

Abstract
H-ras, N-ras, and K-ras are canonical ras gene family members frequently activated by point mutation in human cancers and coding for 4 different, highly related protein isoforms (H-Ras, N-Ras, K-Ras4A, and K-Ras4B). Their expression is nearly ubiquitous and broadly conserved across eukaryotic species, although there are quantitative and qualitative differences of expression depending on the tissue and/or developmental stage under consideration. Extensive functional studies have determined during the last quarter century that these Ras gene products are critical components of signaling pathways that control eukaryotic cell proliferation, survival, and differentiation. However, because of their homology and frequent coexpression in various cellular contexts, it remained unclear whether the different Ras proteins play specific or overlapping functional roles in physiological and pathological processes. Initially, their high degree of sequence homology and the observation that all Ras isoforms share common sets of downstream effectors and upstream activators suggested that they were mostly redundant functionally. In contrast, the notion of functional specificity for each of the different Ras isoforms is supported at present by an increasing body of experimental observations, including 1) the fact that different ras isoforms are preferentially mutated in specific types of tumors or developmental disorders; 2) the different transforming potential of transfected ras genes in different cell contexts; 3) the distinct sensitivities exhibited by the various Ras family members for modulation by different GAPs or GEFs; 4) the demonstration that different Ras isoforms follow distinct intracellular processing pathways and localize to different membrane microdomains or subcellular compartments; 5) the different phenotypes displayed by genetically modified animal strains for each of the 3 ras loci; and 6) the specific transcriptional networks controlled by each isoform in different cellular settings.
Keywords: Ras, Ras isoforms, canonical Ras, functional specificity, redundancy
Introduction
The Ras Subfamily
The 3 canonical members of the Ras gene family (H-ras, N-ras, and K-ras) were identified more than a quarter century ago because of their frequent oncogenic activation in human tumors. They are the founding members of the wider Ras superfamily including more than 150 small GTPases, divided into at least 5 distinct subfamilies (Ras, Rho/Rac, Rab, Arf, and Ran) on the basis of primary sequence relationships. In particular, the Ras subfamily encompasses the H-ras, N-ras, and K-ras genes together with the closely related R-Ras/TC21, Ral, and Rap loci.1-5
All Ras superfamily proteins share very similar molecular structures and a common ability to bind and hydrolyze guanine nucleotides. The Ras proteins are continually cycling between active (GTP bound) and inactive (GDP bound) conformational states dependent on structural changes occurring mostly in the 2 motile switch I and switch II regions, which are also responsible for the functional interactions of these proteins with negative (GAP) and positive (GEF) cellular regulators.2,6-13 The binary behavior aspects of these proteins enable them to function as molecular switches in a broad range of signaling processes related to the transduction of extracellular signals to the interior of cells. Oncogenic mutations at positions 12, 13, or 61 of the H-ras, N-ras, and K-ras genes are among the most common genetic lesions in mammalian tumors.14-16 These mutations result in significant impairment of the overall GTPase activity of the carrier Ras proteins and lock them into a constitutively activated state in which they signal to downstream effectors, even in the absence of extracellular stimuli.
Expression of the H-ras, N-ras, and K-ras genes is nearly ubiquitous and broadly conserved across species, although there are specific differences of expression levels depending on the tissue and the developmental stage under study.17-22 In particular, these 3 loci are known to code for 4 different protein isoforms (H-Ras, N-Ras, K-Ras4A, and K-Ras4B), the latter 2 resulting from alternative splicing of exon 4 of the K-ras locus.14,23-26 These 4 Ras isoform proteins are highly homologous regarding their primary amino acid sequence (~80%), and the differences among them concentrated in the so-called hypervariable region (HVR) of their C-terminal domains.4,23,26 These mammalian ras genes are expressed in all cell lineages and organs, although there are differences in expression through prenatal and postnatal development, and certain adult tissues preferentially express one or other member of the family.19,27,28
The mammalian Ras subfamily proteins (H-Ras, N-Ras, K-Ras4A, and K-Ras4B) are highly conserved across different species and play functionally significant roles in numerous cellular processes, including proliferation, differentiation, and cell death. The high number of Ras activators and effectors identified in mammalian cells places the Ras proteins at the crossroads of a staggering number of cellular signaling networks. Such a central role of Ras gene products in normal cell signaling is also consistent with the high frequency of oncogenic activation of ras genes in human cancers. The importance of Ras signaling in tumor initiation and maintenance is emphasized not only by the prevalence of ras mutations but also by the deregulation of many of its activator or effector pathways, thus affecting Ras pathway activity.4,15,26,29-32 Indeed, the study of the contribution of Ras signaling to tumor development has greatly improved our current understanding of the molecular basis for the pathogenesis of many human cancers.33-37
Emerging Notions of Specificity
Historically, the high degree of sequence identity coupled to the early reports describing the nearly identical ability of mutated H-ras, N-ras, and K-ras oncogenes to cause transformation of NIH3T3 and other cell types and to activate the same, shared downstream cellular effectors supported for a long time the idea of functional in vivo overlapping for the protein products of these 3 distinct ras genes.15,19,27,38 Indeed, because of these preconceived notions of functional redundancy, a majority of the earlier Ras studies were done using H-Ras only, with the underlying assumption that the different Ras proteins were functionally overlapping and interchangeable. However, the accumulation of additional experimental data soon began to suggest otherwise, supporting the possibility of distinct functional roles for each Ras family member.3,5,26,39 Initial suspicions of the possibility of functional specificity for each distinct Ras isoform were raised by observations such as 1) the prevalent presence of specific ras oncogenes in particular forms of human tumors15,40-44; 2) the high conservation across mammalian species of the distinct amino acid sequences of the different Ras isoforms in their C-terminal HVRs; or 3) the different patterns of expression, intracellular processing, and subcellular location displayed by the fully processed, mature protein products of the different ras gene isoforms.
It was apparent from the early beginning studies that different human tumor types showed preferential oncogenic activation of specific Ras isoforms. Thus, K-ras mutations occur at very high frequency in pancreatic, colon, or lung cancer, whereas N-ras and H-ras mutations are extremely uncommon in those tumors; conversely, N-ras mutations occur in a high percentage of acute leukemias, whereas H-ras and K-ras mutations are much less common there40-49 (Fernandez-Medarde and Santos, current journal issue). The simplest interpretation for such observations would be that the oncogenic H-Ras, N-Ras, and K-Ras proteins display different biological specificities.
The notion of functional specificity for the different Ras subfamily protein isoforms was also consistent with and supported by the distinct, unique patterns of expression, intracellular processing, and subcellular location described for each of the different Ras isoforms in various types of cells, tissues, or organisms. Although N-ras, H-ras, and K-ras are nearly ubiquitously expressed, the quantitative ratios among their expressed isoforms may vary widely depending on the cell lineage, tissue, or even developmental stage under analysis. Thus, whereas K-Ras4B is the most frequently expressed K-ras isoform under normal conditions, expression of K-Ras4A is also significantly induced during differentiation of pluripotent embryonic stem cells in vitro.50 In mice, the level of H-ras transcripts is highest in the brain, muscle, and skin and lowest in the liver; K-ras transcripts are more abundant in the gut, lung, and thymus and are rare in skin and skeletal muscle; finally, N-ras transcripts are more prevalent in the testis and thymus.19 Differential expression of these 3 ras genes has also been observed during mouse embryonic development, with N-ras expression being highest at day 10 of gestation and K-ras expression being lowest toward the end of gestation.19,51 In any case, despite the variations in relative expression levels, all 3 ras genes are concurrently expressed in most mouse and human tissues.27,28
Comparison of the highly conserved primary sequences of these 4 highly homologous Ras protein isoforms shows that their differences are concentrated in the HVR of their C-terminal domains4,23,52 (Fig. 1). Interestingly, the primary amino acid sequence of the HVR of the different Ras gene products is very well conserved among vertebrate species as distant as the human and carp, suggesting that such variability reflects the possible functional specificity of each specific HVR protein domain instead of being just a random phenomenon.53 In addition, the specific differences observed in the processes of intracellular processing and localization of the different Ras protein isoforms newly synthesized in ribosomes may provide further biological basis for isoform-specific function.17,18,20,21,54-60 Indeed, although the highly conserved domains of these Ras isoforms include the same effector-binding loops and regulatory regions, the access to their potential interacting partners may be governed to a large extent through their differing localizations within cells.61 The tissue-specific requirements of the various Ras isoforms may be explained by the contrasting abundance of their protein products in different cell types.19,27,28 These requirements may also be explained by the disparity in the distribution and the activation outputs of these isoforms within intracellular membrane compartments of different cell types. For example, plasma membrane–tethered K-Ras can induce transformation, whereas mitochondrial K-Ras induces apoptosis.62 Moreover, although activated H-Ras is associated with both the Golgi and the endoplasmic reticulum (ER), only the ER-associated form can activate Raf1-Erk signaling.61 In contrast to the ER-tethered H-Ras,61 the Golgi-associated forms do not induce transformation,56 suggesting that the subcellular distribution of Ras effectors determines their activation by Ras and, in turn, regulates Ras functions.
Figure 1.
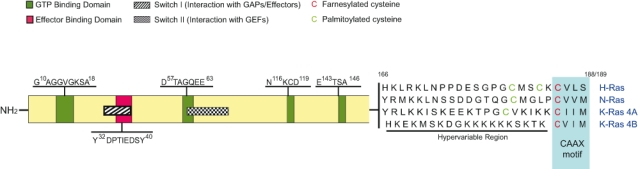
Primary structure of Ras proteins. The structure of Ras proteins includes highly conserved domains responsible for binding and hydrolysis of guanine nucleotides, functional interaction with activators and effectors, and attachment to membranes. Green boxes refer to the 4 epitopes responsible for interaction with guanine nucleotides. The red box represents the area of interaction with downstream effectors. The areas represented by the striped box (switch I) and the squared box (switch II) undergo conformational changes depending on Ras binding to GDP or GTP. The unique cysteine residue located in the CAAX box (red) is farnesylated, whereas the immediately upstream cysteine residues (green) located in the hypervariable region may be palmitoylated.
In summary, despite the enormous body of experimental knowledge accumulated over more than 30 years on different aspects of the Ras gene subfamily members, there still remain many fundamental, unanswered questions regarding the biology and functional significance of the different Ras isoforms. Among these, a critical issue is the complete clarification, and quantitative characterization, of the degree of functional specificity or overlapping exhibited by the different Ras isoforms in the various cell lineages and tissues where they are all present and simultaneously expressed.
We will focus the following sections on reviewing experimental evidence from various Ras research areas, providing significant clues to address the issue of functional specificity or redundancy of the different isoforms of the Ras subfamily. In particular, we will discuss 1) studies of Ras function in the context of different pathological conditions associated with Ras mutations, including cancer and hereditary developmental syndromes; 2) studies of the functional role of Ras proteins in various physiological cellular contexts; 3) specific aspects of the expression, biosynthesis, and intracellular processing of different Ras isoforms; 4) analysis of distinct phenotypes displayed by various transgenic and knockout strains for each Ras family member; and 5) analysis of transcriptional networks dependent on the presence of specific Ras isoforms.
Specificity of Ras Isoforms in Pathological Contexts
The mitogenic potency of the ras gene products has been very widely documented in the scientific literature. The mutationally activated forms of H-ras, K-ras, and N-ras can efficiently transform cells in vivo and in vitro and have also been detected in a broad spectrum of human tumors and developmental syndromes. The preferential association of particular ras oncogene isoforms with specific forms of sporadic tumors or hereditary syndromes is a powerful argument favoring the notion of functional specificity of the different Ras isoforms in each of those different pathological settings.
