suggests that the culture environment is producing DNA damage in these cells. This conclusion is strengthened by the premature cellular senescence found in mouse cells deficient in a variety of DNA repair functions such as ATM, Brca2, Ku80, XPG, and ligase IV, (Wright and Shay 2000), and the fact that these premature growth arrests are not observed when combined with ARF deficiencies that mediate p53-dependent checkpoints. Although ARF does not appear to be involved in the acute DNA damage response, it has been implicated in sustaining the ensuing growth arrest (Kahn et al. 2000).
The INK4a/ARF locus encodes two proteins, and there is considerable controversy over their relative contributions to the premature growth arrest of mouse cells in culture versus their tumor suppressor function in human cancers. Although the ARF/p53 pathway appears to be the primary mediator of the murine cultured growth arrest, p16Ink4a levels also increase progressively in mouse cultures (Zindy et al. 1997). The level of p16Ink4a may be a general indicator of stress, as it increases as a function of aging in multiple mouse organs including postmitotic tissues such as the brain (Zindy et al. 1997). Free radicals produced by amine oxidase in serum may be one component that contributes to the stress experienced by mouse fibroblasts in culture (Parchment et al. 1990), and it has been reported that under appropriate serum-free conditions mouse cells are immortal and remain diploid for hundreds of doublings (Loo et al. 1987). Mouse cells are much more sensitive to a variety of stresses than human cells (Kapahi et al. 1999), and it is likely that culture conditions that are adequate for human fibroblasts do not support the long-term proliferation of mouse fibroblasts.
The ability to produce a controlled environment in tissue culture has produced enormous advances in our understanding of the cell cycle, checkpoint controls, responses to stress and DNA damage, and replicative aging. A careful analysis of the contribution of different molecules to these various processes will be required for the development of sophisticated therapies. It is unambiguous that p16Ink4A is an important tumor suppressor involved in cell cycle regulation. The demonstration that inadequate growth conditions can induce an arrest associated with elevated levels of p16Ink4A, focuses attention on its role in mediating stress responses rather than in counting cell divisions. This will permit a more effective analysis of telomeres in replicative aging, their role in tumor prevention and the possibilities of manipulating telomere biology for the treatment of cancer.
Materials and methods
Cell culture
Neonatal human foreskin keratinocytes were cocultured (∼1–3 × 106 cells/100 mm dish) with mitomycin-C treated (10 μg/mL for 2 h) ATCC Swiss 3T3 cells in DMEM/F12 (3:1) media (GIBCO) containing 5% bovine fortified calf serum (Hyclone Laboratories, Logan, UT), hydrocortisone (0.4 μg/mL), epidermal growth factor (20 ng/mL), transferrin (5 μg/mL), insulin (5 μg/mL), cholera toxin (10−10 M), triiodothyronine (2 × 10−11 M), adenine (180 μM), penicillin/streptomycin solution (0.1% v/v, penicillin 10,000 IU/mL and streptomycin 10,000 μg/mL) (Rheinwald 1989). After removing mitomycin-treated 3T3 cells with EDTA (0.02% in 1× PBS), 80% confluent keratinocytes were trypsinized and replated on 3T3 feeder layers at 1:16 split ratios. Some cultures were transferred to a chemically defined medium (KGM 2 medium; Clonetics, Walkersville, MD) on plastic substrates as of the first passage.
Dermal fibroblasts from the same foreskins were cultured in DMEM/medium 199 (4:1) containing 10% fortified bovine calf serum (Hyclone Laboratories), and gentamicin (50 μg/mL). Some cultures were transferred to chemically defined FGM medium (Clonetics). Because the cells grew very poorly in the completely defined medium, it was supplemented with the minimal amount of serum (0.25%) found to produce sustained growth of ∼1.5 doublings/wk.
Normal human mammary epithelial (HME) cells from a 33-year-old patient with no history of breast cancer were obtained and cultured as described (Stampfer et al. 1980). Some cultures were transferred to mitomycin-C treated 3T3 feeder layers in MCDB170 medium plus growth factors but also supplemented with 1% serum to permit the survival of the feeder layers.
