

Introduction
Metal binding proteins are essential to many life-sustaining processes. The non-thermodynamic binding selectivity and specificity of metals to proteins in living systems has captured the focus of much research effort.2 Equally essential to life-sustaining processes are the biomolecular mechanisms by which tightly bound metals are selectively transferred2b,3 or released4 from a thermodynamically strong coordination sphere within a protein. As mechanistic knowledge of these metal mobilizing biological processes is uncovered, occasions may arise for translating the biological processes into ones useful in chemical catalysis in the laboratory. In particular, the biomolecular mechanisms by which Nature releases catalytically important thiophilic metals like Cu from high-sulfur binding sites within proteins should be useful points of inspiration from which to develop novel preparative protocols where the catalytic turnover of a thiophilic metal in a sulfur-containing environment is essential. Opportunities for productive metal catalysis in sulfur-rich systems are found in energy-related research (metal-catalyzed desulfurization of carbon-based fuels),5 detoxification of nerve gas agents (metal-catalyzed transformations of phosphonothioate and phosphorothioates),6 as well as in the synthesis of fine chemicals through desulfitative transformations.7
As a case in point, consider the metallothioneins, ubiquitous small proteins capable of binding up to 7 equivalents of mono and divalent metals such as Cu(I) and Zn(II), among others.8 Significant insights into the triggered release of Zn and Cu from the cysteine-rich coordination spheres resident within metallothioneins have been obtained.4a,b,9 Although positioned within a tightly binding thiol/thiolate-rich ligand environment, the thiophilic metal is rapidly released when a metallothionein is exposed to an exogenous disulfide4f,10 or to related reagents,4b,4d or to NO.4c,11 Following one of these leads we describe herein a novel Cu(I)-catalyzed reaction system that finds its specific inspiration in the mild S-centered oxidative release of metal from the high cysteine coordination sphere of the metallothioneins upon exposure to an exogenous disulfide.
Copper is an essential element in biology, and it also plays a central role in many modern metal-mediated organic transformations.12 In particular, by virtue of its thiophilicity and accessible multiple oxidation states, it is a key metal in new pH-neutral, desulfitative carbon-carbon bond forming processes that take place between thioorganic substrates and boronic acids (Figure 1). The “first-generation” of the copper-mediated, desulfitative reaction systems proceeds under anaerobic conditions, and is catalyzed by Pd and enabled by stoichiometric quantities of a Cu(I) carboxylate.7a,13 A newer 2nd-generation system takes place under aerobic reaction conditions, is palladium-free, and uses only catalytic quantities of Cu.14 Organostannanes are also effective partners in desulfitative couplings.7a,15
Figure 1.

1st and 2nd Generation Desulfitative Coupling of Thioorganics and Boronic Acids
While the anaerobic and aerobic desulfitative coupling protocols superficially appear to be quite different, they share two common aspects that are essential to Cu-catalysis in a thiophilic environment. In each case (1) the thiolate is fully scavenged from the reaction system, and (2) the boronic acid –B(OH)2 moiety is provided with a thermodynamically strongly bonding “oxygenate” ligand for its 3rd valence. These characteristics of the desulfitative coupling reactions are jointly satisfied in the 1st generation versions of the desulfitative chemistry through the use of stoichiometric quantities of the copper carboxylate source:7a,15a the CuI scavenges the thiolate and the carboxylate serves as the 3rd valence for –B(OH)2. The 2nd generation aerobic reactions mentioned above proceed with only catalytic quantities of Cu, but they require a sacrificial second equivalent of the boronic acid. Under the aerobic reaction conditions this second equivalent of the boronic acid serves to scavenge the thiolate from the reaction cycle as a non-perturbing thio ether. The aerobic reaction parameters of the 2nd generation desulfitative couplings also provide the “oxygenate” moiety, derived from O2, that pairs with the –B(OH)2 moiety.
This mechanistic understanding of the 1st and 2nd generation thioorganic-boronic acid desulfitative coupling reactions raises an interesting challenge: “Can the desulfitative coupling of a thioorganic and a boronic acid be achieved using only catalytic quantities of Cu and without squandering an extra equivalent of the boronic acid?” The biological metallothionein system mentioned above suggests a solution to the catalytic challenge of the chemical system. Within a metallothionein, exposure of a metal-bound thiolate ligand to an exogenous disulfide converts the strongly binding thiolate to a weakly binding disulfide ligand,16 thereby liberating the metal from the protein through an S-centered oxidation (Scheme 1).
Scheme 1.
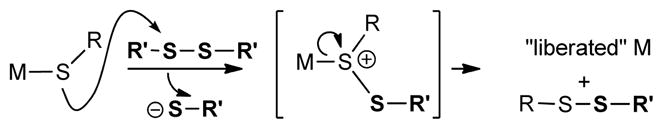
To render Cu(I) catalytically viable in a desulfitative chemical transformation without sacrificing a second equivalent of the boronic acid coupling agent, a small molecule analog of the metallothionein system was designed. The “MT-mimic” is shown generically in Scheme 2. While serving the same conceptual purpose, the analog replaces the S—S reactant of the biological system with an N—O moiety. Through its oxime N—O bond the MT-mimic was designed to internally provide a mild S-centered oxidation of a Cu(I) thiolate thereby converting the strongly bonding thiolate to a weakly bonding disulfide equivalent (in this case the S—N bond of the benzoisothiazole shown). This mild oxidative trapping of thiolate was also intended to continuously regenerate a catalytically viable “oxygenate” form of Cu(I) (and a stoichiometric oxygenate residue to pair with –B(OH)2) so that a useful Cu(I)-catalyzed desulfitative carbon-carbon bond forming reaction with boronic acids can ensue.
