

Abstract
Trichoderma reesei (Hypocrea jecorina) is nowadays the most important industrial producer of cellulase and hemicellulase enzymes, which are used for pretreatment of cellulosic biomass for biofuel production. In this study, we introduce a novel component, GRD1 (glucose-ribitol dehydrogenase 1), which shows enzymatic activity on cellobiose and positively influences cellulase gene transcription, expression, and extracellular endo-1,4-β-d-glucanase activity. grd1 is differentially transcribed upon growth on cellulose and the induction of cellulase gene expression by sophorose. The transcription of grd1 is coregulated with that of cel7a (cbh1) under inducing conditions. GRD1 is further involved in carbon source utilization on several carbon sources, such as those involved in lactose and d-galactose catabolism, in several cases in a light-dependent manner. We conclude that GRD1 represents a novel enhancer of cellulase gene expression, which by coregulation with the major cellulase may act via optimization of inducing mechanisms.
INTRODUCTION
Plant cell walls, which predominantly consist of cellulosic material, represent one of the most abundant renewable energy sources on earth. Natural recycling processes, which involve fungi as essential degraders of cellulosic biomass, are crucial for the environmental carbon cycle. One of the most efficient fungi in this respect is Trichoderma reesei (anamorph of Hypocrea jecorina), which is of high industrial importance because of this very characteristic. This ascomycete is able to produce and secrete up to 100 g of extracellular protein per liter (7), an attribute which is mainly exploited for the production of highly efficient cellulase mixtures. These enzymes are widely used in industry (3, 19, 20). The most promising application nowadays, however, is the use of cellulases for the production of second-generation biofuels. Since T. reesei is the most prolific producer of cellulases, the components of its cellulase system have been subject to extensive research on the structure, induction, expression, and regulation of the production of these enzymes (32, 53).
Cellulases are induced by cellulose, but by the late 1950s, it was already found that soluble carbon sources can also induce cellulases (39). It has been reported that sophorose (37, 61), cellobiose (38), lactose (39), l-sorbose (29), l-arabitol, and a few additional substrates (40) can act as inducers of cellulase gene expression in T. reesei. Sophorose (2-O-β-glucopyranosyl-d-glucose), a transglycosylation product of cellobiose, is a very powerful inducer of cellulases (37, 61) which is converted from cellobiose by beta-glucosidase (68). However, upon induction by sophorose and lactose, only an incomplete array of cellulases is produced (42, 62). Additionally, it was reported that despite seemingly similar conditions of cellulase induction, differential gene expression occurs on cellulose and sophorose (52).
While the regulation of cellulase gene expression on different carbon sources and in the presence of numerous inducing compounds has been studied for decades now, the influence of light on this process was only discovered a few years ago (51). Although the initially detected effect of light on the transcription of cellulase genes was only about 50%, later studies involving different signaling components revealed up to 10-fold differences between parental and mutant strains in dependence on light (54, 57). Important components involved in light-modulated cellulase gene transcription are ENV1 (51) and the photoreceptors BLR1 and BLR2 (6). These considerable effects of light require controlled light conditions, i.e., the elimination of random light or dark pulses during cultivation, for the analysis of cellulase gene expression in T. reesei, which we also routinely apply.
In this study, we characterize a novel dehydrogenase which is strongly upregulated upon induction by sophorose versus growth on cellulose and is coregulated with cel7a (cbh1). GRD1 (glucose-ribitol dehydrogenase 1) exhibits enzymatic activity on cellobiose and positively affects cellulase gene transcription, expression as well as extracellular endo-1,4-β-d-glucanase activity. The finding that the effects of GRD1 are in part dependent on the light status is in agreement with light-modulated regulation of cellulase gene expression.
MATERIALS AND METHODS
Strains, plasmids, and culture conditions.
T. reesei strain QM9414 (ATCC 26921) and a T. reesei Δtku70 strain, which is defective in the nonhomologous end-joining pathway (24), were used in the present study. For Northern analyses, T. reesei was grown in liquid culture in 200 ml Mandels Andreotti minimal medium (36) supplemented with 0.1% (wt/vol) peptone to induce germination and with 1% (wt/vol) microcrystalline cellulose (catalog no. 14204; Serva, Heidelberg, Germany) as a carbon source under controlled light conditions. For cultivation in light, daylight neon lamps were used and set to 1,800 lx (25 μmol photons m−2 s−1). Harvesting of dark-grown cultures was done under safe red light (red, E27, 15-W Philips PF712E darkroom lamp) in order to prevent random light pulses. After cultivation of QM9414 in darkness for analysis on glycerol (24 h), glucose (17 h), or lactose (20 h) and harvesting of the dark control sample at time point 0 (18), strains were grown in light for 15, 30, 60, and 120 min and harvested by filtration. The duration of precultivation in darkness (24, 17, or 20 h, respectively) was chosen according to the growth of the strains in order to ensure comparable biomass production. For the replacement experiments, T. reesei was grown as described above on glycerol. After 24 h of cultivation in darkness, the mycelia were harvested, washed, and transferred into fresh Mandels Andreotti minimal medium containing 1.5 mM sophorose (no. 35208.01 Serva, Heidelberg, Germany) as the carbon source. After 2, 4, and 5 h of cultivation in constant light or constant darkness, mycelia were harvested, washed with tap water, and frozen in liquid nitrogen.
Escherichia coli JM109 was used for the propagation of vector molecules and DNA manipulations (71).
Rapid subtraction hybridization for analysis of differential expression.
Isolation of total RNA, preparation of cDNA, comparative cDNA hybridization, and reverse Northern blotting were performed as described earlier (27, 52, 56). Briefly, parental strain QM9414 was grown on Mandels Andreotti minimal medium with 1% (wt/vol) cellulose as carbon source for 48 h or pregrown on 1% (wt/vol) glycerol for 24 h, replaced into medium containing 1.5 mM sophorose for cellulase induction, and harvested after 5 h of cultivation as described earlier (52). After isolation of total RNA, cDNA was prepared using the Creator SMART cDNA library construction kit (Clontech, TakaraBio United States), digested with EcoRII (all restriction enzymes used were purchased from Fermentas, Vilnius, Lithuania), and purified. Amounts of 100 ng of tester cDNA (QM9414/sophorose) and 3 μg of driver cDNA (QM9414/cellulose) were used for rapid subtraction hybridization. Putatively differentially expressed cDNA fragments were cloned and transformed into E. coli JM109. PCR fragments amplified from these clones were tested for differential expression by reverse Northern blotting.
Construction of the T. reesei (Δtku70) Δgrd1 strain.
To construct a Δgrd1 strain of T. reesei, the 3′ region of grd1 was amplified using primers pDelGRD13F (sequences of all oligonucleotides used in this study are listed in Table 1) and pDelGRD13R, and the PCR product was digested with SalI and EcoRV. The 5′ region was amplified using primers pDelGRD15F and pDelGRD15R, and the PCR product was digested with Acc65I and SalI. The pBluescript vector (Stratagene, La Jolla, CA) was digested with EcoRV and Acc65I. The prepared 3′ and 5′ fragments were purified and ligated simultaneously into the pBluescript vector to obtain pBgrd135. This plasmid was digested with SalI, dephosphorylated using shrimp alkaline phosphatase (Fermentas), and ligated to a pyr4 fragment excised from pBpyr4 (pBluescript containing the pyr4 fragment excised from pFG1 integrated into the SalI site [22]) with SalI to obtain pDELgrd1P (see Fig. S1A in the supplemental material). For transformation, pDELgrd1P was linearized with EcoRV and 10 μg of linearized vector was used for the transformation of protoplasts of the Δtku70 strain as described previously (23).
Table 1.
