

Abstract
Tuberous sclerosis is a human disease caused by mutations in the TSC1 or the TSC2 tumor suppressor gene. Previous studies of a Drosophila TSC2 homolog suggested a role for the TSC genes in maintaining DNA content, with loss of TSC2 leading to polyploidy and increased cell size. We have isolated mutations in the Drosophila homolog of the TSC1 gene. We show that TSC1 and TSC2 form a complex and function in a common pathway to control cellular growth. Unlike previous studies, our work shows that TSC1– or TSC2– cells are diploid. We find that, strikingly, the heterozygosity of TSC1 or TSC2 is sufficient to rescue the lethality of loss-of-function insulin receptor mutants. Further genetic analyses suggest that the TSC genes act in a parallel pathway that converges on the insulin pathway downstream from Akt. Taken together, our studies identified the TSC tumor suppressors as novel negative regulators of insulin signaling.
Keywords: Cell size, tumor suppressor, insulin signaling, Akt
During metazoan development, cell-intrinsic and cell-extrinsic factors must act coordinately to specify the characteristic size of diverse cell types (for review, see Su and O'Farrell 1998; Stocker and Hafen 2000). An intrinsic factor that contributes to cell size is DNA content, and cell size correlates with ploidy in various species (Su and O'Farrell 1998). On the other hand, it has long been appreciated that cell-extrinsic factors such as hormones, growth factors, and nutrition play important roles in growth control at the organismal level (for review, see Conlon and Raff 1999). Thus a challenge is to understand how cell-intrinsic and cell-extrinsic factors coordinately control cellular growth and how changes in such regulation lead to pathological conditions such as cancer.
One of the hormone-mediated pathways that plays a pivotal role in cellular growth involves insulin or insulin-like growth factors (IGF) (for review, see Proud and Denton 1997). Biochemical studies have shown that on binding of insulin or IGFs, insulin receptor (InR) or IGF receptors recruit phosphoinositide 3-kinase (PI3K) to the membrane, either directly or through insulin receptor substrates (IRS) as intermediates. Phosphorylation of the membrane lipid phosphatidylinositol 4,5-biphosphate (PIP2) by PI3K produces the second messenger phosphatidylinositol 3,4,5-triphosphate (PIP3), which activates Akt, a Ser/Thr kinase. Akt and another Ser/Thr kinase encoded by the TOR (target of rapamycin) gene ultimately control the phosphorylation of two downstream effectors, p70 S6 kinase (S6K) and 4E-binding protein (4E-BP), which are involved in translational control (for review, see Thomas and Hall 1997; Sonenberg and Gingras 1998; Schmelzle and Hall 2000). Phosphorylation of S6K increases its kinase activity toward the ribosome S6 subunit, leading to increased translation of 5′ terminal oligopyrimidine tract (TOP) mRNAs that largely encode components of the translational apparatus such as ribosomal proteins. Phosphorylation of 4E-BP releases the eukaryotic initiation factor 4E (eIF4E) from the inactive eIF4E/4E-BP complex, permitting eIF4E to function in translation initiation. A well-known negative regulator of this pathway is the PTEN tumor suppressor, which functions as a phosphatase to convert PIP3 to PIP2 (for review, see Cantley and Neel 1999). The importance of insulin signaling in growth control has been elegantly demonstrated by recent genetic studies from Drosophila. Loss-of-function mutations in Drosophila homologs of components of the insulin pathway, including InR, IRS, PI3K, Akt, TOR, and S6K, all result in decreased cell size, and overexpression of PI3K, Akt, and S6K, or loss of the negative regulator PTEN, result in increased cell size (Chen et al. 1996; Böhni et al. 1999; Goberdhan et al. 1999; Huang et al. 1999; Montagne et al. 1999; Verdu et al. 1999; Weinkove et al. 1999; Gao et al. 2000; Oldham et al. 2000; Zhang et al. 2000).
Tuberous sclerosis (TSC) is an autosomal dominant disorder that affects 1 in 6000 individuals (for review, see Young and Povey 1998). This disease is characterized by the widespread development of benign tumors termed harmatomas, which frequently lead to skin rashes, seizures, and mental retardation. TSC is caused by a mutation in either the TSC1 or TSC2 tumor suppressor gene. TSC2 encodes a putative GTPase-activating protein (GAP), and TSC1 encodes a novel protein containing two coiled-coil domains (The European Chromosome 16 Tuberous Sclerosis Consortium 1993; van Slegtenhorst et al. 1997). The TSC1 and TSC2 proteins have been shown to form a complex in mammalian cells (van Slegtenhorst et al. 1998; Nellist et al. 1999) and have been proposed to control various cellular functions including cell cycle (Soucek et al. 1997, 1998), endocytosis (Xiao et al. 1997), cell adhesion (Lamb et al. 2000), and transcription (Henry et al. 1998). However, it is not clear how these potential functions are used during normal development and how mutations of the TSC genes result in benign tumors.
Although animal models lacking TSC1 have not been reported until now, studies of animal models lacking the TSC2 gene have provided insight into the function of the TSC genes in development. The Eker rat strain contains a germline insertion mutation in the rat TSC2 gene, which causes a premature truncation of the TSC2 protein (Kobayashi et al. 1997; Rennebeck et al. 1998). Murine models lacking TSC2 have also been generated by gene targeting (Kobayashi et al. 1999; Onda et al. 1999). In these models, homozygous TSC2 mutants are embryonic lethal, whereas heterozygous carriers are prone to tumor formation. Recently, Drosophila homologs of TSC1 and TSC2 were reported (Ito and Rubin 1999). Ito and Rubin showed that cells lacking gigas, the Drosophila homolog of TSC2, are increased in cell size. They further suggested that the increased size of the gigas mutant cells is due to a failure to enter M phase at the end of imaginal disc development, resulting in polyploidy and, consequently, increased cell size (Ito and Rubin 1999). Given that such an increase in DNA content has not been reported in any TSC benign tumors in vertebrates, the described gigas mutant phenotype might potentially reflect the uniqueness of Drosophila development. In that aspect, studies of Drosophila mutants lacking the TSC1 gene could complement the analysis of the gigas mutants. For simplicity, TSC1 and TSC2 are used throughout the rest of the paper to refer to the Drosophila TSC1 and TSC2 homologs unless otherwise specified.