Acknowledgments
This work was supported by grant AG01228 from the National Institute on Aging to W.E.W. and Texas ATP010019 to C.P.M. We thank Kenia Gandia for technical assistance. J.W.S. is a senior scholar of the Ellison Medical Foundation.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL Woodring.Wright@utsouthwestern.edu; FAX (214) 648-8694.
E-MAIL Jerry.Shay@UTSouthwestern.edu; FAX (214) 648-8694.
Article and publication are at www.genesdev.org/cgi/doi/10.1101/gad.859201.
References
- Blasco MA, Lee HW, Hande MP, Samper E, Lansdorp PM, DePinho RA, Greider CW. Telomere shortening and tumor formation by mouse cells lacking telomerase RNA. Cell. 1997;91:25–34. doi: 10.1016/s0092-8674(01)80006-4. [DOI] [PubMed] [Google Scholar]
- Bodnar AG, Ouellette M, Frolkis M, Holt SE, Chui C-P, Morin GB, Harley CB, Shay JW, Lichsteiner S, Wright WE. Extension of life-span by introduction of telomerase into normal human cells. Science. 1998;279:349–352. doi: 10.1126/science.279.5349.349. [DOI] [PubMed] [Google Scholar]
- Dickson MA, Hahn WC, Ino Y, Ronfard V, Wu JY, Weinberg RA, Louis DN, Li FP, Rheinwald JG. Human keratinocytes that express hTERT and also bypass a p16(INK4a)-enforced mechanism that limits life span become immortal yet retain normal growth and differentiation characteristics. Mol Cell Biol. 2000;20:1436–1447. doi: 10.1128/mcb.20.4.1436-1447.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimri GP, Lee X, Basile G, Acosta M, Scott G, Roskelley C, Medrano EE, Linskens M, Rubelj I, Pereira-Smith O, et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci. 1995;92:9363–9367. doi: 10.1073/pnas.92.20.9363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farwell DG, Shera KA, Koop JI, Bonnet GA, Matthews CP, Reuther GW, Coltrera MD, McDougall JK, Klingelhutz AJ. Genetic and epigenetic changes in human epithelial cells immortalized by telomerase. Am J Pathol. 2000;156:1537–1547. doi: 10.1016/S0002-9440(10)65025-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster SA, Wong DJ, Barrett MT, Galloway DA. Inactivation of p16 in human mammary epithelial cells by CpG island methylation. Mol Cell Biol. 1998;18:1793–1801. doi: 10.1128/mcb.18.4.1793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green H, Rheinwald JG, Sun TT. Properties of an epithelial cell type in culture: The epidermal keratinocyte and its dependence on products of the fibroblast. Prog Clin Biol Res. 1977;17:493–500. [PubMed] [Google Scholar]
- Greider CW, Blackburn EH. Identification of a specific telomere terminal transferase activity in Tetrahymena extracts. Cell. 1985;43:405–413. doi: 10.1016/0092-8674(85)90170-9. [DOI] [PubMed] [Google Scholar]
- Harley CB. Telomere loss: Mitotic clock or genetic time bomb? Mutat Res. 1991;256:271–282. doi: 10.1016/0921-8734(91)90018-7. [DOI] [PubMed] [Google Scholar]
- Harley CB, Fletcher AB, Greider CW. Telomeres shorten during aging. Nature. 1990;345:458–460. doi: 10.1038/345458a0. [DOI] [PubMed] [Google Scholar]
- Hayflick L. Cell Aging. In: Eisdorfer C, editor. Annual Review of Gerontology and Geriatrics. Vol. 1. New York, NY: Springer; 1980. pp. 26–67. [Google Scholar]
- Hayflick L, Moorhead PS. The limited in vitro lifetime of human diploid cell strains. Exp Cell Res. 1961;25:585–621. doi: 10.1016/0014-4827(61)90192-6. [DOI] [PubMed] [Google Scholar]