Oligonucleotides used in this study
| Primer | Sequencea |
|---|---|
| Primer | Sequencea |
| pDelGRD13F | 5′-ATGTCGACTTCATCTCTCAACCCAAC-3′ |
| pDelGRD13R | 5′-ATGATATCACACATCCGTCAGCTCAG-3′ |
| pDelGRD15F | 5′-ATGGTACCGCACTTGTGAATCTCCGAAC-3′ |
| pDelGRD15R | 5′-ATGTCGACTGAAGCGTGAGAGGTAAGATG-3′ |
| grd1cDNAF | 5′-GTCGAGGTTGTCAATGTCAG-3′ |
| grd1cDNAR | 5′-TTGATTCGTTGGTTGGTAGAG-3′ |
| grd1anti1F | 5′-ATCTGCAGAGGCCTGGCTTGACACAATGGCAC-3′ |
| grd1anti1R | 5′-ATGCTAGCACTAGTGTCGACTTTAAAGATCTAGGCCTTACGCACCAGGCAGAAG-3′ |
| GRD1GSTF | 5′-ATGGATCCATGGCACCGCCACCGCTCTC-3′ |
| GRD1GSTR | 5′-ATCTCGAGCGGTCATACCAGCTTCTTAC-3′ |
| tef1F | 5′-AGAAGGTCGGCTTCAACC-3′ |
| tef1R | 5′-GGTAGTCGGTGAAAGCCTC-3′ |
Restriction sites introduced to facilitate cloning are underlined.
Deletion strains were tested for integration of the construct by PCR using primers grd1cDNAF and grd1cDNAR, which bind outside the region to be deleted. Successful deletion resulted in the amplification of a longer fragment (2,805 bp) than amplification from DNA of the parental strain (1,303 bp) (data not shown).
For complementation of Δgrd1 with the grd1 wild-type gene, we amplified the entire gene, including flanking sequences, using primers pDelgrd15F and pDelgrd13R. This PCR product was ligated into pGEMT (Promega, Madison, WI) to obtain grd1pGEMT. The fragment was excised from grd1pGEMT using NotI and ligated into the NotI site of the vector pBSamds (55) to obtain pBgrd1Re. After retransformation of Δgrd1, integration was confirmed using primers grd1cDNAF and grd1cDNAR as described above.
Construction of a T. reesei grd1 overexpression strain.
To construct a grd1 overexpression strain of T. reesei, plasmid pLH1hph (26) was used as the backbone vector. The hph open reading frame was excised by digestion with NsiI and XbaI to obtain pLH1. A fragment was amplified using primers grd1anti1F and grd1anti1R, which were used to introduce a multiple cloning site and digested with PstI and NheI and ligated into the NsiI-XbaI (for creation of corresponding ends to PstI and NheI)-digested pLH1 vector to obtain pLH1mcs. Primer grd1anti1R contains five additional restriction sites (SpeI, SalI, DraI, BglII, and StuI) for the introduction of a new multiple cloning site. From pBSXH (51), the hph (pki1p::hph::cbh2t) cassette was excised by digestion with XhoI, Klenow fill-in was applied to create a blunt end, and the resulting fragment was digested with BamHI. The vector pLH1mcs was digested with BamHI and SmaI and ligated with the digested hph fragment to obtain pLH1mcsHPH. This vector was digested with StuI, purified, and religated in order to remove the grd1 antisense fragment and leave the multiple cloning site intact. The resulting vector (paGPDhph) contains a gpd1 (glycerol 3-phosphate) promoter and terminator, hygromycin B resistance gene (hph), and a multiple cloning site (see Fig. S1B in the supplemental material). The open reading frame of grd1 was amplified using Phusion polymerase (Finnzymes, Espoo, Finland) and primers grd1cDNAF and grd1cDNAR. The PCR product was ligated into the StuI site of paGPDhph to obtain pagrd1hph (see Fig. S1B in the supplemental material). After confirmation of the correct orientation of the inserted fragment, the resulting vector was linearized by digestion with NarI and used for transformation into the genome of parental strain QM9414 as described previously (23). Mutant strains were tested for integration of the construct by PCR using primers grd1cDNAF and TgpdR (26) or grd1cDNAR and PgpdF (26). These primers bind inside the gpd1 promoter or terminator region or in the grd1 region. Successful integration resulted in a specific band (1,824 bp or 2,195 bp, respectively), and no amplification was possible with DNA from the parental strain (data not shown).
Biolog Phenotype MicroArray technique.
T. reesei strains were grown on 2% (wt/vol) malt extract agar plates at 28°C for 1 week until sporulation. Inocula were extracted from these plates. The assay was performed as described previously (12, 13). The Biolog Phenotype MicroArray microplates were incubated for 72 h in constant light or constant darkness. Analyses were repeated at least twice for each strain.
Phylogenetic analysis.
Clustal X 2.0.7 (33) was used for the alignment of amino acid sequences. MEGA 4.0.2 (64) was used for phylogenetic analysis, using the minimum evolution method and 500 bootstrap replications as a test of phylogeny. Protein sequences were obtained from the Joint Genome Institute T. reesei, Trichoderma viride, and Trichoderma atroviride genome databases (http://genome.jgi-psf.org/). Sequences of proteins encoded by the genomes of Neurospora crassa, Aspergillus spp., Fusarium spp., and Magnaporthe grisea were retrieved from their respective genome databases at the Broad Institute (http://www.broadinstitute.org/science/data).
Nucleic acid isolation and hybridization.
Fungal mycelia were harvested by filtration, washed with tap water, frozen, and ground in liquid nitrogen. For extraction of DNA, mycelium powder was suspended in buffer A (1.2 M NaCl, 5 mM EDTA, 0.1 M Tris-HCl, pH 8.0), incubated for 20 min at 65°C, cooled on ice, mixed with 1 volume phenol/chloroform/isoamyl alcohol (49:49:2 [vol/vol/vol]), and centrifuged (21,000 × g for 15 min). Following an extraction with 1 volume of chloroform/isoamyl alcohol (24:1 [vol/vol]), DNA was precipitated with 1 volume of isopropanol and washed with 70% (vol/vol) ethanol. Total RNA was isolated by the guanidinium thiocyanate method (10). Standard methods (50, 56) were used for electrophoresis, blotting, and hybridization of nucleic acids.
cDNA preparation and RT-PCR.
Total RNA was treated with DNase I (Fermentas, Vilnius, Lithuania). cDNA was prepared using a RevertAID H minus first-strand cDNA synthesis kit (Fermentas). GoTaq polymerase (Promega, Madison, WI) was used for subsequent PCR amplification. Primers tef1F and tef1R were used for amplification of tef1 for 25 cycles, and primers grd1cDNAF and grd1cDNAR for amplification of grd1 for 35 cycles.
Biomass determination.
Parental strain QM9414 and the Δgrd1 strain were grown in liquid medium with cellulose as the carbon source as described above. Mycelia were ground in liquid nitrogen, and the powder was suspended in 0.1 N NaOH. This suspension was sonicated three times for 1 min and incubated for 3 h at room temperature. Samples were centrifuged for 10 min at 10,000 rpm, and the supernatants were transferred to new reaction tubes. The protein concentration reflecting biomass content was measured using a Bio-Rad protein assay (Bio-Rad Laboratories, Hercules, CA).
Determination of cellulase activity in culture filtrates.
The parental strain QM9414 and the Δgrd1 and grd1S (constitutively expressing GRD1) strains were grown as described above. Culture filtrates were used for the analysis of endo-1,4-β-d-glucanase activity with the aid of an azo-CM-cellulose kit (no. S-ACMC, Megazyme, Wicklow, Ireland), which was used according to the manufacturer's instructions.
Protein recovery from culture filtrates and Western blotting.