Ras Isoform Mutations in Human Tumors
The most frequent mechanism of oncogenic activation involves point mutations affecting the interaction of Ras with guanine nucleotides. Mutations detected in naturally occurring ras oncogenes affect codons 12, 13, 59, and 61. These mutations result in inhibition of GTP hydrolysis, either by diminishing GTPase activity or (for codon 59) by modulating the rate of guanine nucleotide exchange.26
Oncogenic ras mutations are found in a great variety of human cancers, although their incidence varies considerably with tumor type.29,43,63 Qualitatively, H-ras mutations have been reported in melanoma, bladder, thyroid, and mammary carcinoma; K-ras mutations have been found in bladder, ovarian, thyroid, lung, colon and rectum, and pancreatic carcinoma; neuroblastoma; rhabdomyosarcoma; and acute nonlymphocytic leukemia. Finally, N-ras mutations were also described in melanoma, thyroid carcinoma, teratocarcinoma, fibrosarcoma, neuroblastoma, rhabdomyosarcoma, Burkitt lymphoma, acute promyelocytic leukemia, T cell leukemia, and chronic myelogenous leukemia45-49 (Fernandez-Medarde and Santos, current journal issue). Quantitative analysis demonstrates the preferential association of some of the ras oncogenes with specific forms of human tumors. Thus, K-ras–activating missense mutations are frequently detected in non–small cell lung cancer (15%-20%),40 colon adenomas (40%),41 and pancreatic adenocarcinomas (95%),42 making it the single most common mutationally activated human oncoprotein. Likewise, N-ras mutations are frequently present in hematological malignancies (20%-30%) such as acute myeloblastic leukemia.43,44 In contrast, other tumor types do not show any significant preference for a specific ras oncogene isoform. For example, more than half of malignant thyroid tumors (poorly differentiated or undifferentiated) harbor a mutation in K-ras, H-ras, or N-ras.64 Furthermore, mutations in all 3 ras isoforms may occur within the same tumor in some thyroid adenomas and carcinomas, suggesting that each isoform may contribute to different aspects of tumoral growth. Simultaneous mutations in K-ras and N-ras have also been detected in multiple myeloma.65 Finally, although ras mutations are rare in breast cancer, point mutations in H-ras or K-ras have been detected in primary carcinomas and in some mammary tumor-derived cell lines.47,66
Ras Isoform Mutations in Developmental Disorders
The implication of aberrant Ras signaling in other noncancerous, pathogenic disorders including different human developmental defect syndromes is also demonstrated by the discovery in patients of germline mutations in different members of the ras gene family or in other components of Ras signaling pathways.46,48 The developmental disorders associated with Ras pathway mutations frequently share phenotypic features including facial abnormalities, heart defects, impaired growth and development, and in some instances, a predisposition to specific cancers.45,48
Germline mutations in the H-ras and K-ras genes have been found, respectively, in patients suffering from Costello syndrome (CS) or Noonan syndrome (NS)48,67-71 (Fernandez-Medarde and Santos, current journal issue). CS and NS share some phenotypic features but also display specific phenotypes of their own. The NS patients are characterized by short stature, distinct facial anomalies, a typical spectrum of congenital heart defects including pulmonic stenosis, hypertrophic cardiomyopathy, and septal defect and developmental delays.48 The CS patients display some Noonan-like characters, along with other unique phenotypic features including nasal papillomata, loose skin, and strong predisposition to tumors, mainly rhabdomyosarcoma, ganglioneuroblastoma, and bladder cancer.67,68
Different reports have identified K-ras germline mutations in 2% to 4% of affected NS individuals.69-71 The K-ras mutations described in NS patients are spread through different domains of the primary sequence of K-Ras, including the C-terminal region.69-71 On the other hand, about 90% of CS patients harbor germline mutations in the H-ras gene. These germline H-ras mutations affect the same structural domains that are mutated in cancer.72-76 Indeed, a majority of the H-ras alleles identified in CS patients introduce amino acid substitutions in codons 12 and 13 that also occur as somatic mutations in tumors. However, the most common substitution identified in CS (G12S) is uncommon in cancer.72,73 Interestingly, some of the H-ras and K-ras germline mutations detected in NS and CS patients fall out of the usual sites of ras oncogenic activation and affect amino acid residues located in the C-terminal regions of these Ras proteins. For example, significant numbers of amino acid substitutions in the α-5 helix of K-Ras4B (including V152G, D153V, and F156I) have been detected in NS patients.69-71 Likewise, H-ras germline mutations causing substitutions such as K117R and A146T have also been reported in CS patients.75,76 Structural and functional analyses of this type of mutations in the C-terminal region suggest that the resulting mutant Ras proteins exhibit increased rate of guanine nucleotide dissociation favoring the active, GTP-bound Ras conformation.30,72,76 Finally, germline mutations in N-ras have not yet been associated with the so-called neurocardiofaciocutaneous (NCFC) syndromes. However, heterozygous germline activating mutations in N-ras (G13D) have been found that are linked to selective immune abnormalities, such as autoimmune lymphoproliferative syndrome and hematological malignancies.77
Mechanistic Aspects Underlying Functional Specificity/Redundancy of Ras Isoforms
An important, still unresolved question concerns our understanding of the biological basis for the preferential presence of particular ras isoform mutations in the context of specific pathological conditions such as particular tumors or other developmental syndromes. Considering that mutant ras isoforms often display higher biological potency than their wild-type counterparts, an interesting hypothesis is that the specific pathological phenotypes are the net result of exacerbated cellular signaling resultant from superimposing the signaling outcomes mediated by the actual cohort of wild-type and mutated Ras isoforms present in the affected cell types. It is very likely that 1) the intrinsically different biological potency of the different ras isoforms, together with 2) the different cellular contexts in which these isoforms are expressed, are the essential factors contributing to determining the functional specificity or redundancy observed in each particular case.
Different biological potency of Ras isoforms
A number of separate reports have documented that the H-Ras, N-Ras, and K-Ras isoforms display very distinct transforming potential when transfected into different, specific cell lines.78-80 For example, it has been reported that the H-ras oncogene exhibits consistently greater transforming ability than oncogenic N-ras or K-ras when tested in a range of fibroblast-based transformation assays.78,79 In contrast, the N-ras isoform showed greater transforming potential than the other 2 in the human hemopoietic cell line TF-1.79 As the distinct transforming potential of oncogenic Ras isoforms does not seem to result from differences in expression level or stability of the encoded Ras proteins, these observations suggest that there are tissue-specific components in the different biological potencies shown by each of the 3 ras oncogenes.79 Consistent with this notion, a separate report has described the differential impact of the effector loop mutation P34G on the biological activity of the 3 Ras isoforms in the context of a hyperactive mutation (G12V).81 Interestingly, although the P34G mutation maps to a region showing almost complete homology among the 3 Ras isoforms, very different biological phenotypes and downstream signaling outcomes were observed depending on the particular ras isoform under consideration. Thus, whereas the H-RasV12G34 mutant retained the ability to cause morphological transformation of fibroblasts, the N-RasV12G34 and K-RasV12G34 mutants were completely devoid of transforming activity. Furthermore, whereas the actin cytoskeleton of fibroblasts overexpressing H-RasV12G34 was identical to that of RasV12-transformed fibroblasts, cells overexpressing the N- or K-RasV12G34 mutants showed an actin cytoskeleton typical of untransformed cells.81
Further studies in other biological settings confirm the different biological potency of the 3 Ras isoforms.82-85 For example, using a bone marrow transduction/transplantation model, it has been shown that all H-Ras, N-Ras, and K-Ras have the potential to induce myeloid leukemia in mice, but they differ in terms of their potency and the resulting disease phenotype.82 Thus, oncogenic N-Ras can induce acute myeloid leukemia (AML)– or chronic myelomonocytic leukemia (CMML)–like disease in mice, whereas expression of oncogenic K-Ras invariably induces a CMML-like disease, and oncogenic H-Ras always induces an AML-like disease in the same mouse model.82 Finally, there is also experimental evidence indicating that, besides their mutant, activated counterparts, wild-type Ras may also contribute to transformation.32,86 In support of this notion, it has also been demonstrated that oncogenic H-Ras requires wild-type N-Ras for transformation.80,87 Other reports have described the convergence, in wild-type cells, of separate signals originated from different Ras isoforms to generate a common final biological output. Thus, K-Ras and N-Ras were reported to jointly exert their influence on the cytoskeleton by affecting migration, invasion, and anchorage-independent growth via a mechanism in which N-Ras influences adhesion through Raf and RhoA, whereas K-Ras coordinates motility by signaling through Akt and Cdc42.86
Effect of cellular context on the functional properties of Ras isoforms
It is clearly apparent that the biological and cellular context in which the different Ras isoforms are expressed plays also a significant role in determining the observable, functional properties displayed by those isoforms. For example, there are clear phenotypic differences associated with oncogenic activation of either endogenous K-ras or N-ras in vivo in the colonic epithelium.88 Thus, K-ras activation in this epithelium stimulated hyperproliferation in a MEK-dependent manner and, in the context of an Apc mutant colonic tumor, led to defects in terminal differentiation and expansion of putative stem cells within the tumor epithelium. In contrast, oncogenic N-ras did not alter the growth properties of the epithelium but was able to confer resistance to apoptosis, suggesting that these functional differences may account, at least in part, for the high frequency of K-ras mutations detected in colon cancer.88
Ras protein isoforms appear also to play a significant role in tumor progression and the metastatic processes as the result of their differential involvement in regulating the turnover of focal adhesions and cell movement.89 For example, some reports have suggested that K-Ras is more effective than H-Ras in stimulating cell motility.90,91 One contributing factor might be the preferential activation of Rac by K-Ras because cell movement requires the coordinated assembly and disassembly of stress fibers.92 Furthermore, in colon epithelial cells, K-Ras but not H-Ras disrupts basolateral polarity by altering the expression of different intercellular adhesion proteins, such as β1 integrin and N-cadherin among others.93 The preferential ability of K-Ras to induce loss of cell-cell and cell-substratum adherence and to stimulate cell motility could account for the highly invasive and metastatic phenotype of K-Ras–derived tumor cells.94,95
It is clearly established that the frequency of K-ras mutations in human malignancies is considerably higher as compared with the other Ras isoforms. It has also been shown that K-Ras displays reduced capacity to induce apoptosis presumably because of its ability to activate more efficiently the antiapoptotic cascade.91 As the induction of apoptosis or cell cycle arrest are thought to represent safeguard mechanisms limiting tumor growth, the compromised ability of K-Ras to induce apoptosis might explain, at least in part, the selective growth advantage of tumor cells harboring K-Ras mutations.91
Finally, the embryological origin of the hosting cells or tissues may also be a determining factor influencing the actual phenotypes caused by different Ras isoforms. A number of reports have described that activation of different Ras isoforms promotes distinct phenotypes depending upon the cell type in which they are expressed. Consistent with this notion is the reported ability of activated H-Ras to induce adipocytic or neuronal differentiation.96-98 Oncogenic H-Ras was also reported to promote tumorigenic transformation of the bladder and salivary gland,99,100 2 tissues arising from the transitional zone, a region where endoderm and ectoderm meet. On the other hand, myeloid malignancies, germ cell tumors, congenital melanocytic nevi, and cutaneous melanomas, derived from the neural crest (but not mucosal melanomas, which are not derived from the neural crest), are frequently associated with activated N-Ras.101-103 Finally, K-Ras mutations are detected at highest frequency in tissues derived from the endodermal germ layer, such as pancreas, lung, and colorectal. In this regard, it has been recently suggested that the preponderance of K-Ras in tumors might be explained by the ability of this isoform, but not of H-Ras or N-Ras, to promote the expansion of a stem/progenitor cell population, which subsequently acquires additional mutations to promote tumor progression.104,105 In fact, activated H-ras induces differentiation, leading to the cessation of proliferation, and eventual death, which could explain the absence of H-ras mutation in those tissues derived from the endoderm. Activated N-ras exerts no detectable biological effects in endodermal progenitor cells.104,106-108
Specificity of Ras Isoforms in Physiological Settings
There are also a significant number of experimental observations supporting the notion that the different members of the Ras family play specific cellular roles in a variety of different physiological settings.1,4,109 These include 1) the distinct sensitivities exhibited by individual Ras isoforms or particular subsets of the Ras family for functional interactions with the specific regulators of the Ras GTPase cycle (GAPs110 and GEFs111,112) or with different downstream effectors participating in various cellular Ras signaling pathways.17,25,91,113-118 Furthermore, 2) the differences observed among different Ras isoforms regarding their intracellular processing pathways and their differential compartmentalization to specific intracellular compartments or plasma membrane microdomains17,20,56,58,90,113,115,119-122 provide additional strong evidence in favor of the notion of functional specificity.
