

Abstract
Bacteroidales are attractive as water quality indicators because of their potential to discern sources of fecal pollution, and it is presumed that these bacteria do not multiply outside their host organisms. The persistence of a fecal Bacteroidales marker was monitored over 14 days in river water microcosms that varied in temperature from 10°C to 30°C and salinity from 0‰ to 30‰ by quantitative PCR (qPCR). Decay rates were estimated and compared to the results of other studies examining the survival and persistence of Bacteroidales markers by converting decay rates from other studies to a common decay rate unit. The log-linear decay rates estimated in this work ranged from −0.18 to −1.31 ln(CT/C0) day−1, where CT is the threshold cycle and C0 is the concentration of cells at time zero, which is comparable to findings in previous studies. Salinity had a positive effect on Bacteroidales marker persistence, while decay was more rapid at higher temperatures. Comparison of 16S rRNA gene clone libraries generated from microcosm samples indicated that most of the operational taxonomic unit (OTU) and phylogenetic diversity was found within samples and not between samples, indicating at least qualitatively that diverse lineages persist and likely have similar survival characteristics under most of the conditions examined. It was noted that the samples at higher salinities also had the smallest amount of diversity between samples as well as the lowest decay rates. This research also highlights the need for a repository of raw survival and persistence data if more sophisticated models of decay are to be employed and compared between different studies.
INTRODUCTION
Fecal pollution has negative impacts from both environmental and economic perspectives. The presence of traditional fecal indicator bacteria (FIB), namely, Escherichia coli and Enterococcus, is the standard by which the extent of fecal contamination and potential health hazard is currently assessed for recreational waters. Determining the presence of FIB is a relatively easy task, but determining the source(s) is a considerably more complex problem. Molecular methods based on the identification of 16S rRNA gene markers of fecal Bacteroidales have been successfully applied to delineate the sources of fecal pollution based on differences in host species intestinal community compositions (9, 20, 34). Determination of the mere presence of different sources of fecal pollution by using host-specific Bacteroidales markers was a significant advance for water quality analysis. The next problem that presented itself was quantifying the relative contributions of fecal pollution from respective sources. Since the first Bacteroidales host-specific molecular marker detection systems were published, studies have followed describing quantitative PCR (qPCR) methods for quantifying the abundance of specific markers (15, 21, 25, 28) in natural samples. Although enumeration of the specific markers is currently possible, it is clear that host-specific marker quantities in natural waters do not directly correspond to the relative contributions of fecal contamination from respective host sources due to differences in marker abundances within hosts, but this can potentially be overcome through surveys of marker abundances within host species (35). Even with knowledge of marker distributions and quantities in hosts, it remains questionable if accurate source apportionment can be realized due to unresolved environmental factors that may influence host marker populations (36) and survival. Studies are needed to elucidate the variance and survival effects as a precursor to understanding how observed distributions in natural waters occur. For example, a point measurement in a water body is the result of not just host marker variance and survival, but also mixing and dilution of potentially numerous sources. The different potential sources may also be of various ages (transport time) and may have experienced different routes (environmental conditions). In summary, the ability to correctly identify and apportion fecal sources is dependent on the variance of marker concentrations in different hosts and the survival characteristics of these markers in relation to environmental conditions to account for the possible range of environmental conditions the markers may have encountered before being sampled.
Surveys of the gut microflora demonstrated not just interspecific variation in Bacteroidales community composition but also intraspecific variation (14, 26). Variability in marker concentrations was also found for the human-specific HF183 marker (33), with the host harboring anywhere from nondetectable concentrations to >109 markers g−1 wet feces. In contrast, a study utilizing different host-specific Bacteroidales markers found that each respective marker (human, dog, cow, and horse) made up a relatively consistent percentage of the total Bacteroidales (35). While these results are somewhat mixed, depending upon methodology, the results give promise that the intraspecific variability is not so great as to preclude the use of Bacteroidales as a quantitative tool. In most situations where nonpoint source fecal pollution is responsible for exceeding regulatory standards, it can safely be assumed that the fecal pollution is a mixture of many individual host contributions (e.g., fecal pollution sourced from cattle farming is the result of many individual cow contributions). This assumption is safe as long as the total load is greater than that which could come from a small sample of individuals. If this assumption is met, then the relative concentration of a respective Bacteroidales marker should approach the overall mean which can be inferred from survey studies. Systematic surveys of the host-specific marker abundances found in fecal samples have been undertaken (35), but the need exists for broader surveys from different populations of host organisms. The relationship of a marker's concentration to its host species' fecal load is, as mentioned before, only half of the problem that needs to be solved before quantification can be asserted. The second dilemma for quantification, survival characteristics of host-specific markers, poses more complex problems. Bacteroidales markers can encounter a wide array of physical conditions once released into the environment from their respective hosts, and differential persistence of these markers can mislead researchers if left unaccounted. Microcosm survival experiments, no matter how well designed, do not fully match what happens in reality; however, they do provide parameter estimates for models that can serve as hypotheses to be tested against observations made in natural environment.
Efforts have focused on determining survival characteristics of both general and host-specific markers in the environment with respect to environmental factors. The effects of temperature on Bacteroidales survival have been investigated (8, 29), and a general positive relationship between temperature and decay rates has been found, as expected. Several studies have also examined the persistence of Bacteroidales in the presence or absence of light (4, 39, 40), which have had mixed results. The results of Bae and Wuertz did not suggest large differences in decay rates due to light at a salinity of ∼33‰ (4), which is similar to the findings of Walters and Field (39) in freshwater microcosms, although the reported decay rates between these two studies were substantially different. In contrast, Walters et al. (40) found a large increase in the decay rate due to light exposure in seawater microcosms. The difference in decay rates due to salinity was examined systematically by Okabe and Shimazu at salinities ranging from 0‰ to 30‰ at 10°C, and the salinity effect was negligible compared to the effects of temperature, but decay rates were consistently lower in the higher-salinity microcosms (29). More recently, direct comparisons have been made between Bacteroidales decay rates and that of E. coli (16). Aside from methodological differences in microcosm design and qPCR primers, differences in the decay rate constant units reporting also exist, and consequently decay rates have not been directly comparable between these studies without prior conversion of rates to the same convention.
