

Abstract
Gram-negative bacterial pathogens have developed specialized secretion systems to transfer bacterial proteins directly into host cells. These bacterial effectors are central to virulence and reprogram host cell processes to favor bacterial survival, colonization, and proliferation. Knowing the complete set of effectors encoded by a particular pathogen is the key to understanding bacterial disease. In addition, the identification of the molecular assemblies that these effectors engage once inside the host cell is critical to determining the mechanism of action of each effector. In this work we used stable isotope labeling of amino acids in cell culture (SILAC), a powerful quantitative proteomics technique, to identify the proteins secreted by the Salmonella pathogenicity island-2 type three secretion system (SPI-2 T3SS) and to characterize the host interaction partners of SPI-2 effectors. We confirmed many of the known SPI-2 effectors and were able to identify several novel substrate candidates of this secretion system. We verified previously published host protein-effector binding pairs and obtained 11 novel interactions, three of which were investigated further and confirmed by reciprocal co-immunoprecipitation. The host cell interaction partners identified here suggest that Salmonella SPI-2 effectors target, in a concerted fashion, cellular processes such as cell attachment and cell cycle control that are underappreciated in the context of infection. The technology outlined in this study is specific and sensitive and serves as a robust tool for the identification of effectors and their host targets that is readily amenable to the study of other bacterial pathogens.
Keywords: Bacteria, Mass Spectrometry (MS), Protein-Protein Interactions, Proteomics, Secretion, Host-Pathogen Interactions, Type Three Secretion System
Introduction
Bacterial pathogens have evolved specialized secretion systems to transfer bacterial virulence proteins, also called effectors, from the bacterium to the host cell cytoplasm. The type three secretion system (T3SS)9 is a large macromolecular machine consisting of three transmembrane complexes: an inner membrane ring, an outer membrane ring, and a translocon pore that forms within the host cell membrane. Effectors are transferred from bacteria to the host cell through a needle-shaped structure that connects the outer membrane ring and the translocon (1). Effectors translocated by the T3SS are central to pathogenesis, as mutations that prevent proper assembly of the secretion machinery result in significant virulence defects and often completely hinder bacterial colonization of the host. Consequently, technology to identify and characterize effectors is the key to our understanding of bacterial disease.
Salmonella enterica serovar typhimurium (S. typhimurium) is an intracellular pathogen that causes gastroenteritis in humans and a systemic infection resembling typhoid fever in mice (2). S. typhimurium virulence depends on two T3SSs that are encoded on Salmonella pathogenicity islands 1 and 2 (SPI-1 and -2) (2, 3). The SPI-1 T3SS is key during the early stages of infection and effectors translocated by this secretion system mediate entry of S. typhimurium into nonphagocytic cells (4, 5). This is accomplished by commandeering the ability of the host to rearrange its cytoskeleton, resulting in the generation of membrane ruffles that permit uptake of the S. typhimurium cells. Accordingly, many SPI-1 effectors have biochemical activities that facilitate this specific cellular reprogramming, such as the guanine-nucleotide exchange factor SopE/SopE2 and the GTPase-activating protein SptP that act upon the host cell Rho family GTPase proteins that modulate cytoskeletal dynamics. Another facet of SPI-1 effector function is that several effectors target related processes to facilitate bacterial entry into host cells. In addition to SopE/SopE2 and SptP, which manipulate Rho protein signaling, the SipA and SipC effectors directly influence actin dynamics through interaction with actin or its regulatory proteins (6).
Once inside the cell, S. typhimurium resides within a membrane-bound compartment, the Salmonella-containing vacuole (SCV). The SPI-2 T3SS translocates a separate set of effectors into the host cell cytosol through the SCV membrane to ensure survival and replication of intracellular bacteria and is critical for S. typhimurium virulence (7–9). However, unlike with several SPI-1 effectors, the predicted or demonstrated biochemical activities of SPI-2 effectors do not offer much insight into their biological functions. For example, analogous to the SPI-1 effectors SopE and SptP, the SPI-2 effectors SseL and SspH2 also possess opposing biochemical activities, catalyzing deubiquitination and E3 ubiquitin ligation reactions, respectively (10–12). However, knowledge of the specific biological functions of these SPI-2 effectors is largely lacking and is not obviously inferred from their enzymatic activities. In general, the mechanisms of action and the host cell targets of many SPI-2 effectors are still poorly understood (13).
The genomic context and sequence information are often insufficient to accurately identify T3SS substrates. For example, many bona fide effectors are situated distally from the loci that encode their cognate T3SS (14). As a complementary approach to identifying T3SS substrates, proteomics offers direct detection of these proteins, as high sensitivity mass spectrometry can readily identify proteins that are secreted into culture supernatants. A combination of the annotated genome sequence, the judicious use of T3SS mutants, inducible secretion conditions, and quantitative mass spectrometry provides a powerful platform to directly measure the SPI-2 secretome.
In this study we employed stable isotope labeling of amino acids in cell culture (SILAC) to identify and characterize S. typhimurium effectors secreted by the SPI-2 T3SS. SILAC utilizes selective metabolic labeling of proteins by growing cells in the presence of differentially labeled amino acid isotopologs, allowing different experimental conditions (e.g. specific versus nonspecific interactors) to be mass encoded. After liquid chromatography-coupled tandem mass spectrometry, the relative abundances of proteins in the original sample are inferred from the isotopic ratios of the labeled peptides. Thus, the peptide mass also indicates its origin from either the control or experimental samples, allowing for the elimination of unspecific background that is equally abundant under control and experimental conditions and thereby reliably identifying proteins specific to the experimental sample (15, 16).