Differential Interactions with Regulators and Effectors of Ras Signaling Pathways
A number of reports have described the varying degrees of sensitivity exhibited by different Ras isoforms in their functional interactions with positive and negative regulators of the Ras cycle, including various mammalian RasGAP110 and RasGEF isoforms.111,112 For example, some studies have reported that Ras-GRF1 is able in vivo to activate H-Ras and R-Ras but not N-Ras or K-Ras.123 Other reports have shown the ability of RasGRP2 to activate N-Ras and K-Ras but not H-Ras.111 Finally, the ubiquitously expressed Sos GEF factor has been reported to be able to induce GDP/GTP exchange on all 3 N-, H-, and K-Ras isoforms, but with different degrees of potency (hierarchy H-Ras > N-Ras > K-Ras).124
Regarding their participation in cellular signaling, most Ras isoforms appear to be able to qualitatively activate the same effector pathways, but there are multiple reports documenting the occurrence of quantitative differences in their ability to activate particular downstream effector pathways.17,25,91,113-118 For example, it is clearly apparent that the different Ras isoforms vary in their ability to activate Raf-1 and phosphoinositide 3-kinase. Consistent with this notion, K-Ras is reportedly more potent than H-Ras to induce in vivo activation of Raf-1, based on the higher efficiency displayed by K-Ras to cause recruitment of Raf-1 to the plasma membrane and subsequent activation of its kinase activity.118 In contrast, similar experiments show that H-Ras is a considerably more potent PI3K activator than K-Ras.118 Other studies have similarly described quantitative variations in the efficiency of activation of downstream effects by different Ras isoforms in a number of other cellular contexts.90,124,125 Thus, K-Ras was also reported to be a much more potent and efficient activator of Rac-dependent signaling than H-Ras.91 The distinct C-terminal HVRs of H-Ras and K-Ras appear to be the primary determinants conditioning their differential accessibility to the activators of Rac in the specific, membrane-bound subcellular compartments where they are localized.126-129 Likewise, H-Ras and K-Ras have been reported to induce higher activation of NF-κB than N-Ras.130 It has also been described that H-Ras can become activated by the cyclopentenone 15-deoxy-Δ12,14-prostaglandin J2 through the formation of a covalent adduct that does not occur with N-Ras or K-Ras.131
In summary, activation of the different Ras isoforms induces a plethora of cellular responses that depend on 1) the particular set of effectors that are preferentially activated in each case and 2) on the intensity and amplitude of such an activation, which may also itself undergo positive or negative differential modulation. For example, calmodulin has been reported to downregulate Ras-ERK signaling.116 Interestingly, only K-Ras (but not H-Ras or N-Ras) is able to bind to calmodulin and to inhibit downstream Raf and ERK1/2 activation, thus demonstrating the existence of differential mechanisms of negative regulation among different Ras isoforms.116
Distinct Intracellular Processing and Subcellular Location of Ras Isoforms
Attachment of Ras proteins to cellular membranes is indispensable for them to display their full biological functionality. For this purpose, the newly synthesized, cytosolic, inactive Ras precursors need to undergo a series of postransductional modifications in order to attain full biological activity59,132-136 (Fig. 2). The first set of modifications increases the hydrophobicity at the C-terminus of the Ras isoforms through farnesylation of Cys186 in the CAAX box, which targets the Ras proteins to the surface of the ER. This is followed by proteolytic cleavage of the –AAX motif and carboxymethylation of the resulting C-terminal cysteine residue.54,137-139 Interestingly, K-Ras appears to be more efficiently methylated than H-Ras or N-Ras, although the underlying mechanism is unknown.17 From this point on, at least 2 separate routes of intracellular processing are specifically followed by the different Ras isoforms in order to reach their final cell membrane destinations. In the case of H-Ras, N-Ras, and K-Ras4A, a second set of lipid-modifying processing events is required that involves palmitoylation of Cys residues immediately adjacent to the Cys186 of the CAAX box. The fully processed, palmitoylated H-Ras, N-Ras, and K-Ras4A proteins are thus enabled to follow the secretory pathway, trafficking via vesicles budded from the Golgi body towards the plasma membrane.21 These 3 Ras isoforms display subtle, specific differences when undergoing this second processing step. Whereas H-Ras is palmitoylated in both Cys181 and Cys184, N-Ras and K-Ras4A are only monopalmitoylated, respectively, in Cys181 and Cys180.21 Interestingly, the 2 palmitoyl groups of H-Ras are not equally significant for intracellular trafficking: Palmitoylated Cys181 support sell surface localization, whereas monopalmitoylation on Cys184 confines H-Ras to the Golgi apparatus.21 Palmitoylated H-Ras and N-Ras can be found on the plasma membrane, the Golgi, and at least transiently, on the ER.137,140-142 Palmitoylated Ras has also been found on recycling endosomes derived from the plasma membrane.143
Figure 2.
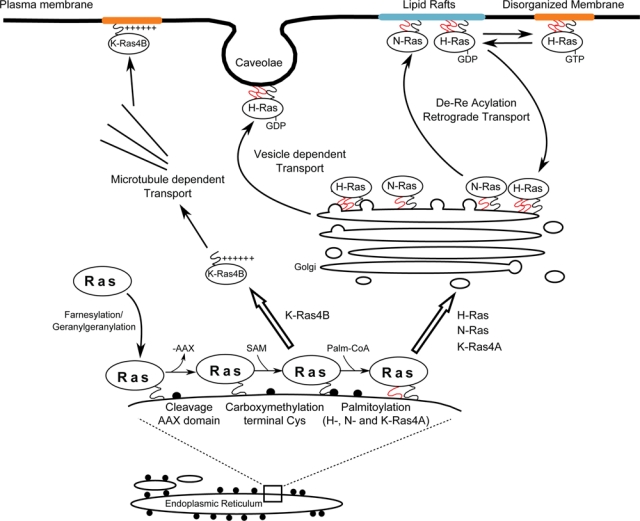
Processing, transport, and localization of Ras proteins. Differences of primary structure and posttranslational modifications are responsible for the differences of intracellular transport and subcellular localization observed among Ras isoforms. Farnesylation (black squiggle) and palmitoylation (red squiggle) of newly synthesized H-Ras, N-Ras, and K-Ras4A are the essential signals controlling their traffic via the ER-Golgi secretory pathway to their specific final plasma membrane destinations. In contrast, farnesylated K-Ras4B follows a different processing pathway and is directly shuttled to the plasma membrane through a mechanism dependent on the presence of a specific, polybasic lysine-rich sequence at its C-terminal region.
Unlike farnesylation, palmitoylation is a reversible process, and the half-life of palmitoyl groups on the Ras proteins is very short (20-60 minutes) in comparison to that of their host protein moieties (over 20 hours).122,144 The resulting cycle of acylation-deacylation enables Ras proteins to shuttle between the plasma membrane and the Golgi, thus providing an alternative regulatory mechanism to control the specific subcellular compartmentalization of different Ras isoforms.145,146
Unlike the other isoforms, K-Ras4B does not undergo palmitoylation, and its intracellular processing does not follow the secretory pathway but involves direct shuttling from the ER to the plasma membrane. The mechanism by which prenylated K-Ras4B bypasses the conventional Golgi secretory pathway to reach the plasma membrane is still poorly understood. The specific polybasic, lysine-rich region in its HVR is clearly involved in controlling trafficking and membrane anchorage through electrostatic interaction with anionic phospholipids of the inner side of the plasma membrane.17 There is also experimental evidence supporting a role of microtubules in the process of K-Ras4B intracellular trafficking to the plasma membrane.147,148
In summary, the specific amino acid sequence of the C-terminal HVRs of the different Ras isoforms determines differences of posttranslational modifications and intracellular trafficking that result in the different subcellular localizations that likely account for differences of biological activity observed as a result of their functional interactions with colocalized upstream regulators or downstream effectors.17,25,56,90,119,120,122
As a result of the differences in their posttranslational modifications, the palmitoylated and polybasic-targeted Ras isoform proteins are directed to different localizations in the plasma membrane, where they incorporate into nanoclusters that potentially facilitate Ras-dependent signaling by concentrating the components of specific effector cascades142 (Fig. 2).
The palmitoyl groups of H-Ras preferentially target this isoform to cholesterol-rich microdomains (lipid rafts and caveolae), thus rendering H-Ras– dependent signaling potentially sensitive to perturbations of plasma membrane cholesterol.20 The localization of Ras proteins within different membrane subdomains is dynamic and largely depends on the activation state of these proteins. Thus, H-Ras, in its active GTP conformation, is known to redistribute from lipid rafts to bulk plasma membrane through a mechanism requiring the HVR, and this change of membrane domain localization is necessary for efficient activation of downstream effectors.21,58,149 N-Ras also localizes into the lipid rafts in the plasma membrane, but it is never associated to caveolae.123 In addition, N-Ras appears to move in the opposite direction than H-Ras when activated.21 At present, it is not known whether the H- and N-Ras nanoclusters are identical or share just a limited number of molecular markers.
K-Ras is normally localized outside of lipid rafts, irrespective of its bound nucleotide state.149 The absence of hydrophobic acyl groups on K-Ras facilitates its cytosolic shuttling between the cell surface and intracellular organelles. In addition, the K-Ras polybasic domain confers to this particular isoform the capacity to aggregate the anionic lipid phosphatidylinositol-4,5-bisphosphate, a substrate for PI3K, which is itself a key Ras effector.150 The membrane localization of this isoform can also be altered through phosphorylation of amino acid residues located within the polybasic region, which is known to release K-Ras from the plasma membrane. For example, PKC-mediated phosphorylation of this region was reported to promote localization to mitochondria, consequently triggering apoptosis.62 In neurons, the polybasic-prenyl motif of K-Ras has also been reported to act as a reversible Ca2+/CaM-regulated molecular switch releasing K-Ras from the plasma membrane and partially redistributing it to internal sites.151
The occupation of specific subcellular “niches” by different Ras isoforms, or just their preferential enrichment at those locations, is bound to result also in differential contributions of these different Ras proteins to the biological outcomes of the common, shared cellular signaling pathways in which they all are able to participate. Such a variety of differential signaling responses attributable to different Ras isoforms in different cellular settings allows for a wide range of possibilities to modulate the intensity of Ras signaling as well as the spatial and temporal distribution of those signals. For example, activation of receptor tyrosine kinases (RTKs) is known to result in the internalization of a large proportion of the surface receptor population that may thus be targeted for lysosomal degradation.152 It remains to be determined the extent to which each different Ras isoform is able to cotraffic with specific, activated receptor complexes, thus facilitating signaling from the surface of early/recycling endosomes. Although both H- and K-Ras can recruit Raf to the plasma membrane, high-resolution microscopy has revealed that the activated K-Ras nanoclusters retain Raf stably, whereas the activated H-Ras–Raf interactions are transient.153 These observations are consistent with separate reports indicating that K-Ras is a better activator of the Raf-MAPK cascade than H-Ras.118
The differential subcellular localization of the Ras isoforms determines also their preferential interaction with colocalized regulatory proteins (GEFs and GAPs) and effectors, thus facilitating the generation of distinct functional effects as a consequence of this compartmentalized downstream Ras signaling.154 For example, Ras-MAPK activation driven by H-Ras and N-Ras displays a delayed and sustained profile when these isoforms are located at the Golgi, but it is transient when the isoforms are located at the plasma membrane. The functional differences caused by different activation kinetics of the Ras-MAPK pathway are clearly illustrated in PC12 cells by the contrasting effects of EGF (transient activation) or NGF (sustained activation), leading to proliferation or differentiation, respectively.155 Ras signaling from endosomes and the so-called rasosomes can also provide an increased intensity of signal.156
A number of reports have also noted the presence of specific Ras isoforms in less canonical subcellular locations, as the mitochondria or the nucleus. Thus, p19, a H-Ras isoform lacking the C-terminal HVR, has been reported to localize to the cytosol and nucleus of cells, where it apparently functions as a regulator of the activity of the p73 tumor suppressor.157 Other studies have also reported the occurrence of N-Ras– and K-Ras–mediated signaling in mitochondria.62,158,159 K-Ras phosphorylation at the HVR was reported to destabilize its electrostatic interactions with the plasma membrane, thus promoting its redistribution to mitochondria.62 Mitochondrial N-Ras and K-Ras have been described to play a functional role in the maintenance of normal mitochondrial morphology and function.158
A recent report suggests that deubiquitinating enzymes can also influence Ras isoform subcellular localization and activation. It was reported that EGF-induced expression of USP17 leads to inhibition of H-Ras and N-Ras localization to the plasma membrane, leaving K-Ras unaffected. However, USP17 seems not to affect N-Ras localization to the ER and the Golgi.160
In summary, a multitude of studies indicate that the subcellular localization of the Ras isoforms is in a constant state of spatiotemporal flux, regulated by a variety of reversible posttranslational modifications that may greatly impact on their signaling capacity. Although all isoforms have been found on a variety of organelles, the key difference determining functional specificities might be the relative proportions of each isoform present in each location.