Scheme 2.

The Metallothionein Mimic Concept
Results
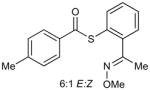
The catalysis concept described above was explored through an efficient, Cu-catalyzed desulfitative coupling of MT-mimic thiol esters with boronic acids and organostannanes to give ketones. Since the synthesis of ketones from thiol esters and boronic acids/organostannanes is well-known,14b,15a,17 the significance of the work described here lies not in the specific synthetic demonstration, but rather in the underlying principles of catalysis by Cu in sulfur-rich systems that are revealed by the study. The requisite thiol ester substrates (3), which bear an MT-mimicking sulfur pendant, were easily prepared by standard procedures from the stable and storable O-methyl oxime of 2-mercaptoacetophenone, 2, which, in turn, was generated from commercially available mercaptosalicylic acid, 1, in a straightforward fashion (Scheme 3). Although the choice of the acetophenone derivative brings with it the issue of E/Z oxime stereoisomerism, the structurally simpler all-E-benzaldehyde oximes were ineffective substrates because of their tendency to eliminate MeOH and generate the corresponding nitriles under the reaction conditions.
Scheme 3.

Construction of the Metallothionein Mimic
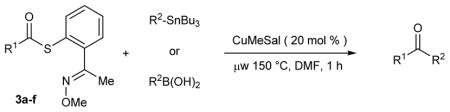
Exploratory studies of Cu-only catalysis using the MT-mimic 3a were carried out (Table 1). A mixture of thiol ester 3a and either (4-methoxyphenyl)tri-n-butylstannane or 4-(formylphenyl) boronic acid were exposed to varying amounts of CuI-3-methylsalicylate (CuMeSal18) in DMF at various temperatures under argon. In most cases the boron and the tin reagents showed similar reactivity trends. Desulfitative coupling performed at 60 °C is facile for the E-oxime stereoisomer (Table 1, entries 1 – 4), but the process at this temperature is strictly stoichiometric in Cu (recovery of the Z-oxime reflects the mass balance). Catalytic turnover of Cu ensues at 100 °C, although recovery of the unreactive Z-oxime at this temperature still minimizes the reaction efficiency (Table 1, entries 5 and 6). Reactions were noticeably faster using microwave irradiation (Table 1, entries 7–10), with the fastest reactions and full conversion of both the E- and the Z-oxime stereoisomers taking place within 1 h at 150 °C. Under these conditions the desired ketone and the anticipated benzoisothiazole were obtained in a roughly 1:1 ratio.
Table 1.
Control Experiments. Cu-Catalyzed Desulfitative Couplinga
|
| |||||||
|---|---|---|---|---|---|---|---|
| thiol ester | M | R′ | CuMeSal % | rxn conditions | ketone % | S-N trap % | |
| 1 |
|
n-Bu3Sn | OMe | 20 | 60 °C, 24 h | 22 | 17 |
| 2 | (HO)2B | CHO | 20 | 60 °C, 24 h | 16 | 0b | |
| 3 | n-Bu3Sn | OMe | 120 | 60 °C, 12 h | 83c | 51d | |
| 4 | (HO)2B | CHO | 120 | 60 °C, 12 h | 85c | 44d | |
| 5 | n-Bu3Sn | OMe | 20 | 100 °C, 12 h | 80 | 72 | |
| 6 | (HO)2B | CHO | 20 | 100 °C, 12 h | 76 | 60 | |
| 7 | n-Bu3Sn | OMe | 20 | μw, 100 °C, 2 h | 82 | 70 | |
| 8 | (HO)2B | CHO | 20 | μw, 100 °C, 2 h | 72 | 65 | |
| 9 | n-Bu3Sn | OMe | 20 | μw, 150 °C, 1 h | 95 | 87 | |
| 10 | (HO)2B | CHO | 20 | μw, 150 °C, 1 h | 80 | 78 | |
| 11 | n-Bu3Sn | OMe | 0 | μw, 150 °C, 1 h | 0 | trace | |
| 12 | (HO)2B | CHO | 0 | μw, 150 °C, 1 h | 0 | trace | |
| 13 | --- | --- | 20 | μw, 150 °C, 1 h | NA | trace | |
| 14 |
|
n-Bu3Sn | OMe | 20 | μw, 150 °C, 1h | 17 | NA |
| 15 | (HO)2B | CHO | 20 | μw, 150 °C, 1h | 0 | NA | |
A Schlenk tube (entry 1–6) or microwave tube (entry 7–15) was equipped with a magnetic stir bar. To the tube was added the thiol ester (0.1 mmol), CuMeSal (0.02 mmol) and organostanane (0.11 mmol) or boronic acid (0.12 mmol). After flushing with argon, anhydrous and degassed DMF (1 mL) was added. The reaction mixture was stirred under the indicated conditions. After cooling, ethyl ether (10 mL) was added to the mixture. The reaction mixture was washed with water, brine, dried over MgSO4 and evaporated. The residue was purified by preparative plate silica chromatography using hexanes/EtOAc as the eluent
In contrast to the analogous control experiment with the organostannane (entry 1), benzoisothiazoline is not formed in this reaction. Rather, 20% of the S-arylation product (4-[2-(1-methoxyimino-ethyl)-phenylsulfanyl]-benzaldehyde) is formed. However, when the reaction was quenched under argon prior to the work up, neither the thioether nor the benzoisothiazoline was generated.