A 500-μl volume of culture filtrate from cultivation on Mandels Andreotti minimal medium with 1% (wt/vol) microcrystalline cellulose in constant light and constant darkness for 72 h and 96 h was precipitated by adding 2 ml methanol, 500 μl chloroform, and 1.5 ml water and mixed well. After centrifugation for 5 min at 8,000 rpm, the upper phase was discarded. Thereafter, 1.5 ml methanol was added and mixed well and the mixture centrifuged again for 5 min. Then the supernatant was discarded, and the pellet was dried at room temperature and redissolved in 50 μl sterile water.
SDS-PAGE and Western blotting were performed according to standard protocols (50). Proteins were blotted on a nitrocellulose membrane (Hybond-C Extra, Amersham Biosiences, United Kingdom) by semidry electroblotting. For analysis of the expression of cellulases, antibodies against the major cellulase CEL7A (46) and a horseradish peroxidase-conjugated anti-mouse IgG (Promega, Madison, WI) were used.
Production and purification of a GST-GRD1 fusion protein.
To construct a glutathione S-transferase (GST) fusion expression vector, the grd1 open reading frame was amplified from cDNA prepared from QM9414 grown on lactose by PCR using primers GRD1GSTR and GRD1GSTF. The resulting PCR product was cloned into the BamHI-XhoI sites of the pGEX-4T2 (Stratagene) vector to obtain GRD1GSTpGex. This vector was transformed into E. coli BL21 (Stratagene). The resulting E. coli strain was grown on selective LB medium to an optical density at 600 nm (OD600) of 0.600. Production of the heterologous protein was induced using 0.1 mM (final concentration) isopropyl-β-d-thiogalactopyranoside (IPTG; Fermentas), and the culture was harvested by centrifugation after 3 h of growth under inducing conditions. The heterologous protein was purified using glutathione-Sepharose 4B according to the manufacturer's protocol (GE Bioscience, Uppsala, Sweden). Purification and production of the GST fusion protein was confirmed by SDS-PAGE. The concentration of the resulting recombinant protein was determined with the Bio-Rad protein assay (Bio-Rad).
Determination of enzyme specificity.
GRD1 enzymatic activity was determined spectrophotometrically essentially as described earlier (48). Briefly, we measured the change in absorbance at 340 nm for NADH and NADPH oxidation or NAD+ and NADP+ reduction of the respective substrate by GRD1 at 30°C. Concentrations of 100 mM substrate and 0.25 mM NADH/NADPH and 100 μg of purified GST protein were used in the assay. Activities are given as specific activities in nkat/mg protein.
For purification of the enzyme assay, we used Vivaspin 2 columns with a 3,000 molecular-mass-cutoff polyethersulfone (PES) membrane (Sartorius Stedim Biotech, Cedex, France). Amounts of 1 ml of reaction mixture were added to the columns and centrifuged for 10 min at 10,000 rpm in a fixed-angle rotor at 20°C. For high-performance liquid chromatography (HPLC) analysis, we used an Aminex HPX-87H column (Bio-Rad) with 10 mM H2SO4 as the mobile phase at a flow rate of 0.5 ml min−1 at 40°C. The concentration of substrate was calculated from a calibration curve established with 1 mM, 10 mM, 50 mM, and 100 mM concentrations of the respective substrate.
RESULTS
Characterization of a putative cellulase induction-associated gene.
A cDNA screening experiment comparing genes differentially transcribed during growth on cellulose or after the induction of cellulase gene expression by sophorose (52) revealed that these two conditions are indeed not entirely comparable. Several genes were found to be differentially regulated on these two carbon sources, which is in agreement with earlier studies showing that the cellulase mixtures secreted upon induction by these carbon sources are not identical (42, 62). We therefore conducted a similar experiment aimed at identifying genes specifically transcribed in the presence of sophorose, using cDNA from cultures grown in the presence of sophorose (5 h after replacement) as the tester and cDNA resulting from cellulose-grown mycelia as the driver. This analysis revealed several transcripts to be more abundant upon growth in the presence of sophorose than with cellulose as the carbon source (see Table S1 in the supplemental material). Among those, the gene represented by STCD21 and STCD55, a putative glucose-ribitol dehydrogenase (TR_123946), was found most frequently (9 fragments targeted this gene). Enzymes of this group (EMBL-EBI InterPro domain IPR002347) are tetrameric and catalyze the oxidation of d-glucose without prior phosphorylation to d-β-gluconolactone. Because of its presumed function as a glucose-ribitol dehydrogenase, we named this gene grd1 (glucose-ribitol dehydrogenase 1).
Grd1 comprises a 1,205-bp open reading frame interrupted by one intron and encodes a protein of 304 amino acids. It has no N-terminal signal peptide and is predicted to be located in the cytoplasm (PSORTII; http://www.psort.org/).
GRD1 is a conserved but not ubiquitous dehydrogenase in fungi.
Searching the T. reesei genome for genes encoding proteins with characteristics similar to those of GRD1, we found nine additional models comprising the same domains (IPR002198, IPR002347, and IPR016040), hence indicating that the T. reesei genome comprises 10 putative glucose-ribitol dehydrogenases (Fig. 1). However, although GRD1 seems to be conserved in several fungal species, we could not find a characterized orthologue of GRD1 in fungi. Nevertheless, BLAST searches against several fungal genome databases (http://www.broad.mit.edu/annotation/fungi, http://genome.jgi-psf.org/, and http://www.aspergillusgenome.org) showed that this protein is well conserved. Bidirectional blast search suggested that only the putative orthologues found in Aspergillus nidulans, Aspergillus clavatus, Aspergillus oryzae, Aspergillus terreus, and Trichoderma virens were true homologues to GRD1. Phylogenetic analysis of predicted proteins from several fungi confirmed this result and showed that GRD1 is not ubiquitously present in other fungi (Fig. 2; also see Fig. S2 in the supplemental material). We found that GRD1 clusters with predicted proteins of different Aspergillus spp. and has a homologue in T. virens, but no orthologue was detected in N. crassa, Aspergillus niger, M. grisea, or Fusarium spp. Surprisingly, T. atroviride also seems to have no orthologue of GRD1.
Fig. 1.

Phylogenetic analysis of short-chain dehydrogenases using the minimum evolution method and 500 bootstrap replications as test of phylogeny. Phylogenetic tree obtained by MEGA 4.0.2. The sequences were obtained from the JGI T. reesei genome database, version 2.0 (http://genome.jgi-psf.org/Trire2/Trire2.home.html). The scale bar reflects evolutionary distance.
Fig. 2.

Phylogenetic analysis of short-chain dehydrogenases using the minimum evolution method and 500 bootstrap replications as test of phylogeny. Phylogenetic tree obtained by MEGA 4.0.2. The sequences were obtained from the JGI T. reesei genome database, version 2.0 (http://genome.jgi-psf.org/Trire2/Trire2.home.html), and the BROAD database (http://www.broadinstitute.org/scientific-community/data). F. oxysporum, Fusarium oxysporum; F. graminearum, Fusarium graminearum; F. verticillioides, Fusarium verticillioides; A. flavus, Aspergillus flavus. The scale bar reflects evolutionary distance.
Grd1 is located within a putative secondary metabolism cluster in the genome.
Analysis of the grd1 locus within the T. reesei genome (Fig. 3 and Table 2) showed that grd1 is flanked by two uncharacterized genes (TR_111739 and TR_111742) and a predicted polyketide synthase (TR_81964) further downstream. In T. virens also, orthologues of the four neighbors of TV_216312 (homologue of T. reesei grd1) are present (TV_69355, TV_223624, TV_49693, and TV_49800), albeit the order of genes is different and the area contains two additional predicted genes. One encodes an uncharacterized protein with a molecular function as a tetracycline resistance protein (TV_213302), and the second gene encodes an uncharacterized protein (TV_223622).
Fig. 3.