Bacteroidales distributions among differing hosts are governed largely by diet-digestive system type (26) and therefore can serve as fecal pollution indicators that can discriminate among sources (i.e., human versus ruminant). The Bacteroidales are a deeply divergent and diverse group of microorganisms based on 16S rRNA phylogeny; if the survival characteristics of the different Bacteroidales clades are also divergent, then these differences must be considered. It is first important to understand just how specific a 16S marker is because the markers used are essentially clusters or more loosely associated groups of Bacteroidales based upon 16S primer-probe hybridization. While 16S rRNA gene primer-probe combinations are often designed to target a clade, there is no guarantee that this clade will have cohesive survival characteristics in the environment. A more likely scenario is that Bacteroidales markers cover a wide range of strains, species, genera, and families, depending on specificity, and the likelihood that they all share similar survival characteristics is currently unjustified. Studies have already concluded that some ruminant markers persist longer than human markers (39). In addition, differential survival characteristics have been observed between strains of E. coli (2); these observations cast serious doubt on any presumptions that distantly related—or even closely related—Bacteroidales should have similar survival characteristics.
The objective of this study was first to experimentally determine decay rates of fecal Bacteroidales at various temperatures and salinities. We do note that salinity and temperature are not the only factors influencing Bacteroidales survival, but serve as a useful reference point. Second, Bacteroidales decay rates from this study and that of previous studies were compared by normalizing the decay estimate(s) for temperature, salinity, and decay rate formulation (model differences), when possible. The final objective was to determine the most abundant Bacteroidales population structure and potential changes in this structure with respect to time, salinity, and temperature variations.
MATERIALS AND METHODS
Sample collection and microcosm inocula.
River water for the microcosm experiments was collected the morning of the experiment setup (as all samples were) from the Tangipahoa River, southeast Louisiana. River water had a salinity of 0 ‰ at the sample site on the day of sampling. Sewage influent was collected during peak morning flow at the City of Hammond south sewage treatment plant in a sterile 1-liter wide-mouth, high-density polyethylene (HDP) bottle and stored on ice for transport. Runoff from a dairy production facility was collected at the point of discharge into a holding pond in the same manner as the sewage sample. The sample collected at the dairy facility was a mixture of manure from a facility with greater than 100 head of cattle. The sewage and dairy production waste was mixed in a 1:1 ratio in a new sterile container and diluted to 10−3 with river water in a 20-liter sterile HDP Nalgene container that was physically agitated for mixing. Solids were allowed to settle for 1 h before microcosm setup.
Microcosm design.
Approximately 19 liters of the mixed-sewage sample in river water was decanted to a new sterile 20-liter HDP Nalgene container that had a spigot for sample dispersal. The zero-salinity microcosms (three different temperatures × replicates = 6 total) were dispersed into prewashed and sterile 1-liter HDP Nalgene bottles to a volume of approximately 950 ml. The salinity treatments of 5‰ and 30‰ were prepared by the addition of Instant Ocean sea salts incrementally to the 20-liter container to obtain the desired salinity and dispersed in the same manner as the zero-salinity treatment. Microcosms were incubated statically with the sample lid loosely placed on the Nalgene bottles to allow for gas exchange. Samples were kept in the dark at the designated temperatures in air-circulated, temperature-controlled environments.
Microcosm sampling and DNA extraction.
Two initial samples from the 20-liter container were taken to represent the beginning concentration and composition. Replicate microcosms were sampled at 0, 24, 48, 96, 144, 192, 264, and 336 h (0 to 14 days). Treatments included salinities of 0, 5, and 30‰ at 10, 20, and 30°C, all in the dark, for a total of 18 microcosms and 144 discrete samples. The sample naming scheme was as follows: time (days)_salinity (‰)_temperature (°C) replicate (A or B), such that a sample collected at day 4, 30‰ salinity, and 20°C, replicate A, would be named 4S30T20A. Temperature was monitored daily in each microcosm before opening of incubators and did not vary greater than ±0.5°C. For each sample, 100 ml of sample was removed and filtered with a 0.45-μm-pore MF-Millipore mixed-cellulose-ester hydrophilic 47-mm filter (Millipore Corp., Billerica, MA) by vacuum filtration using an alcohol-flamed glass filtration device. DNA was extracted essentially as described previously (15), with slight modification. Filters were folded aseptically and placed in 15-ml sterile capped polypropylene tubes along with 0.5 ml of guanidine isothiocyanate buffer (5 M guanidine isothiocyanate buffer, 100 mM EDTA, pH 8.0, and 0.5% sodium lauroyl sarcosinate), inverted several times to ensure complete wetting of the filter, and then placed at −20°C until further DNA extraction (after the last sample was taken 14 days later). The DNeasy tissue kit (Qiagen, Valencia, CA) was used to extract DNA as described by the DNeasy manual. Five hundred milliliters of Qiagen AL buffer was added to the 15-ml tube and vortexed for 1 min. The tube was then incubated at 70°C for 10 min with 1 min of vortexing at high speed immediately afterward. This process was repeated three times. Next, 400 ml ethanol was added to sample and mixed for 1 min by vortexing. This mixture was then pipetted into the DNeasy mini-spin column and centrifuged for 1 min at 8,000 rpm (two loads per tube); flowthrough was discarded. Columns were placed in new collection tubes and washed twice with 500 ml buffer AW1 at 8,000 rpm for 1 min. Columns were placed in a new collection tube and washed once with 500 ml buffer AW2 at 14,000 rpm for 1 min, flowthrough was discarded, and the columns were spun again at 14,000 rpm for 3 min. The columns were then placed in 1.5-ml collection tubes, and 200 ml buffer AE (Qiagen) was added directly to column membrane and allowed to incubate for 2 min at room temperature before centrifugation at 8,000 rpm for 1 min to collect DNA. DNA was stored at −20°C.
Creation of quantitative PCR standards.