We used SILAC technology to explore two important aspects of S. typhimurium effectors in this study: identification and function within the host cell environment. We developed a straightforward screen to rapidly and comprehensively analyze the entire complement of S. typhimurium effectors secreted by the SPI-2 T3SS. This screen did not require extensive genetic manipulation and in one run successfully identified many of the known S. typhimurium SPI-2 effectors. Furthermore, using this approach, we generated a strong candidate list of novel SPI-2 T3SS-secreted proteins, including the L21 protein of the large ribosomal subunit. We also used SILAC methodology to screen for host interaction partners of effectors translocated by the SPI-2 T3SS. This interaction partner screen identified previously published host protein-effector pairs and revealed 11 novel interactions, three of which were further confirmed by reciprocal co-immunoprecipitation. The newly identified interactions point toward a set of biological processes, such as host cell attachment, innate immunity, and cell cycling, in which these effectors fulfill their respective functions. Some of these processes have been understudied in the context of S. typhimurium infection, and our results suggest that they merit future investigation, further underscoring the utility of SILAC technology. The technology outlined in this study proved to be specific and sensitive and provides a tool for the identification of bacterial effectors and the study of their function within the host cell.
EXPERIMENTAL PROCEDURES
Strain Construction
Deletions of the lysA, argH, and fliF genes were performed using allelic exchange. Briefly, 1 kb of flanking sequence for each gene was amplified and cloned into pRE112 using compatible restriction enzyme sites designed to include a 14-amino acid scar of the deleted gene between the flanks. Deletion constructs were transformed into SM10λpir and conjugated into recipient S. typhimurium SL1344 wild type or ssaR strains. Plasmid integration was driven by antibiotic selection, and subsequent plasmid excision was driven by sucrose counterselection and verified by antibiotic sensitivity. Gene deletion was verified by PCR, auxotrophy was verified by growth in limiting medium, and flagellar impairment was verified by motility assay.
SPI-2 Secretome Preparation
S. typhimurium lysA argH fliF derivatives were inoculated at 1:100 from an overnight culture into 500 ml of modified low phosphate, low magnesium (LPM) medium pH 5.8 (17). Modifications to LPM medium included increasing MgCl2 to 24 μm and replacing casamino acids with individual amino acids at the following concentrations: Ala, 2.8%; Asp, 6.3%; Cys, 0.3%; Glu, 21.1%; Gly, 2.2%; His, 2.7%; Ile, 5.6%; Leu, 8.4%; Met, 2.7%; Phe, 4.6%; Pro, 9.9%; Ser, 5.6%; Thr, 4.2%; Trp, 1.1%; Tyr, 6.1%; Val, 5.0%; Arg, 3.6%; and Lys, 7.5%). The SPI-2 secretion-competent strain was inoculated into LPM medium containing [4,4,5,5-D4]lysine and [U-13C6]arginine, and the SPI-2 secretion mutant (ssaR) strain was inoculated into LPM medium containing natural abundance, isotopically labeled arginine and lysine. Cultures were grown for 8 h at 37°C with shaking at 225 rpm. Bacteria were harvested, and culture supernatants were passed through a 0.22-μm filter. Filtered supernatants from the two cultures were pooled in an equivalent ratio based upon optical density measurement of the culture at the time of harvest. Supernatants were concentrated successively using a nitrogen-stirring cell (Amicon) and centrifugal concentrators (Amicon) with 10,000-MWCO (molecular weight cut-off) filters. Filters were washed with TBS, and the washes were pooled with the supernatant and further concentrated. Secreted proteins were precipitated overnight using three volumes of 100% EtOH, 50 mm sodium acetate, and 20 μg of glycogen. Precipitated proteins were separated by SDS-PAGE or isoelectric focusing and subjected to in-gel digestion as indicated below.
Secretion Analysis of STM2949 and L21
STM2949 and rplU including 1 kb of upstream sequence was amplified from S. typhimurium SL1344 genomic DNA and cloned using SalI and BglII restriction sites to replace gogB in the pWSK129 derivative pgogB-HA, which encodes a double hemaggluttinin epitope tag (18). These plasmids, renamed pWSK129ptpS-2xHA and pWSK129-L21–2xHA, were transformed into wild type S. typhimurium SL1344- and SPI-2-deficient strains. In vitro SPI-2 secretion assays were performed essentially as described previously (18). Secreted or total protein fractions were separated by SDS-PAGE, transferred to PVDF, and probed with rat anti-HA (Roche Applied Science), mouse anti-DnaK (Stressgen Biotechnologies), and anti-SseB (laboratory-raised) antibodies.
Cloning
For expression of effectors in human embryonic kidney (HEK) 293T cells (ATCC), effector genes were amplified from S. typhimurium SL1344 genomic DNA and inserted into a pcDNA3 expression vector engineered to include an N-terminal tandem hemagglutinin (HA) tag (pcDNA3(2HA)) using SrfI and NotI restriction sites, as described previously (16). A bacterial expression plasmid was engineered based on the pGEX-6P-3 vector (Amersham Biosciences) to carry an N-terminal triple HA tag by insertion of three consecutive hemagglutinin sequences between the BamHI and EcoRI restriction sites. Effector genes were amplified from S. typhimurium SL1344 genomic DNA and inserted into pGEX(3HA) using either the XhoI and NotI or SalI and NotI restriction sites. The gene encoding oxysterol-binding protein 1 was amplified from cDNA clone ATCC 10659005 (ImageID 4560111) and inserted into the unmodified pcDNA3 vector (Invitrogen) using the HindIII and BamHI restriction sites.