Here we report the first animal model lacking the TSC1 gene. We show that TSC1 and TSC2 form a complex in Drosophila cells and function in a common pathway to control cellular growth. We further show that the TSC genes act in a parallel pathway that converges on the insulin pathway downstream from Akt. Our studies identified the TSC tumor suppressors as novel negative regulators of insulin signaling.
Results
Isolation of TSC1 mutants
We used the FRT/FLP recombination system (Xu and Rubin 1993) to screen the Drosophila genome for mutations that affect cell size. Two lethal mutations were recovered on the right arm of the third chromosome that produced enlarged cells in mutant clones in the eye and wing. Both mutants die around embryo–larval transition without gross morphological defects. These mutations were localized to the 95D7–11; 95E8 region by deficiency mapping. This region contains a Drosophila homolog of the mammalian TSC1 gene (Ito and Rubin 1999). Given that mutations of the Drosophila TSC2 homolog gigas lead to a similar increase in cell size and that mutations in TSC1 or TSC2 result in the same disease in humans, we investigated whether our mutations at 95D-E disrupted the Drosophila TSC1 homolog. DNA sequencing analysis revealed point mutations in both alleles (Fig. 1B). TSC112 has a single-base deletion that causes a frameshift, truncating the protein at amino acid 896. This mutation is predicted to delete part of the coiled-coil domains of the TSC1 protein, which have been implicated in binding to TSC2 (van Slegtenhorst et al. 1998). Another allele, TSC129, contains a single-base nonsense mutation (C to T) that replaces amino acid 61 (Gln) with a stop codon. This mutation is predicted to be a null allele because it truncates most of the protein. A 4.7-kb construct containing only the TSC1 genomic DNA was able to fully rescue TSC112and TSC129 homozygotes to viable and phenotypically wild-type animals, confirming that the two mutations indeed disrupted TSC1 (Fig. 1). TSC112 and TSC129 are indistinguishable in their cell-size phenotype. All the subsequent analyses were performed by using the TSC129 allele.
Figure 1.

Structure of the TSC1 locus and predicted structure of the TSC proteins. (A) Genomic structure of the TSC1 locus. The TSC1 gene is located between CG6129 and sec10 on 3R. The exons are indicated with thick lines and the introns are shown as thin lines connecting adjacent exons. The transcription start site is indicated with a single line with an arrow at the end. The translation start site is indicated with double lines with an arrow at the end. The genomic DNA fragment used in the rescue construct is indicated. (B) Schematic structures of the Drosophila TSC1 and TSC2 proteins. TSC1 is a predicted 1100 aa polypeptide containing two coiled-coil domains (CC1 and CC2). The molecular lesions in TSC112 and TSC129 are indicated. TSC2 is a predicted 1847 aa polypeptide containing a GAP domain.
TSC1 autonomously controls cell and organ size
Scanning electron microscopy revealed that the unit eyes (ommatidia) in TSC1 mutant clones are larger than the surrounding wild-type ommatidia (Fig. 2A). Eye sections show that the rhabdomeres of mutant photoreceptors are approximately 1.8 times the size of neighboring wild-type counterparts in area (n = 48). However, the organization of ommatidia and the differentiation of various cell types are nearly normal, with occasional loss or gain of photoreceptor cells (Fig. 2B). Careful examination of mosaic ommatidia consisting of genetically marked TSC1 mutant and nonmutant cells revealed that the effect of TSC1 mutation on cell size is strictly cell autonomous (Fig. 2B). A similar autonomous effect on cell size is also observed in the wing (Fig. 2C). Therefore, TSC1 autonomously controls cell size. To examine whether TSC1 regulates organ size, we selectively removed TSC1 function in the eye imaginal disc by using the eyeless-FLP technique (Newsome et al. 2000). This technique allowed us to consistently generate eye discs in which >90% of the cells are mutant for TSC1. Such eye discs are 2.9 times the size of wild-type discs in area (n = 19; Fig. 2D,E). Thus, TSC1 autonomously controls organ size.
Figure 2.

TSC1 autonomously controls cell and organ size. (A) Scanning electron micrograph of a compound eye carrying a clone of homozygous TSC129 cells. The mutant clone occupies the upper half of the eye in the picture. Note that the TSC1 mutant ommatidia are larger than their wild-type counterparts. (B) Section through a TSC129 clone in the adult eye. The mutant clone is marked by the absence of pigment. The rhabdomeres of mutant photoreceptor cells are increased in area by about 80% compared with heterozygous photoreceptor cells (measured by Axiovision software). At the clone border, mosaic ommatidia containing normal-sized heterozygous cells (arrowhead) and enlarged homozygous TSC1 mutant cells (arrow) can be seen, indicating that TSC1 controls cell size autonomously. (C) Wing-margin bristles containing a TSC129 mutant clone. Note that the TSC1 mutant bristles (marked by y-, indicated by a line above the wing margin) are thicker and longer than the wild-type bristles, which have a dark color. (D–E) Images of a wild-type eye-antenna disc (D) and an eye-antenna disc in which TSC1 function was selectively removed in the eye by using the ey-Flp technique (E). The discs were also stained for glass-lacZ that is expressed in retinal cells. These images were taken under the same magnification. Note that the TSC1– eye disc is much larger than the wild-type disc.