The primers Bac32F and Bac708R (9) were used to generate PCR fragments with iTaq DNA polymerase (Bio-Rad, Hercules, CA), and these fragments were subsequently cloned with the TOPO TA cloning kit and TOP10 chemically competent cells (Invitrogen, Carlsbad, CA), as per the manufacturer's instructions. Presumptive recombinants were grown overnight, and plasmid DNA was extracted with the Eppendorf FastPlasmid minikit (Eppendorf, Hamburg, Germany). Recombinants were tested for presence of suitable template DNA by using the quantitative fecal Bacteroidales primers 5′GCTCAGGATGAACGCTAGCT (forward) and 5′CCGTCATCCTTCACGCTACT (reverse) (15). A recombinant that was positive with the PCR was selected as a quantitative standard. The concentration of plasmid DNA with the insert was quantified with a Nanodrop ND1000 UV/visible spectrophotometer (NanoDrop Products, Wilmington, DE). The DNA concentration was used to estimate copy number and to create serial dilutions for standard curves.
Quantitative PCR enumeration of fecal Bacteroidales markers.
Previously described quantitative fecal Bacteroidales PCR primers and probes were used: forward primer 5′GCTCAGGATGAACGCTAGCT, reverse primer 5′CCGTCATCCTTCACGCTACT, and 6-carboxyfluorescein/6-carboxytetramethylrhodamine (FAM/TAMRA) probe 5′CAATATTCCTCACTGCTGCCTCCCGTA (15), corresponding to Bacteroides fragilis NCTC 9343 16S positions 24 to 43 (forward primer), 404 to 423 (reverse primer), and 349 to 375 (probe), respectively. Each 50-μl quantitative real-time PCR mixture contained 25 μl iQ supermix (Bio-Rad, Hercules, CA), 2 ml template DNA, 400 ng ml−1 bovine serum albumin (BSA), 0.4 μM each primer, and 0.2 μM probe. The cycling conditions consisted of an initial denaturation step of 95°C for 5 min, followed by 40 cycles of 95°C for 15 s, 52°C for 30 s, and 72°C for 30 s. All quantitative reactions were performed using a Bio-Rad iCycler system (Hercules, CA). All quantitative PCRs were performed in triplicate, and average values are reported.
Clone library construction.
Two to 4 ng of DNA was used as a template for reactions, and Bacteroidales-specific primers 32F and 708R (9) were used to amplify 16S rRNA gene fragments for sequencing. Each 50-μl reaction mixture contained 25 μl iQ supermix (Bio-Rad, Hercules, CA), 0.4 μM each primer, and 400 ng μl−1 BSA. Precautions were taken to minimize PCR artifacts (1) by reducing the number of cycles to a minimum. An initial denaturing step of 95°C for 5 min, followed by 20 cycles of 94°C for 30s, 53°C for 1 min, and 72°C for 2 min, was used for all reactions. Reconditioning of PCR mixtures (38) was done by using 2 μl of the initial PCR mixture in an identical PCR that was reduced to 3 cycles and included a final elongation step of 5 min at 72°C. Four microliters of reconditioned PCR mixture was used for each cloning reaction, using PCR 2.1 TOPO kits (Invitrogen, Carlsbad, CA) as per the manufacturer's instructions. Transformants were screened, and then glycerol stocks were prepared in 96-well plates and sequenced at SeqWright (Houston, TX).
Phylogenetic analysis and statistical analysis.
All sequences were trimmed of vector, primer sequences, and any poor-quality regions near the ends, leaving partial 16S sequences of approximately 600 bp. Nucleotide sequences were aligned with the NAST server (12). Potential chimeric sequences were identified with Mallard (3) and removed from further analysis. Pairwise distances were computed with the Kimura two-parameter model of nucleotide substitution (22) in the DNADIST package of PHYLIP (17). Operational taxonomic units (OTUs) were calculated by using the farthest-neighbor-clustering algorithm and a 95% similarity cutoff with mothur (32). The phylogenetic tree was inferred by the neighbor-joining method (31), using pairwise distances calculated by the Kimura two-parameter method (22) with MEGA4 (37). OTUs containing less than two sequences were removed from further analysis. Diversity indices, nonmetric multidimensional scaling (NMS), and multiresponse permutation procedures (MRPP) was performed with PC-ORD v.5 (MJM Software).
Statistical analysis of decay curves was performed with either the decay model of Chick (10) for comparison with previously reported decay rates or the Baranyi-Robert model (5–7), a model that is frequently used in predictive microbiology to model microbial growth or death under different environmental conditions. We calculated decay rates with the model of Chick, using the relationship ln(C/C0) = −kt, where C is equal to the concentration of cells at time t and k is the decay constant. Decay rate k was estimated from the slope of the regression line (39). The Baranyi-Robert model parameters were estimated with DMFit, available at ComBase (http://www.combase.cc/default.html). DMFit fits the raw data to the equation
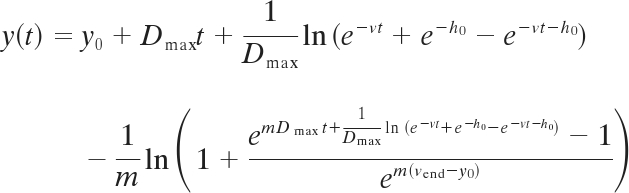 |
where y(t) = ln[x(t)] with x(t) equal to the Bacteroidales marker concentration 100 ml−1, y0 = ln(x0), yend = ln(xmin), with x0 being the initial point and xmin the asymptote found at the end of the decay curve (if present). Dmax is the maximum decay rate, m is a curvature parameter that is used to define the switch from exponential decay, and v is a curvature parameter that is used to define the switch to exponential decay (log linear). The parameter h0 is a dimensionless parameter and characterizes the initial physiological state of the cells before entering exponential decay; using h0, a shoulder time (h) can be calculated by . The shoulder time is the time preceding log-linear decay and is not always observed in decay studies. By default, the parameter v is set to Dmax and m is set to 10. Survival curves that do not have a shoulder are reduced in parameters and do not include h. More detailed discussions on the Baranyi-Robert model are available in references 5 to 7.