- Huffman KE, Levene SD, Tesmer VM, Shay JW, Wright WE. Telomere shortening is proportional to the size of the G-rich telomeric 3′-overhang. J Biol Chem. 2000;275:19719–19722. doi: 10.1074/jbc.M002843200. [DOI] [PubMed] [Google Scholar]
- Jarrard DF, Sarkar S, Shi Y, Yeager TR, Magrane G, Kinoshita H, Nassif N, Meisner L, Newton MA, Waldman FM, et al. p16/pRb pathway alterations are required for bypassing senescence in human prostate epithelial cells. Cancer Res. 1999;59:2957–2964. [PubMed] [Google Scholar]
- Jiang XR, Jimenez G, Chang E, Frolkis M, Kusler B, Sage M, Beeche M, Bodnar AG, Wahl GM, Tlsty TD, et al. Telomerase expression in human somatic cells does not induce changes associated with a transformed phenotype. Nat Genet. 1999;21:111–114. doi: 10.1038/5056. [DOI] [PubMed] [Google Scholar]
- Jones CJ, Kipling D, Morris M, Hepburn P, Skinner J, Bounacer A, Wyllie FS, Ivan M, Bartek J, Wynford-Thomas D, et al. Evidence for a telomere-independent “clock” limiting RAS oncogene-driven proliferation of human thyroid epithelial cells. Mol Cell Biol. 2000;20:5690–5699. doi: 10.1128/mcb.20.15.5690-5699.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahn SH, Moritsugu J, Wahl GM. Differential requirement for p19ARF in the p53-dependent arrest induced by DNA damage, microtubule disruption, and ribonucleotide depletion. Proc Natl Acad Sci. 2000;97:3266–3271. doi: 10.1073/pnas.050560997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapahi P, Boulton ME, Kirkwood TB. Positive correlation between mammalian life span and cellular resistance to stress. Free Radic Biol Med. 1999;26:495–500. doi: 10.1016/s0891-5849(98)00323-2. [DOI] [PubMed] [Google Scholar]
- Kiyono T, Foster SA, Koop JI, McDougall JK, Galloway DA, Klingelhutz AJ. Both Rb/p16INK4a inactivation and telomerase activity are required to immortalize human epithelial cells. Nature. 1998;396:84–88. doi: 10.1038/23962. [DOI] [PubMed] [Google Scholar]
- Loo DT, Fuquay JI, Rawson CL, Barnes DW. Extended culture of mouse embryo cells without senescence: Inhibition by serum. Science. 1987;236:200–202. doi: 10.1126/science.3494308. [DOI] [PubMed] [Google Scholar]
- Mendrysa SM, Perry ME. The p53 tumor suppressor protein does not regulate expression of its own inhibitor, MDM2, except under conditions of stress. Mol Cell Biol. 2000;20:2023–2030. doi: 10.1128/mcb.20.6.2023-2030.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morales CP, Holt SE, Ouellette M, Kaur KJ, Yan Y, Wilson KS, White MA, Wright WE, Shay JW. Absence of cancer-associated changes in human fibroblasts immortalized with telomerase. Nat Genet. 1999;21:115–118. doi: 10.1038/5063. [DOI] [PubMed] [Google Scholar]
- Ouellette MM, McDaniel LD, Wright WE, Shay JW, Schultz RA. The establishment of telomerase-immortalized cell lines representing human chromosome instability syndromes. Hum Mol Genet. 2000;9:403–411. doi: 10.1093/hmg/9.3.403. [DOI] [PubMed] [Google Scholar]