The Z-oxime stereoisomer of the thiol ester was recovered.
The benzoisothiazole is formed in less than a one to one ratio relative to the ketone because, at 60° C, the formation of the benzoisothiazole from the intermediate Cu thiolate is slower than the rate of formation of the ketone.
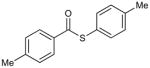
Control experiments demonstrated that both the Cu and the internal O-methyl oxime moiety are essential for catalytic turnover. In the absence of Cu under microwave heating at 150 °C the thiol ester 3a did not generate ketone from either the stannane or the boronic acid reactant (Table 1, entries 11 and 12). Furthermore, the thiol ester 3a was surprisingly robust under microwave heating at 150 °C in the absence of the boron or tin reactant, even in the presence of the Cu catalyst (Table 1, entry 13).
The importance of the internal O-methyl oxime moiety for catalytic turnover was highlighted through a series of experiments. Exposure of the simple thiol ester p-tolyl(CO)S-p-tolyl to (4-methoxyphenyl)tri-n-butylstannane in the presence of 20 mol % CuMeSal under microwave heating at 150 °C generated a small amount (17%) of the ketone (Table 1, entry 14), while an analogous reaction with 4-(formylphenyl) boronic acid showed no evidence of reaction with simple thiol esters (Table 1, entry 15). The former result demonstrates, at least at the elevated reaction temperatures under study, that the stannane system does not require the presence of the internal oxime function to facilitate coupling with the thiol ester, but under the conditions explored the transformation to keone appears to be stoichiometric, not catalytic in Cu. As a control experiment to assess the importance of the internal oxime moiety as both a Cu-thiolate trapping agent and an oxygenate partner for Cu(I) in the catalytic cycle, p-tolyl(CO)S-p-tolyl and both (4-methoxyphenyl)tri-n-butylstannane and 4-(formylphenyl) boronic acid were exposed to 20 mol % CuMeSal in the presence of the external O-methyl oxime C6H5(C=NOMe)CH3 (Scheme 4). The stannane reaction led to less than 20% of the ketone, another transformation that appears to be stoichiometric, but not catalytic in Cu. The boronic acid again showed no evidence of reaction. The mechanistic implications of the data presented in Table 1 and Scheme 4 are discussed below.
Scheme 4.

Control Experiments with a Simple Thiol Ester
Different commercially available CuIX sources including X = Cl, I, and thiophene-2-carboxylate were all effective in this new desulfitative cross-coupling; however, CuI-3-methylsalicylate provided the best yields, in general. No reaction took place in the absence of a CuIX catalyst. Interestingly, and for reasons not currently understood, CuII(OAc)2 was effective in promoting catalysis with the boronic acids, but not with the organostannanes.
Two somewhat more reactive oxime variants (bearing O-carboxylate and O-phenyl moieties) were also prepared and studied, but neither system was viable for the production of ketones from thiol esters via the copper-catalyzed chemistry. This is most likely a function of the easy reductive cleavage of the more reactive =N—O(CO)R and =N—OPh bonds by Cu(I),19 while the less reactive N—O bond of the O-methyl oximes remains untouched by Cu under the reaction conditions.
Having confirmed the value of the metallothionein mimic for catalytic turnover of Cu under anaerobic conditions, the generality of this Cu-catalyzed desulfitative coupling was studied. Table 2 shows that a series of organoboron and organotin reagents are excellent reaction partners in Cu-catalyzed couplings with different MT-mimic thiol esters. Near stoichiometric quantities of either the boron or tin coupling partner suffice to generate good to excellent yields of ketones. Electron-deficient (entries 1–6, 10, 12) and electron-rich (entries 7, 8, 13) aryl boronic acids participated efficiently in the reaction. The coupling of an alkenyl (entry 9) and a heteroaryl (entry 11) boronic acid was also possible. For the boronic acid system, variation in the nature of the acyl moiety was well accommodated; chloro, cyano, ester, ketone, aldehyde and phenolic functional groups were tolerated. Loadings of Cu less than 20 mol % were acceptable in some cases, but 20 mol % CuMeSal was suitable in all cases explored.
Table 2.