Tissue Culture
HEK293T cells (ATCC) were grown in Dulbecco's modified eagle medium (DMEM) containing 4500 mg/liter glucose and 4 mm l-glutamine and supplemented with 1% non-essential amino acids, 1% GlutaMax, and 10% fetal bovine serum (all from Invitrogen). To screen for interaction partners using effectors expressed in HEK293T (see Fig. 4, strategy A), cells were transferred into arginine- and lysine-free DMEM (Caisson Laboratories Inc.) supplemented with 10% dialyzed FBS (Invitrogen) and either 36.5 mg/liter [4,4,5,5-D4]lysine and 21 mg/liter [U-13C6]arginine (Cambridge Isotope Laboratories), for heavy control cells, or expanded in regular growth medium for effector transfection as described previously (16). To screen for interaction partners using effectors expressed in Escherichia coli (Fig. 4, strategy B), cells were transferred into arginine- and lysine-free DMEM (Caisson Laboratories Inc.) supplemented with 10% dialyzed FBS (Invitrogen) and either 36.5 mg/liter [4,4,5,5-D4]-lysine and 21 mg/liter [U-13C6]-arginine (Cambridge Isotope Laboratories) for heavy cells or equimolar amounts of normal isotopic abundance arginine and lysine (Sigma-Aldrich) for light cells and maintained for at least five cell divisions in these media prior to immunoprecipitation experiments.
FIGURE 4.

Schematic overview of the interaction partner screen. For strategy A, effector genes were inserted into a mammalian expression vector as tandem HA epitope fusions (A); HA-tagged effectors were expressed in HEK293T grown in regular medium (B); lysates from these cells, as well as untransfected control cells labeled with heavy isotopes, were subjected to immunoprecipitation with an anti-HA antibody (C); bound protein was eluted, samples were pooled, proteins were digested with trypsin (D); and tryptic peptides were analyzed by LC-MS/MS (E). For strategy B, effector genes were inserted into a bacterial expression vector as GST tandem HA epitope fusions, and effectors were expressed in E. coli and purified (F); purified effectors were added to lysates from HEK293T cells grown in the presence of heavy isotopes, and these lysates, as well as control lysates, were subjected to immunoprecipitation with an anti-HA antibody (C); the following steps were common to strategy A (D and E). G, Western blot stained with anti-HA showing expression of effectors in HEK293T. In this example, expression levels of SseI and SseL were classified as “high,” SseG as “intermediate,” and SseD and SsaB as “low” (see Table 2). *, nonspecific band. H, SDS-PAGE of effectors purified from E. coli.
Transfection
HEK293T cells (ATCC) were seeded at a density of ∼1 × 106 cells/dish in 10-cm dishes and transfected the following day with 8 μg/dish Ca3(PO4)2-complexed DNA. Cell lysates were harvested 48 h after transfection.
Protein Expression and Purification
E. coli BL21 DE3 was transformed with pGEX-6P-3 derivatives containing cloned effectors and grown at 30°C with shaking (225 rpm) until an A600 of ∼0.4 was reached. Isopropyl-β-d-thiogalactopyranoside was added to 1 mm, and growth was continued for 3 or 16 h at 37 or 16°C, respectively. Cells were harvested and resuspended in lysis buffer (PBS, pH 7.4, 1 mm EDTA, 1 mm DTT, 10 μg/ml DNase, 10 μg/ml RNase, and EDTA-free complete protease inhibitor mixture (Roche Applied Science)). Cells were lysed by passage through a French pressure cell at 16,000 psi, and lysate was clarified by successive centrifugation steps at 8,000 and 30,000 × g for 10 and 30 min, respectively. All purification steps were performed on an AKTApurifier (GE Healthcare). Clarified lysate was applied to a GSTrap column (GE Healthcare) and eluted in a single step with GST elution buffer (50 mm Tris, pH 8, 10 mm glutathione, 1 mm EDTA, and 1 mm DTT). Appropriate fractions were pooled and loaded onto a preparative S200 column (GE Healthcare) that was run using S200 buffer (50 mm Tris, pH 8, 100 mm NaCl, 1 mm EDTA, and 1 mm DTT. Appropriate fractions were pooled and diluted 3-fold with MonoQ buffer A (50 mm Tris, pH 8, 25 mm NaCl, 1 mm EDTA, and 1 mm DTT) prior to loading onto a MonoQ anion exchange column (GE Healthcare). Proteins were eluted using a linear gradient of MonoQ buffer B (50 mm Tris, pH 8, 1 m NaCl, 1 mm EDTA, and 1 mm DTT) applied over 20 column volumes. Appropriate fractions were pooled, concentrated if necessary, and frozen at −80°C. Protein concentration was determined using Coomassie Plus Bradford assay reagent (Thermo Scientific) following the manufacturer's instructions.
Immunoprecipitation for Mass Spectrometry
N-hydroxysuccinimide (NHS)-activated Sepharose beads (GE Healthcare) were activated by treatment with ice-cold HCl and incubated with 1 mg/ml normal mouse IgG (Jackson ImmunoResearch Laboratories) or mouse-α-HA (clone 2C16) at 4 °C overnight. HEK293T cells were washed with PBS and harvested in lysis buffer (20 mm Tris/HCl, pH 7.5, 150 mm NaCl, 1% Nonidet P-40, 10 mm sodium pyrophosphate, 50 mm NaF, and 1 mm Na3VO4 (all from Sigma)) completed with EDTA-free protease inhibitor mixture (Roche Applied Science). Debris was pelleted at 16,000 × g and 4 °C for 30 min, and total protein concentration in the supernatants was determined as indicated above; 15 mg of total protein were used for each immunoprecipitation. For strategy B (see Fig. 4), ∼100 pmol of purified effector was added to lysates labeled with heavy isotopes for each immunoprecipitation. Lysates containing effector protein and control lysates were independently precleared with 150 μl of IgG-coated Sepharose beads (30 min, 4 °C), and subsequent immunoprecipitations were performed with 75 μl of anti-HA antibody-coated Sepharose beads (2 h, 4 °C). Beads were washed three times with lysis buffer, bound protein was eluted with 6 m urea/2 m thiourea, and eluates from both samples were pooled. Protein mixtures were supplemented with 20 μg of glycogen and precipitated with a 5-volume excess of ethanol and a final concentration of 100 mm sodium acetate, pH 5.0. Protein were pelleted at 16,000 × g for 10 min.