Calculation of decay rates from previous studies following Chick's law was done with only the linear portion of the reported decay curves. For instance, if a previous report utilized a different method to report the decay rate, the decay model reported was employed with a starting concentration of 108 copies ml−1 (arbitrary) to recreate a death curve. The resulting death curve was the used to calculate a death rate following Chick's law. The use of this method avoided unnecessary algebraic conversions between models, especially under circumstances in which appropriate algebraic conversions were not apparent. Previous survival studies were performed at different temperatures, and temperature corrections for decay rate constants were performed with temperature-decay relationships provided by different authors (see Table 2). The data generated are included in the supplemental material.
Table 2.
Relative decay rates and conversion to ln(CT/C0) from previously reported studies and this studya
| Target | Temp (°C) | Salinity (‰) | Relative decay rate with constant light | Decay rate conversion to ln(CT/C0) day−1 in the dark | Reference |
|---|---|---|---|---|---|
| Fecal Bacteroidetes | 13b | 0 | −0.9701 | This study | |
| Human | 13 | Freshwater | −1.7 | −1.4 | 39 |
| Human | 13 | Freshwater | −1.4 | −1.2 | 39 |
| Cow | 13 | Freshwater | −0.8 | −0.6 | 39 |
| Cow | 13 | Freshwater | −1 | −0.99 | 39 |
| All Bacteroides | 13c | Freshwater | −0.4176 | 8 | |
| Bac-Pre | 13d | 0 | −0.9441 | 29 | |
| Human-Bac | 13d | 0 | −0.8672 | 29 | |
| Cow-Bac2 | 13d | 0 | −0.9998 | 29 | |
| Bacteroidales | ∼12 | ∼33 | −0.3693 | −0.4673 | 4 |
| Human | ∼12 | ∼33 | −0.3269 | −0.543 | 4 |
| Cow | ∼12 | ∼33 | −0.3311 | 4 | |
| Fecal Bacteroidetes | 12e | 30 | −0.2331 | This study | |
| Human | ∼17 | 34.2 | −1.3 | −0.264 | 40 |
| Fecal Bacteroidetes | 17e | 30 | −0.3611 | This study | |
| qHF183 | 25 | Freshwater | −2.6931 | −2.132 | 16 |
| BacHum | 25 | Freshwater | −2.9904 | −2.6466 | 16 |
| AllBac | 25 | Freshwater | −1.8059 | −1.404 | 16 |
| Fecal Bacteroidetes | 25b | 0 | −1.2425 | This study | |
| qHF183 | 15 | Freshwater | −1.8202 | 16 | |
| BacHum | 15 | Freshwater | −1.9596 | 16 | |
| AllBac | 15 | Freshwater | −1.6869 | 16 | |
| Fecal Bacteroidetes | 15b | 0 | −1.0155 | This study | |
| qHF183 | 25 | Freshwater/sediment | −2.4496 | 16 | |
| BacHum | 25 | Freshwater/sediment | −2.214 | 16 | |
| AllBac | 25 | Freshwater/sediment | −1.0372 | 16 |
Decay rates were converted to ln(CT/C0) day−1 when necessary.
Temperature effect adjusted by 3-point linear regression at 0‰ salinity: y = −0.0227 × −0.675; R2 = 0.8913.
Temperature effect adjusted by the linear temperature model of Bell et al. (8).
Temperature effect adjusted by using the original decay rates of log10 copies day−1 before conversion to ln(CT/C0) with temperature-dependent data presented by Okabe and Shimazu in Table 1 in reference 29 showing the following results: Bac-Pre, y = 0.2693 ln(x) − 0.3141, R2 = 0.9936; Human-Bac, y = 0.3247 ln(x) − 0.3986, R2 = 0.9901; and Cow-Bac2, y = 0.3356ln(x) − 0.4463, R2 = 0.9622.
Temperature adjusted by using a 2-point linear fit at 30‰ salinity with y = −0.0256x + 0.0741.
Nucleotide sequence accession numbers.
Sequences determined in this study have been deposited in GenBank under accession no. HM442128 to HM443067.
RESULTS
The mean initial concentration of Bacteroidales markers in microcosms was 1.02 × 107 copies 100 ml−1 (standard deviation [SD], 0.59 × 107 100 ml−1; n = 18). Decay rates calculated by using Chick's law ranged from −1.31 to −0.18 in this study (Table 1). The Baranyi-Robert model was utilized to estimate maximum decay rates (Dmax), shoulder time in hours (h), and ending concentration (yend) (Table 1 and Fig. 1). Treatments that have a yend value have reached the limit of detection, considering the DNA concentration and the qPCR conditions of this study. Because of the subjective nature of visually selecting the linear region of copy number decline to estimate decay rates, the Baranyi-Robert decay model provided a more objective method to determine maximum decay rates for comparison between treatments. The maximum decay rates estimated by the Baranyi-Robert model varied with both temperature and salinity (Fig. 2), as did the persistence of Bacteroidales (Fig. 1). Bacteroidales markers had slower decay rates at higher salinities and at lower temperatures (see Fig. S1 in the supplemental material). Decay rates estimated by the Baranyi-Robert model compared to maximum decay rates estimated by using Chick's law had a Pearson's correlation of 0.91, suggesting good overall agreement. The shoulder time estimates of the Baranyi-Robert model were brief (<2 days) for 10 and 20°C at 0‰ salinity and long at 10 and 20°C at 30‰ (<10 and <4 days, respectively), and no shoulder time was detected for all other treatments. The 30, 5, and 0‰ treatments across all temperatures had mean decay rates of −0.01, −0.02, and −0.05 log10 copies h−1 (SD, 0.005, −0.02, and −0.05, respectively).
Table 1.