Cu-Catalyzed Desulfitative Coupling using a “Metallothionein Mimic”
 | |||
|---|---|---|---|
| entry | R1 | R2 | ketone (%) |
| Boronic Acid Couplings using 1.2 equiv of R2B(OH)2a | |||
| 1 | p-tolyl | 4-chlorophenyl | 91 |
| 2 | p-tolyl | 4-cyanophenyl | 88 |
| 3 | p-tolyl | 4-(methoxycarbonyl)phenyl | 86 |
| 4 | p-tolyl | 4-(formyl)phenyl | 80 |
| 5 | p-tolyl | 3-acetylphenyl | 71 |
| 6 | p-tolyl | 3-formylphenyl | 69 |
| 7 | p-tolyl | 3-hydroxyphenyl | 70 |
| 8 | p-tolyl | 2,5-dimethoxyphenyl | 68 |
| 9 | p-tolyl | trans-2-(4-chlorophenyl)vinyl | 52 |
| 10 | n-propyl | 4-(methoxycarbonyl)phenyl | 82 |
| 11 | CH2OAc | 3-furyl | 73 |
| 12 | CH2OAc | 4-(methoxycarbonyl)phenyl | 78 |
| 13 | (E)-1-propenyl | 4-methoxyphenyl | 80 |
|
| |||
| Organostannane Couplings using 1.1 equiv of R2SnBu3b | |||
| 14 | p-tolyl | 4-methoxyphenyl | 95 |
| 15 | p-tolyl | 4-fluoro-3-methylphenyl | 81 |
| 16 | p-tolyl | 4-iodophenyl | 68 |
| 17 | thienyl | 4-methoxyphenyl | 86 |
| 18 | n-propyl | 2-methoxypyridin-2-yl | 70 |
| 19 | Cyclohexyl | 4-methoxyphenyl | 62 |
Typical experimental procedure: A dry microwave tube (10 mL) was equipped with a magnetic stir bar. To the tube was added the corresponding thiol ester (0.1 mmol), boronic acid (0.12 mmol) and CuMeSal (0.02 mmol). The reaction tube was flushed with argon and sealed. Through the septum anhydrous and degassed DMF (1 mL) was added. The mixture was subsequently heated in a microwave reactor at 150 °C for 1 h. After cooling, ethyl ether (10 mL) was added to the mixture. The reaction mixture was washed with water (2×5 mL), brine (5 mL), dried over MgSO4 and evaporated. The residue was purified by preparative plate silica chromatography using hexanes/EtOAc as the eluent.
Typical experimental procedure: A dry microwave tube (10 mL) was equipped with a magnetic stir bar. To the tube were added the corresponding thiol ester (0.1 mmol) and CuMeSal (0.02 mmol). The reaction vessel was flushed with argon and sealed. Through the septum the organostanane (0.11 mmol) dissolved in anhydrous and degassed DMF (1 mL) was added. The mixture was subsequently heated in a microwave reactor at 150 °C for 1 h. After cooling, ethyl ether (10 mL) was added to the mixture. The reaction mixture was washed with water, brine, dried over MgSO4 and evaporated. The residue was purified by preparative plate silica chromatography using hexanes/EtOAc as the eluent.
Organostannanes were also suitable reaction partners for a variety of MT-mimic thiol esters. Thiol esters 3a–f were treated with 1.1 equiv of an organostannane in the presence of 20 mol % CuMeSal in DMF for 1 h under microwave heating (150 °C). As shown in Table 2, aromatic, heteroaromatic, and aliphatic thiol esters all coupled efficiently with a variety of electron-rich and electron-poor aromatic and of heteroaromatic organostannanes. A 4-iodophenyl moiety (entry 16) was tolerated as was a 2-pyridylstannane (entry 18).
Although the full scope and limitations of this new Cu-catalyzed reaction are yet to be tested, the catalytic system has been extended to include preliminary examples of aliphatic transfer from boron as well as preliminary examples of the construction of peptidic ketones. Early probe reactions for both systems are depicted in Figure 2. Aliphatic boron reagents were suitable reaction partners in this chemistry: Et3B and B-2-phenylethyl-9-BBN each reacted with thiol ester 3a to produce the corresponding ketones in 80 and 71% yields, respectively. And, in exploratory reactions, both a boronic acid and an organostannane coupled with the high enantiopurity phenylalanine derived thiol ester 4 at 90 °C and afforded the corresponding peptidly ketones in good yields. No racemization was observed: the configuration of the amino acid stereogenic center was fully preserved. Exploratory reactions with potassium alkyltrifluoroborates, arylboronate esters and arylsilanes were not successful.
Figure 2.

Cu-Catalyzed Desulfitative Couplings: Aliphatic Transfer Reagents and Peptidyl Ketone Synthesis
Discussion
By way of the discrete examples presented within, the current study establishes a conceptual framework upon which to design new processes that are catalyzed by thiophilic metals and proceed through metal thiolates. We suggest the general mechanism depicted in Figure 3 for the anaerobic, Cu-catalyzed desulfitative coupling of the ‘metallothionein mimic’ thiol esters with boron or tin reagents.
Figure 3.