Sample Preparation for Mass Spectrometry
Culture supernatants of the secretome analysis were highly complex, and thus proteins were fractionated by SDS-PAGE or isoelectric focusing and subjected to in-gel digestion prior to mass spectrometric analysis essentially as described previously (19). Briefly, gel slices were minced, washed to remove stain using NH4HCO3/EtOH, and dehydrated with EtOH. Proteins were reduced with DTT, and free sulfhydryl groups were blocked with iodoacetamide. Gel pieces were subjected to repeated washing/dehydration steps as outlined above, and excess EtOH was removed by vacuum centrifugation. Proteins were digested with trypsin, and the remaining gel pieces were successively extracted with MeCN/TFA/AcOH, MeCN/AcOH, and MeCN. The organic solvents from all extracts were removed by vacuum centrifugation.
Samples immunoprecipitated from solution in the interaction partner screen were less complex and were therefore subjected to liquid chromatography-tandem mass spectrometry (LC-MS/MS) without additional SDS-PAGE fractionation. Immunoprecipitates were resolubilized in a small volume of 6 m urea/2 m thiourea and reduced with 1 μg of DTT for 30 min at room temperature, and cysteine residues were acetylated with 5 μg of iodoacetamide for 20 min at room temperature. Proteins were then treated with 1 μg of LysC (3 h at room temperature), diluted with 4 volumes of 50 mm NH4HCO3, and incubated with 1 μg of trypsin at 37 °C overnight. Tryptic peptides were diluted in 3% acetonitrile, 1% trifluoroacetic acid, and 0.5% acetic acid and bound to a C18 resin. The resin was washed with 0.5% acetic acid, and peptides were eluted with 0.5% acetic acid, 80% acetonitrile for mass spectrometric analysis.
Mass Spectrometry and Data Analysis
Mass spectrometry, data analysis, and database searches were performed as described previously (16). Briefly, digested peptides were analyzed by LC-MS/MS on a LTQ-OrbitrapXL (Thermo Fisher Scientific, Bremen, Germany). The LTQ-OrbitrapXL was coupled on-line to Agilent 1100 series nanoflow HPLC instruments using a nanospray ionization source (Proxeon Biosystems) holding columns packed into 15-cm-long, 75-μm-inner diameter fused silica emitters (8-μm-diameter opening pulled on a P-2000 laser puller from Sutter Instruments) using 3-μm-diameter ReproSil Pur C18 beads. Buffer A consisted of 0.5% acetic acid, and buffer B consisted of 0.5% acetic acid and 80% acetonitrile. Gradients were run from 6% B to 30% B over 60 min and then 30% B to 80% B in the next 10 min, held at 80% B for 5 min, and then dropped to 6% B for another 15 min to recondition the column. The LTQ-OrbitrapXL was set to acquire a full-range scan at 60,000 resolution from 350 to 1500 Thomson units (Th) in the Orbitrap and to simultaneously fragment the top five peptide ions in each cycle in the LTQ. For immunoprecipitates, proteins were identified using Mascot (v2.2, Matrix Science) to search against the international protein index (IPI) human database with S. typhimurium effectors and common contaminants added along with concatenated reversed sequences (v3.69, 154,798 sequences), using the following criteria: electrospray ionization-ion trap fragmentation characteristics and tryptic specificity with up to one missed cleavage; ±10 ppm and ±0.6 Da accuracy for MS and MS/MS measurements, respectively; cysteine carbamidomethylation as a fixed modification; N-terminal protein acetylation, methionine oxidation, and duplex ([2H4]Lys, [13C6]Arg) SILAC as appropriate. Peptides matching these criteria with ion scores greater than 28 were determined to meet a false discovery rate of ∼1%, so we considered proteins identified if at least two such peptides were observed. Quantitation of SILAC data for immunoprecipitations was performed using MSQuant (PMID 19888749). The analytical variability of SILAC data in the types of experiments performed here is typically <30%, and biological variability was addressed in these experiments by performing at least three independent replicates of each experiment. For the interaction partner screen, each effector was analyzed in triplicate, and binding partners were listed here only if they gave ratios >3 and were detected with more than two unique peptides in at least two replicates. For the secretome data, all identification and quantification was done using MaxQuant (PMID 19029910) with the default settings for Orbitrap data.
Immunoprecipitation for Western Blotting
Cell lysates were generated as described above. For oxysterol-binding protein 1 (OSBP)-SseL co-immunoprecipitations, HEK293T lysates containing 3 mg of total protein were mixed with 2 μg of purified HA3-SseL. Mixtures were precleared with 70 μl of protein G-Sepharose 4 Fast Flow (GE Healthcare) for 1 h at 4 °C, and supernatants were incubated with either 5 μg of goat α-OSBP (Abcam) or mouse α-HA (clone 2C16) for 1 h at 4 °C. Immune complexes were precipitated with 40 μl of protein G-Sepharose 4 Fast Flow by incubation for 30 min at 4 °C and subsequent centrifugation at 4000 × g for 5 min. After three washes in lysis buffer, beads were resuspended in 30 μl of SDS-PAGE loading buffer for Western blot analysis. Sgt1-SspH2 and Bub3-SspH2 co-immunoprecipitations were performed using empty vector (pcDNA3(2HA)) or SspH2 (pcDNA3(2HASspH2))-transfected HEK293T lysate containing 2 mg of total protein. Lysates were precleared as described above and then incubated with 1–5 μg of either mouse α-HA (clone 2C16), mouse IgG, mouse α-Sgt1, or mouse α-Bub3 (BD Biosciences) antibodies. Immune complex capture and washing were performed essentially as outlined above.