Phenotypes of Genetically Modified Ras Mouse Models
The study of genetically engineered mouse models has been very instrumental in order to assess the degree of functional specificity or redundancy exhibited by different Ras family members in various physiological or pathological contexts. The earlier Ras animal models focused mostly on analyzing the specific contributions of different Ras oncogene isoforms to various tumoral pathologies. In particular, since the development of the Ras oncomouse in 1987 (harboring a transgenic MMTV–H-ras oncogene construct),161 a very extensive collection of different mouse models including constitutive or inducible transgenic, knockout or knockin strains has been generated and studied for the purpose of characterizing the specific functional contributions of different Ras isoforms to the initiation or progression of various tumor types88,162-171 or developmental disorders.172,173 The collection of animal models analyzing the contribution of K-ras genes to the development of different forms of tumors is particularly extensive174,175 and is also the subject of an accompanying review in this journal issue (O’Hagan and Heyer, current journal issue).
In contrast to the studies of Ras-related pathologies, the generation and analysis of animal models of normal, wild-type Ras function started only in the late 1990s. An initial report showed in 1995 the dispensability of the N-Ras wild-type locus for normal mouse growth development.176 In contrast, 2 later reports described that K-Ras4B is essential for embryogenesis,177,178 as the embryos carrying homozygous null mutations in the K-ras locus died at midgestation (between E12 and E14) and presented anemia and fetal liver defects177 as well as increased motoneuronal cell death and thinned heart ventricular walls.178 On the other hand, mouse strains harboring homozygous null mutants of the H-ras 177,179 or K-ras4A 180,181 loci showed normal growth rates and were indistinguishable from wild-type control animals, indicating that these individual genes are also dispensable for normal mouse development. Simultaneous removal of H-ras and N-ras resulted also in viable, fertile mice that did not show any obvious phenotypic abnormalities.179 However, in this case, the number of adult, double knockout animals resulting from crosses between heterozygous N-ras/H-ras animals was lower than expected according to Mendelian ratios, suggesting the possibility of partial functional overlapping between N-Ras and H-Ras in their contribution to full embryo viability.179 Further insight into the functional relationships between the 3 different Ras isoforms is now available through the study of newly developed mouse strains carrying null H-Ras and N-Ras alleles along with a floxed K-Ras locus and a knocked-in inducible Cre recombinase, which can be rendered Ras-less, devoid of Ras proteins, by exposure to 4-hydroxytamoxifen (4OHT) or by infection with adenoviruses expressing a Cre recombinase.182
The joint analysis of all available reports on Ras knockout animal models indicates that, among all Ras isoforms, only K-Ras4B is necessary and sufficient for development of mice to the adult stage. This notion would be consistent with K-Ras4B performing specific cellular functions that are not shared by H-Ras, N-Ras, or K-Ras4A. Alternatively, the selective requirement of K-Ras4B for normal mouse development might reflect a requirement for expression of this isoform in a specific cell type(s) during a critical developmental stage(s).23 This second possibility is supported by the observation that mice in which the H-ras coding sequences are knocked into the K-ras locus (where native K-Ras protein expression is abolished and substituted by H-Ras, expressed under the control of the K-ras regulatory regions) were born at the expected Mendelian frequency, although the adult animals frequently presented with cardiomyopathies.183 These observations suggest that H-Ras may be able to fully replace the essential function(s) of K-Ras during embryogenesis, when its expression is controlled by the K-ras promoter, but not during adult life. More importantly, these findings suggest that the mortality of the K-ras knockout mice may not derive from intrinsic inability of the other isoforms to compensate for K-Ras4B function but rather from their inability to be expressed in the same embryonic compartments, or with the same timing, as K-Ras4B.
Transcriptomic Patterns Associated with Specific Ras Isoforms
As the H-Ras, N-Ras, and K-Ras proteins are very similar regarding their catalytic and effector-binding properties, most scientific data discussed so far in this review support the notion that their functional specificity in processes of cell growth, development, or cancer is largely the consequence of their differential spatiotemporal compartmentalization in the different cell contexts where they are expressed. An additional factor that can also potentially contribute to confer functional specificity to specific Ras isoforms is their proven ability to modulate distinct transcriptomic profiles in the particular cell lineages in which they are expressed. The development of microarray-based technologies allowing single-step examination of the complete genome and proteome landscape of a cell has allowed us to test and support such a hypothesis. In particular, these techniques have been instrumental to characterize the different patterns of genomic and proteomic expression arising in cells as a consequence of 1) the presence of particular Ras isoforms in various oncogenic contexts or 2) the absence of specific Ras genes in the context of knockout cellular models lacking specific ras genes.184-192 The results of these analyses support also the notion that the different Ras genes may condition the expression of particular transcriptional programs in the cells where they are expressed.
Genomic and Proteomic Expression Patterns Associated with the Presence of Specific Ras Oncogenes
Early studies using suppression subtractive hybridization (SSH) techniques in immortalized, nontumorigenic rat embryo fibroblasts and in H-ras–transformed cells identified differential gene expression profiles that were also instrumental to categorize common and distinct targets in cells transformed by mutant H-ras, N-ras, or K-ras oncogenes.187 The cells transformed by oncogenic H-ras showed overexpression of genes linked to invasion and metastasis and downregulation of genes related to antiproliferative, anti-invasive, or antiangiogenic processes. A majority (>90%) of the transcripts sensitive to H-ras transformation showed also similar expression profiles in cells transformed by the other Ras oncogenes. However, a shorter list of transcripts could still be identified that appeared to be differentially regulated by each of the different Ras oncogene isoforms.187 The H-ras oncogene can also induce specific transcriptional profiles in other cellular settings. Thus, microarray-based analysis of PB-3c mast cells stably transfected with oncogenic H-ras revealed a transcriptional profile characterized by increased expression of genes specifically affecting tumorigenesis such as cell adhesion, signaling, or transcriptional regulation and parallel downregulation of a set of interferon-inducible genes.193
The existence of differential gene expression programs specifically driven by different Ras oncogene isoforms was also supported by separate studies in which constitutively active K-ras and H-ras oncogenes were introduced into the intermediate-stage colon adenoma cell line Caco-2.186 In this case, analysis of the resulting transcriptional patterns indicated that these 2 Ras oncogenes regulate different biological processes, which may separately impact onto the overall process of colon carcinogenesis. Thus, oncogenic K-Ras preferentially modulated expression of genes involved in cytokine signaling, cell adhesion, and colonic development, whereas oncogenic H-Ras was mostly involved with regulation of genes controlling cell morphology, an observation consistent with the epithelial-mesenchymal transition observed in these cells. The available data suggest that, in colorectal tumors, H-RasV12 may be preferentially involved with regulation of cell transformation (facilitating mesenchymal morphology, anchorage-independent proliferation, and tumor growth in SCID mice), whereas the contribution of K-RasV12 appears to be more preferentially focused on eliciting phenotypic changes in the colonic epithelium that create an environment conducive to further mutational events and tumor progression. Separate reports have also described gene expression profiling in pancreatic cell lines infected with antisense K-ras or in mouse tumors derived from RasV12/E1A-transformed MEFs.189,190
A separate study comparing the transcriptional profiles caused by oncogenic K-ras in a mouse model of lung cancer to those of corresponding human lung tumors harboring K-ras mutations188 has described significant coincidences between the gene expression patterns of these 2 distinct biological systems.188 Furthermore, whereas a gene expression signature of K-Ras activation could not be identified when analyzing human tumors alone, the integration of data from the mouse and human systems uncovered a gene expression signature of oncogenic K-ras that may be specifically linked to the development of adenocarcinomas.188
Gene expression profiles associated to the expression of oncogenic N-ras in myeloma cells have also been characterized.192 Interestingly, IL6 is a potent mitogen and activator of Ras signaling in various myeloma cells and cell lines.194 However, transfection of these cells with N-ras oncogenes specifically induced expression of a distinct set of genes that were not inducible by IL6 treatment or stromal interactions. This N-Ras–dependent genes set includes components of Ras signaling pathways such as ETV5 and DUSP6,195,196 suggesting that the N-Ras oncogene may activate different downstream targets than IL6 or, alternatively, prolong activation of shared signaling pathways, thus resulting in a different gene expression outcome.
Massive proteomic analyses have also been useful to characterize differential expression profiles associated to specific Ras oncogenes.191,197 For example, study of the MALDI-TOF MS proteomic profiles of mouse embryo fibroblasts independently transformed by H-Ras, K-Ras, or N-Ras oncogenes allowed identification of a significant number of distinct, differentially regulated protein spots in comparison to the normal, control cells.191 A majority of the individual dysregulations of protein expression detected in the transformed fibroblasts were specifically attributable to the presence of one particular Ras oncogene. Indeed, of 204 different dysregulations identified, only 42 were shared in all the fibroblast lines transformed by any of the 3 Ras oncogenes assayed, suggesting that each particular Ras oncogene controls a specific transcriptional program contributing to the process of malignant transformation. Further studies will be needed to ascertain how the different, specific transcriptional programs triggered by each individual Ras oncogene contribute to generating the shared, final tumoral phenotype of malignant transformation.182,198
Genomic/Proteomic Expression Patterns Associated with the Absence of Specific Ras Isoforms
A complementary approach to identifying specific gene expression programs controlled by the different Ras isoforms is the characterization of the transcriptomic profiles of cells devoid of different, normal Ras genes. The availability of single and double knockout mouse strains for the H-ras and N-ras genes has been particularly instrumental in this regard. Analysis of the transcriptional networks of fibroblasts harboring single or double null mutations in the H-ras and N-ras loci clearly indicates that these 2 isoforms control different transcriptional networks and supports the notion of different functional roles for H-Ras and N-Ras in the cells.184,185 Interestingly, actively growing, unsynchronized cultures of H-Ras and N-Ras knockout fibroblasts displayed rather antagonistic transcriptional profiles, and the transcriptome of cells lacking H-Ras was significantly closer to that of wild-type fibroblasts than to that of N-Ras knockout cells. Likewise, parallel specific alterations of the cellular transcriptomic profile were also detected in association with the absence of H-Ras and/or N-Ras during early stages of the cell cycle, in cells that had been serum starved and subsequently stimulated with FBS.184,185 In particular, functional characterization of the different sets of differentially expressed genes identified in those studies indicated that lack of H-Ras in cells was consistently associated to impairment of transcriptional programs driving processes of cell growth and proliferation.184,185 On the other hand, similar functional analyses uncovered the specific involvement of N-Ras with control of immune modulation/host defense and apoptotic responses, observations consistent with a number of previous, separate studies.199-202 Mechanistic analysis indicates that Stat1 is an essential transcriptional activator mediating the control of N-Ras over inflammatory and immune responses, whereas the modulation of apoptosis exerted by N-Ras involves direct regulation of Bax and Perp expression.184,185
The analysis of transcriptional networks from Ras knockout cell lines has also yielded a better understanding of the contribution of different Ras isoforms to cell cycle progression.1,4,67,203 The absolute requirement for Ras activity at different points between G0 and S phase of the cell cycle had been documented in a number of early reports.204-209 The available experimental evidence indicates that the contribution of Ras activity is absolutely needed for both the initial entry into the cell cycle (G0/G1 transition) and for the subsequent G1 progression, in a process to which multiple Ras effector pathways can contribute.210-219 However, the exact mechanisms underlying the participation of Ras proteins in cell cycle activation and progression are still largely undefined, and it is also unknown whether the different Ras isoforms play specific or redundant functional roles in such processes. Comparison of the transcriptional profiles of cells lacking H-ras and N-ras, alone or in combination, during quiescence (serum starved), G0/G1 transition (1 hour serum stimulation poststarvation), or G1 progression (8 hours serum stimulation poststarvation) illustrates the differential contributions of these 2 Ras isoforms regarding their contribution to cell cycle progression. Interestingly, absence of N-Ras results in significant impairment of the transcriptional response to serum during G0/G1 transition, whereas absence of H-Ras had an ever more pronounced effect on a later wave of serum-induced transcriptional activation corresponding to mid-G1 progression, raising the interesting possibility of preferential involvement of N-Ras with the immediate-early cellular responses to serum stimulation and of H-Ras with the cellular responses related to growth and proliferation during mid-G1 progression.184,185 Analysis of triple knockout Ras-less MEFs182 has confirmed the capital role of Ras proteins in control of the cell cycle. Interestingly, the Ras-less MEFs have normal levels of cyclin D1/Cdk4 and cyclin E/Cdk2, and these complexes are inactive and unable to phosphorylate pRB in vitro, suggesting that, in contrast to current hypotheses, Ras signaling does not induce proliferation in these cells by inducing expression of D-type cyclins.