TSC1 controls cellular growth and proliferation during imaginal disc development
To examine when the cell size change occurs in TSC1 mutant cells, we examined mutant clones in the imaginal discs. Larger nuclei and increased cell size in the mutant clones were evident in the third-instar imaginal discs (Fig. 3A–F), suggesting that cell size change occurred during larval development. In addition, TSC1 mutant (-/-) clones contained more cells than did their +/+ twin spots (for example, see Fig 3A–C). To directly examine the cell proliferation defects of TSC1− cells, we induced clones at 26 h, 50 h, and 74 h after egg deposition (AED) and quantitated cell numbers in the mutant clones and the twin spots at 119 h AED. The average ratio of cell numbers in TSC1 mutant clones versus the twin spots was 3.1, 1.6, and 1.2 for clones induced at 26 h, 50 h, and 74 h AED, respectively (n > 40). Because the mutant clones and twin spots originate from mitotic sister cells born at the same developmental time, these results suggest that lack of TSC1 affects cell proliferation, not just cell size. However, we cannot formally exclude the possibility that the increased cell number in the mutant clones is caused by decreased cell death of the mutant cells.
Figure 3.

TSC1 controls cellular growth and proliferation during imaginal disc development. In all panels, TSC1 mutant clones were generated by FRT/FLP and marked by the absence of Ubi-GFP signal (green). (A–C) Confocal images of a third-instar eye disc containing a large TSC1− clone (arrow). The adjacent area of brighter green staining represents a +/ + twin spot (arrowhead). The disc was stained for the neuronal specific nuclear Elav protein (red). Three images are shown, one of GFP (A), one of Elav staining (B), and one of superimposed GFP and Elav staining (C). Note the increased size of the mutant cell nuclei and the dramatically increased area of the mutant clone as compared with its twin spot. (D–F) Confocal images of a portion of a third-instar eye disc containing TSC1 mutant clones (arrows). The disc was stained with phalloidin (red), which highlights the outlines of the cells. Three images are shown, one of GFP (D), one of phalloidin staining (E), and one of superimposed GFP and phalloidin staining (F). Note the increased size of the mutant cells. (G–H) Confocal images of a portion of a third-instar wing disc containing a TSC1 mutant clone (arrow). The disc was stained with fibrillarin (red), which labels the nucleolus of the cells. Three images are shown, one of GFP (G), one of fibrillarin staining (H), and one of superimposed GFP and fibrillarin staining (H). Note the increased nucleolar size of the TSC1– cells.
The nucleolus is the major site of ribosome assembly within the cell, and its size has been shown to correlate with protein synthesis and proliferation rate (Derenzini et al. 1998). We used an antibody against the nucleolar protein fibrillarin (Aris and Blobel 1988) to examine nucleolar size in TSC1− cells. We found that the nucleolar area of TSC1− cells in the wing imaginal disc was 2.1 times (n = 70) that of neighboring wild-type cells (Fig. 3G–I), consistent with a role for TSC1 in cellular growth and proliferation.
Loss of TSC genes does not increase ploidy
Studies of the Drosophila TSC2 gene suggested that TSC2− cells undergo endoreplication, resulting in polyploidy and consequently increased cell size (Ito and Rubin 1999). To address whether a similar mechanism is responsible for the increased size of the TSC1− cells, we examined DNA content in TSC1− cells by using several different techniques: quantitative measurement of DNA dye Hoechst 33342 or propidium iodide staining in intact imaginal discs with confocal or fluorescent light microscopy, and FACS analysis of dissociated imaginal disc cells. We found that, surprisingly, TSC1− cells were diploid in DNA content (Fig. 4A,B). Although TSC1 mutant nuclei are larger than their wild-type siblings, the relative intensity of DNA staining is weaker in the mutant cells, and the total fluorescence in the nuclei is comparable between the mutant and the wild-type cells (Fig. 4A,B). This observation prompted us to reexamine the DNA content of TSC2− cells. Similar to the results from the TSC1− cells, we observed total fluorescence of DNA dye staining in the TSC2− cells that was comparable with the wild-type cells (Fig. 4C,D). Consistent with microscopic measurement of DNA dye staining, FACS analysis of dissociated imaginal disc cells revealed similar DNA content in TSC1− or TSC2− cells as compared with the wild-type cells (Fig. 4E,F). These results suggest that loss of the TSC genes does not increase ploidy, and thus the increased size of TSC1− or TSC2− cells is not due to increased DNA content.
Figure 4.

TSC1− and TSC2− cells are diploid. In A–D, mutant clones were generated by FRT/FLP and marked by the absence of Ubi-GFP signal (green). (A–B) Confocal images of a portion of a third-instar eye disc containing two TSC1− clones (arrows). The disc was stained with DNA dye propidium iodide (PI, shown in red). Two images are shown, one of GFP (A) and one of PI staining (B). Note that the intensity of PI staining is weaker in the mutant cells. Quantitation of Z series by using LSM 510 software showed that the total fluorescence in the nuclei is comparable between the mutant and the wild-type cell (data not shown). (C–D) Fluorescent light microscopy images of a portion of a third-instar eye disc containing a TSC2 (gigas) mutant clone (arrow). The disc was stained with DNA dye Hoechst 33342 (blue). Note that the intensity of Hoechst 33342 staining is weaker in the mutant cells. Quantitation by using the Axiovision software showed that the total fluorescence in the nuclei is comparable between the mutant and the wild-type cell (data not shown). (E–F) Flow cytometric analysis of dissociated wing imaginal discs containing TSC1− (E) and TSC2− (F) clones. The profiles of mutant and wild-type (WT) cells are indicated by heavy and light traces, respectively. No signal was detected beyond the DNA content of cycling diploid cells (data not shown). Note that the DNA content of TSC1− or TSC2− cells is similar to that of wild-type cells.