Western Blotting
Proteins were separated on 10–12% acrylamide gels and transferred to PVDF membranes (Bio-Rad). Membranes were blocked with 5% w/v skim milk powder, 0.1% v/v Tween in PBS, or TBS and incubated in the same buffer with primary and secondary antibodies as follows: mouse α-HA (clone 2C16) 1:3,000; rat α-HA (clone 3F10; Roche Applied Science) 1:2,500; goat α-OSBP (Abcam) 1:1,000; mouse α-SGT1 (BD Biosciences) 1:5,000; mouse α-Bub3 (BD Biosciences) 1:1,000; goat α-mouse HRP (Jackson ImmunoResearch Laboratories) 1:10,000; and rabbit α-goat HRP (Santa Cruz Biotechnology) 1:5,000. Proteins were detected with ECL detection reagent (GE Healthcare) and BioMax light film for chemiluminescence (Kodak).
RESULTS
SILAC Technology to Identify Novel SPI-2-secreted Proteins
We designed a quantitative proteomic screen to identify the repertoire of proteins secreted by the S. typhimurium SPI-2 T3SS (Fig. 1). Isogenic strains of S. typhimurium SL1344 were created containing deletions of argH, lysA, and fliF. Deletion of fliF prevents secretion of flagellar proteins and was included because preliminary experiments had detected excessive amounts of the flagellin protein FliC that was masking the detection of lower abundance proteins. Initial SILAC experiments in S. typhimurium indicated that even when fed arginine and lysine, the bacteria would still produce about 5% of these amino acids, so deletions in the arginine and lysine biosynthetic pathways (argH and lysA) generated the requisite auxotrophies to allow differential labeling of the bacterial proteome. The lysA and argH genes were specifically targeted for deletion to avoid impingements on alternate pathways with shared precursor metabolites in the arginine and lysine biosynthetic pathways, e.g. diaminopimelic acid biosynthesis for cell wall assembly. To differentiate between general secreted proteins and those secreted via the SPI-2 T3SS, an additional deletion of the SPI-2 structural protein encoded in the ssaR gene was employed in one strain (20).
FIGURE 1.
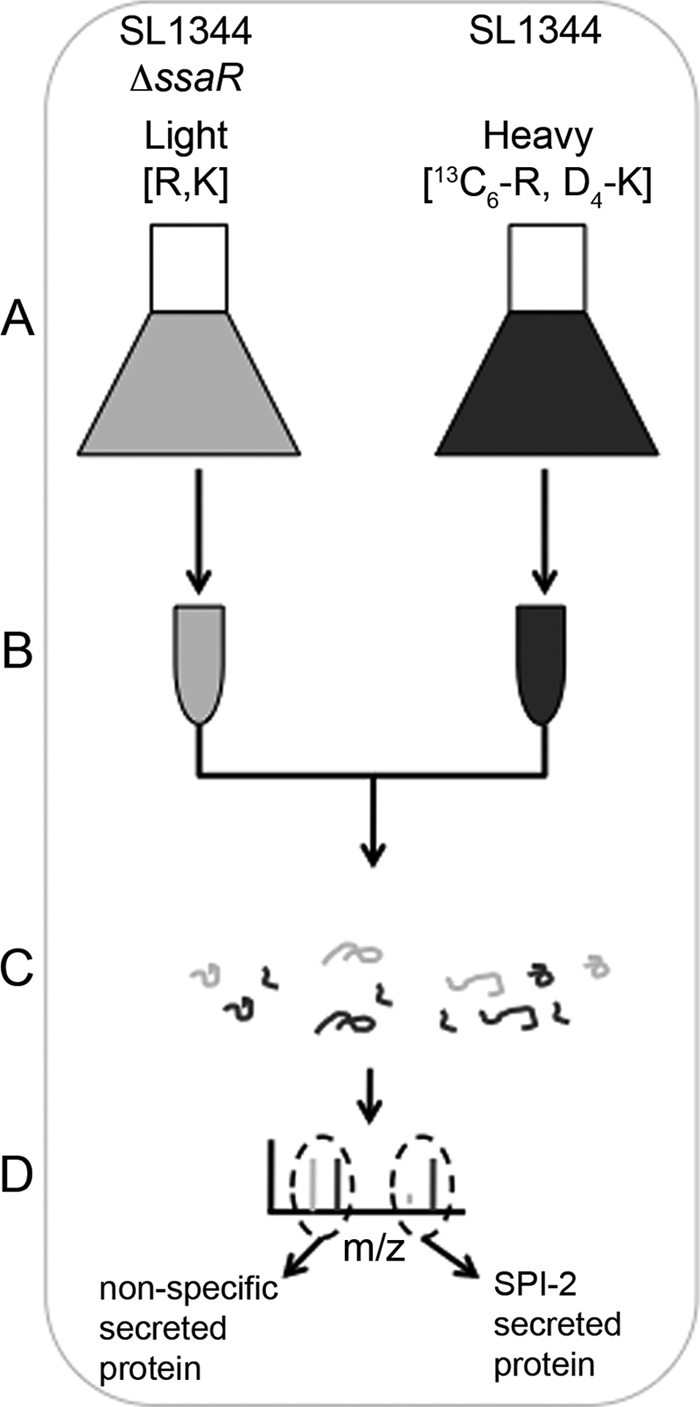
Schematic overview of the secretome analysis. A, S. typhimurium SL1344 ssaR were grown in defined minimal medium (LPM) containing regular abundance arginine (R) and lysine (K); SL1344 wild type were grown in the same medium containing Arg and Lys labeled with heavy isotopes. (Both bacterial strains were also deleted for their argH, lysA, and fliF genes; see text under “Results.”) B, bacterial cells were pelleted and culture supernatants filtered, pooled, and concentrated. C, proteins from concentrated supernatants were precipitated and subjected to in-gel digestion. D, tryptic peptides were analyzed by LC-MS/MS. Proteins secreted by the SPI-2 T3SS are characterized by enrichment of peptides containing heavy isotopes.