Acknowledgments
The authors apologize to other authors of original reports not included in the references section because of space constraints.
Footnotes
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
This work was supported by the Instituto de Salud Carlos III (ISCIII) [grant number FIS PS09/01979]; Junta de Castilla y León (JCyL) [grant numbers SA044A08, GR93]; and Red Temática de Investigación Cooperativa en Cáncer (RTICC) [grant number RD06/ 0020/000].
References
- 1. Abankwa D, Gorfe AA, Hancock JF. Mechanisms of Ras membrane organization and signalling: Ras on a rocker. Cell Cycle. 2008;7:2667-73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bourne HR, Sanders DA, McCormick F. The GTPase superfamily: a conserved switch for diverse cell functions. Nature. 1990;348:125-32 [DOI] [PubMed] [Google Scholar]
- 3. Colicelli J. Human RAS superfamily proteins and related GTPases. Sci STKE. 2004;2004:RE13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Karnoub AE, Weinberg RA. Ras oncogenes: split personalities. Nat Rev Mol Cell Biol. 2008;9:517-31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Reuther GW, Der CJ. The Ras branch of small GTPases: Ras family members don’t fall far from the tree. Curr Opin Cell Biol. 2000;12:157-65 [DOI] [PubMed] [Google Scholar]
- 6. Field J, Broek D, Kataoka T, Wigler M. Guanine nucleotide activation of, and competition between, RAS proteins from Saccharomyces cerevisiae. Mol Cell Biol. 1987;7:2128-33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hall BE, Yang SS, Boriack-Sjodin PA, Kuriyan J, Bar-Sagi D. Structure-based mutagenesis reveals distinct functions for Ras switch 1 and switch 2 in Sos-catalyzed guanine nucleotide exchange. J Biol Chem. 2001;276:27629-37 [DOI] [PubMed] [Google Scholar]
- 8. Herrmann C. Ras-effector interactions: after one decade. Curr Opin Struct Biol. 2003;13:122-9 [DOI] [PubMed] [Google Scholar]
- 9. Ma J, Karplus M. Molecular switch in signal transduction: reaction paths of the conformational changes in ras p21. Proc Natl Acad Sci U S A. 1997;94:11905-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Satoh T, Nakamura S, Kaziro Y. Induction of neurite formation in PC12 cells by microinjection of proto-oncogenic Ha-ras protein preincubated with guanosine-5’-O-(3-thiotriphosphate). Mol Cell Biol. 1987;7:4553-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wittinghofer A, Pai EF. The structure of Ras protein: a model for a universal molecular switch. Trends Biochem Sci. 1991;16:382-7 [DOI] [PubMed] [Google Scholar]
- 12. Milburn MV, Tong L, deVos AM, et al. Molecular switch for signal transduction: structural differences between active and inactive forms of protooncogenic ras proteins. Science. 1990;247:939-45 [DOI] [PubMed] [Google Scholar]
- 13. Schlichting I, Almo SC, Rapp G, et al. Time-resolved X-ray crystallographic study of the conformational change in Ha-Ras p21 protein on GTP hydrolysis. Nature. 1990;345:309-15 [DOI] [PubMed] [Google Scholar]
- 14. Barbacid M. ras genes. Annu Rev Biochem. 1987;56:779-827 [DOI] [PubMed] [Google Scholar]
- 15. Bos JL. ras oncogenes in human cancer: a review. Cancer Res. 1989;49:4682-9 [PubMed] [Google Scholar]
- 16. Broach JR, Deschenes RJ. The function of ras genes in Saccharomyces cerevisiae. Adv Cancer Res. 1990;54:79-139 [DOI] [PubMed] [Google Scholar]
- 17. Hancock JF. Ras proteins: different signals from different locations. Nat Rev Mol Cell Biol. 2003;4:373-84 [DOI] [PubMed] [Google Scholar]
- 18. Hancock JF, Cadwallader K, Paterson H, Marshall CJ. A CAAX or a CAAL motif and a second signal are sufficient for plasma membrane targeting of ras proteins. EMBO J. 1991;10:4033-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Leon J, Guerrero I, Pellicer A. Differential expression of the ras gene family in mice. Mol Cell Biol. 1987;7:1535-40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Roy S, Luetterforst R, Harding A, et al. Dominant-negative caveolin inhibits H-Ras function by disrupting cholesterol-rich plasma membrane domains. Nat Cell Biol. 1999;1:98-105 [DOI] [PubMed] [Google Scholar]
- 21. Roy S, Plowman S, Rotblat B, et al. Individual palmitoyl residues serve distinct roles in H-ras trafficking, microlocalization, and signaling. Mol Cell Biol. 2005;25:6722-33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Su AI, Wiltshire T, Batalov S, et al. A gene atlas of the mouse and human protein-encoding transcriptomes. Proc Natl Acad Sci U S A. 2004;101:6062-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bar-Sagi D. A Ras by any other name. Mol Cell Biol. 2001;21:1441-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lowy DR, Willumsen BM. Function and regulation of ras. Annu Rev Biochem. 1993;62:851-91 [DOI] [PubMed] [Google Scholar]
- 25. Plowman SJ, Hancock JF. Ras signaling from plasma membrane and endomembrane microdomains. Biochim Biophys Acta. 2005;1746:274-83 [DOI] [PubMed] [Google Scholar]
- 26. Rojas JM, Santos E. ras genes and human cancer: different implications and different roles. Curr Genomics. 2002;3:295-311 [Google Scholar]
- 27. Furth ME, Aldrich TH, Cordon-Cardo C. Expression of ras proto-oncogene proteins in normal human tissues. Oncogene. 1987;1:47-58 [PubMed] [Google Scholar]
- 28. Fiorucci G, Hall A. All three human ras genes are expressed in a wide range of tissues. Biochim Biophys Acta. 1988;950:81-3 [DOI] [PubMed] [Google Scholar]
- 29. Bos JL. The ras gene family and human carcinogenesis. Mutat Res. 1988;195:255-71 [DOI] [PubMed] [Google Scholar]
- 30. Der CJ, Finkel T, Cooper GM. Biological and biochemical properties of human rasH genes mutated at codon 61. Cell. 1986;44:167-76 [DOI] [PubMed] [Google Scholar]
- 31. Downward J. Targeting RAS signalling pathways in cancer therapy. Nat Rev Cancer. 2003;3:11-22 [DOI] [PubMed] [Google Scholar]
- 32. Raptis L, Brownell HL, Corbley MJ, Wood KW, Wang D, Haliotis T. Cellular ras gene activity is required for full neoplastic transformation by the large tumor antigen of SV40. Cell Growth Differ. 1997;8:891-901 [PubMed] [Google Scholar]
- 33. Hirakawa T, Ruley HE. Rescue of cells from ras oncogene-induced growth arrest by a second, complementing, oncogene. Proc Natl Acad Sci U S A. 1988;85:1519-23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Land H, Parada LF, Weinberg RA. Tumorigenic conversion of primary embryo fibroblasts requires at least two cooperating oncogenes. Nature. 1983;304:596-602 [DOI] [PubMed] [Google Scholar]
- 35. Ruley HE. Adenovirus early region 1A enables viral and cellular transforming genes to transform primary cells in culture. Nature. 1983;304:602-6 [DOI] [PubMed] [Google Scholar]
- 36. Land H, Parada LF, Weinberg RA. Cellular oncogenes and multistep carcinogenesis. Science. 1983;222:771-8 [DOI] [PubMed] [Google Scholar]
- 37. Ward JM, Perantoni AO, Santos E. Comparative immunohistochemical reactivity of monoclonal and polyclonal antibodies to H-ras p21 in normal and neoplastic tissues of rodents and humans. Oncogene. 1989;4:203-13 [PubMed] [Google Scholar]
- 38. Chesa PG, Rettig WJ, Melamed MR, Old LJ, Niman HL. Expression of p21ras in normal and malignant human tissues: lack of association with proliferation and malignancy. Proc Natl Acad Sci U S A. 1987;84:3234-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Shields JM, Pruitt K, McFall A, Shaub A, Der CJ. Understanding Ras: ’it ain’t over ’til it’s over’. Trends Cell Biol. 2000;10:147-54 [DOI] [PubMed] [Google Scholar]
- 40. Mitsuuchi Y, Testa JR. Cytogenetics and molecular genetics of lung cancer. Am J Med Genet. 2002;115:183-8 [DOI] [PubMed] [Google Scholar]
- 41. Grady WM, Markowitz SD. Genetic and epigenetic alterations in colon cancer. Annu Rev Genomics Hum Genet. 2002;3:101-28 [DOI] [PubMed] [Google Scholar]
- 42. Jaffee EM, Hruban RH, Canto M, Kern SE. Focus on pancreas cancer. Cancer Cell. 2002;2:25-8 [DOI] [PubMed] [Google Scholar]
- 43. Almoguera C, Shibata D, Forrester K, Martin J, Arnheim N, Perucho M. Most human carcinomas of the exocrine pancreas contain mutant c-K-ras genes. Cell. 1988;53:549-54 [DOI] [PubMed] [Google Scholar]
- 44. Rodenhuis S, Slebos RJ. Clinical significance of ras oncogene activation in human lung cancer. Cancer Res. 1992;52:2665s-9s [PubMed] [Google Scholar]
- 45. Bentires-Alj M, Kontaridis MI, Neel BG. Stops along the RAS pathway in human genetic disease. Nat Med. 2006;12:283-5 [DOI] [PubMed] [Google Scholar]
- 46. Cichowski K, Jacks T. NF1 tumor suppressor gene function: narrowing the GAP. Cell. 2001;104:593-604 [DOI] [PubMed] [Google Scholar]
- 47. Rochlitz CF, Scott GK, Dodson JM, et al. Incidence of activating ras oncogene mutations associated with primary and metastatic human breast cancer. Cancer Res. 1989;49:357-60 [PubMed] [Google Scholar]
- 48. Tartaglia M, Gelb BD. Noonan syndrome and related disorders: genetics and pathogenesis. Annu Rev Genomics Hum Genet. 2005;6:45-68 [DOI] [PubMed] [Google Scholar]
- 49. Watzinger F, Lion T. Ras family. Atlas Gent Cytogenet Oncol Haematol. 1999 Available at: http://atlasgeneticsoncology.org/Deep/Ras.html
- 50. Pells S, Divjak M, Romanowski P, et al. Developmentally-regulated expression of murine K-ras isoforms. Oncogene. 1997;15:1781-6 [DOI] [PubMed] [Google Scholar]
- 51. Muller R, Slamon DJ, Adamson ED, et al. Transcription of c-onc genes c-rasKi and c-fms during mouse development. Mol Cell Biol. 1983;3:1062-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Thapar R, Williams JG, Campbell SL. NMR characterization of full-length farnesylated and non-farnesylated H-Ras and its implications for Raf activation. J Mol Biol. 2004;343:1391-408 [DOI] [PubMed] [Google Scholar]
- 53. Ellis CA, Clark G. The importance of being K-Ras. Cell Signal. 2000;12:425-34 [DOI] [PubMed] [Google Scholar]
- 54. Hancock JF, Magee AI, Childs JE, Marshall CJ. All ras proteins are polyisoprenylated but only some are palmitoylated. Cell. 1989;57:1167-77 [DOI] [PubMed] [Google Scholar]
- 55. Hancock JF, Paterson H, Marshall CJ. A polybasic domain or palmitoylation is required in addition to the CAAX motif to localize p21ras to the plasma membrane. Cell. 1990;63:133-9 [DOI] [PubMed] [Google Scholar]
- 56. Matallanas D, Sanz-Moreno V, Arozarena I, et al. Distinct utilization of effectors and biological outcomes resulting from site-specific Ras activation: Ras functions in lipid rafts and Golgi complex are dispensable for proliferation and transformation. Mol Cell Biol. 2006;26:100-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Prior IA, Hancock JF. Compartmentalization of Ras proteins. J Cell Sci. 2001;114:1603-8 [DOI] [PubMed] [Google Scholar]
- 58. Prior IA, Muncke C, Parton RG, Hancock JF. Direct visualization of Ras proteins in spatially distinct cell surface microdomains. J Cell Biol. 2003;160:165-70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Willumsen BM, Christensen A, Hubbert NL, Papageorge AG, Lowy DR. The p21 ras C-terminus is required for transformation and membrane association. Nature. 1984;310:583-6 [DOI] [PubMed] [Google Scholar]