Schematic representation of the genomic locus of grd1. Scaffold or position numbers are represented as specified in the respective genome databases (http://genome.jgi-psf.org/Trire2/Trire2.home.html and http://www.aspgd.org/). Genes encoding orthologues of GPD1 are shown in white, and orthologues of the polyketide synthase are shown in gray. Protein identification numbers are given in Table 2. The scheme is drawn to scale.
Table 2.
Proteins encoded by genes found around the genomic locus of GRD1 and its orthologues
| No.a | Identification no. of proteinb2 in: |
|||
|---|---|---|---|---|
| T. reesei | T. virens | T. atroviride | A. nidulans | |
| 1 | 111733 | 41120 | ||
| 2 | 23332 | 292229 | ||
| 3 | 69650 | 69355 | AN7075 | |
| 4 | 111739 | 223624 | AN7072 | |
| 5 | GRD1 | 216312 | AN7074 | |
| 6 | 111742 | 49693 | AN7073 | |
| 7 | 81964 | 49800 | 80219 | AN7071 |
| 8 | 223622 | |||
| 9 | 213302 | |||
See Fig. 2.
Protein identification numbers are from the respective genome databases (http://genome.jgi-psf.org/Trire2/Trire2.home.html and http://www.aspgd.org/).
Using the Aspergillus genome database (http://www.aspergillusgenome.org), we found an orthologue of grd1 in the A. nidulans genome (AN7074). Furthermore, we also detected orthologues of four genomic neighbors of grd1 in A. nidulans (AN7075, AN7072, AN7073, and AN7071). Again, the order of genes in the A. nidulans genomic locus differed from that in T. reesei (Fig. 3). This slightly altered assembly of orthologous genes within the genomic area around grd1 in different fungi indicates that they form a cluster of functionally related genes. The finding of a predicted polyketide synthase among the members of this cluster suggests a function in secondary metabolism. However, in the T. reesei genome, several carbohydrate-active enzyme (CAZyme) clusters (comprising genes encoding carbohydrate-degrading enzymes) were found to include genes related to secondary metabolism (41).
We could not find an orthologue of grd1 in N. crassa, and we therefore extended the genomic area for the blast search. We found only two genes (TR_111733 and TR_69652) which have orthologues in N. crassa. The two genes (NCU04798.4 and NCU05192.4) are on different contigs/scaffolds in the genome database. Also, no orthologue for the polyketide synthase (TR_81964) was detected. Consequently, the genomic area of grd1 is not present in the genome of N. crassa.
A similar result was obtained for T. atroviride (http://genome.jgi-psf.org/Triat2/Triat2.home.html). To find traces of the presumably lost grd1 locus in T. atroviride, we used the polyketide synthase as an anchor. Using the sequence of TR_81964, we found an orthologue in T. atroviride (TA_80219). This polyketide synthase is located on contig 23 (1677486 to 1683274) in the genome database. We extended the upstream area of grd1's neighbors in T. reesei and found two genes (an α-1,2-mannosidase gene, TR_111733, and a gene encoding a hypothetical protein, TR_23332) which are also present in the T. atroviride genome (TA_41120 and TA_292229). Both T. atroviride genes were also located on contig 23. The distance between TR_111733 and TR_81964 in the T. reesei genome is around 24,000 bp, but in T. atroviride, the distance between the orthologues TA_41120 and TA_80219 is around 8,2000 bp. We conclude that the genomic area of grd1 is missing in T. atroviride.
Transcription of grd1 correlates with transcription of cel7a (cbh1) on sophorose.
grd1 was isolated in our cDNA screening because of its predominant transcription upon the induction of cellulase gene expression in the presence of sophorose. We therefore tested grd1 transcription under these conditions in more detail. Since cellulase gene expression is modulated by light and by the light regulatory protein ENV1 (51), we used controlled light conditions for our experiments and included the ENV1 nonfunctional strain env1PAS− in our analysis (Fig. 4).
Fig. 4.
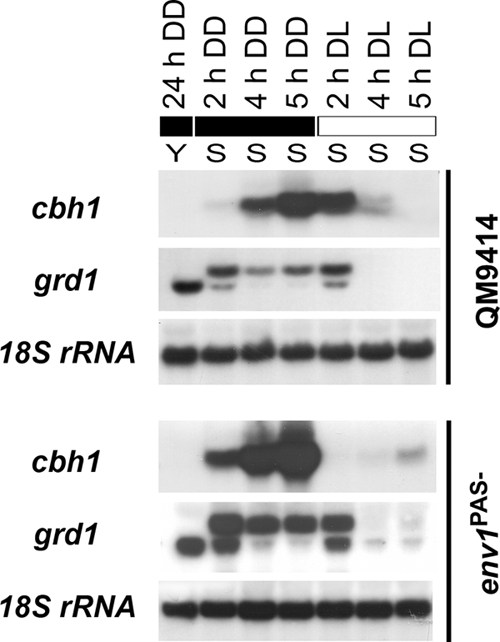
Northern analysis of cel7a (cbh1) and grd1 transcription. Strains were grown on Mandels Andreotti minimal medium with 1% (wt/vol) glycerol (Y) as carbon source for 24 h in constant darkness (DD) (18) and then transferred to fresh medium with 1.5 mM sophorose (S) for cellulase induction in darkness (18) or after illumination (1,800 lx, 25 μmol photons m−2 s−1) for 2, 4, or 5 h (DL). A hybridization with 18S rRNA is shown below the respective series as a control. Under the same conditions but without the addition of sophorose as an inducing compound, no expression of cellulase genes occurs (21).
We found that grd1 is not transcribed exclusively under cellulase-inducing conditions but is also transcribed upon growth on glycerol (preculture). Interestingly, the transcript abundance of grd1 clearly correlates with that of cel7a (cbh1) in both light and darkness and is enhanced in the absence of functional ENV1. Notably, we observed a significant shift in transcript sizes upon the onset of cellulase induction after replacement into sophorose-containing medium. This reaction to changing nutrient conditions strongly suggests a function of GRD1 in the pathway of nutrient signal perception and/or transmission. Since coregulation of genes indicates functions in similar pathways (14, 25, 28, 30), a function of GRD1 in the regulation of cellulase gene expression can be assumed.
Nevertheless, the most intriguing result of this analysis was the strikingly different transcription patterns in light and darkness. Not only is cellulase gene transcription enhanced in darkness compared to the transcription in light, which is in contrast to earlier findings (51), we also found that in light, induction of cellulase gene transcription is transient on sophorose. Although these findings are surprising, they are in accordance with earlier results showing that the influence of light on growth is dependent on the carbon source (5, 67). Hence, the expression of hydrolytic enzymes can also be expected to be altered. Additionally, a recent study of Aspergillus nidulans showed that even an alteration of the concentration of the carbon source can reverse light-dependent gene expression (1).
grd1 is predominantly transcribed on soluble carbon sources.
The appearance of a grd1 transcript upon growth on the cellulase-noninducing carbon source glycerol prompted us to screen for transcription of grd1 on further carbon sources and to test for potential light responsiveness. We therefore cultivated T. reesei QM9414 on glycerol and, additionally, on lactose (cellulase-inducing carbon source) and glucose (cellulase-repressing carbon source) in constant darkness and then harvested mycelia at several time points after illumination was introduced (Fig. 5 A). Transcript analysis confirmed the above-mentioned correlation of transcript size with conditions of cellulase induction. Upon growth on the cellulase-inducing carbon source lactose, we detected the greater transcript abundance, while under conditions not inducing cellulase gene expression (glucose and glycerol), the lesser amount of transcript was present. Additionally, grd1 shows a certain response to light. However, we could not detect a grd1 transcript on cellulose using Northern blots, which confirms the validity of our initial cDNA screening approach for genes transcribed more abundantly on sophorose than on cellulose. We therefore applied RT-PCR, which revealed transcription of grd1 also upon growth on cellulose for 72 h in light and darkness, albeit at very low levels (Fig. 5B).
Fig. 5.