Mimicking Metallothioneins for Chemical Catalysis: The Proposed Catalytic Cycle
The catalytic cycle is assumed to begin with coordination of CuI to the thiol ester-oxime. This S,N-chelation, when followed by a transmetalation from boron or tin to CuI (transmetalation from boron and tin to copper is an assumed essential step in a wide variety of Cu-catalyzed bond-forming processes),12d,14,20 allows the Cu to position the thiol ester carbonyl carbon and the organocopper R2 in close proximity. That proximity effect along with a coordination-induced electrophilic activation of the thiol ester can be anticipated to lead to ketone formation through a directed reaction of the in situ generated organocopper reagent.21
Subsequent to the carbon-carbon bond forming step, a CuI-thiolate is generated. At this point the catalytic cycle would falter in the absence of an effective means to scavenge the thiolate from the Cu and to regenerate a suitable “oxygenate” for pairing with the boron (or tin). However, with the designed MT-mimic, direct reaction of the Cu-thiolate with the internal oxime functionality effectively traps the thiolate as the benzoisothiazole and an active Cu oxygenate catalyst is released. 22 Therefore, the internal coordinating oxime group not only lowers the barrier to reaction through pre-association of reactants, but also serves as a ‘metallothionein mimic’ to oxidatively scavenge the thiolate and regenerate an active metal catalyst. The thiolate scavenging motif used within this study was modeled after the known thermal dealkylative cyclization of 2-alkylmercapto aryl oximes to benzoisothiazoles.23
The use of the acetophenone rather than a benzaldehyde oxime moiety to trap an internal thiolate raises the question of the influence of the oxime stereochemistry on the reaction dynamics. Although the O-methyl oximes exist as mixtures of E and Z stereoisomers, the stereoisomerism was not problematic, at least at the higher reaction temperatures explored, because the oxime stereoisomers are easily interconverted under the reaction conditions (related oxime equilibrations in metal-catalyzed reactions are known).24 At lower reaction temperatures the E-oxime was transformed cleanly to the desired ketone and the benzoisothiazole leaving the Z-oxime stereoisomer untouched (determined by 1H NMR analysis of the crude reaction product prior to purification). The corresponding 2-mercaptobenzaldehyde oximes were also briefly investigated. Although the benzaldehyde-derived oximes exist exclusively in the E-configuration, their effectiveness in the designed reaction system was compromised by their competitive elimination to aryl cyanides.
Figure 4 provides an explanation for the different behavior of the boron and tin reagents in their control experiment reactions with the simple thiol ester p-tolyl(CO)S-p-tolyl. Only the tin reagent, (4-methoxyphenyl)tri-n-butylstannane, generates the corresponding ketone from the simple thiol ester in a reaction that is stoichiometric in Cu. The boronic acid, 4-(formylphenyl) boronic acid, returns the thiol ester untouched. Support for a rapid Cu-mediated proto-deborylation of the boronic acid in the absence of a suitable coordinating thiol ester reactant as suggested in Figure 4 is found in earlier studies of boronic acid-based desulfitative transformations.17i The control experiments suggest that the organocopper intermediate generated from the organostannane reaction variant, which lacks the active OH residues of the boronic acid, possesses a sufficient lifetime to allow reaction with the simple thiol ester.
Figure 4.

Divergent Behavior of Boron and Tin Reagents Exposed to Copper
Conclusions
To date three fundamentally different metal-mediated methods have been developed for the pH-neutral desulfitative reaction of boronic acids and organostannanes with thioorganic substrates. The original “1st-generation process” takes place under anaerobic conditions and requires a palladium catalyst partnered with a stoichiometric quantity of a Cu(I) carboxylate cofactor.7a,13 The thioorganic reactant spans a variety of sp2 and sp systems; organostannanes also function as suitable reaction partners. In pursuing similar transformations that would require only catalytic quantities of the Cu(I) carboxylate, a “2nd-generation” pH-neutral, desulfitative coupling of boronic acids with thiol esters was developed. This reaction system takes place aerobically and requires only catalytic quantities of Cu.14 The aerobic 2nd generation desulfitative reaction therefore circumvents two limitations of the 1st generation family of reactions: it requires no Pd and it turns over using only catalytic quantities of Cu. However, this mechanistically different aerobic reaction system (which currently is limited to thiol ester reactants) imposes a new limitation by requiring a second, sacrificial equivalent of the boronic acid in order to complete the catalytic cycle.
The chemistry described within this paper demonstrates an alternative and mechanistically unique desulfitative reaction system that is catalyzed by Cu-only. It was designed to be catalytic in Cu and proceed at neutral pH like the aerobic 2nd generation reaction system, but, in contrast, it uses a single equivalent of the boronic acid or organostannanes reaction partner and functions anaerobically, not aerobically.
The mechanistic relationships among the three different Cu-based desulfitative couplings of thioorganics with boronic acids can be understood globally by recognition of the two key criteria that are required for efficient catalytic turnover of Cu in a thiophilic environment: (1) the strongly ligating thiolate must be trapped or scavenged so that the catalytically active Cu is liberated, free to participate in the reaction cycle, and (2) the reaction system must be designed in order to provide the boron reaction partner with a thermodynamically suitable replacement moiety that will occupy the valence vacated by the “R” group that transfers to the thioorganic.