Baranyi model parameters, maximum decay rates, maximum decay rates transformed to ln(CT/C0) · day−1, and decay rates
| Temp (°C) | Salinity (‰) | Dmax (log10 copies h−1) | ha | y0 | yendb | SE (fit) | R2 | Dmax converted to log-linear decay rate [ln(CT/C0) day−1]c | Decay rate [ln(CT/C0) day−1]d | R2 |
|---|---|---|---|---|---|---|---|---|---|---|
| 10 | 0 | −0.029 | 43.041 | 7.242 | 2.878 | 0.279 | 0.978 | −1.575 | −0.856 | 0.872 |
| 20 | 0 | −0.050 | 26.270 | 6.835 | 2.897 | 0.269 | 0.976 | −2.785 | −1.221 | 0.815 |
| 30 | 0 | −0.059 | 7.028 | 3.027 | 0.398 | 0.932 | −3.255 | −1.310 | 0.790 | |
| 10 | 5 | −0.013 | 7.299 | 0.356 | 0.948 | −0.730 | −0.759 | 0.958 | ||
| 20 | 5 | −0.031 | 7.205 | 3.225 | 0.489 | 0.912 | −1.702 | −0.864 | 0.822 | |
| 30 | 5 | −0.026 | 6.628 | 2.822 | 0.419 | 0.929 | −1.426 | −0.714 | 0.913 | |
| 10 | 30 | −0.015 | 235.487 | 7.007 | 0.319 | 0.713 | −0.829 | −0.182 | 0.938 | |
| 20 | 30 | −0.008 | 79.686 | 6.913 | 0.292 | 0.871 | −0.464 | −0.438 | 0.927 | |
| 30 | 30 | −0.006 | 7.011 | 0.426 | 0.705 | −0.321 | −0.261 | 0.986 |
h is the shoulder time (see Materials and Methods for description). When no shoulder time is observed, there is no h, and the model is reduced by this parameter.
yend is the ending aysmptote (see Materials and Methods for description). When no final asymptote is observed, there is no yend, and the model is reduced by this parameter.
Maximum decay rate estimated by the Baranyi-Roberts model converted to log-linear decay rate day−1.
Decay rate calculated by Chick's law by linear regression; only the linear portions of decay curves were utilized.
Fig. 1.

Bacteroidales marker decay curves at salinities of 0, 5, and 30‰ at 10, 20, and 30°C, respectively. Temperature (T) and salinity (S) treatment are indicated in the top right of each plot, with temperature indicated first in parentheses, followed by salinity. The lines are the Baranyi-Robert model fit (5).
Fig. 2.

Combined effects of salinity and temperature on Bacteroidales marker decay rates. The data points represent the mean maximum decay rates (log10 copies h−1) estimated with the Baranyi-Robert model (5).
Decay rates that were relevant to other studies are presented using Chick's law in Table 2, derived by extrapolation in some cases (see Materials and Methods). Freshwater microcosms incubated in the dark at 13°C (with some rates adjusted for temperature differences) had decay rates ranging from −0.42 to −1.4 and a mean of −0.94 (SD, 0.29) across all studies. These values included studies using different primer-probe combinations, yet six of the nine estimated decay rates were between a narrow range of −0.87 and −1.2. Decay rates from dark microcosms with salinities ranging from 30‰ to 34.2‰ and temperatures ranging from 12°C to 17°C had decay rates ranging from −0.23 to −0.54 and a mean of −0.37 (SD, 0.12). Although there were multiple primer-probe combinations used across these studies, the mean difference between dark decay rates from freshwater to saline (at least 30‰ salinity) was on average 0.57 greater in freshwater microcosms. Results were generally similar for comparable environmental conditions (temperature, salinity, and light treatments) to previously published results. The final concentrations of Bacteroidales copy numbers did not drop below ∼103 copies 100 ml−1 (approximately equivalent to 10 copies per PCR); therefore, this value can be viewed as the limit of detection with the sample volumes and reaction conditions used, consistent with the limit of detection reported for this assay (15).
A total of 940 Bacteroidales partial 16S rRNA gene sequences (approximately 600 bp after primer regions were removed) recovered from the microcosms were aligned by using the Greengenes NAST server (12) after potential chimeras were removed, and subsequently 46 OTUs containing at least two representatives were identified at 95% similarity by using mothur (32). All subsequent community-based analysis was based on a 95% OTU definition. Several samples at 0‰ salinity and 5‰ salinity past day 2 failed to produce useable sequences. Samples that contained less than 20 sequences were removed from further analyses, leaving a total of 21 samples. Shannon's diversity (H′) appeared to decrease with time, while evenness (E) remained fairly constant through time (Fig. 3a and b), with the exception of samples 4S520CB and 8S030CA; further data collection is needed to assess if these trends are significant. The more prevalent OTUs were labeled in order of number of occurrences (OTU1 was the most frequently observed, followed by OTU2, etc.), and the most prevalent OTUs were found across most samples. OTUs 1 to 10 contained >75% of all sequences recovered. OTU1 was found in all samples and was the only OTU that was present throughout, although OTUs 2 to 4 were also present in most samples. NMS ordination of samples (Fig. 4) did not reveal any time-related clustering, but did show clustering of 30‰ salinity samples (average within group Bray-Curtis dissimilarity of 0.46) compared to the 0 and 5‰ salinity treatments (0.71 and 0.57 dissimilarity, respectively). Comparison of different salinity and temperature treatments by multiresponse permutation procedures (MRPP) revealed a significant difference only between the 5 and 30‰ salinity treatments (see Table S1 in the supplemental material). In general, Bray-Curtis dissimilarity between initial samples and subsequent samples tended to increase with time (Fig. 5) from day 0 to day 4.
Fig. 3.

Changes in Shannon's diversity (H′) with time (A) and evenness with time (B) across all microcosm conditions.
Fig. 4.

NMS ordination of microcosm samples. Individual samples are labeled with the day of sampling indicated first, followed by salinity and temperature. An OTU definition of 95% similarity and Sorensen (Bray-Curtis) distance were used for ordination. A total of 112 iterations of the data were used to generate a final two-dimensional solution with a stress of 15.65 and a final instability of <0.00000.
Fig. 5.

Changes in Bray-Curtis dissimilarity (95% similarity of OTU) through time. Initial dissimilarity was measured between replicate samples.