Transcript analysis of grd1 upon growth on different carbon sources. (A) Wild-type QM9414 was grown on Mandels Andreotti minimal medium with 1% (wt/vol) glycerol as carbon source in constant darkness (DD) (18) and then exposed to constant light (1,800 lx, 25 μmol photons m−2 s−1) for 15, 30, 60, and 120 min. A hybridization with 18S rRNA is shown below the respective series as a control. (B) RT-PCR of transcription of grd1 and tef1 in wild-type strain QM9414 after 72 h of growth in Mandels Andreotti medium with 1% (wt/vol) cellulose as carbon source in constant light and constant darkness. tef1 (encoding translation elongation factor 1 alpha) was used as a control.
Carbon source utilization varies between parental strain and Δgrd1 strain in light and in darkness.
In order to investigate the function of GRD1 in T. reesei, we constructed a deletion mutant of grd1 in a strain defective in the nonhomologous-end-joining pathway (24). The growth of the Δgrd1 strain on plates is indistinguishable from the growth of the parental strain QM9414. Since grd1 is not transcribed only under cellulase-inducing conditions, we intended to gain further insight as to the putative function of this dehydrogenase by the use of Biolog Phenotype MicroArrays (13), which allow the analysis of growth on 95 carbon sources. We tested the growth of the Δgrd1 strain and the parental strain in light and darkness after 72 h of cultivation (Fig. 6; also see Fig. S3 in the supplemental material).
Fig. 6.

Light stimulation of growth of T. reesei on selected carbon sources and the effect of GRD1. (A) Growth of the wild-type QM9414 or Δgrd1 strain was analyzed by using the Biolog microplate assay. Biomass equivalents (OD750) after 72 h of growth are shown. This time was chosen because then both strains were still in the phase of active growth on most carbon sources tested. The y axis shows values obtained under constant darkness, whereas the x axis show those obtained in constant light (1,800 lx). All experiments were done in triplicates; standard deviations are indicated by error bars. (B) Data table detailing results for the selected carbon sources.
Besides an only minor difference in growth on the cellulase inducer cellobiose (38), we found that GRD1 positively influences growth on α-d-lactose. Interestingly, we also found differences in growth between the parental strain and the mutant on several intermediates of lactose and d-galactose catabolism (32, 44, 58), specifically, d-galactose, d-fructose, d-xylose, xylitol, d-mannitol, and l-arabinose. In some cases, these differences were dependent on the light status (Fig. 6). Since lactose is an important inducer of cellulase gene expression (59), this influence of GRD1 on growth on these intermediates is in accordance with the hypothesis that GRD1 is involved in inducer modification at an as-yet-unknown metabolic level.
GRD1 shows enzymatic activity with cellobiose.
The putative function of GRD1 prompted us to investigate the substrate specificity of GRD1 in more detail. We used a GRD1-GST fusion protein expressed for this purpose from cDNA in E. coli without trypsin digestion, because it was shown that cleavage of the GST moiety of the fusion protein could lead to an inactivation of the protein (58).
The enzymatic activity was tested on d-glucose, d-sorbitol, d-xylose, xylitol, d-galactose, l-arabinose, maltose, d-galactitol, l-arabitol, d-arabitol, d-mannitol, d-ribitol, cellobiose, and sophorose as substrates and with NADH, NAD+, NAD(P)H, or NAD(P)+ as the cofactor. Specific activity was only detected on cellobiose (52.9 nkat/mg protein, with NADPH as cofactor). The reaction mixtures from the assays with a positive result (cellobiose), as well as those with d-glucose as the negative control, were purified to remove high-molecular-weight compounds and monitored by HPLC analysis. We found that the retention time of glucose was 12 min and that of cellobiose was 10 min. As a negative control for our samples, we used a reaction mixture similar to that used in the assay but without incubation. This control was immediately purified to remove the GST fusion protein and, hence, prevent the enzymatic reaction.
For d-glucose, as expected, no conversion was detected. In contrast, we could detect a consumption of cellobiose. After incubation, the substrate concentration was decreased from the reference concentration of 8.71 mM ± 0.26 mM (mean ± standard deviation) to 7.66 mM ± 0.22 mM cellobiose. This small (12.1%) but significant difference (P value, 0.05) is in accordance with the low activity of GRD1 detected on cellobiose as described above.
GRD1 is involved in the regulation of cellulase gene expression.
The above-described results indicate that GRD1 could play a role in the induction or regulation of cellulase gene expression. Although grd1 was identified as being upregulated on sophorose versus cellulose, it can be assumed that it influences cellulase gene expression on cellulose as well. In order to test this hypothesis, we investigated the transcription of the major cellulase of T. reesei, cel7a (cbh1), upon growth on cellulose in constant light and constant darkness. We found that the deletion of grd1 does not abolish the induction of cellulase gene transcription and does not lead to severe growth defects upon cultivation on cellulose. Nevertheless, the lack of GRD1 causes considerably decreased transcript levels of cel7a (cbh1), but only in light (Fig. 7 A). Consequently, the function of GRD1 is of light-dependent relevance for cellulase gene transcription in T. reesei, a phenomenon which was also observed earlier (21, 54, 57).
Fig. 7.

(A) Northern analysis of cel7a (cbh1) transcription. Strains were grown on Mandels Andreotti minimal medium with 1% (wt/vol) microcrystalline cellulose as carbon source for 72 h and 96 h in constant light (LL) (1,800 lx, 25 μmol photons m−2 s−1) or constant darkness (DD). Representative hybridizations with 18S rRNA are shown below the respective series. (B) Western blot analysis of CEL7A from culture filtrates of wild-type QM9414 and Δgrd1 strains during growth in liquid cultures on Mandels Andreotti minimal medium with 1% (wt/vol) microcrystalline cellulose as carbon source for 72 h and 96 h in constant light (1,800 lx, 25 μmol photons m−2 s−1) or constant darkness. Equal amounts of culture filtrate were used.
Investigation of the abundance of CEL7A (CBH1) secreted into the medium confirmed the involvement of GRD1 in the regulation of cellulase gene expression. We detected clearly decreased levels of CEL7A (CBH1) secreted into the medium in both light and darkness (Fig. 7B). Surprisingly, this Western analysis also showed that the abundance of CEL7A (CBH1) is lower in light than in darkness in the parental strain, which is contrary to transcription data reported earlier (51) and also observed in this study. Consequently, the hypothesis of a strict correlation of transcription with secreted amounts of cellulases is no longer supported. Additional levels of regulation of cellulase gene expression and secretion must be considered operative and warrant further investigation.
Deletion of grd1 leads to decreased cellulase activity.
The results described above indicate a function of GRD1 in the regulation of the expression of cellulose-degrading enzymes. We therefore analyzed the effect of GRD1 on extracellular enzyme activity. In accordance with our results on the transcription of cel7a (cbh1) in the Δgrd1 strain, cellulase activity was clearly decreased in culture filtrates of this strain in light after 96 h of growth (Fig. 8). However, this negative effect of the lack of GRD1 on cellulase activity was much more severe in darkness, hence confirming the decreased abundance of CEL7A (CBH1) detected by Western blotting. In order to further confirm the effect of GRD1 on cellulase gene expression, we constructed a strain that constitutively expresses GRD1 (grd1S). The overexpression of GRD1 in grd1S reversed the effect and consequently corroborates the positive function of GRD1 in the regulation of cellulase gene expression. Moreover, this result shows that in light, cel7a (cbh1) is likely to be one major target of the regulatory output exerted by GRD1, although an effect of additional enzymes involved in cellulose degradation can be assumed. GRD1 seems to affect the transcription of cel7a (cbh1) only in light, while in darkness, further posttranscriptional mechanisms are likely to be involved.
Fig. 8.