While the O-methyloxime moiety of the current reaction system serves to trap the thiolate and provide a full equivalent of oxygenate, in principle, the thermodynamically strong tin-sulfur bond should permit an organostannane reactant to participate in desulfitative carbon-carbon bond formation with simple thiol esters using only catalytic quantities of Cu sources in the absence of a full equivalent of an oxygenate 4th valence. The key to catalytic turnover of the organostannane system with simple thiol esters will thus be regeneration of a catalytically active CuX species through an effective metathesis between Cu-SR′ and Bu3SnX as shown in the Figure 5. Studies to probe this aspect of Cu-catalyzed desulfitative transformations are currently underway.
Figure 5.

Organostannane Desulfitative Coupling with Cu Only: A Catalytic Cycle
In closing we reiterate that knowledge of the biomolecular principles by which metals are liberated from their binding to proteins can be used to inspire the design of effective systems for chemical catalysis in the laboratory. In the specific case described within, a thiophilic metal (CuI) was liberated from a strongly binding thiolate by mimicking the mobilization of thiophilic metals from the tightly binding tetracysteinate environment of the metallothioneins. Additional novel and useful catalytic chemical discoveries may well result from further pursuits of this concept. This new anaerobic reaction system constitutes an alternative approach to known Cu-only catalysis in the presence of thiolates that is based upon aerobic Cu-thiolate chemistry.14
Methods
Preparation of 1-(2-Mercaptophenyl)ethanone O-methyloxime (2)
To a dry and argon-flushed 100 mL round-bottomed flask equipped with a magnetic stirring bar was added 2′-mercaptoacetophenone25 (3.04 g, 20 mmol), O-methylhydroxylamine hydrochloride (2.51 g, 30 mmol) and MeOH (60 mL). Pyridine (2.77 g, 35 mmol) was then slowly added via syringe. After stirring the reaction mixture at room temperature overnight, the solvent was evaporated. The residue was dissolved in diethyl ether (20 mL) and the organic phase was washed with 1 M aqueous HCl (2 × 10 mL), water (10 mL) and brine (5 mL). After drying over anhydrous MgSO4 and filtering, the solvent was evaporated. The product was purified by flash chromatography (silica gel, 20:1 hexanes:EtOAc) to afford the title compound as a yellow oil (6:1 E/Z mixture of isomers, 3.11 g, 86%). Major Isomer: 1H NMR (400 MHz, CDCl3) δ 7.32-7.27 (m, 2H), 7.16-7.14 (m, 2H), 4.01 (s, 3H), 3.97 (s, 1H), 2.22 (s, 3H); Characteristic signals for minor isomer: 1H NMR δ 3.84 (s, 3H), 2.16 (s, 3H); Major Isomer: 13C NMR (100 MHz, CDCl3) δ 155.8, 135.8, 131.2, 129.2, 128.9, 125.7, 62.2, 15.3; IR (neat, cm−1): 3061 (m), 2937 (s), 2548 (m), 1613 (m); HRMS (FAB) Calcd for C9H12ONS (M+H+): 182.0634. Found: 182.0632.
Preparation of S-2-(1-(Methoxyimino)ethyl)phenyl 4-methylbenzothioate (3a)
The preparation of 3a is representative for the synthesis of the other thiol esters 3b–f. To a dry and argon-flushed 50 mL round-bottomed flask equipped with a magnetic stirring bar was added 1-(2-mercaptophenyl)ethanone O-methyloxime (2) (181 mg, 1.0 mmol), p-toluoyl chloride (162 mg, 1.05 mmol) and THF (20 mL). Pyridine (87 mg, 1.1 mmol) was added slowly via syringe and after stirring at room temperature for 4 h the white precipitate was removed by filtration. The filtrate was washed with water (2×10 mL), brine (5 mL) and then dried over anhydrous MgSO4. The solvent was evaporated to give a yellow oil that was purified by flash chromatography (silica gel, 10:1 hexanes:EtOAc) to afford the title compound as a clear oil (6:1 mixture of E/Z isomers, 293 mg, 98%). Characterization details are provided in the Supporting Information.
Microwave Irradiation Experiments
Microwave irradiation experiments were carried out using a Discover microwave reactor from CEM Corporation. All experiments were performed in sealed tubes (capacity 10 mL) under an argon atmosphere utilizing microwave irradiation of 300 W. The temperature was ramped from room temperature to 150 °C in 1 minute. Once this temperature was reached, the reaction mixture was held at 150 °C for 60 minutes.
Representative Procedure for Cu-Catalyzed Desulfitative Couplings
To a dry microwave tube (10 mL) equipped with a stirring bar was added S-2-(1-(methoxyimino)ethyl)phenyl 4-methylbenzothioate (3a) (30 mg, 0.1 mmol), 4-chlorophenyl boronic acid (19 mg, 0.12 mmol) and CuMeSal (4 mg, 0.02 mmol). The reaction tube was flushed with argon and sealed with a septum. Anhydrous and degassed DMF (1 mL) was added and the mixture was subsequently heated in a microwave reactor at 150 °C for 1 h. After cooling the reaction mixture was diluted with diethyl ether (10 mL). The reaction mixture was washed with water (2×5 mL), brine (3 mL) and dried over anhydrous MgSO4. The solvent was removed at reduced pressure and the crude product was purified by preparative plate silica gel chromatography using hexanes/EtOAc (4:1) as the eluent. 4-Chloro-4′-methylbenzophenone was obtained as a white solid (21 mg, 91%). Characterization details are provided in the Supporting Information.