- Parchment RE, Lewellyn A, Swartzendruber D, Pierce GB. Serum amine oxidase activity contributes to crisis in mouse embryo cell lines. Proc Natl Acad Sci. 1990;87:4340–4344. doi: 10.1073/pnas.87.11.4340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rheinwald JG. Methods for clonal growth and serial cultivation of normal human epidermal keratinocytes and mesothelial cells. In: Baserga R, editor. Cell growth and division: A practical approach. Oxford, UK: IRL; 1989. pp. 81–94. [Google Scholar]
- Robles SJ, Adami GR. Agents that cause DNA double strand breaks lead to p16INK4a enrichment and the premature senescence of normal fibroblasts. Oncogene. 1998;16:1113–1123. doi: 10.1038/sj.onc.1201862. [DOI] [PubMed] [Google Scholar]
- Rubin H. Cell aging in vivo and in vitro. Mech Aging Dev. 1997;98:1–35. doi: 10.1016/s0047-6374(97)00067-5. [DOI] [PubMed] [Google Scholar]
- ————— Telomerase and cellular life span: Ending the debate? Nat Biotechnol. 1998;16:396–397. doi: 10.1038/nbt0598-396. [DOI] [PubMed] [Google Scholar]
- Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell. 1997;88:593–602. doi: 10.1016/s0092-8674(00)81902-9. [DOI] [PubMed] [Google Scholar]
- Shelton DN, Chang E, Whittier PS, Choi D, Funk WD. Microarray analysis of replicative senescence. Curr Biol. 1999;9:939–945. doi: 10.1016/s0960-9822(99)80420-5. [DOI] [PubMed] [Google Scholar]
- Sherr CJ, DePinho RA. Cellular senescence: Mitotic clock or culture shock? Cell. 2000;102:407–410. doi: 10.1016/s0092-8674(00)00046-5. [DOI] [PubMed] [Google Scholar]
- Stampfer M, Hallowes RC, Hackett AJ. Growth of normal human mammary cells in culture. In Vitro. 1980;16:415–425. doi: 10.1007/BF02618365. [DOI] [PubMed] [Google Scholar]
- van Steensel B, Smogorzewska A, de Lange T. TRF2 protects human telomeres from end-to-end fusions. Cell. 1998;92:401–413. doi: 10.1016/s0092-8674(00)80932-0. [DOI] [PubMed] [Google Scholar]
- Vaziri H, Benchimol S. Reconstitution of telomerase activity in normal human cells leads to elongation of telomeres and extended replicative life span. Curr Biol. 1998;8:279–282. doi: 10.1016/s0960-9822(98)70109-5. [DOI] [PubMed] [Google Scholar]
- Wellinger RJ, Ethier K, Labrecque P, Zakian VA. Evidence for a new step in telomere maintenance. Cell. 1996;85:423–433. doi: 10.1016/s0092-8674(00)81120-4. [DOI] [PubMed] [Google Scholar]
- Wright WE, Shay JW. The two-stage mechanism controlling cellular senescence and immortalization. Exp Gerontol. 1992;27:383–389. doi: 10.1016/0531-5565(92)90069-c. [DOI] [PubMed] [Google Scholar]
- ————— Telomere dynamics in cancer progression and prevention: Fundamental differences in human and mouse telomere biology. Nat Med. 2000;6:849–851. doi: 10.1038/78592. [DOI] [PubMed] [Google Scholar]
- Wright WE, Piatyszek MA, Rainey WE, Byrd W, Shay JW. Telomerase activity in human germline and embryonic tissues and cells. Dev Genet. 1996;18:173–179. doi: 10.1002/(SICI)1520-6408(1996)18:2<173::AID-DVG10>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- Yang J, Chang E, Cherry AM, Bangs CD, Oei Y, Bodnar A, Bronstein A, Chiu CP, Herron GS. Human endothelial cell life extension by telomerase expression. J Biol Chem. 1999;274:26141–26148. doi: 10.1074/jbc.274.37.26141. [DOI] [PubMed] [Google Scholar]
- Zindy F, Quelle DE, Roussel MF, Sherr CJ. Expression of the p16INK4a tumor suppressor versus other INK4 family members during mouse development and aging. Oncogene. 1997;15:203–211. doi: 10.1038/sj.onc.1201178. [DOI] [PubMed] [Google Scholar]