TSC1 and TSC2 function in a common signaling pathway and form a stable complex in Drosophila cells
Based on their identical disease phenotypes, the human TSC1 and TSC2 genes have been proposed to function in a common signaling pathway. The Drosophila TSC1 and TSC2 mutants provided us with an opportunity to address this issue genetically. If TSC1 and TSC2 act in a common pathway, we might expect TSC1 TSC2 double-mutant cells to have an identical phenotype to that of the TSC1 or TSC2 single mutant. Indeed, we observed that TSC1 TSC2 double-mutant cells show a cell size increase that is identical to that of TSC1 or TSC2 single-mutant cells (Fig. 5A–C), suggesting that these genes act in a common pathway that controls cellular growth. Human TSC1 and TSC2 proteins can associate with each other (van Slegtenhorst et al. 1998; Nellist et al. 1999). Given the identical phenotype of TSC1−, TSC2−, and TSC1− TSC2− cells, we suspected that Drosophila TSC1 and TSC2 proteins might also associate with each other. To investigate this possibility, we performed coimmunoprecipitation assays in Drosophila S2 cells that expressed TSC1 and TSC2 proteins tagged with Myc and V5 epitopes, respectively (Fig. 5D). These experiments revealed a specific interaction between TSC1 and TSC2 (Fig. 5E). Furthermore, we mapped the interaction domain to the N-terminal half of TSC2 (Fig. 5E), consistent with studies of the human TSC2 protein (van Slegtenhorst et al. 1998). Taken together, these results suggest that TSC1 and TSC2 function in a protein complex to regulate cellular growth in Drosophila.
Figure 5.

Interactions between TSC1 and TSC2. (A–C) Wing margins containing TSC1− (A), TSC2− (B), and TSC1− TSC2− (C) clones, respectively. Note that TSC single- or double-mutant bristles (marked by y−, indicated by a line above the wing margin) are similar in size. (D) Schematic representation of epitope-tagged TSC1 and TSC2 proteins that were expressed in S2 cells. The TSC1 construct corresponds to the full-length protein and migrates as an ∼150-kD protein. Three TSC2 constructs were generated, including the full-length (TSC2, >200 kD), N-terminal half (TSC2N, ∼130 kD), and C-terminal half (TSC2C, ∼130 kD). (E) S2 cells expressing TSC1/TSC2, TSC1/TSC2N, and TSC1/TSC2C constructs were lysed and total cell lysate was immunoprecipitated (IP) with α-MYC and immunoblotted with α-V5 (left panel). TSC2 or TSC2N, but not TSC2C, can be immunoprecipitated with TSC1. The same blot was stripped and reprobed with α-MYC to show that TSC1 was expressed at comparable levels in all lanes (middle panel). As an additional control, TSC2, TSC2N, and TSC2C were precipitated from the same cell lysate by using Ni-NTA agarose (α-His) and probed with α-V5. These proteins were expressed at comparable levels (right panel). (F–H) Wings expressing MS1096/UAS-TSC1 (F), MS1096/UAS-TSC2 (G), and MS1096/UAS-TSC1; UAS-TSC2, respectively. These images were taken under the same magnification. Wings overexpressing both TSC1 and TSC2 are ∼28% the size of wings expressing TSC1 or TSC2 alone.
Coexpression of Drosophila TSC1 and TSC2, but not either gene alone, reduces cell and organ size
We used the GAL4-UAS system (Brand and Perrimon 1993) to assess the effect of overexpressing TSC1 and TSC2 on cellular growth. Drosophila TSC1, TSC2, or a combination of both gene products, was expressed from UAS transgenes under the control of the GAL4 driver line MS1096, which expresses high levels of GAL4 near uniformly in the wing-pouch region (Capdevila and Guerrero 1994). Although overexpression of TSC1 or TSC2 alone did not result in any visible phenotype, coexpression of both genes resulted in a dramatic reduction in wing size (Fig. 5F–H). Quantitation of wing size and wing-hair density revealed a 72% reduction in wing size and a 65% reduction in cell size (n = 20). Thus, most of the wing-size reduction can be accounted for by the reduction in cell size. We suggest that TSC1 and TSC2 function as a complex in vivo and are present at rate-limiting concentrations as a protein complex. Thus only simultaneous overexpression of both proteins, but not either protein alone, can increase the concentration of the TSC1–TSC2 protein complex, which in turn results in cell growth suppression that is opposite of the loss-of-function phenotype.
TSC genes act in a parallel pathway that converges on the insulin pathway downstream from Akt
The insulin pathway is known to play a central role in the control of cellular growth in Drosophila (for review, see Stocker and Hafen 2000). The loss-of-function and gain-of-function phenotypes of the TSC genes are strikingly similar to those of PTEN, a negative regulator of the InR-PI3K-Akt pathway (Goberdhan et al. 1999; Huang et al. 1999; Gao et al. 2000). This prompted us to investigate the relationship between the TSC genes and components of the insulin pathway. Previous studies have shown that loss of inr or Akt leads to decreased cell size. To investigate the relationship between inr, Akt, and the TSC genes, we examined TSC1 Akt and TSC1 inr double-mutant clones. Cells homozygous for a strong allele of inr (inr35; Fernandez et al. 1995), or a null allele of Akt (Akt1q; Staveley et al. 1998) are smaller, and are rarely recovered in adult eye clones because of cell competition during development (Fig. 6A,B; also see Verdu et al. 1999). However, TSC1 inr35or TSC1 Akt1q double-mutant cells showed a similar cell size increase as that observed in TSC1− cells (Fig. 6A–E). Furthermore, the competitive disadvantage of inr and Akt mutant cells was also rescued in the TSC1 inr35 or TSC1 Akt1q double-mutant clones, resulting in larger clones that contained more cells (Fig. 6A–D and data not shown). This result suggests that TSC1 acts genetically downstream from Akt. This observation is compatible with either TSC1 acting molecularly downstream from Akt in the linear InR-PI3K-Akt pathway, or TSC1 acting in a parallel pathway that converges on the insulin pathway downstream from Akt.
Figure 6.

The TSC genes act in a parallel pathway that converges on the insulin pathway downstream from Akt. Eye sections of various mutant clones. The genotype of the mutant clone is labeled below each section, as well as the relative size of the mutant rhabdomeres as compared with the wild-type counterparts (X value). At least 50 ommatidia were measured in each genotype. Mutant clones were marked by the absence of pigment, and the borders of mutant clones were outlined with red lines. Although inr− or akt− clones contain few cells, TSC1− inr−, or TSC1− akt− clones contain many more cells (only portions of the mutant clones are shown in C and D). (A) inr−. (B) akt−. (C) TSC1− inr−. (D) TSC1− akt−. (E) TSC1−. (F) PTEN−. (G) TSC1− PTEN−.