Biomass-specific cellulase activity of wild-type QM9414 strain and Δgrd1 and grd1S strains during growth in liquid cultures on Mandels Andreotti minimal medium with 1% (wt/vol) microcrystalline cellulose as carbon source for 72 h and 96 h in constant light (1,800 lx, 25 μmol photons m−2 s−1) or constant darkness. Data from at least two independent experiments were combined for each value. Error bars show standard deviations.
DISCUSSION
In this study, we characterize a novel component of the cellulase regulon of T. reesei, the dehydrogenase GRD1. Interestingly, although N. crassa also possesses a potent cellulolytic machinery (66), no orthologue of GRD1 is present in the genome of this fungus, while an orthologue is present in the phylogenetically more distant aspergilli. In this respect, the locus of grd1 in the genomes of Trichoderma spp. and Aspergillus nidulans could provide a clue to the reason for this finding. The variable orientations yet comparable compositions of genes found in the genomic area around grd1 in these fungi, including a polyketide synthase, suggest the presence of a cluster involved in secondary metabolism. The aspergilli are known for their potent and well-studied secondary metabolism (70, 72), and in many cases, genes involved in this process are clustered (2, 8, 9, 63). In contrast, N. crassa was not even known to possess secondary metabolism, except for carotene and melanin biosynthesis, prior to sequencing of the genome (18). The genome sequence, however, revealed the presence of several polyketide synthase and nonribosomal peptide synthase genes, albeit the inventory in N. crassa is considerably smaller than that of T. reesei (41). In this respect, it is interesting that in T. reesei, 5 of the 25 genomic regions of high CAZyme gene density also comprise genes encoding proteins involved in secondary metabolism (41). However, we also want to note here that the homologue of grd1 in the Sordariomycete Thielavia terrestris (http://genome.jgi-psf.org/Thite2/Thite2.home.html) is not located in a cluster of genes involved in secondary metabolism. Hence, we assume that the proximity of the gene encoding GRD1 to a polyketide synthase-encoding gene reflects an evolutionary benefit of the interconnection of genes involved in carbon metabolism and secondary metabolism. Nevertheless, the occurrence of grd1 in a different genomic environment (in T. terrestris) may support a function independent of secondary metabolism.
Our results clearly show a function of GRD1 in the regulation of cellulase gene expression. The reaction product of this polyol dehydrogenase likely is cellobiitol. This compound has not yet been reported to induce cellulase expression if used as a carbon source (4, 38). However, cellobiitol has been shown to inhibit beta-glucosidase in Sporotrichum thermophile (45). While beta-glucosidase activity is essential for cellulase induction in T. reesei (31) and the beta-glucosidase bgl1 has a positive effect on cellulase gene expression (16, 35), it is known that a decreased beta-glucosidase activity leads to higher cellulase activities by enabling the maintenance of a larger pool of the inducing disaccharide (58). On the other hand, the transglycosylation product of cellobiose, sophorose, induces an intracellular beta-glucosidase (34). Thus, GRD1 could act as a regulator of beta-glucosidase activity in order to prevent inhibitory glucose concentrations in the cell.
The coregulation of grd1 with cel7a (cbh1), together with the enzymatic activity of grd1 on the degradation product of CBH1, cellobiose, could be responsible for intracellular sensing of cellulase efficiency and adjustment of cellulase levels and beta-glucosidase activity to optimize energy-efficient substrate utilization. In this respect also, the expression of GRD1 under noninducing conditions is useful, since the different transcript size under these conditions indicates activation upon reception of the cellulose-derived signal. Most of the hydrolysis of cellobiose to glucose occurs outside the cell, but nevertheless, an uptake mechanism for cellobiose has been confirmed in T. reesei (17). These findings corroborate a specific signaling function of this compound for the presence of cellulose outside the cell. The lack of GRD1 would thus cause increasing glucose and decreased cellobiose levels (reflecting decreased amounts of cellulose) in the medium and, hence, in the cell (as produced by beta-glucosidase), which would cause decreased cellulase gene expression. We therefore propose a model in which GRD1 represents an intracellular sensor of extracellular cellulolytic activity that adjusts beta-glucosidase activity and, hence, inducer (sophorose) and/or repressor (glucose) abundance.
The regulation of cellulase gene expression in T. reesei has been subject to extensive research for decades. Especially after the cloning of the major cellulase of T. reesei, cbh1 (60, 65), elucidation of the mechanism of cellulase induction and regulation was studied at the molecular level. A few years thereafter, comparison of mRNA and transcript levels in several Trichoderma strains and under different conditions led to the well-founded assumption that the regulation of cellulase gene expression occurs at a pretranslational level (15, 43, 47). Until recently, light was not considered an influencing factor in cellulase gene expression. In this study, we report the surprising discovery that the assumption of strictly transcriptional regulation of cellulase gene expression does not hold true for the application of different light conditions. Several reasons for this phenomenon are conceivable. On one hand, light-dependent splicing has been shown in fungi (49, 69), but if this mechanism were at work, additional bands resulting from (probably nonfunctional) truncated versions of CBH1 should have been detected on Western blots for light-grown cultures, which was not the case. However, clock regulation of ribosome biogenesis (but not transcriptional regulation of ribosome genes) has been shown and, hence, provides a means for posttranscriptional regulation of clock-controlled genes (11). Because of the crucial importance of light for resetting of the circadian clock, such a mechanism is likely to also be affected by light, which would provide a reasonable explanation for posttranscriptional regulation of cellulase gene expression by light.
Interestingly, this effect does not seem to be limited to the cellulase genes themselves. From our data, the question also arises as to how the effect of GRD1 on cellulase gene transcription and, obviously, extracellular cellulase activity can be dependent on the light status. The beneficial effect of GRD1 on cellulase gene transcription in light is very clear; the light-dependent differences in the abundance of secreted CEL7A (CBH1) in the parental strain and the Δgrd1 strain are less pronounced. Moreover, decreased abundance of CEL7A (CBH1) is seen not only in light but also in darkness. Consequently, the mechanisms regulating cellulase gene expression at a posttranscriptional level are likely to be influenced by upstream signaling pathways, one of which signals the presence (or increased abundance) of the product of the enzymatic reaction of GRD1.
We conclude not only that components of signal transduction pathways are responsible for the maintenance of balanced light responses but that metabolic enzymes, such as GRD1, can also have a light-dependent effect. The lack of GRD1 leads to altered light responsiveness of cel7a (cbh1) transcription and, hence, affects the interpretation and output of signals transmitted under the conditions tested. Consequently, a distinct effect of light pulses on cellulase gene expression should also be considered, with experiments aimed at the analysis of metabolic genes not supposed to be part of a signal transduction cascade.
Supplementary Material
ACKNOWLEDGMENTS
Our work was supported by grants from the Austrian Science Fund (FWF; P-20004, P-21072, and V152-B20) to M.S.
Footnotes
Supplemental material for this article may be found at http://aem.asm.org/.
Published ahead of print on 20 May 2011.
The authors have paid a fee to allow immediate free access to this article.