The SPI-2 secretion-competent strain was inoculated into defined SPI-2-inducing medium (LPM) containing [4,4,5,5-D4]lysine and [U-13C6]arginine, and the SPI-2 secretion mutant (ssaR) strain was inoculated into the same medium containing natural abundance isotopically labeled arginine and lysine (Fig. 1) (21, 22). Following growth, culture supernatants were filtered, combined, concentrated, and precipitated. Precipitated proteins were separated in one dimension using either SDS-PAGE or solution-based isoelectric focusing. Proteins were digested to peptides and analyzed by LC-MS/MS (Fig. 1). The ensuing spectra were searched against the S. typhimurium data base, identifications were assigned, and peptide abundances were quantified. SPI-2-secreted effectors in this screen are expected to show high abundance of heavy peptides and therefore high heavy/light SILAC ratios (Fig. 1).
This analysis identified 758 total proteins, although relatively few had high ratios of heavy/light peptides (see inset in Fig. 2 and supplemental Table S1). As shown in Fig. 2, we were able to detect 11 known SPI-2 translocated proteins, including effectors and translocon components, each of which showed a ratio of >2, suggesting that the screen does indeed have specificity for the SPI-2 secretome (see Table 1). Another protein with a ratio of >10 was the L21 protein of the 50S ribosomal subunit. Other candidate proteins that warrant further analysis are indicated in Table 1.
FIGURE 2.

SILAC ratios of proteins detected in culture supernatants. Ratios of MS peak intensities under SPI-2 secretion versus nonspecific conditions (SPI-2/nSp) are plotted for each protein. A total of 758 proteins were identified in the analysis (inset). Most hits with a ratio of 3 or higher corresponded to known SPI-2 T3SS effectors (black bars) or translocon components (gray bars).
TABLE 1.
Secreted proteins with a heavy/light ratio of >2
| ID | Unique peptides | Protein descriptions | PEPa | Ratio H/Lb normalized |
|---|---|---|---|---|
| PSLT037 | 11 | SpvD | 1.85E-98 | 11 |
| PSLT038 | 5 | SpvC | 1.23E-26 | 11 |
| STM0469 | 4 | 50S L21 | 4.15E-20 | 11 |
| STM0972 | 9 | SopD2 | 3.81E-31 | 11 |
| STM1224 | 9 | SifA | 2.32E-53 | 11 |
| STM1398 | 14 | SseB | 1.55E-175 | 11 |
| STM1400 | 26 | SseC | 0 | 11 |
| STM1401 | 6 | SseD | 6.96E-101 | 11 |
| STM1631 | 4 | SseJ | 1.91E-26 | 11 |
| STM1698 | 10 | SteC | 3.01E-26 | 11 |
| STM1583 | 13 | SteA | 1.38E-178 | 3.9662 |
| STM1540 | 14 | Putative secreted hydrolase | 2.10E-296 | 2.9847 |
| STM2817 | 3 | S-Ribosylhomocysteinase | 3.01E-22 | 2.8741 |
| STM2444 | 12 | Thiosulphate-binding protein precursor | 1.27E-58 | 2.7342 |
| STM2287 | 2 | SseL | 2.52E-05 | 2.7283 |
| STM2780 | 3 | PipB2 | 1.87E-21 | 2.6272 |
| STM1263 | 9 | Conserved hypothetical protein | 2.55E-56 | 2.6224 |
| STM0781 | 8 | Molybdate-binding periplasmic protein precursor | 8.03E-79 | 2.5248 |
| STM3053 | 31 | Glycine dehydrogenase (decarboxylating) | 0 | 2.4934 |
| STM1119 | 6 | Trp repressor-binding protein | 1.57E-62 | 2.4744 |
| STM1891 | 14 | High affinity zinc uptake system periplasmic-binding protein | 2.33E-193 | 2.449 |
| STM4561 | 7 | Putative periplasmic protein | 2.96E-70 | 2.4445 |
| STM1033 | 4 | ATP-dependent clp protease | 4.37E-07 | 2.3654 |
| STM0366 | 2 | Probable secreted protein | 1.10E-74 | 2.2539 |
| STM1088 | 4 | PipB | 2.61E-19 | 2.2375 |
| STM2892 | 5 | Surface presentation of antigens protein (associated with type III secretion and virulence) | 5.98E-82 | 2.1548 |
| STM3159 | 2 | Biopolymer transport ExbB protein | 8.91E-28 | 2.1371 |
| STM1408 | 2 | SsaI | 3.22E-08 | 2.0995 |
| STM1055 | 1 | GtgE | 1.85E-02 | 8.5383 |
| STM2585 | 1 | SteE | 2.40E-04 | 5.8228 |
| STM1123 | 1 | Putative secreted protein | 3.17E-02 | 2.3579 |
| STM1503 | 1 | YnfB | 1.45E-11 | 2.1629 |
| STM1720 | 1 | Conserved hypothetical protein | 1.06E-09 | 2.0423 |
a PEP, posterior error probability.
b H/L, heavy/light.