To distinguish between these two possibilities, we generated cells that are doubly mutant for null alleles of PTEN and TSC1. PTEN is a negative regulator of the InR-PI3K-Akt pathway, and loss of PTEN results in increased Akt activity and cellular growth. We reasoned that if TSC1 acts downstream from Akt within the InR-PI3K-Akt pathway, we might expect PTEN TSC1 double-mutant cells to show a similar cell-size phenotype to either single mutant. However, if TSC1 acts parallel to the InR-PI3K-Akt the pathway, we might expect PTEN TSC1 double-mutant cells to show additive effects on cell size as compared with each single mutant. We observed that PTEN TSC1 double-mutant photoreceptors are 2.9 times the size of wild-type cells, as compared with 1.9 for PTEN− and 1.8 for TSC1− (n = 50). This result strongly suggests that the TSC genes function in a parallel pathway that converges on the insulin pathway at a point downstream from Akt.
Heterozygosity of TSC1 or TSC2 rescues the lethality of loss-of-function InR mutants
In the course of our studies, we observed a striking genetic interaction between the TSC genes and inr mutations. Flies homozygous for inr35, a strong loss-of-function allele, are larval lethal (Fernandez et al. 1995). However, homozygous inr35 flies that are heterozygous for TSC1 can survive to adults (Table 1), suggesting that a mere 50% reduction in the dosage of the TSC1 gene can rescue the developmental arrest of an inr mutant.
Table 1.
Heterozygosity of TSC1 or TSC2 rescues the lethality of loss-of-function insulin-receptor mutants
| Genotype
|
Animals surviving to pupal stage (%)
|
Animals surviving to adult stage (%)
|
|
|---|---|---|---|
|
0 | 0 | |
|
56 | 6.6 | |
|
ND | 0 | |
|
ND | 39 |
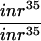
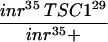
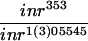
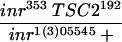
In the cross between inr35 TSC129 and inr35, 191 flies were scored.
In the cross between inr353 TSC2192 and inr1(3)05545, 110 flies were scored.
(ND) Not determined.
Similarly, heterozygosity of TSC2 is sufficient to rescue the lethality of another inr mutant (Table 1). Flies carrying the allelic combination inr353/inrl(3)05545 are 100% lethal (Fernandez et al. 1995). However, approximately 39% of inr353/inrl(3)05545 flies that are heterozygous for TSC2 can survive to adults (Table 1). Taken together, these results provide convincing in vivo evidence that the TSC genes are negative regulators of insulin signaling in development.
Discussion
The mechanisms of how body and organ size are regulated are largely unknown (for review, see Conlon and Raff 1999). Recent genetic studies in Drosophila suggest that the insulin pathway may coordinately control both cell growth and cell proliferation and in turn regulate organ size. Here, we have provided evidence that the TSC tumor suppressor genes also play an essential role in the control of cell size and organ size. Although an increase in cell size has been observed in human tumors carrying TSC1 or TSC2 mutations (for review, see Young and Povey 1998), the underlying mechanisms are not clear. Our results suggest that the TSC genes and the insulin pathway act antagonistically in the control of cellular growth. Loss of TSC genes resulted in a cell-size phenotype that is almost identical to that of PTEN. Similarly, co-overexpression of the TSC genes reduces cell size as PTEN overexpression. The results from our double-mutant analyses suggest that the TSC genes act in a pathway parallel to the insulin pathway, and the TSC pathway converges on the insulin pathway downstream from Akt. Several proteins are known to function directly downstream from Akt, or to converge on the insulin pathway downstream from Akt. These include S6K (Thomas and Hall 1997), 4E-BP (Sonenberg and Gingras 1998), and Ser/Thr kinase TOR (Schmelzle and Hall 2000). Our genetic analysis is consistent with the TSC pathway regulating any of these candidate proteins. Alternatively, the TSC genes may regulate unknown regulators of cell growth. We cannot distinguish between these models at present.
Our most convincing evidence for a functional link between the TSC genes and insulin signaling came from the observation that heterozygosity of TSC1 or TSC2 is sufficient to rescue the lethality of loss-of-function InR mutants. This argues that the TSC genes are intimately linked to insulin signaling, rather than functioning in a totally independent cell-growth pathway. These results suggest that the TSC tumor suppressor genes are novel negative regulators of insulin signaling, and modulating the activities of the TSC genes might provide a potential way to correct insulin signaling defects in certain diseases such as diabetes and obesity.
An important challenge in the future is to understand the molecular mechanism by which the TSC tumor suppressors regulate the insulin pathway. The predicted structures of the TSC1 (containing coiled-coil domains) and the TSC2 protein (containing a GAP domain) offer clues in that regard. The TSC2 protein has been shown to possess GAP activity toward two small GTPases, Rab5 and Rap1 (Wienecke et al. 1995; Xiao et al. 1997). Rab5 is a rate-limiting component of the endocytic pathway (Bucci et al. 1992). It has been speculated that loss of the TSC tumor suppressors could lead to missorting of internalized growth factor receptors or other signal-mediated membrane-bound molecules that would otherwise undergo lysosomal degradation, thus leading to a constitutive activation of certain growth-promoting pathways (Xiao et al. 1997). The in vivo function of Rap1 is largely unknown, and Rap1 can function both as positive and negative regulator of cell proliferation under different conditions (Kitayama et al. 1989; Yoshida et al. 1992). Further studies of the Drosophila TSC genes may provide insights into the relative importance of Rab5GAP and Rap1GAP activities in growth suppression.
Materials and methods
Molecular biology
Genomic DNA was isolated from the TSC1 mutant embryos and amplified with PCR. The PCR products were directly sequenced by using primers spanning the TSC1 locus. This analysis revealed point mutations in the TSC112 and the TSC129 allele (Fig. 1B). A 4.7-kb genomic fragment containing just the TSC1 transcription unit was cloned into Casperhs-1, a modified Casperhs vector (Pan and Rubin 1997), for the rescue experiment.
A full-length TSC1 cDNA clone, LD24327, was obtained from Research Genetics, and used to generate UAS-TSC1 construct.