Supplementary Material
Acknowledgments
The National Institutes of General Medical Sciences, DHHS, supported this investigation through Grant No. GM066153. Dr. Gary Allred of Synthonix provided the boronic acids, organostannanes and Cu salts used in our studies. Dr. Ethel Garnier-Amblard and Dr. Hao Li provided critical proofreading and suggestions.
Footnotes
Supporting Information Available: Experimental procedures, synthesis and characterization of all new compounds and scanned spectra. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 2.(a) Waldron KJ, Robinson NJ. Nature Reviews. 2009;6:25–35. doi: 10.1038/nrmicro2057. [DOI] [PubMed] [Google Scholar]; (b) Tottey S, Harvie DR, Robinson NJ. Acc Chem Res. 2005;38:775–783. doi: 10.1021/ar0300118. [DOI] [PubMed] [Google Scholar]
- 3.Huffman DL, O’Halloran TV. Annu Rev Biochem. 2001;70:677–701. doi: 10.1146/annurev.biochem.70.1.677. [DOI] [PubMed] [Google Scholar]
- 4.(a) Maret W. Biochemistry. 2004;43:3301–3309. doi: 10.1021/bi036340p. [DOI] [PubMed] [Google Scholar]; (b) Chen Y, Irie Y, Keung WM, Maret W. Biochemistry. 2002;41:8360–8367. doi: 10.1021/bi020030+. [DOI] [PubMed] [Google Scholar]; (c) Hartmann HJ, Weser U. BioMetals. 2000;13:153–156. doi: 10.1023/a:1009275122084. [DOI] [PubMed] [Google Scholar]; (d) Jacob C, Maret W, Vallee BL. Biochem Biophys Res Commun. 1998;248:569–573. doi: 10.1006/bbrc.1998.9026. [DOI] [PubMed] [Google Scholar]; (e) Jacob C, Maret W, Vallee BL. Proc Nat Acad Sci. 1998;95:3489–3494. doi: 10.1073/pnas.95.7.3489. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Maret W. Proc Natl Acad Sci USA. 1994;91:237–241. doi: 10.1073/pnas.91.1.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Song C. Catalysis Today. 2003;86:211–263. [Google Scholar]
- 6.Smith BM. Chem Soc Rev. 2008;37:470–478. doi: 10.1039/b705025a. [DOI] [PubMed] [Google Scholar]
- 7.(a) Prokopcová H, Kappe OC. Angew Chem, Int Ed. 2009;48:2276–2286. doi: 10.1002/anie.200802842. [DOI] [PubMed] [Google Scholar]; (b) Dubbaka SR, Vogel P. Angew Chem, Int Ed. 2005;44:7674–7684. doi: 10.1002/anie.200463007. [DOI] [PubMed] [Google Scholar]
- 8.Coyle P, Philcoxa JC, Carey LC, Rofea AM. Cell Mol Life Sci. 2002;59:627–647. doi: 10.1007/s00018-002-8454-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Maret W, Vallee BL. Proc Nat Acad Sci. 1998;95:3478–3482. doi: 10.1073/pnas.95.7.3478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Maret W. Neurochem Int. 1995;27:111–117. doi: 10.1016/0197-0186(94)00173-r. [DOI] [PubMed] [Google Scholar]
- 11.Zangger K, Öz G, Haslinger E, Kunert O, Armitage IM. FASEB. 2001;15:1303–1305. doi: 10.1096/fj.00-0641fje. [DOI] [PubMed] [Google Scholar]
- 12.(a) Phipps RJ, Gaunt MJ. Science (Washington, DC) 2009;323:1593–1597. doi: 10.1126/science.1169975. [DOI] [PubMed] [Google Scholar]; (b) Evano G, Blanchard N, Toumi M. Chem Rev. 2008;108:3054–3131. doi: 10.1021/cr8002505. [DOI] [PubMed] [Google Scholar]; (c) Alexakis A, Bäckvall JE, Krause N, Pàmies O, Diéuez M. Chem Rev. 2008;108:2796–2823. doi: 10.1021/cr0683515. [DOI] [PubMed] [Google Scholar]; (d) Beletskaya IP, Cheprakov AV. Coord Chem Rev. 2004;248:2337–2364. [Google Scholar]; (e) Ley SV, Thomas AW. Angew Chem, Int Ed Engl. 2003;42:5400–5449. doi: 10.1002/anie.200300594. [DOI] [PubMed] [Google Scholar]
- 13.Musaev DG, Liebeskind LS. Organometallics. 2009;28:4639–4642. doi: 10.1021/om900602b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.(a) Villalobos JM, Srogl J, Liebeskind LS. J Am Chem Soc. 2007;129:15734–15735. doi: 10.1021/ja074931n. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Liebeskind LS, Yang H, Li H. Angew Chem, Int Ed. 2009;48:1417–1421. doi: 10.1002/anie.200804524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.(a) Li H, Yang H, Liebeskind LS. Org Lett. 2008;10:4375–4378. doi: 10.1021/ol8018456. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Egi M, Liebeskind LS. Org Lett. 2003;5:801–802. doi: 10.1021/ol0273497. [DOI] [PubMed] [Google Scholar]; (c) Alphonse FA, Suzenet F, Keromnes A, Lebret B, Guillaumet G. Org Lett. 2003;5:803–805. doi: 10.1021/ol027453o. [DOI] [PubMed] [Google Scholar]