A phylogenetic tree was constructed with sequences generated in this study and nearest matches found in the Greengenes (13) aligned database (see Fig. S2 in the supplemental material). A single representative for each OTU cluster was included to simplify the tree. The most abundant OTUs recovered were distantly related among the Bacteroidales and have a mean pairwise distance of 0.218 for OTUs 1 to 10. The overall mean pairwise sequence diversity for all 46 OTUs was 0.216, and it was 0.17 when a mask was applied to hypervariable regions (24). The mean pairwise distance (calculated by the Kimura two-parameter model of nucleotide substitution) within samples was 0.158 (SD, 0.007). The most abundant OTU recovered was associated with the genus Prevotella: in total, 278 clones (30%) were associated with Prevotella, and the rest of the clones were associated with the diverse genus Bacteroides, with the exceptions of four clones related to Paraprevotella and five clones related to an uncultivated clade. The clones recovered were most closely related to many different animal source clones, confirming that the sewage-mixture inoculum sufficiently represented a diverse sample of Bacteroidales.
DISCUSSION
A primary goal was to compare the decay rates observed in this work with those from similar studies. Knowledge of decay rates of Bacteroidales is essential for interpretation of results from environmental surveys, especially if Bacteroidales markers are to be employed as fecal pollution indicators. Given the large diversity of Bacteroidales observed both within and between different host organisms (14, 18, 26), multiple studies examining the decay rates of Bacteroidales are needed to determine the general applicability of this indicator method(s).
Decay rate estimates from multiple studies were transformed to the log-linear decay model of Chick for direct comparison. When decay rate estimates from this study and other studies were next adjusted for microcosm conditions (temperature and salinity), good agreement was observed between rate estimates in most cases. The study with the lowest dark freshwater decay rate used an inoculum coming from a single healthy horse (8). The study with the highest dark freshwater decay rates had inocula coming from either strictly human feces from a mixture of eight individuals or strictly cow feces from 10 patties (39). The study by Walters and Field (39) also used primer-probe combinations targeting human or cow markers, which may explain some the variability observed. In contrast to these studies, our study used mixed-source sewage samples representing much larger populations. The study by Walters, Yamahara, and Boehm (40) used similar inocula to our study and also observed similar decay rates, perhaps due to the diverse sample inocula used. The comparisons presented here are ignoring the primer-probe sets used in the studies, but despite this, still point to the possibility of an inoculum effect, as mentioned in previous work (40). This inoculum effect may be an artifact of small sample size or differences in the physiological states of Bacteroidales coming from different hosts at different times. Until now, studies have discussed the general observations between different studies, but the data have not been transformed for more direct comparisons. These findings taken together highlight two specific needs for future studies. First, larger population samples (sample inocula consisting of contributions from many hosts), which would be better representative of natural pollution scenarios, need to be used to for estimating decay of Bacteroidales.
Sampling of individual hosts and enumeration of the relative concentrations of Bacteroidales markers are certainly worthwhile endeavors, but from a microbial source tracking viewpoint, it is far more effective to sample large populations of markers to arrive at meaningful estimates of mean marker concentrations. Second, a database-type approach, in which raw data from survival studies are available, would afford researchers the opportunity to statistically compare decay rates from different studies. More specifically, making the raw data available would allow individual researchers to create or utilize different decay models of their own choice. It is clear that a simple linear decay model is inadequate, yet without raw data available, researchers will be constrained by this limitation when comparing results.
In dark microcosm studies, decay rates were inversely related to salinity. The underlying mechanism for enhanced survival due to salinity increases was not elucidated in our experiment, but results from other studies shed some light on possible reasons for these observations. It is possible that increased salinity may have inhibited or killed predators in the microcosm design of this study and that of other studies, causing the decrease in decay rates relatied to decreased predation activity, which has been shown to significantly impact Bacteroides marker persistence (23). Using filtered and unfiltered surface waters, the differential survival or persistence due to predation or other biological actions has been shown previously to affect Bacteroidales (8). In the study by Okabe and Shimazu (29), the use of unfiltered seawater increased persistence of Bacteroidales markers in higher-salinity samples, and this was indirectly attributed to salinity controls on predation. Temperature, on the other hand, had a positive effect on decay rates. This pattern is consistent across all studies examining temperature effects, with the exception that Bell et al. (8) noticed a decrease in decay rates between 30 and 35°C, although temperatures around 35°C are not likely to occur often in most natural waters. In our study, in 10°C microcosms, Bacteroidales always persisted longer than in higher-temperature microcosms at any salinity (Fig. 1). This is in contrast to comparisons of decay rate estimates using a single-parameter estimate of decay rate: either Chick's law k or Baranyi-Robert model Dmax (Fig. 2). Namely, the single-parameter estimate of decay would lead one to believe that decay is faster at 10°C and 30‰ than at the other two higher temps. Visual inspection of the survival curves (Fig. 1) shows that this is because the 10°C and 30‰ salinity treatment has an extended shoulder time before reaching the exponential decay phase. This discrepancy is not fully captured when only k or Dmax is used. Previous work has shown that the use of a single-parameter model, such as the model of Chick, to describe bacterial decay is insufficient (4, 11, 19, 27, 30, 41). Given the disparity in comparison of maximum decay rates, log-linear decay rates, and actual persistence of Bacteroidales markers as illustrated in Fig. 1, as well as Table 2, the use of single-parameter decay models to describe decay of Bacteroidales is inadequate. The differences in decay rates as estimated by two different techniques, namely, with the Baranyi-Robert model compared to Chick's model of decay, pronounce this finding. Both the shapes of the decay curves and the decay rate estimates demonstrate the large variance that can be introduced to decay rate estimates solely from the methodology used to estimate decay and persistence. We have compared decay rate estimates from multiple studies using Chick's model (log-linear model), although the shape of the decay curves observed in some cases does not justify the use of a log linear model. Care was taken to convert decay rates for comparison, yet in the absence of all raw data, these rate estimates are not truly directly comparable due to the possible differences in the method initially used to estimate decay rates. A clear example of incomparability is when decay curves deviate from linearity, yet linear approximations are still utilized and reported, this study included. Despite this, the comparison of log-linear decay rates is not completely invalid. The high correlation between log-linear decay (Chick's model) rates and maximum decay rate (Baranyi-Robert model) suggests useful information is gained from these comparisons.