Myc-tagged TSC1 and V5/His-tagged TSC2 constructs were made by using pAc5.1/V5-HisB vector (Invitrogen). Sequences encoding the N-terminal Myc epitope (MEQKLISEEDLNE) was added by PCR in place of the first Met codon of a full-length TSC1 cDNA.
Drosophila strains
TSC2192 (gig192), a null TSC2 allele, and UAS-TSC2 flies were kindly provided by Naoto Ito (Ito and Rubin 1999). inr35and inr 353 (strong alleles) were gifts from Manfred Frasch (Fernandez et al. 1995). Aktq , a kinase-dead null allele of Akt, was kindly provided by Armen Manoukian (Staveley et al. 1998). PTENdj189 is a null allele (Gao et al. 2000). Genotypes for generating clones are as follows.
TSC1 mutant clones:
y w hsp-flp; TSC129 FRT82B/FRT82B, w + for adult eyes y w hsp-flp; TSC129 FRT82B/FRT82B, y + for adult wings yw hsp-flp; TSC129 FRT82B/FRT82B, Ubi-GFP + for imaginal discs
ey-Flp TSC1 :
y w ey-flp glass-lacZ; TSC129 FRT82B/FRT82B, w+cl3R3
TSC2 mutant clones:
yw hsp-flp; TSC2192 FRT80B/FRT80B, Ubi-GFP + for imaginal discs
TSC1 TSC2 double-mutant clones (marked by y-):
y w hsp-flp; TSC2192 FRT80B TSC129/y + P[TSC1 +] FRT80B TSC129 for adult wings
TSC1 Akt double-mutant clones (marked by w-):
y w hsp-flp; FRT82B Aktq TSC129/FRT82B w + for adult eyes
TSC1 inr double-mutant clones (marked by w-):
y w hsp-flp; FRT82B TSC129 inr35/FRT82B w + for adult eyes
PTEN mutant clones (marked by w-):
y w hsp-flp; PTENdj189 FRT40A/FRT40A, y +w + for adults
TSC1 PTEN double-mutant clones (marked by w-):
y w hsp-flp; PTENdj189/PTENdj189; FRT82B TSC129/FRT82B w + P[PTEN +] for adult eyes
Cell transfection and immunoprecipitation
S2 cells are transfected by using the Calcium Phosphate method (Invitrogen). Cells were lysed in cold immunoprecipitation buffer (1% Triton X-100, 0.5% NP-40, 150mM NaCl, 10mM Tris at pH 7.4, 1mM EDTA, 1mM EGTA, 0.2mM PMSF). Immunoprecipitation was performed by using anti-Myc antibody (Santa Cruz Biotechnology) and protein G Sepharose according to manufacturer's instruction.
Flow cytometry
FACS analysis of dissociated imaginal wing disc cells was performed as previously described (Neufeld et al. 1998). TSC1 or TSC2 homozygous mutant clones induced by FLP/FRT-mediated recombination were marked by their lack of GFP expression by using ubiquitin-GFP FRT chromosomes (donated to the Bloomington stock center by Bruce Edgar). FACS sorting was performed on a FACStar machine and analyzed with CellQuest program.
Histology and microscopy
Clones of mutant cells were generated by FLP/FRT-mediated mitotic recombination (Xu and Rubin 1993). Immunostaining of imaginal discs was performed as described by Xu and Rubin (1993). Antifibrillarin antibody was a gift of John Aris (Aris and Blobel 1988). Confocal images were collected by using a Zeiss LSM 510 confocal microscope and analyzed by using LSM 510 software. Light-microscopy images were acquired on a Zeiss Axioplan microscope equipped with an AxioCam digital camera and analyzed by using the Axiovision software.
Acknowledgments
We thank Elizabeth Chen, Jin Jiang, and Keith Wharton for critical reading of the manuscript. We also thank the Bloomington Stock Center, Developmental Studies Hybridoma Bank, Manfred Frasch, Naoto Ito, and John Aris for fly stocks and reagents, and the UT Southwestern Molecular and Cellular Imaging Facility for assistance with eye sections. D.J.P. is Virginia Murchison Linthicum Endowed Scholar in Medical Science, and is supported by NIH (GM62323) and AHA (0130222N).
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL dpan@mednet.swmed.edu; FAX (214) 648-8885.
Article and publication are at www.genesdev.org/cgi/doi/10.1101/gad.901101.