In E. coli, L21 is thought to interact with the 23S rRNA in conjunction with L20, although L21 does not appear to be essential for translation (23). S. typhimurium L21 is a 103-amino acid protein encoded by the rplU gene. Although we were not expecting L21 to obtain a high ratio in the secretion screen analysis, the N terminus of L21 showed hallmarks of a T3SS signal sequence. As shown in supplemental Fig. 1A, the first 25 residues of L21 can be modeled to an amphipathic helix with a positively charged face. Furthermore, there is an overrepresentation of serine and glutamine residues that also localize to one face of the modeled helix. These properties are consistent with those of canonical T3SS signal sequences, although the latter do not contain negatively charged residues, whereas L21 has two glutamate residues that again localize to one face of a modeled helix (24). Thus, it was plausible that L21 was capable of acting as a substrate for the SPI-2 T3SS, and we tested this possibility.
Accordingly, S. typhimurium L21 was cloned along with its native promoter in-frame and upstream of a tandem HA epitope tag. We noted that cloning rplU was not trivial in E. coli, perhaps owing to the fact that overexpression of L21 was detrimental to translation. Nevertheless, we were able to obtain a clone that expressed abundant L21 in both E. coli and S. typhimurium as judged by immunoblot analysis for the HA epitope. This construct was introduced into S. typhimurium SL1344 WT and ssaR strains, and an in vitro SPI-2 secretion assay was performed. As shown in supplemental Fig. 1B, we were able to detect expression of L21 in the bacterial pellets of both strains; however, we were unable to detect any significant signal from L21 in the secreted fraction. In contrast, we were able to readily detect secretion of SteA under the same conditions. We also probed for endogenous SseB to confirm that the L21-HA-expressing strain was secretion-competent. We were able to detect SseB in the secreted fraction of all WT strains but not in the ssaR strain. Immunodetection of DnaK showed that proteins detected in the secreted fraction were not a result of cell lysis.
To ensure that our inability to detect L21 in secreted fractions was not because of an inability to precipitate the protein using trichloroacetic acid, we performed the same analysis, except we incorporated an immunoprecipitation of the secreted fraction with an anti-HA antibody. As shown in supplemental Fig. 1C, we were again unable to detect L21 in the SPI-2 T3SS-secreted fraction, although SteA was readily detected, confirming that the immunoprecipitation was successful. Finally, we verified that the expression level of L21 was similar between S. typhimurium WT and ssaR strains. These strains, expressing epitope-tagged L21, were grown in LPM over a period of 8 h. Samples were harvested every 2 h and normalized by optical density, and lysates were immunoblotted with anti-HA antibody. We did not observe appreciable differences in the amount of L21 present between the WT and ssaR strains (data not shown).
To further assess the accuracy of proteins identified in the secretion screen, we chose the candidate protein STM2949, with a ratio of 1.08, indicative of a protein that was secreted equally in SPI-2 secretion-competent (WT) and defective (ssaR) strains. STM2949 was chosen for further analysis because it encodes an ortholog of PtpS (6-pyruvoyl tetrahydrobiopterin synthase) that catalyzes the conversion of H2NTP (7,8 dihydroneopterin 3-triphosphate) to PTP (6-pyruvoyl tetrahydropterin), an intermediate reaction in the synthesis of BH4 (tetrahydrobiopterin). BH4 is not widely synthesized in bacteria, but it is a potent inducer of nitric-oxide synthase in mammalian cells, potentially linking PtpS to host antimicrobial defense responses (25, 26). Furthermore, homologs of STM2949 are highly conserved among pathogenic bacteria, and PtpS has been reported to be a virulence factor for a parasite pathogen (27).
We amplified STM2949 from S. typhimurium SL1344 genomic DNA and cloned it in-frame upstream of a tandem HA epitope tag analogous to L21. We assessed the secretion of STM2949 taking advantage of the epitope tag to perform immunoblot analysis of secreted and bacterial fractions in vitro under SPI-2 secretion conditions in SPI-2 secretion-competent and -deficient strains. As shown in Fig. 3, we were able to detect STM2949 in the secreted fraction under SPI-2 inducing conditions; however this secretion was independent of ssaR, indicating that STM2949 secretion is not via the SPI-2 T3SS. Because the cytosolic marker, DnaK, was not detected in the supernatant fraction, the presence of STM2949 in this fraction was attributed to secretion and not cell lysis (Fig. 3). Furthermore, as indicated in Fig. 3, the impairment in SPI-2 secretion was confirmed by our ability to detect the SPI-2 translocon protein, SseB, in the supernatant fraction derived from wild type but not ssaR strains. Thus, our SILAC-based screen correctly identified the secretion of STM2949 in a SPI-2-independent manner, i.e. with a ratio of ∼1. This analysis yielded further confidence in the application of SILAC technology toward the identification of secretion substrates.
FIGURE 3.

Secretion of PtpS (STM2949). Shown is a Western blot analysis of bacterial pellets and culture supernatants of wild type SL1344 and ssaR transformed with either ptpS-2xHA or a control plasmid, grown in minimal medium. Western blots (IB) were probed with antibodies directed against HA (for detection of PtpS), DnaK (pellet control), and SseB (secretion control). *, nonspecific band.
SILAC Technology to Probe for Host Interaction Partners of Bacterial Effectors
We furthermore took advantage of SILAC technology to identify host proteins that are specifically bound by secreted bacterial effectors. Genes encoding all published SPI-2 effectors, including those confirmed in our secretome analysis, were inserted into a mammalian cell culture expression vector to generate N-terminal tandem HA fusion proteins. These constructs were used to transfect HEK293T cells grown in the presence of arginine and lysine that contain regular abundance isotopes. Protein complexes were immunoprecipitated using an antibody directed against the HA tag from lysates of these cells, as well as untransfected control cells grown in medium containing arginine and lysine labeled with heavy isotopes. Following immunoprecipitation, protein complexes from both samples were pooled and treated with trypsin, and tryptic fragments were subjected to LC-MS/MS (Fig. 4, strategy A). Proteins that nonspecifically interacted with either the antibody or the resin showed equal peak intensities of their heavy and light forms, giving SILAC ratios of ∼1, whereas proteins that specifically interacted with the effector were significantly enriched in their light forms, resulting in high light:heavy SILAC ratios (Fig. 4).