- 16.Sigel H, Scheller KH, Rheinberger VM, Fischer BE. J Chem Soc, Dalton Trans. 1980:1022–1028. [Google Scholar]
- 17.(a) Liebeskind LS, Yang H, Li H. Angew Chem, Int Ed Engl. 2009;48:1417–1421. doi: 10.1002/anie.200804524. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Yang H, Liebeskind LS. Org Lett. 2007;9:2993–2995. doi: 10.1021/ol070991m. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Yang H, Li H, Wittenberg R, Egi M, Huang W, Liebeskind LS. J Am Chem Soc. 2007;129:1132–1140. doi: 10.1021/ja0658719. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Rodríguez-Cendejas CG, Liebeskind LS, Peña-Cabrera E. ARKIVOC. 2005;6:250–256. [Google Scholar]; (e) Fausett BW, Liebeskind LS. J Org Chem. 2005;70:4851–4853. doi: 10.1021/jo050110u. [DOI] [PubMed] [Google Scholar]; (f) Yu Y, Liebeskind LS. J Org Chem. 2004;69:3554–3557. doi: 10.1021/jo049964p. [DOI] [PubMed] [Google Scholar]; (g) Wittenberg R, Srogl J, Egi M, Liebeskind LS. Org Lett. 2003;5:3033–3035. doi: 10.1021/ol034962x. [DOI] [PubMed] [Google Scholar]; (h) Savarin C, Srogl J, Liebeskind LS. Org Lett. 2000;2:3229–3331. doi: 10.1021/ol000231a. [DOI] [PubMed] [Google Scholar]; (i) Liebeskind LS, Srogl J. J Am Chem Soc. 2000;122:11260–11261. [Google Scholar]
- 18.Savarin C, Srogl J, Liebeskind LS. Org Lett. 2001;3:91–93. doi: 10.1021/ol006807d. [DOI] [PubMed] [Google Scholar]
- 19.Liu S, Yu Y, Liebeskind LS. Org Lett. 2007;9:1947–1950. doi: 10.1021/ol070561w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.(a) Falck JR, Bhatt RK, Ye J. J Am Chem Soc. 1995;117:5973–5982. [Google Scholar]; (b) Allred GD, Liebeskind LS. J Am Chem Soc. 1996;118:2748–2749. [Google Scholar]; (c) Chan DMT, Lam PYS. In: Boronic Acids - Preparation, Applications in Organic Synthesis and Medicine. Hall DG, editor. Wiley-VCH; Weinheim: 2005. pp. 205–240. [Google Scholar]; (d) Falck JR, Patel PK, Bandyopadhyay A. J Am Chem Soc. 2007;129:790–793. doi: 10.1021/ja064948q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Breit B, Schmidt Y. Chem Rev. 2008;108:2928–2951. doi: 10.1021/cr078352c. [DOI] [PubMed] [Google Scholar]
- 22.As a control experiment, the 2-mercaptoacetophenone O-methyloxime was treated with 1 equiv of Cu(I) 3-methylsalicylate in dry, degassed EtOAc under argon. The resulting yellow precipitate was filtered and dried under vacuum. This yellow solid was placed in a flame-dried test tube with dry, degassed DMF under argon and monitored by TLC for formation of the benzoisothiazole. No product was observed after 2 h at room temperture nor after 2 h at 40 °C. After 2 h at 60 °C a trace of product was noted. The reaction was then warmed to 80 °C where product formation ensued. After 24 h at 80 °C the reaction was diluted with water, extracted into diethyl ether and the product residue was purified by preparative silica gel chromatography. The benzoisothiazole was isolated in 81% yield.
- 23.(a) Crawford RJ, Woo C. J Org Chem. 1966;31:1655–1656. [Google Scholar]; (b) Meth-Cohn O, Tarnowski B. Synthesis. 1978:58–60. [Google Scholar]; (c) Lawson AJ. Phosphorus, Sulfur Silicon Relat Elem. 1982;12:357–367. [Google Scholar]; (d) McKinnon DM, Kingsley RL. Can J Chem. 1988;66:1405–1409. [Google Scholar]; (e) Känel H-R, Wegmann A, Neff D. 5,527,917. Process for Preparation of 1,2-Benzisothiazoles. 1996; (f) Kagano H, Goda H, Yamamoto M, Sakaue S, Toudou M. 5,856,504. Process for Producing Isothiazole Derivatives. 1999
- 24.Desai LV, Hull KL, Sanford MS. J Am Chem Soc. 2004;126:9542–9543. doi: 10.1021/ja046831c. [DOI] [PubMed] [Google Scholar]
- 25.Topolski M. J Org Chem. 1995;60:5588–5594. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.