REFERENCES
- 1. Atoui A., et al. 2010. Cross-talk between light and glucose regulation controls toxin production and morphogenesis in Aspergillus nidulans. Fungal Genet. Biol. 47: 962–972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Brown D. W., Adams T. H., Keller N. P. 1996. Aspergillus has distinct fatty acid synthases for primary and secondary metabolism. Proc. Natl. Acad. Sci. U. S. A. 93: 14873–14877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Buchert J., et al. 1998. Applications of Trichoderma reesei enzymes in the pulp and paper industry, p. 343–363. In Harman G. E., Kubicek C. P. (ed.) Trichoderma and Gliocladium, vol. 2 Taylor and Francis, London, United Kingdom [Google Scholar]
- 4. Canevascini G., Coudray M., Southgate R., Meier H. 1979. Induction and catabolite repression of cellulase synthesis in the thermophilic fungus Sporotrichum thermophile. Microbiology 110: 291–303 [Google Scholar]
- 5. Carlile M. J. 1965. The photobiology of fungi. Annu. Rev. Plant Physiol. Plant Mol. Biol. 16: 175–202 [Google Scholar]
- 6. Castellanos F., et al. 2010. Crucial factors of the light perception machinery and their impact on growth and cellulase gene transcription in Trichoderma reesei. Fungal Genet. Biol. 47: 468–476 [DOI] [PubMed] [Google Scholar]
- 7. Cherry J. R., Fidantsef A. L. 2003. Directed evolution of industrial enzymes: an update. Curr. Opin. Biotechnol. 14: 438–443 [DOI] [PubMed] [Google Scholar]
- 8. Chiang Y. M., et al. 2009. A gene cluster containing two fungal polyketide synthases encodes the biosynthetic pathway for a polyketide, asperfuranone, in Aspergillus nidulans. J. Am. Chem. Soc. 131: 2965–2970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chiang Y. M., et al. 2008. Molecular genetic mining of the Aspergillus secondary metabolome: discovery of the emericellamide biosynthetic pathway. Chem. Biol. 15: 527–532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chomczynski P., Sacchi N. 1987. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal. Biochem. 162: 156–159 [DOI] [PubMed] [Google Scholar]
- 11. Dong W., et al. 2008. Systems biology of the clock in Neurospora crassa. PLoS One 3: e3105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Druzhinina I. S., Komon-Zelazowska M., Atanasova L., Seidl V., Kubicek C. P. 2010. Evolution and ecophysiology of the industrial producer Hypocrea jecorina (Anamorph Trichoderma reesei) and a new sympatric agamospecies related to it. PLoS One 5: e9191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Druzhinina I. S., Schmoll M., Seiboth B., Kubicek C. P. 2006. Global carbon utilization profiles of wild-type, mutant, and transformant strains of Hypocrea jecorina. Appl. Environ. Microbiol. 72: 2126–2133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Eisen M., Spellman P., Brown P., Botstein D. 1998. Cluster analysis and display of genome-wide expression patterns. Proc. Natl. Acad. Sci. U. S. A. 95: 14863–14868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. el-Gogary S., Leite A., Crivellaro O., Eveleigh D., El-Dorry H. 1989. Mechanism by which cellulose triggers cellobiohydrolase I gene expression in Trichoderma reesei. Proc. Natl. Acad. Sci. U. S. A. 86: 6138–6141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Fowler T., Brown R. D. 1992. The bgI1 gene encoding extracellular β-glucosidase from Trichoderma reesei is required for rapid induction of the cellulase complex. Mol. Microbiol. 6: 3225–3235 [DOI] [PubMed] [Google Scholar]
- 17. Fritscher C., Messner R., Kubicek C. P. 1990. Cellobiose metabolism and cellobiohydrolase I biosynthesis by Trichoderma reesei. Exp. Mycol. 14: 405–415 [Google Scholar]
- 18. Galagan J. E., et al. 2003. The genome sequence of the filamentous fungus Neurospora crassa. Nature 422: 859–868 [DOI] [PubMed] [Google Scholar]
- 19. Galante Y., De Conti A., Monteverdi R. 1998. Application of Trichoderma enzymes in the food and feed industries, p. 327–342. In Harman G. E., Kubicek C. P., (ed.) Trichoderma and Gliocladium, vol. 2 Taylor and Francis, London, United Kingdom [Google Scholar]
- 20. Galante Y., De Conti A., Monteverdi R. 1998. Application of Trichoderma enzymes in the textile industry, p. 311–326. In Harman G. E., Kubicek C. P., (ed.) Trichoderma and Gliocladium, vol. 2 Taylor and Francis, London, United Kingdom [Google Scholar]
- 21. Gremel G., Dorrer M., Schmoll M. 2008. Sulphur metabolism and cellulase gene expression are connected processes in the filamentous fungus Hypocrea jecorina (anamorph Trichoderma reesei). BMC Microbiol. 8: 174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gruber F., Visser J., Kubicek C., Graaff L. 1990. Cloning of the Trichoderma reesei pyrG gene and its use as a homologous marker for a high-frequency transformation system. Curr. Genet. 18: 447–451 [Google Scholar]
- 23. Gruber F., Visser J., Kubicek C. P., de Graaff L. H. 1990. The development of a heterologous transformation system for the cellulolytic fungus Trichoderma reesei based on a pyrG-negative mutant strain. Curr. Genet. 18: 71–76 [DOI] [PubMed] [Google Scholar]
- 24. Guangtao Z., et al. 2009. Gene targeting in a nonhomologous end joining deficient Hypocrea jecorina. J. Biotechnol. 139: 146–151 [DOI] [PubMed] [Google Scholar]
- 25. Harmer S., et al. 2000. Orchestrated transcription of key pathways in Arabidopsis by the circadian clock. Science 290: 2110–2113 [DOI] [PubMed] [Google Scholar]
- 26. Hartl L., Kubicek C. P., Seiboth B. 2007. Induction of the gal pathway and cellulase genes involves no transcriptional inducer function of the galactokinase in Hypocrea jecorina. J. Biol. Chem. 282: 18654–18659 [DOI] [PubMed] [Google Scholar]
- 27. Jiang H., Kang D., Alexandre D., Fisher P. 2000. RaSH, a rapid subtraction hybridization approach for identifying and cloning differentially expressed genes. Proc. Natl. Acad. Sci. U. S. A. 97: 12684–12689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kasuga T., et al. 2005. Long-oligomer microarray profiling in Neurospora crassa reveals the transcriptional program underlying biochemical and physiological events of conidial germination. Nucleic Acids Res. 33: 6469–6485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kawamori M., Morikawa Y., Takasawa S. 1985. Inductive formation of cellulases by L-sorbose in Trichoderma reesei. Appl. Microbiol. Biotechnol. 22: 235–236 [Google Scholar]
- 30. Kim S., et al. 2001. A gene expression map for Caenorhabditis elegans. Sci. STKE 293: 2087–2092 [DOI] [PubMed] [Google Scholar]
- 31. Kubicek C. 1987. Involvement of a conidial endoglucanase and a plasma-membrane-bound β-glucosidase in the induction of endoglucanase synthesis by cellulose in Trichoderma reesei. Microbiology 133: 1481–1487 [DOI] [PubMed] [Google Scholar]
- 32. Kubicek C. P., Mikus M., Schuster A., Schmoll M., Seiboth B. 2009. Metabolic engineering strategies for the improvement of cellulase production by Hypocrea jecorina. Biotechnol. Biofuels 2: 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Larkin M., et al. 2007. Clustal W and Clustal X version 2.0. Bioinformatics 23: 2947–2948 [DOI] [PubMed] [Google Scholar]
- 34. Loewenberg J. R. 1984. Sophorose induction of an intracellular b-glucosidase in Trichoderma. Arch. Microbiol. 137: 53–57 [DOI] [PubMed] [Google Scholar]
- 35. Mach R., et al. 1995. The bgl1 gene of Trichoderma reesei QM9414 encodes an extracellular, cellulose-inducible-glucosidase involved in cellulase induction by sophorose. Mol. Microbiol. 16: 687–697 [DOI] [PubMed] [Google Scholar]
- 36. Mandels M., Andreotti R. E. 1978. Problems and challenges in the cellulose to cellulase fermentation. Process Biochem. 13: 6–13 [Google Scholar]