A second goal of this work was to determine if phylogenetically distinct groups of Bacteroidales differed in their persistence under different environmental conditions. As time progressed through this study, diversity appeared to decrease slightly while evenness tended to remain consistent. The apparent decrease in diversity may only be an artifact of sampling methodology since more clones were recovered from the initial samples. We cannot attest to the significance of these findings with the current data set; a much larger sampling is therefore warranted. The initial decrease from day 0 to day 4 was the only large decrease, with the exception of a few samples that had a richness of less than 10. Although relatively small clone libraries were used, most samples had greater than 10 OTUs present, suggesting that no dominant OTU had greater survival characteristics in this study. Comparison of the levels of OTU diversity showed that the 30‰ salinity samples had less diversity between samples. This helps explain the clustering of 30‰ samples when NMS ordination was used. When this finding is viewed in conjunction with the increased persistence observed for the 30‰ salinity samples, it would appear to be a related phenomenon: specifically, if strains of Bacteroidales are surviving longer, the expected outcome would be that community composition would change less through time.
While OTU diversity serves as one useful measure, the phylogenetic differences are also very informative. The mean nucleotide diversity within and between samples indicates that much of the overall diversity is found within each sample. The diversity found throughout the sampling in this study demonstrates at least semiquantitatively that very diverse (based on 16S rRNA sequences) Bacteroidales clusters have similar survival characteristics examined here. Although diversity indices trended downward, as would be expected under any circumstances when populations are decreasing in size, the fact that diversity was only observed to have dropped greatly in two samples (samples 4S5T20B and 8S0T30A) lends support to the idea that several Bacteroidales populations are able to endure for similar lengths of time. Alternatively, when diversity indices did drop, this may be an observation due to longer persistence of a few OTUs relative to the rest of the population. We have also opted to use the term “Bacteroidales” in this work. This emphasizes the diverse nature of the organisms studied here, as well as how little is known. It is equally important when qPCR studies are undertaken using markers for members of the Bacteroidales that researchers take care to define the phylogenetic specificity of the primer-probe combination used. For example, a well-designed and executed study referred to the target marker as “Bacteroides” (25); however, a query of the AllBac primers and probes used in this study (only including 100% matches of all three primer-probe targets in conjunction within the Bacteroidales; RDP release 10, 30 August 2010) retrieved >36,000 sequences of both Bacteroidaceae and Prevotellaceae. This doesn't take away from the value of this primer-probe set, but this marker certainly encompasses more than just Bacteroides species. We cannot assume that phylogenetically diverse clades of Bacteroidales will have similar survival characteristics without sufficient justification, and individual researchers should be aware of the specificity of the probes being utilized, especially as our knowledge expands with new database entries.
The results of this study suggest that the use of a larger sample inoculum size in future studies is warranted. Also, a database of survival studies would help researchers compare results more comprehensively. This is not unprecedented, as extensive online databases are available for food pathogens (http://www.combase.cc/). Comparisons of decay rates in our study with those of others show inconsistencies that may be explained in part by the inoculum effect; however, more studies are needed to directly test this hypothesis. It is also clear that use of a single decay rate constant can be misleading, and more complex models may be appropriate. This work did not find evidence suggesting that diverse lineages of Bacteroidales have large differences in their persistence based on 16S rRNA gene clone library analysis. Considering the widespread social, public health, and economic impacts of water quality standards, it is a reasonable expectation that future efforts be more readily comparable and allow for integration of data from multiple studies.
Supplementary Material
Footnotes
Supplemental material for this article may be found at http://aem.asm.org/.
Published ahead of print on 28 January 2011.
REFERENCES
- 1. Acinas S. G., Sarma-Rupavtarm R., Klepac-Ceraj V., Polz M. F. 2005. PCR-induced sequence artifacts and bias: insights from comparison of two 16S rRNA clone libraries constructed from the same sample. Appl. Environ. Microbiol. 71:8966–8969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Anderson K. L., Whitlock J. E., Harwood V. J. 2005. Persistence and differential survival of fecal indicator bacteria in subtropical waters and sediments. Appl. Environ. Microbiol. 71:3041–3048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ashelford K. E., Chuzhanova N. A., Fry J. C., Jones A. J., Weightman A. J. 2006. New screening software shows that most recent large 16S rRNA gene clone libraries contain chimeras. Appl. Environ. Microbiol. 72:5734–5741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bae S., Wuertz S. 2009. Rapid decay of host-specific fecal Bacteroidales cells in seawater as measured by quantitative PCR with propidium monoazide. Water Res. 43:4850–4859 [DOI] [PubMed] [Google Scholar]
- 5. Baranyi J., Roberts T. A. 1994. A dynamic approach to predicting bacterial growth in food. Int. J. Food Microbiol. 23:277–294 [DOI] [PubMed] [Google Scholar]
- 6. Baranyi J., Roberts T. A. 1995. Mathematics of predictive food microbiology. Int. J. Food Microbiol. 26:199–218 [DOI] [PubMed] [Google Scholar]
- 7. Baranyi J., Roberts T. A., McClure P. J. 1993. A non-autonomous differential equation to model bacterial growth. Food Microbiol. 10:43–59 [Google Scholar]
- 8. Bell A., et al. 2009. Factors influencing the persistence of fecal Bacteroides in stream water. J. Environ. Qual. 38:1224–1232 [DOI] [PubMed] [Google Scholar]