References
- Aris J P, Blobel G. Identification and characterization of a yeast nucleolar protein that is similar to a rat liver nucleolar protein. J Cell Biol. 1988;107:17–31. doi: 10.1083/jcb.107.1.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Böhni R, Riesgo-Escovar J, Oldham S, Brogiolo W, Stocker H, Andruss BF, Beckingham K, Hafen E. Autonomous control of cell and organ size by CHICO, a Drosophila homolog of vertebrate IRS1–4. Cell. 1999;97:865–875. doi: 10.1016/s0092-8674(00)80799-0. [DOI] [PubMed] [Google Scholar]
- Brand AH, Perrimon N. Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development. 1993;118:401–415. doi: 10.1242/dev.118.2.401. [DOI] [PubMed] [Google Scholar]
- Bucci C, Parton RG, Mather IH, Stunnenberg H, Simons K, Hoflack B, Zerial M. The small GTPase rab5 functions as a regulatory factor in the early endocytic pathway. Cell. 1992;70:715–728. doi: 10.1016/0092-8674(92)90306-w. [DOI] [PubMed] [Google Scholar]
- Cantley LC, Neel BG. New insights into tumor suppression: PTEN suppresses tumor formation by restraining the phosphoinositide 3-kinase / AKT pathway. Proc Natl Acad Sci. 1999;96:4240–4245. doi: 10.1073/pnas.96.8.4240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capdevila J, Guerrero I. Targeted expression of the signaling molecule decapentaplegic induces pattern duplications and growth alterations in Drosophila wings. EMBO J. 1994;13:4459–4468. doi: 10.1002/j.1460-2075.1994.tb06768.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, Jack J, Garofalo RS. The Drosophila insulin receptor is required for normal growth. Endocrinology. 1996;137:846–856. doi: 10.1210/endo.137.3.8603594. [DOI] [PubMed] [Google Scholar]
- Conlon I, Raff M. Size control in animal development. Cell. 1999;96:235–244. doi: 10.1016/s0092-8674(00)80563-2. [DOI] [PubMed] [Google Scholar]
- Derenzini M, Trere D, Pession A, Montanaro L, Sirri V, Ochs RL. Nuclear function and size in cancer cells. Am J Pathol. 1998;152:1291–1297. [PMC free article] [PubMed] [Google Scholar]
- The European Chromosome 16 Tuberous Sclerosis Consortium. Identification and characterization of the tuberous sclerosis gene on chromosome 16. Cell. 1993;75:1305–1315. doi: 10.1016/0092-8674(93)90618-z. [DOI] [PubMed] [Google Scholar]
- Fernandez R, Tabarini D, Azpiazu N, Frasch M, Schlessinger J. The Drosophila insulin receptor homolog: A gene essential for embryonic development encodes two receptor isoforms with different signaling potential. EMBO J. 1995;14:3373–3384. doi: 10.1002/j.1460-2075.1995.tb07343.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao X, Neufeld TP, Pan D. Drosophila PTEN regulates cell growth and proliferation through PI3K-dependent and -independent pathways. Dev Biol. 2000;221:404–418. doi: 10.1006/dbio.2000.9680. [DOI] [PubMed] [Google Scholar]
- Goberdhan DCI, Paricio N, Goodman EC, Mlodzik M, Wilson C. Drosophila tumor suppressor PTEN controls cell size and number by antagonizing the Chico / PI3-kinase signaling pathway. Genes & Dev. 1999;13:3244–3258. doi: 10.1101/gad.13.24.3244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henry KW, Yuan X, Koszewski NJ, Onda H, Kwiatkowski DJ, Nordstrom W. Tuberous sclerosis gene 2 product modulates transcription mediated by steroid hormone receptor family members. J Biol Chem. 1998;273:20535–20539. doi: 10.1074/jbc.273.32.20535. [DOI] [PubMed] [Google Scholar]
- Huang H, Potter CJ, Tao W, Li D-M, Brogiolo W, Hafen E, Sun H, Xu T. PTEN affects cell size, cell proliferation and apoptosis during Drosophila eye development. Development. 1999;126:5365–5372. doi: 10.1242/dev.126.23.5365. [DOI] [PubMed] [Google Scholar]
- Ito N, Rubin GM. gigas, a Drosophila homolog of tuberous sclerosis gene product-2, regulates the cell cycle. Cell. 1999;96:529–539. doi: 10.1016/s0092-8674(00)80657-1. [DOI] [PubMed] [Google Scholar]
- Kitayama H, Sugimoto Y, Matsuzaki T, Ikawa Y, Noda M. A ras-related gene with transformation suppressor activity. Cell. 1989;56:77–84. doi: 10.1016/0092-8674(89)90985-9. [DOI] [PubMed] [Google Scholar]
- Kobayashi T, Minowa O, Kuno J, Mitani H, Hino O, Noda T. Renal carcinogenesis, hepatic hemangiomatosis, and embryonic lethality caused by a germ-line Tsc2 mutation in mice. Cancer Res. 1999;59:1206–1211. [PubMed] [Google Scholar]
- Kobayashi T, Mitani H, Takahashi R-I, Hirabayashi M, Ueda M, Tamura H, Hino O. Transgenic rescue from embryonic lethality and renal carcinogenesis in the Eker rat model by introduction of a wild-type Tsc2 gene. Proc Natl Acad Sci. 1997;94:3990–3993. doi: 10.1073/pnas.94.8.3990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamb RS, Roy C, Diefenbach TJ, Vinters HV, Johnson MW, Jay DG, Hall A. The TSC1 tumor suppressor hamartin regulates cell adhesion through ERM proteins and the GTPase Rho. Nature Cell Biol. 2000;2:281–287. doi: 10.1038/35010550. [DOI] [PubMed] [Google Scholar]
- Montagne J, Stewart MJ, Stocker H, Hafen E, Kozma SC, Thomas G. Drosophila S6 kinase: A regulator of cell size. Science. 1999;285:2126–2129. doi: 10.1126/science.285.5436.2126. [DOI] [PubMed] [Google Scholar]
- Nellist M, van Slegtenhorst M, Goedbloed M, van den Ouweland A, Halley D, van der Sluijs P. Characterization of the cytosolic tuberin-hamartin complex. Tuberin is a cytosolic chaperone for hamartin. J Biol Chem. 1999;274:35647–35652. doi: 10.1074/jbc.274.50.35647. [DOI] [PubMed] [Google Scholar]
- Neufeld TP, de la Cruz AFA, Johnston LA, Edgar BA. Coordination of growth and cell division in the Drosophila wing. Cell. 1998;93:1183–1193. doi: 10.1016/s0092-8674(00)81462-2. [DOI] [PubMed] [Google Scholar]