- 60. Hancock JF, Parton RG. Ras plasma membrane signalling platforms. Biochem J. 2005;389:1-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Chiu VK, Bivona T, Hach A, et al. Ras signalling on the endoplasmic reticulum and the Golgi. Nat Cell Biol. 2002;4:343-50 [DOI] [PubMed] [Google Scholar]
- 62. Bivona TG, Quatela SE, Bodemann BO, et al. PKC regulates a farnesyl-electrostatic switch on K-Ras that promotes its association with Bcl-XL on mitochondria and induces apoptosis. Mol Cell. 2006;21:481-93 [DOI] [PubMed] [Google Scholar]
- 63. Kinzler KW, Vogelstein B. Lessons from hereditary colorectal cancer. Cell. 1996;87:159-70 [DOI] [PubMed] [Google Scholar]
- 64. Garcia-Rostan G, Zhao H, Camp RL, et al. ras mutations are associated with aggressive tumor phenotypes and poor prognosis in thyroid cancer. J Clin Oncol. 2003;21:3226-35 [DOI] [PubMed] [Google Scholar]
- 65. Portier M, Moles JP, Mazars GR, et al. p53 and RAS gene mutations in multiple myeloma. Oncogene. 1992;7:2539-43 [PubMed] [Google Scholar]
- 66. Kraus MH, Yuasa Y, Aaronson SA. A position 12-activated H-ras oncogene in all HS578T mammary carcinosarcoma cells but not normal mammary cells of the same patient. Proc Natl Acad Sci U S A. 1984;81:5384-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Schubbert S, Shannon K, Bollag G. Hyperactive Ras in developmental disorders and cancer. Nat Rev Cancer. 2007;7:295-308 [DOI] [PubMed] [Google Scholar]
- 68. Hennekam RC. Costello syndrome: an overview. Am J Med Genet C Semin Med Genet. 2003;117C:42-8 [DOI] [PubMed] [Google Scholar]
- 69. Schubbert S, Zenker M, Rowe SL, et al. Germline KRAS mutations cause Noonan syndrome. Nat Genet. 2006;38:331-6 [DOI] [PubMed] [Google Scholar]
- 70. Carta C, Pantaleoni F, Bocchinfuso G, et al. Germline missense mutations affecting KRAS Isoform B are associated with a severe Noonan syndrome phenotype. Am J Hum Genet. 2006;79:129-35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Zenker M, Lehmann K, Schulz AL, et al. Expansion of the genotypic and phenotypic spectrum in patients with KRAS germline mutations. J Med Genet. 2007;44:131-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Aoki Y, Niihori T, Kawame H, et al. Germline mutations in HRAS proto-oncogene cause Costello syndrome. Nat Genet. 2005;37:1038-40 [DOI] [PubMed] [Google Scholar]
- 73. Estep AL, Tidyman WE, Teitell MA, Cotter PD, Rauen KA. HRAS mutations in Costello syndrome: detection of constitutional activating mutations in codon 12 and 13 and loss of wild-type allele in malignancy. Am J Med Genet A. 2006;140:8-16 [DOI] [PubMed] [Google Scholar]
- 74. Gripp KW, Lin AE, Stabley DL, et al. HRAS mutation analysis in Costello syndrome: genotype and phenotype correlation. Am J Med Genet A. 2006;140:1-7 [DOI] [PubMed] [Google Scholar]
- 75. Kerr B, Delrue MA, Sigaudy S, et al. Genotype-phenotype correlation in Costello syndrome: HRAS mutation analysis in 43 cases. J Med Genet. 2006;43:401-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Zampino G, Pantaleoni F, Carta C, et al. Diversity, parental germline origin, and phenotypic spectrum of de novo HRAS missense changes in Costello syndrome. Hum Mutat. 2007;28:265-72 [DOI] [PubMed] [Google Scholar]
- 77. Oliveira JB, Bidere N, Niemela JE, et al. NRAS mutation causes a human autoimmune lymphoproliferative syndrome. Proc Natl Acad Sci U S A. 2007;104:8953-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Cheng CM, Li H, Gasman S, Huang J, Schiff R, Chang EC. Compartmentalized Ras proteins transform NIH 3T3 cells with different efficiencies. Mol Cell Biol. 2011;31:983-97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Maher J, Baker DA, Manning M, Dibb NJ, Roberts IA. Evidence for cell-specific differences in transformation by N-, H- and K-ras. Oncogene. 1995;11:1639-47 [PubMed] [Google Scholar]
- 80. Pulciani S, Santos E, Long LK, Sorrentino V, Barbacid M. ras gene amplification and malignant transformation. Mol Cell Biol. 1985;5:2836-41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Oliva JL, Zarich N, Martinez N, et al. The P34G mutation reduces the transforming activity of K-Ras and N-Ras in NIH 3T3 cells but not of H-Ras. J Biol Chem. 2004;279:33480-91 [DOI] [PubMed] [Google Scholar]
- 82. Parikh C, Subrahmanyam R, Ren R. Oncogenic NRAS, KRAS, and HRAS exhibit different leukemogenic potentials in mice. Cancer Res. 2007;67:7139-46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Katzav S, Packham G, Sutherland M, Aroca P, Santos E, Cleveland JL. Vav and Ras induce fibroblast transformation by overlapping signaling pathways which require c-Myc function. Oncogene. 1995;11:1079-88 [PubMed] [Google Scholar]
- 84. Ruiz S, Santos E, Bustelo XR. RasGRF2, a guanosine nucleotide exchange factor for Ras GTPases, participates in T-cell signaling responses. Mol Cell Biol. 2007;27:8127-42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Ruiz S, Santos E, Bustelo XR. The use of knockout mice reveals a synergistic role of the Vav1 and Rasgrf2 gene deficiencies in lymphomagenesis and metastasis. PLoS One. 2009;4:e8229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Fotiadou PP, Takahashi C, Rajabi HN, Ewen ME. Wild-type NRas and KRas perform distinct functions during transformation. Mol Cell Biol. 2007;27:6742-55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Hamilton M, Wolfman A. Ha-ras and N-ras regulate MAPK activity by distinct mechanisms in vivo. Oncogene. 1998;16:1417-28 [DOI] [PubMed] [Google Scholar]
- 88. Haigis KM, Kendall KR, Wang Y, et al. Differential effects of oncogenic K-Ras and N-Ras on proliferation, differentiation and tumor progression in the colon. Nat Genet. 2008;40:600-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Nobes CD, Hall A. Rho GTPases control polarity, protrusion, and adhesion during cell movement. J Cell Biol. 1999;144:1235-44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Voice JK, Klemke RL, Le A, Jackson JH. Four human ras homologs differ in their abilities to activate Raf-1, induce transformation, and stimulate cell motility. J Biol Chem. 1999;274:17164-70 [DOI] [PubMed] [Google Scholar]
- 91. Walsh AB, Bar-Sagi D. Differential activation of the Rac pathway by Ha-Ras and K-Ras. J Biol Chem. 2001;276:15609-15 [DOI] [PubMed] [Google Scholar]
- 92. Hall A. Rho GTPases and the actin cytoskeleton. Science. 1998;279:509-14 [DOI] [PubMed] [Google Scholar]
- 93. Yan Z, Deng X, Chen M, et al. Oncogenic c-Ki-ras but not oncogenic c-Ha-ras up-regulates CEA expression and disrupts basolateral polarity in colon epithelial cells. J Biol Chem. 1997;272:27902-7 [DOI] [PubMed] [Google Scholar]
- 94. Finkelstein SD, Sayegh R, Bakker A, Swalsky P. Determination of tumor aggressiveness in colorectal cancer by K-ras-2 analysis. Arch Surg. 1993;128:526-31, discussion 31-2 [DOI] [PubMed] [Google Scholar]
- 95. Suchy B, Zietz C, Rabes HM. K-ras point mutations in human colorectal carcinomas: relation to aneuploidy and metastasis. Int J Cancer. 1992;52:30-3 [DOI] [PubMed] [Google Scholar]
- 96. Bar-Sagi D, Feramisco JR. Microinjection of the ras oncogene protein into PC12 cells induces morphological differentiation. Cell. 1985;42:841-8 [DOI] [PubMed] [Google Scholar]
- 97. Benito M, Porras A, Nebreda AR, Santos E. Differentiation of 3T3-L1 fibroblasts to adipocytes induced by transfection of ras oncogenes. Science. 1991;253:565-8 [DOI] [PubMed] [Google Scholar]
- 98. Qui MS, Green SH. PC12 cell neuronal differentiation is associated with prolonged p21ras activity and consequent prolonged ERK activity. Neuron. 1992;9:705-17 [DOI] [PubMed] [Google Scholar]
- 99. Schulz WA. Understanding urothelial carcinoma through cancer pathways. Int J Cancer. 2006;119:1513-8 [DOI] [PubMed] [Google Scholar]
- 100. Yoo J, Robinson RA. ras gene mutations in salivary gland tumors. Arch Pathol Lab Med. 2000;124:836-9 [DOI] [PubMed] [Google Scholar]
- 101. Bauer J, Curtin JA, Pinkel D, Bastian BC. Congenital melanocytic nevi frequently harbor NRAS mutations but no BRAF mutations. J Invest Dermatol. 2007;127:179-82 [DOI] [PubMed] [Google Scholar]
- 102. Ganguly S, Murty VV, Samaniego F, Reuter VE, Bosl GJ, Chaganti RS. Detection of preferential NRAS mutations in human male germ cell tumors by the polymerase chain reaction. Genes Chromosomes Cancer. 1990;1:228-32 [DOI] [PubMed] [Google Scholar]
- 103. Parikh C, Subrahmanyam R, Ren R. Oncogenic NRAS rapidly and efficiently induces CMML- and AML-like diseases in mice. Blood. 2006;108:2349-57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Quinlan MP, Quatela SE, Philips MR, Settleman J. Activated Kras, but not Hras or Nras, may initiate tumors of endodermal origin via stem cell expansion. Mol Cell Biol. 2008;28:2659-74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Quinlan MP, Settleman J. Isoform-specific ras functions in development and cancer. Future Oncol. 2009;5:105-16 [DOI] [PubMed] [Google Scholar]
- 106. Quinlan MP. The Ad5 12S growth factor induces F9 cell proliferation and differentiation. Oncogene. 1989;4:1051-5 [PubMed] [Google Scholar]
- 107. Verheijen MH, Wolthuis RM, Bos JL, Defize LH. The Ras/Erk pathway induces primitive endoderm but prevents parietal endoderm differentiation of F9 embryonal carcinoma cells. J Biol Chem. 1999;274:1487-94 [DOI] [PubMed] [Google Scholar]
- 108. Yamaguchi-Iwai Y, Satake M, Murakami Y, Sakai M, Muramatsu M, Ito Y. Differentiation of F9 embryonal carcinoma cells induced by the c-jun and activated c-Ha-ras oncogenes. Proc Natl Acad Sci U S A. 1990;87:8670-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Omerovic J, Hammond DE, Clague MJ, Prior IA. Ras isoform abundance and signalling in human cancer cell lines. Oncogene. 2008;27:2754-62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Bollag G, McCormick F. Differential regulation of rasGAP and neurofibromatosis gene product activities. Nature. 1991;351:576-9 [DOI] [PubMed] [Google Scholar]
- 111. Clyde-Smith J, Silins G, Gartside M, et al. Characterization of RasGRP2, a plasma membrane-targeted, dual specificity Ras/Rap exchange factor. J Biol Chem. 2000;275:32260-7 [DOI] [PubMed] [Google Scholar]
- 112. Jones MK, Jackson JH. Ras-GRF activates Ha-Ras, but not N-Ras or K-Ras 4B, protein in vivo. J Biol Chem. 1998;273:1782-7 [DOI] [PubMed] [Google Scholar]
- 113. Liao J, Wolfman JC, Wolfman A. K-ras regulates the steady-state expression of matrix metalloproteinase 2 in fibroblasts. J Biol Chem. 2003;278:31871-8 [DOI] [PubMed] [Google Scholar]
- 114. Mitin NY, Ramocki MB, Zullo AJ, Der CJ, Konieczny SF, Taparowsky EJ. Identification and characterization of rain, a novel Ras-interacting protein with a unique subcellular localization. J Biol Chem. 2004;279:22353-61 [DOI] [PubMed] [Google Scholar]
- 115. Plowman SJ, Muncke C, Parton RG, Hancock JF. H-ras, K-ras, and inner plasma membrane raft proteins operate in nanoclusters with differential dependence on the actin cytoskeleton. Proc Natl Acad Sci U S A. 2005;102:15500-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Villalonga P, Lopez-Alcala C, Bosch M, et al. Calmodulin binds to K-Ras, but not to H- or N-Ras, and modulates its downstream signaling. Mol Cell Biol. 2001;21:7345-54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Vos MD, Ellis CA, Elam C, Ulku AS, Taylor BJ, Clark GJ. RASSF2 is a novel K-Ras-specific effector and potential tumor suppressor. J Biol Chem. 2003;278:28045-51 [DOI] [PubMed] [Google Scholar]