- 9. Bernhard A. E., Field K. G. 2000. Identification of nonpoint sources of fecal pollution in coastal waters by using host-specific 16S ribosomal DNA genetic markers from fecal anaerobes. Appl. Environ. Microbiol. 66:1587–1594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chick H. 1908. An investigation of the laws of disinfection. J. Hyg. 8:92–157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Crane S. R., Moore J. A. 1986. Modeling enteric bacterial die-off: a review. Water Air Soil Pollut. 27:411–439 [Google Scholar]
- 12. DeSantis T. Z., Jr., et al. 2006. NAST: a multiple sequence alignment server for comparative analysis of 16S rRNA genes. Nucleic Acids Res. 34:W394–W399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. DeSantis T. Z., Jr., et al. 2006. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl. Environ. Microbiol. 72:5069–5072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Dick L. K., et al. 2005. Host distributions of uncultivated fecal Bacteroidales bacteria reveal genetic markers for fecal source identification. Appl. Environ. Microbiol. 71:3184–3191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Dick L. K., Field K. G. 2004. Rapid estimation of numbers of fecal Bacteroidetes by use of a quantitative PCR assay for 16S rRNA genes. Appl. Environ. Microbiol. 70:5695–5697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Dick L. K., Stelzer E. A., Bertke E. E., Fong D. L., Stoeckel D. M. 2010. Relative decay of Bacteroidales microbial source tracking markers and cultivated Escherichia coli in freshwater microcosms. Appl. Environ. Microbiol. 76:3255–3262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Felsenstein J. 1993. PHYLIP (Phylogeny Inference Package) version 3.5c. Department of Genetics, University of Washington, Seattle [Google Scholar]
- 18. Fogarty L. R., Voytek M. A. 2005. Comparison of Bacteroides-Prevotella 16S rRNA genetic markers for fecal samples from different animal species. Appl. Environ. Microbiol. 71:5999–6007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gonzalez J. M. 1995. Modelling enteric bacteria survival in aquatic systems. Hydrobiologia 316:109–116 [Google Scholar]
- 20. Gourmelon M., et al. 2007. Evaluation of two library-independent microbial source tracking methods to identify sources of fecal contamination in French estuaries. Appl. Environ. Microbiol. 73:4857–4866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kildare B. J., et al. 2007. 16S rRNA-based assays for quantitative detection of universal, human-, cow-, and dog-specific fecal Bacteroidales: a Bayesian approach. Water Res. 41:3701–3715 [DOI] [PubMed] [Google Scholar]
- 22. Kimura M. 1980. A simple method for estimating evolutionary rate of base substitutions through comparative studies of nucleotide sequences. J. Mol. Evol. 16:111–120 [DOI] [PubMed] [Google Scholar]
- 23. Kreader C. A. 1998. Persistence of PCR-detectable Bacteroides distasonis from human feces in river water. Appl. Environ. Microbiol. 64:4103–4105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lane D. J. 1991. 16S/23S rRNA sequencing. John Wiley and Sons, West Sussex, England [Google Scholar]
- 25. Layton A., et al. 2006. Development of Bacteroides 16S rRNA gene TaqMan-based real-time PCR assays for estimation of total, human, and bovine fecal pollution in water. Appl. Environ. Microbiol. 72:4214–4224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ley R. E., et al. 2008. Evolution of mammals and their gut microbes. Science 320:1647–1651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Muirhead R. W., Collins R. P., Bremer P. J. 2005. Erosion and subsequent transport state of Escherichia coli from cowpats. Appl. Environ. Microbiol. 71:2875–2879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Okabe S., Okayama N., Savichtcheva O., Ito T. 2007. Quanitification of host-specific Bacteroides-Prevotella 16S rRNA genetic markers for assessment of fecal pollution in freshwater. Appl. Microbiol. Biotechnol. 74:890–901 [DOI] [PubMed] [Google Scholar]
- 29. Okabe S., Shimazu Y. 2007. Persistence of host-specific Bacteroides-Prevotella 16S rRNA genetic markers in environmental waters: effects of temperature and salinity. Appl. Microbiol. Biotechnol. 76:935–944 [DOI] [PubMed] [Google Scholar]
- 30. Pachepsky Y. A., et al. 2006. Transport and fate of manure-borne pathogens: modeling perspective. Agric. Water Manag. 86:81–92 [Google Scholar]
- 31. Saitou N., Nei M. 1987. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 4:406–425 [DOI] [PubMed] [Google Scholar]
- 32. Schloss P. D., et al. 2009. Introducing mothur: open source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 75:7537–7541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Seurinck S., Tom D., Willy V., Steven D. S. 2005. Detection and quantification of the human-specific HF183 Bacteroides 16S rRNA genetic marker with real-time PCR for assessment of human faecal pollution in freshwater. Environ. Microbiol. 7:249–259 [DOI] [PubMed] [Google Scholar]
- 34. Shanks O. C., et al. 2006. Basin-wide analysis of the dynamics of fecal contamination and fecal source identification in Tillamook Bay, Oregon. Appl. Environ. Microbiol. 72:5537–5546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Silkie S. S., Nelson K. L. 2009. Concentrations of host-specific and generic fecal markers measured by quantitative PCR in raw sewage and fresh animal feces. Water Res. 43:4860–4871 [DOI] [PubMed] [Google Scholar]
- 36. Stoeckel D. M., Harwood V. J. 2007. Performance, design, and analysis in microbial source tracking studies. Appl. Environ. Microbiol. 73:2405–2415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Tamura K., Dudley J., Nei M., Kumar S. 2007. MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol. Biol. Evol. 24:1596–1599 [DOI] [PubMed] [Google Scholar]
- 38. Thompson J. R., Marcelino L. A., Polz M. F. 2002. Heteroduplexes in mixed-template amplifications: formation, consequence and elimination by ‘reconditioning PCR’. Nucleic Acids Res. 30:2083–2088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Walters S. P., Field K. G. 2009. Survival and persistence of human and ruminant-specific faecal Bacteroidales in freshwater microcosms. Environ. Microbiol. 11:1410–1421 [DOI] [PubMed] [Google Scholar]
- 40. Walters S. P., Yamahara K. M., Boehm A. B. 2009. Persistence of nucleic acid markers of health-relevant organisms in seawater microcosms: implications for their use in assessing risk in recreational waters. Water Res. 43:4929–4939 [DOI] [PubMed] [Google Scholar]
- 41. Whitman R. L., Nevers M. B., Korinek G. C., Byappanahalli M. N. 2004. Solar and temporal effects on Escherichia coli concentration at a Lake Michigan swimming beach. Appl. Environ. Microbiol. 70:4276–4285 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.