- Newsome TP, Asling B, Dickson BJ. Analysis of Drosophila photoreceptor axon guidance in eye-specific mosaics. Development. 2000;127:851–860. doi: 10.1242/dev.127.4.851. [DOI] [PubMed] [Google Scholar]
- Oldham S, Montagne J, Radimerski T, Thomas G, Hafen E. Genetic and biochemical characterization of dTOR, the Drosophila homolog of the target of rapamycin. Genes & Dev. 2000;14:2689–2694. doi: 10.1101/gad.845700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onda H, Lueck A, Marks PW, Warren HB, Kwiatkowski DJ. Tsc2 +/- mice develop tumors in multiple sites that express gelsolin and are influenced by genetic background. J Clin Invest. 1999;104:687–695. doi: 10.1172/JCI7319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan D, Rubin GM. Kuzbanian controls proteolytic processing of Notch and mediates lateral inhibition during Drosophila and vertebrate neurogenesis. Cell. 1997;90:271–280. doi: 10.1016/s0092-8674(00)80335-9. [DOI] [PubMed] [Google Scholar]
- Proud CG, Denton RM. Molecular mechanisms for the control of translation by insulin. Biochem J. 1997;328:329–341. doi: 10.1042/bj3280329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rennebeck G, Kleymenova EV, Anderson R, Yeung RS, Artzt K, Walker CL. Loss of function of the tuberous sclerosis 2 tumor suppressor gene results in embryonic lethality characterized by disrupted neuroepithelial growth and development. Proc Natl Acad Sci. 1998;95:15629–15634. doi: 10.1073/pnas.95.26.15629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmelzle T, Hall MN. TOR, a central Controller of cell growth. Cell. 2000;103:253–262. doi: 10.1016/s0092-8674(00)00117-3. [DOI] [PubMed] [Google Scholar]
- Sonenberg N, Gingras A-C. The mRNA 5′ cap-binding protein elF4E and control of cell growth. Curr Opin Cell Biol. 1998;10:268–275. doi: 10.1016/s0955-0674(98)80150-6. [DOI] [PubMed] [Google Scholar]
- Soucek T, Pusch O, Wienecke R, DeClue JE, Hengstschläger M. Role of the tuberous sclerosis gene-2 product in cell cycle control. Loss of the tuberous sclerosis gene-2 induces quiescent cells to enter S phase. J Biol Chem. 1997;272:29301–29308. doi: 10.1074/jbc.272.46.29301. [DOI] [PubMed] [Google Scholar]
- Soucek T, Yeung RS, Hengstschläger M. Inactivation of the cyclin-dependent kinase inhibitor p27 upon loss of the tuberous sclerosis complex gene-2. Proc Natl Acad Sci. 1998;95:15653–15658. doi: 10.1073/pnas.95.26.15653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staveley BE, Ruel L, Jin J, Stambolic V, Mastronardi FG, Heitzler P, Woodgett JR, Manoukian AS. Genetic analysis of protein kinase B (AKT) in Drosophila. Curr Biol. 1998;8:599–602. doi: 10.1016/s0960-9822(98)70231-3. [DOI] [PubMed] [Google Scholar]
- Stocker H, Hafen E. Genetic control of cell size. Curr Opin Genet Dev. 2000;10:529–535. doi: 10.1016/s0959-437x(00)00123-4. [DOI] [PubMed] [Google Scholar]
- Su TT, O'Farrell PH. Size control: Cell proliferation does not equal growth. Curr Biol. 1998;8:R687–R689. doi: 10.1016/s0960-9822(98)70436-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas G, Hall MN. TOR signalling and control of cell growth. Curr Opin Cell Biol. 1997;9:782–787. doi: 10.1016/s0955-0674(97)80078-6. [DOI] [PubMed] [Google Scholar]
- van Slegtenhorst M, de Hoogt R, Hermans C, Nellist M, Janssen B, Verhoef S, Lindhout D, van den Ouweland A, Halley D, Young J, et al. Identification of the tuberous sclerosis gene TSC1 on chromosome 9q34. Science. 1997;277:805–808. doi: 10.1126/science.277.5327.805. [DOI] [PubMed] [Google Scholar]
- van Slegtenhorst M, Nellist M, Nagelkerken B, Cheadle JP, Snell RG, van den Ouweland A, Reuser A, Sampson JR, Halley D, van der Sluijs P. Interaction between hamartin and tuberin, the TSC1 and TSC2 gene products. Hum Mol Genet. 1998;7:1053–1057. doi: 10.1093/hmg/7.6.1053. [DOI] [PubMed] [Google Scholar]
- Verdu J, Buratovich MA, Wilder EL, Birnbaum MJ. Cell-autonomous regulation of cell and organ growth in Drosophila by Akt/PKB. Nature Cell Biol. 1999;1:500–506. doi: 10.1038/70293. [DOI] [PubMed] [Google Scholar]
- Weinkove D, Neufeld TP, Twardzik T, Waterfield MD, Leevers SJ. Regulation of imaginal disc cell size, cell number and organ size by Drosophila class IA phosphoinositide 3-kinase and its adaptor. Curr Biol. 1999;9:1019–1029. doi: 10.1016/s0960-9822(99)80450-3. [DOI] [PubMed] [Google Scholar]
- Wienecke R, Konig A, DeClue JE. Identification of tuberin, the Tuberous Sclerosis-2 product: tuberin possesses specific Rap1GAP activity. J Biol Chem. 1995;270:16409–16414. doi: 10.1074/jbc.270.27.16409. [DOI] [PubMed] [Google Scholar]
- Xiao G-H, Shoarinejad F, Jin F, Golemis EA, Yeung RS. The tuberous sclerosis 2 gene product, tuberin, functions as a Rab5 GTPase activating protein (GAP) in modulating endocytosis. J Biol Chem. 1997;272:6097–6100. doi: 10.1074/jbc.272.10.6097. [DOI] [PubMed] [Google Scholar]
- Xu T, Rubin GM. Analysis of genetic mosaics in developing and adult Drosophila tissues. Development. 1993;117:1223–1237. doi: 10.1242/dev.117.4.1223. [DOI] [PubMed] [Google Scholar]
- Yoshida Y, Kawata M, Miura Y, Sasaki T, Kikuchi A, Takai Y. Microinjection of smg/rap1/Krev-1 p21 into Swiss 3T3 cells induced DNA synthesis and morphological changes. Mol Cell Biol. 1992;12:3407–3414. doi: 10.1128/mcb.12.8.3407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young J, Povey S. The genetic basis of tuberous sclerosis. Mol Med Today. 1998;4:313–319. doi: 10.1016/s1357-4310(98)01245-3. [DOI] [PubMed] [Google Scholar]
- Zhang H, Stallock JP, Ng JC, Reinhard C, Neufeld TP. Regulation of cellular growth by the Drosophila target of rapamycin dTOR. Genes & Dev. 2000;14:2712–2724. doi: 10.1101/gad.835000. [DOI] [PMC free article] [PubMed] [Google Scholar]