- 37. Mandels M., Parrish F. W., Reese E. T. 1962. Sophorose as an inducer of cellulase in Trichoderma viride. J. Bacteriol. 83: 400–408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Mandels M., Reese E. T. 1960. Induction of cellulase in fungi by cellobiose. J. Bacteriol. 79: 816–826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Mandels M., Reese E. T. 1957. Induction of cellulase in Trichoderma viride as influenced by carbon sources and metals. J. Bacteriol. 73: 269–278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Margolles-Clark E., Ihnen M., Penttilä M. 1997. Expression patterns of ten hemicellulase genes of the filamentous fungus Trichoderma reesei on various carbon sources. J. Biotechnol. 57: 167–179 [Google Scholar]
- 41. Martinez D., et al. 2008. Genome sequencing and analysis of the biomass-degrading fungus Trichoderma reesei (syn. Hypocrea jecorina). Nat. Biotechnol. 26: 553–560 [DOI] [PubMed] [Google Scholar]
- 42. Messner R., Gruber F., Kubicek C. 1988. Differential regulation of synthesis of multiple forms of specific endoglucanases by Trichoderma reesei QM9414. J. Bacteriol. 170: 3689–3693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Messner R., Kubicek-Pranz E., Gsur A., Kubicek C. 1991. Cellobiohydrolase II is the main conidial-bound cellulase in Trichoderma reesei and other Trichoderma strains. Arch. Microbiol. 155: 601–606 [DOI] [PubMed] [Google Scholar]
- 44. Metz B., de Vries R., Polak S., Seidl V., Seiboth B. 2009. The Hypocrea jecorina (syn. Trichoderma reesei) lxr1 gene encodes a D-mannitol dehydrogenase and is not involved in L-arabinose catabolism. FEBS Lett. 583: 1309–1313 [DOI] [PubMed] [Google Scholar]
- 45. Meyer H., Canevascini G. 1981. Separation and some properties of two intracellular β-glucosidases of Sporotrichum (Chrysosporium) thermophile. Appl. Environ. Microbiol. 41: 924–931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Mischak H., et al. 1989. Monoclonal antibodies against different domains of cellobiohydrolase I and II from Trichoderma reesei. Biochim. Biophys. Acta 990: 1–7 [DOI] [PubMed] [Google Scholar]
- 47. Morawetz R., Gruber F., Messner R., Kubicek C. 1992. Presence, transcription and translation of cellobiohydrolase genes in several Trichoderma species. Curr. Genet. 21: 31–36 [Google Scholar]
- 48. Pail M., et al. 2004. The metabolic role and evolution of L-arabinitol 4-dehydrogenase of Hypocrea jecorina. Eur. J. Biochem. 271: 1864–1872 [DOI] [PubMed] [Google Scholar]
- 49. Ruiz-Roldan M., Garre V., Guarro J., Marine M., Roncero M. 2008. Role of the White collar 1 photoreceptor in carotenogenesis, UV resistance, hydrophobicity and virulence of Fusarium oxysporum. Eukaryot. Cell 7: 1227–1230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Sambrook J., Fritsch E. F., Maniatis T. 1989. Molecular cloning: a laboratory manual, 2nd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 51. Schmoll M., Franchi L., Kubicek C. P. 2005. Envoy, a PAS/LOV domain protein of Hypocrea jecorina (anamorph Trichoderma reesei), modulates cellulase gene transcription in response to light. Eukaryot. Cell 4: 1998–2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Schmoll M., Kubicek C. P. 2005. ooc1, a unique gene expressed only during growth of Hypocrea jecorina (anamorph: Trichoderma reesei) on cellulose. Curr. Genet. 48: 126–133 [DOI] [PubMed] [Google Scholar]
- 53. Schmoll M., Kubicek C. P. 2003. Regulation of Trichoderma cellulase formation: lessons in molecular biology from an industrial fungus. A review. Acta Microbiol. Immunol. Hung. 50: 125–145 [DOI] [PubMed] [Google Scholar]
- 54. Schmoll M., Schuster A., Silva Rdo N., Kubicek C. P. 2009. The G-alpha protein GNA3 of Hypocrea jecorina (anamorph Trichoderma reesei) regulates cellulase gene expression in the presence of light. Eukaryot. Cell 8: 410–420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Schmoll M., Seibel C., Tisch D., Dorrer M., Kubicek C. P. 2010. A novel class of peptide pheromone precursors in ascomycetous fungi. Mol. Microbiol. 77: 1483–1501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Schmoll M., Zeilinger S., Mach R., Kubicek C. 2004. Cloning of genes expressed early during cellulase induction in Hypocrea jecorina by a rapid subtraction hybridization approach. Fungal Genet. Biol. 41: 877–887 [DOI] [PubMed] [Google Scholar]
- 57. Seibel C., et al. 2009. Light-dependent roles of the G-protein alpha subunit GNA1 of Hypocrea jecorina (anamorph Trichoderma reesei). BMC Biol. 7: 58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Seiboth B., Gamauf C., Pail M., Hartl L., Kubicek C. P. 2007. The D-xylose reductase of Hypocrea jecorina is the major aldose reductase in pentose and D-galactose catabolism and necessary for beta-galactosidase and cellulase induction by lactose. Mol. Microbiol. 66: 890–900 [DOI] [PubMed] [Google Scholar]
- 59. Seiboth B., Pakdaman B. S., Hartl L., Kubicek C. P. 2007. Lactose metabolism in filamentous fungi: how to deal with an unknown substrate. Fungal Biol. Rev. 21: 42–48 [Google Scholar]
- 60. Shoemaker S., et al. 1983. Molecular cloning of exo-cellobiohydrolase i derived from Trichoderma Reesei strain L27. Nat. Biotechnol. 1: 691–696 [Google Scholar]
- 61. Sternberg D., Mandels G. 1979. Induction of cellulolytic enzymes in Trichoderma reesei by sophorose. J. Bacteriol. 139: 761–769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Sternberg D., Mandels G. 1980. Regulation of the cellulolytic system in Trichoderma reesei by sophorose: induction of cellulase and repression of beta-glucosidase. J. Bacteriol. 144: 1197–1199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Szewczyk E., et al. 2008. Identification and characterization of the asperthecin gene cluster of Aspergillus nidulans. Appl. Environ. Microbiol. 74: 7607–7612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Tamura K., Dudley J., Nei M., Kumar S. 2007. MEGA4: molecular evolutionary genetics analysis (MEGA) software version 4.0. Mol. Biol. Evol. 24: 1596–1599 [DOI] [PubMed] [Google Scholar]
- 65. Teeri T., Salovuori I., Knowles J. 1983. The molecular cloning of the major cellulase gene from Trichoderma reesei. Nat. Biotechnol. 1: 696–699 [Google Scholar]
- 66. Tian C., et al. 2009. Systems analysis of plant cell wall degradation by the model filamentous fungus Neurospora crassa. Proc. Natl. Acad. Sci. U. S. A. 106: 22157–22162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Tisch D., Schmoll M. 2010. Light regulation of metabolic pathways in fungi. Appl. Microbiol. Biotechnol. 85: 1259–1277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Vaheri M., Leisola M., Kauppinen V. 1979. Transglycosylation products of cellulase system of Trichoderma reesei. Biotechnol. Lett. 1: 41–46 [Google Scholar]
- 69. Vargovic P., Pokorný R., Hölker U., Hofer M., Varecka L. 2006. Light accelerates the splicing of srh1 homologue gene transcripts in aerial mycelia of Trichoderma viride. FEMS Microbiol. Lett. 254: 240–244 [DOI] [PubMed] [Google Scholar]
- 70. Wortman J., et al. 2009. The 2008 update of the Aspergillus nidulans genome annotation: a community effort. Fungal Genet. Biol. 46: S2–S13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Yanisch-Perron C., Vieira J., Messing J. 1985. Improved M13 phage cloning vectors and host strains: nucleotide sequences of the M13mp18 and pUC19 vectors. Gene 33: 103–119 [DOI] [PubMed] [Google Scholar]
- 72. Yu J. H., Keller N. 2005. Regulation of secondary metabolism in filamentous fungi. Annu. Rev. Phytopathol. 43: 437–458 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.