- 118. Yan J, Roy S, Apolloni A, Lane A, Hancock JF. Ras isoforms vary in their ability to activate Raf-1 and phosphoinositide 3-kinase. J Biol Chem. 1998;273:24052-6 [DOI] [PubMed] [Google Scholar]
- 119. Ehrhardt A, David MD, Ehrhardt GR, Schrader JW. Distinct mechanisms determine the patterns of differential activation of H-Ras, N-Ras, K-Ras 4B, and M-Ras by receptors for growth factors or antigen. Mol Cell Biol. 2004;24:6311-23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Hamilton M, Wolfman A. Oncogenic Ha-Ras-dependent mitogen-activated protein kinase activity requires signaling through the epidermal growth factor receptor. J Biol Chem. 1998;273:28155-62 [DOI] [PubMed] [Google Scholar]
- 121. Rocks O, Peyker A, Bastiaens PI. Spatio-temporal segregation of Ras signals: one ship, three anchors, many harbors. Curr Opin Cell Biol. 2006;18:351-7 [DOI] [PubMed] [Google Scholar]
- 122. Rocks O, Peyker A, Kahms M, et al. An acylation cycle regulates localization and activity of palmitoylated Ras isoforms. Science. 2005;307:1746-52 [DOI] [PubMed] [Google Scholar]
- 123. Matallanas D, Arozarena I, Berciano MT, et al. Differences on the inhibitory specificities of H-Ras, K-Ras, and N-Ras (N17) dominant negative mutants are related to their membrane microlocalization. J Biol Chem. 2003;278:4572-81 [DOI] [PubMed] [Google Scholar]
- 124. Jaumot M, Yan J, Clyde-Smith J, Sluimer J, Hancock JF. The linker domain of the Ha-Ras hypervariable region regulates interactions with exchange factors, Raf-1 and phosphoinositide 3-kinase. J Biol Chem. 2002;277:272-8 [DOI] [PubMed] [Google Scholar]
- 125. Rosseland CM, Wierod L, Flinder LI, Oksvold MP, Skarpen E, Huitfeldt HS. Distinct functions of H-Ras and K-Ras in proliferation and survival of primary hepatocytes due to selective activation of ERK and PI3K. J Cell Physiol. 2008;215:818-26 [DOI] [PubMed] [Google Scholar]
- 126. Whitehead IP, Campbell S, Rossman KL, Der CJ. Dbl family proteins. Biochim Biophys Acta. 1997;1332:F1-23 [DOI] [PubMed] [Google Scholar]
- 127. Hyvonen M, Macias MJ, Nilges M, Oschkinat H, Saraste M, Wilmanns M. Structure of the binding site for inositol phosphates in a PH domain. EMBO J. 1995;14:4676-85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Lemmon MA, Ferguson KM, O’Brien R, Sigler PB, Schlessinger J. Specific and high-affinity binding of inositol phosphates to an isolated pleckstrin homology domain. Proc Natl Acad Sci U S A. 1995;92:10472-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Rebecchi MJ, Scarlata S. Pleckstrin homology domains: a common fold with diverse functions. Annu Rev Biophys Biomol Struct. 1998;27:503-28 [DOI] [PubMed] [Google Scholar]
- 130. Millan O, Ballester A, Castrillo A, et al. H-Ras-specific activation of NF-kappaB protects NIH 3T3 cells against stimulus-dependent apoptosis. Oncogene. 2003;22:477-83 [DOI] [PubMed] [Google Scholar]
- 131. Oliva JL, Perez-Sala D, Castrillo A, et al. The cyclopentenone 15-deoxy-delta 12,14-prostaglandin J2 binds to and activates H-Ras. Proc Natl Acad Sci U S A. 2003;100:4772-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Lowy DR, Willumsen BM. Protein modification: new clue to Ras lipid glue. Nature. 1989;341:384-5 [DOI] [PubMed] [Google Scholar]
- 133. Ulsh LS, Shih TY. Metabolic turnover of human c-rasH p21 protein of EJ bladder carcinoma and its normal cellular and viral homologs. Mol Cell Biol. 1984;4:1647-52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Laude AJ, Prior IA. Palmitoylation and localisation of RAS isoforms are modulated by the hypervariable linker domain. J Cell Sci. 2008;121:421-7 [DOI] [PubMed] [Google Scholar]
- 135. Casey PJ, Solski PA, Der CJ, Buss JE. p21ras is modified by a farnesyl isoprenoid. Proc Natl Acad Sci U S A. 1989;86:8323-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Gutierrez L, Magee AI, Marshall CJ, Hancock JF. Post-translational processing of p21ras is two-step and involves carboxyl-methylation and carboxy-terminal proteolysis. EMBO J. 1989;8:1093-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Choy E, Chiu VK, Silletti J, et al. Endomembrane trafficking of ras: the CAAX motif targets proteins to the ER and Golgi. Cell. 1999;98:69-80 [DOI] [PubMed] [Google Scholar]
- 138. Dai Q, Choy E, Chiu V, et al. Mammalian prenylcysteine carboxyl methyltransferase is in the endoplasmic reticulum. J Biol Chem. 1998;273:15030-4 [DOI] [PubMed] [Google Scholar]
- 139. Kim E, Ambroziak P, Otto JC, et al. Disruption of the mouse Rce1 gene results in defective Ras processing and mislocalization of Ras within cells. J Biol Chem. 1999;274:8383-90 [DOI] [PubMed] [Google Scholar]
- 140. Apolloni A, Prior IA, Lindsay M, Parton RG, Hancock JF. H-ras but not K-ras traffics to the plasma membrane through the exocytic pathway. Mol Cell Biol. 2000;20:2475-87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Choy E, Philips M. Green fluorescent protein-tagged Ras proteins for intracellular localization. Methods Enzymol. 2001;332:50-64 [DOI] [PubMed] [Google Scholar]
- 142. Omerovic J, Prior IA. Compartmentalized signalling: Ras proteins and signalling nanoclusters. FEBS J. 2009;276:1817-25 [DOI] [PubMed] [Google Scholar]
- 143. Gomez GA, Daniotti JL. H-Ras dynamically interacts with recycling endosomes in CHO-K1 cells: involvement of Rab5 and Rab11 in the trafficking of H-Ras to this pericentriolar endocytic compartment. J Biol Chem. 2005;280:34997-5010 [DOI] [PubMed] [Google Scholar]
- 144. Goodwin JS, Drake KR, Remmert CL, Kenworthy AK. Ras diffusion is sensitive to plasma membrane viscosity. Biophys J. 2005;89:1398-410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Baker TL, Zheng H, Walker J, Coloff JL, Buss JE. Distinct rates of palmitate turnover on membrane-bound cellular and oncogenic H-ras. J Biol Chem. 2003;278:19292-300 [DOI] [PubMed] [Google Scholar]
- 146. Magee AI, Gutierrez L, McKay IA, Marshall CJ, Hall A. Dynamic fatty acylation of p21N-ras. EMBO J. 1987;6:3353-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Thissen JA, Gross JM, Subramanian K, Meyer T, Casey PJ. Prenylation-dependent association of Ki-Ras with microtubules: evidence for a role in subcellular trafficking. J Biol Chem. 1997;272:30362-70 [DOI] [PubMed] [Google Scholar]
- 148. Chen Z, Otto JC, Bergo MO, Young SG, Casey PJ. The C-terminal polylysine region and methylation of K-Ras are critical for the interaction between K-Ras and microtubules. J Biol Chem. 2000;275:41251-7 [DOI] [PubMed] [Google Scholar]
- 149. Prior IA, Harding A, Yan J, Sluimer J, Parton RG, Hancock JF. GTP-dependent segregation of H-ras from lipid rafts is required for biological activity. Nat Cell Biol. 2001;3:368-75 [DOI] [PubMed] [Google Scholar]
- 150. McLaughlin S, Hangyas-Mihalyne G, Zaitseva I, Golebiewska U. Reversible - through calmodulin - electrostatic interactions between basic residues on proteins and acidic lipids in the plasma membrane. Biochem Soc Symp. 2005;72:189-98 [DOI] [PubMed] [Google Scholar]
- 151. Fivaz M, Meyer T. Reversible intracellular translocation of KRas but not HRas in hippocampal neurons regulated by Ca2+/calmodulin. J Cell Biol. 2005;170:429-41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Levkowitz G, Waterman H, Zamir E, et al. c-Cbl/Sli-1 regulates endocytic sorting and ubiquitination of the epidermal growth factor receptor. Genes Dev. 1998;12:3663-74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Plowman SJ, Ariotti N, Goodall A, Parton RG, Hancock JF. Electrostatic interactions positively regulate K-Ras nanocluster formation and function. Mol Cell Biol. 2008;28:4377-85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Mor A, Philips MR. Compartmentalized Ras/MAPK signaling. Annu Rev Immunol. 2006;24:771-800 [DOI] [PubMed] [Google Scholar]
- 155. Traverse S, Gomez N, Paterson H, Marshall C, Cohen P. Sustained activation of the mitogen-activated protein (MAP) kinase cascade may be required for differentiation of PC12 cells: comparison of the effects of nerve growth factor and epidermal growth factor. Biochem J. 1992;288(Pt 2):351-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Kofer-Geles M, Gottfried I, Haklai R, Elad-Zefadia G, Kloog Y, Ashery U. Rasosomes spread Ras signals from plasma membrane ’hotspots’. Biochim Biophys Acta. 2009;1793:1691-702 [DOI] [PubMed] [Google Scholar]
- 157. Guil S, de La Iglesia N, Fernandez-Larrea J, et al. Alternative splicing of the human proto-oncogene c-H-ras renders a new Ras family protein that trafficks to cytoplasm and nucleus. Cancer Res. 2003;63:5178-87 [PubMed] [Google Scholar]
- 158. Wolfman JC, Planchon SM, Liao J, Wolfman A. Structural and functional consequences of c-N-Ras constitutively associated with intact mitochondria. Biochim Biophys Acta. 2006;1763:1108-24 [DOI] [PubMed] [Google Scholar]
- 159. Rebollo A, Perez-Sala D, Martinez AC. Bcl-2 differentially targets K-, N-, and H-Ras to mitochondria in IL-2 supplemented or deprived cells: implications in prevention of apoptosis. Oncogene. 1999;18:4930-9 [DOI] [PubMed] [Google Scholar]
- 160. de la Vega M, Burrows JF, McFarlane C, Govender U, Scott CJ, Johnston JA. The deubiquitinating enzyme USP17 blocks N-Ras membrane trafficking and activation but leaves K-Ras unaffected. J Biol Chem. 2010;285:12028-36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Sinn E, Muller W, Pattengale P, Tepler I, Wallace R, Leder P. Coexpression of MMTV/v-Ha-ras and MMTV/c-myc genes in transgenic mice: synergistic action of oncogenes in vivo. Cell. 1987;49:465-75 [DOI] [PubMed] [Google Scholar]
- 162. Jackson EL, Willis N, Mercer K, et al. Analysis of lung tumor initiation and progression using conditional expression of oncogenic K-ras. Genes Dev. 2001;15:3243-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Meuwissen R, Linn SC, van der Valk M, Mooi WJ, Berns A. Mouse model for lung tumorigenesis through Cre/lox controlled sporadic activation of the K-Ras oncogene. Oncogene. 2001;20:6551-8 [DOI] [PubMed] [Google Scholar]
- 164. Guerra C, Mijimolle N, Dhawahir A, et al. Tumor induction by an endogenous K-ras oncogene is highly dependent on cellular context. Cancer Cell. 2003;4:111-20 [DOI] [PubMed] [Google Scholar]
- 165. Johnson L, Mercer K, Greenbaum D, et al. Somatic activation of the K-ras oncogene causes early onset lung cancer in mice. Nature. 2001;410:1111-6 [DOI] [PubMed] [Google Scholar]
- 166. Dinulescu DM, Ince TA, Quade BJ, Shafer SA, Crowley D, Jacks T. Role of K-ras and Pten in the development of mouse models of endometriosis and endometrioid ovarian cancer. Nat Med. 2005;11:63-70 [DOI] [PubMed] [Google Scholar]
- 167. Aguirre AJ, Bardeesy N, Sinha M, et al. Activated Kras and Ink4a/Arf deficiency cooperate to produce metastatic pancreatic ductal adenocarcinoma. Genes Dev. 2003;17:3112-26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Hingorani SR, Wang L, Multani AS, et al. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell. 2005;7:469-83 [DOI] [PubMed] [Google Scholar]
- 169. Izeradjene K, Combs C, Best M, et al. Kras(G12D) and Smad4/Dpc4 haploinsufficiency cooperate to induce mucinous cystic neoplasms and invasive adenocarcinoma of the pancreas. Cancer Cell. 2007;11:229-43 [DOI] [PubMed] [Google Scholar]
- 170. Tuveson DA, Shaw AT, Willis NA, et al. Endogenous oncogenic K-ras(G12D) stimulates proliferation and widespread neoplastic and developmental defects. Cancer Cell. 2004;5:375-87 [DOI] [PubMed] [Google Scholar]
- 171. Braun BS, Archard JA, Van Ziffle JA, Tuveson DA, Jacks TE, Shannon K. Somatic activation of a conditional KrasG12D allele causes ineffective erythropoiesis in vivo. Blood. 2006;108:2041-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. Chen X, Mitsutake N, LaPerle K, et al. Endogenous expression of Hras(G12V) induces developmental defects and neoplasms with copy number imbalances of the oncogene. Proc Natl Acad Sci U S A. 2009;106:7979-84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Schuhmacher AJ, Guerra C, Sauzeau V, Canamero M, Bustelo XR, Barbacid M. A mouse model for Costello syndrome reveals an Ang II-mediated hypertensive condition. J Clin Invest. 2008;118:2169-79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Singh M, Lima A, Molina R, et al. Assessing therapeutic responses in Kras mutant cancers using genetically engineered mouse models. Nat Biotechnol. 2010;28:585-93 [DOI] [PubMed] [Google Scholar]
- 175. Perez-Mancera PA, Tuveson DA. Physiological analysis of oncogenic K-ras. Methods Enzymol. 2006;407:676-90 [DOI] [PubMed] [Google Scholar]
- 176. Umanoff H, Edelmann W, Pellicer A, Kucherlapati R. The murine N-ras gene is not essential for growth and development. Proc Natl Acad Sci U S A. 1995;92:1709-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. Johnson L, Greenbaum D, Cichowski K, et al. K-ras is an essential gene in the mouse with partial functional overlap with N-ras. Genes Dev. 1997;11:2468-81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. Koera K, Nakamura K, Nakao K, et al. K-ras is essential for the development of the mouse embryo. Oncogene. 1997;15:1151-9 [DOI] [PubMed] [Google Scholar]
- 179. Esteban LM, Vicario-Abejon C, Fernandez-Salguero P, et al. Targeted genomic disruption of H-ras and N-ras, individually or in combination, reveals the dispensability of both loci for mouse growth and development. Mol Cell Biol. 2001;21:1444-52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180. Plowman SJ, Williamson DJ, O’Sullivan MJ, et al. While K-ras is essential for mouse development, expression of the K-ras 4A splice variant is dispensable. Mol Cell Biol. 2003;23:9245-50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. Plowman SJ, Arends MJ, Brownstein DG, et al. The K-Ras 4A isoform promotes apoptosis but does not affect either lifespan or spontaneous tumor incidence in aging mice. Exp Cell Res. 2006;312:16-26 [DOI] [PubMed] [Google Scholar]
- 182. Drosten M, Dhawahir A, Sum EY, et al. Genetic analysis of Ras signalling pathways in cell proliferation, migration and survival. EMBO J. 2010;29:1091-104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183. Potenza N, Vecchione C, Notte A, et al. Replacement of K-Ras with H-Ras supports normal embryonic development despite inducing cardiovascular pathology in adult mice. EMBO Rep. 2005;6:432-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184. Castellano E, De Las Rivas J, Guerrero C, Santos E. Transcriptional networks of knockout cell lines identify functional specificities of H-Ras and N-Ras: significant involvement of N-Ras in biotic and defense responses. Oncogene. 2007;26:917-33 [DOI] [PubMed] [Google Scholar]
- 185. Castellano E, Guerrero C, Nunez A, De Las Rivas J, Santos E. Serum-dependent transcriptional networks identify distinct functional roles for H-Ras and N-Ras during initial stages of the cell cycle. Genome Biol. 2009;10:R123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186. Roberts ML, Drosopoulos KG, Vasileiou I, et al. Microarray analysis of the differential transformation mediated by Kirsten and Harvey Ras oncogenes in a human colorectal adenocarcinoma cell line. Int J Cancer. 2006;118:616-27 [DOI] [PubMed] [Google Scholar]
- 187. Zuber J, Tchernitsa OI, Hinzmann B, et al. A genome-wide survey of RAS transformation targets. Nat Genet. 2000;24:144-52 [DOI] [PubMed] [Google Scholar]
- 188. Sweet-Cordero A, Mukherjee S, Subramanian A, et al. An oncogenic KRAS2 expression signature identified by cross-species gene-expression analysis. Nat Genet. 2005;37:48-55 [DOI] [PubMed] [Google Scholar]
- 189. Vasseur S, Malicet C, Calvo EL, et al. Gene expression profiling by DNA microarray analysis in mouse embryonic fibroblasts transformed by rasV12 mutated protein and the E1A oncogene. Mol Cancer. 2003;2:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190. Ohnami S, Aoki K, Yoshida K, et al. Expression profiles of pancreatic cancer cell lines infected with antisense K-ras-expressing adenoviral vector. Biochem Biophys Res Commun. 2003;309:798-803 [DOI] [PubMed] [Google Scholar]
- 191. Kim S, Lee YZ, Kim YS, Bahk YY. A proteomic approach for protein-profiling the oncogenic ras induced transformation (H-, K-, and N-Ras) in NIH/3T3 mouse embryonic fibroblasts. Proteomics. 2008;8:3082-93 [DOI] [PubMed] [Google Scholar]
- 192. Croonquist PA, Linden MA, Zhao F, Van Ness BG. Gene profiling of a myeloma cell line reveals similarities and unique signatures among IL-6 response, N-ras-activating mutations, and coculture with bone marrow stromal cells. Blood. 2003;102:2581-92 [DOI] [PubMed] [Google Scholar]
- 193. Brem R, Certa U, Neeb M, Nair AP, Moroni C. Global analysis of differential gene expression after transformation with the v-H-ras oncogene in a murine tumor model. Oncogene. 2001;20:2854-8 [DOI] [PubMed] [Google Scholar]
- 194. Kawano M, Hirano T, Matsuda T, et al. Autocrine generation and requirement of BSF-2/IL-6 for human multiple myelomas. Nature. 1988;332:83-5 [DOI] [PubMed] [Google Scholar]
- 195. Janknecht R, Monte D, Baert JL, de Launoit Y. The ETS-related transcription factor ERM is a nuclear target of signaling cascades involving MAPK and PKA. Oncogene. 1996;13:1745-54 [PubMed] [Google Scholar]
- 196. Muda M, Theodosiou A, Rodrigues N, et al. The dual specificity phosphatases M3/6 and MKP-3 are highly selective for inactivation of distinct mitogen-activated protein kinases. J Biol Chem. 1996;271:27205-8 [DOI] [PubMed] [Google Scholar]
- 197. Blackstock WP, Weir MP. Proteomics: quantitative and physical mapping of cellular proteins. Trends Biotechnol. 1999;17:121-7 [DOI] [PubMed] [Google Scholar]
- 198. Malumbres M, Pellicer A. RAS pathways to cell cycle control and cell transformation. Front Biosci. 1998;3:d887-912 [DOI] [PubMed] [Google Scholar]
- 199. Perez de Castro I, Diaz R, Malumbres M, et al. Mice deficient for N-ras: impaired antiviral immune response and T-cell function. Cancer Res. 2003;63:1615-22 [PubMed] [Google Scholar]
- 200. Perez de Castro I, Bivona TG, Philips MR, Pellicer A. Ras activation in Jurkat T cells following low-grade stimulation of the T-cell receptor is specific to N-Ras and occurs only on the Golgi apparatus. Mol Cell Biol. 2004;24:3485-96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201. Wolfman JC, Palmby T, Der CJ, Wolfman A. Cellular N-Ras promotes cell survival by downregulation of Jun N-terminal protein kinase and p38. Mol Cell Biol. 2002;22:1589-606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202. Eskandarpour M, Kiaii S, Zhu C, Castro J, Sakko AJ, Hansson J. Suppression of oncogenic NRAS by RNA interference induces apoptosis of human melanoma cells. Int J Cancer. 2005;115:65-73 [DOI] [PubMed] [Google Scholar]
- 203. Pruitt K, Der CJ. Ras and Rho regulation of the cell cycle and oncogenesis. Cancer Lett. 2001;171:1-10 [DOI] [PubMed] [Google Scholar]
- 204. Durkin JP, Whitfield JF. Characterization of G1 transit induced by the mitogenic-oncogenic viral Ki-ras gene product. Mol Cell Biol. 1986;6:1386-92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205. Cai H, Szeberenyi J, Cooper GM. Effect of a dominant inhibitory Ha-ras mutation on mitogenic signal transduction in NIH 3T3 cells. Mol Cell Biol. 1990;10:5314-23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206. Dobrowolski S, Harter M, Stacey DW. Cellular ras activity is required for passage through multiple points of the G0/G1 phase in BALB/c 3T3 cells. Mol Cell Biol. 1994;14:5441-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207. Feig LA, Cooper GM. Inhibition of NIH 3T3 cell proliferation by a mutant ras protein with preferential affinity for GDP. Mol Cell Biol. 1988;8:3235-43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208. Mulcahy LS, Smith MR, Stacey DW. Requirement for ras proto-oncogene function during serum-stimulated growth of NIH 3T3 cells. Nature. 1985;313:241-3 [DOI] [PubMed] [Google Scholar]
- 209. Stacey DW, Kung HF. Transformation of NIH 3T3 cells by microinjection of Ha-ras p21 protein. Nature. 1984;310:508-11 [DOI] [PubMed] [Google Scholar]
- 210. Gille H, Downward J. Multiple ras effector pathways contribute to G(1) cell cycle progression. J Biol Chem. 1999;274:22033-40 [DOI] [PubMed] [Google Scholar]
- 211. Satoh T, Endo M, Nakafuku M, Akiyama T, Yamamoto T, Kaziro Y. Accumulation of p21ras.GTP in response to stimulation with epidermal growth factor and oncogene products with tyrosine kinase activity. Proc Natl Acad Sci U S A. 1990;87:7926-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212. Taylor SJ, Shalloway D. Cell cycle-dependent activation of Ras. Curr Biol. 1996;6:1621-7 [DOI] [PubMed] [Google Scholar]
- 213. Jones SM, Kazlauskas A. Growth-factor-dependent mitogenesis requires two distinct phases of signalling. Nat Cell Biol. 2001;3:165-72 [DOI] [PubMed] [Google Scholar]
- 214. Kumar A, Marques M, Carrera AC. Phosphoinositide 3-kinase activation in late G1 is required for c-Myc stabilization and S phase entry. Mol Cell Biol. 2006;26:9116-25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215. Stacey D, Kazlauskas A. Regulation of Ras signaling by the cell cycle. Curr Opin Genet Dev. 2002;12:44-6 [DOI] [PubMed] [Google Scholar]
- 216. Takuwa N, Takuwa Y. Regulation of cell cycle molecules by the Ras effector system. Mol Cell Endocrinol. 2001;177:25-33 [DOI] [PubMed] [Google Scholar]
- 217. Sa G, Hitomi M, Harwalkar J, Stacey AW, Gc GC, Stacey DW. Ras is active throughout the cell cycle, but is able to induce cyclin D1 only during G2 phase. Cell Cycle. 2002;1:50-8 [PubMed] [Google Scholar]
- 218. Marshall CJ. Small GTPases and cell cycle regulation. Biochem Soc Trans. 1999;27:363-70 [DOI] [PubMed] [Google Scholar]
- 219. Marshall C. How do small GTPase signal transduction pathways regulate cell cycle entry? Curr Opin Cell Biol. 1999;11:732-6 [DOI] [PubMed] [Google Scholar]