Expression of bacterial effectors in HEK293T cells was analyzed by immunoblot, and effector expression was classified as high, intermediate, or low based on the intensity of the specific HA epitope signal (Fig. 4G). About 60% of all effectors tested showed intermediate to high levels of expression in tissue culture and were analyzed using this experimental approach (Table 2). Effectors that were expressed at low or undetectable levels in HEK293T were instead subjected to a second line of analysis. Genes encoding these effectors were inserted into a bacterial expression vector to generate N-terminal glutathione S-transferase (GST), and triple HA fusions and proteins were purified from E. coli. Recombinant purified effectors were then incubated with tissue culture lysates containing heavy isotopes, and effector complexes were immunoprecipitated using an antibody against the HA tag. Immunoprecipitates were mixed with control immunoprecipitates from cell lysates containing natural abundance labeled proteomes and subjected to trypsin digest and mass spectrometry (Fig. 4, strategy B). Using this experimental strategy, we characterized specific interactors by the high abundance of their heavy peptides and consequently high heavy/light SILAC ratios.
TABLE 2.
List of effectors and expression levels in HEK293T and E. coli
| Effector | Expression level in HEK293Ta | Expression level in E. colib |
|---|---|---|
| GogB | Low | Intermediate |
| PipB | High | NAc |
| PipB2 | Low | Low |
| SifA | Intermediate | NA |
| SifB | Intermediate | NA |
| SL1928 | High | NA |
| SlrP | Low | High |
| SopB/SigD | High | High |
| SopD | Low | Intermediate |
| SopD2 | High | NA |
| SpvC | NA | High |
| SsaB/SpiC | Low | High |
| SseC | Low | Low |
| SseD | Low | Low |
| SseF | High | NA |
| SseG | Intermediate | NA |
| SseI | Intermediate | NA |
| SseJ | Intermediate | NA |
| SseK1 | Low | High |
| SseK2 | Low | Intermediate |
| SseL | Intermediate | NA |
| SspH1 | Intermediate | NA |
| SspH2 | Intermediate | NA |
| SteA | NA | High |
a The descriptors of high, intermediate, and low for the column “Expression level in HEK293T” refer to the intensity of the specific HA epitope signal detected by immunoblot in the lysate of HEK293T cells following transfection (see Fig. 4G for specific examples).
b The descriptors high, intermediate, and low for the column “Expression level in E. coli” refer to the relative recombinant protein levels detected by Coomassie Blue staining in purified protein extracts as depicted in Fig. 4H.
c NA, not analyzed.
A total of 24 S. typhimurium SPI-2-secreted effectors were studied in this screen (Table 2). 13 effectors (PipB, SifA, SifB, SL1928, SopB/SigD, SopD2, SseF, SseG, SseI, SseJ, SseL, SspH1, and SspH2) were analyzed using expression in tissue culture; and 11 effectors (GogB, SlrP, SopB/SigD, SopD, SpvC, SsaB/SpiC, SseK1, SseK2, SseL, and SteA) were expressed in E. coli. Only three of the effectors analyzed (PipB2, SseC, and SseD) could not be expressed in either HEK293T or E. coli and hence were not subjected to co-immunoprecipitation experiments. SseC proved toxic to E. coli cells when induced for protein overexpression, and SseD, although overexpressed, could not be retained on the affinity resin.
Identification of New Host Proteins Targeted by S. typhimurium Effectors
The experimental procedures described above confirmed previously and independently described interactions. Binding of S. typhimurium SopB, also referred to as SigD, by the small host GTPase Cdc42 is one example of a well established host protein-effector interaction (16, 28). We therefore chose to use SopB as a model to test both experimental strategies outlined above, involving either direct expression of effector in tissue culture (Fig. 4, strategy A) or recombinant expression and purification in E. coli (Fig. 4, strategy B). Indeed, both strategy A and B confirmed Cdc42 as a binding partner of SopB with 5 and 8 unique peptides and SILAC ratios of >4 and >5, respectively (Table 3).
TABLE 3.
Interaction partners identified in this study
| Effector | Interaction partner(s) | Unique peptides | Specific/nonspecific ratio | References |
|---|---|---|---|---|
| SopB/SigD | Cdc42a | 5 | >4 | Ref. 16 |
| Cdc42b | 8 | >5 | Ref. 28 | |
| SseF | Junction Plakoglobina | 3 | ∞ | |
| SseG | Desmoplakina | 2 | ∞ | |
| Caprin-1a | 2 | ∞ | ||
| SseJ | RhoAa | 9 | ∞ | Ref. 30 |
| RhoCa | 6 | ∞ | ||
| SseL | Oxysterol-binding protein 1a | 3 | 7.1 ± 0.5 | |
| Talinb | 4 | ∞ | ||
| SspH1 | PKN1a | 19 | ∞ | Ref. 29 |
| SspH2 | Sgt1a | 10 | ∞ | |
| AIPa | 3 | ∞ | ||
| Bub3a | 2 | ∞ | ||
| 14-3-3γa | 2 | ∞ | ||
| BAG regulator 2a | 2 | ∞ |
a Interactor identified using effector expressed in HEK293T.
b Interactor identified using effector expressed in E. coli.
Further validating the methodology, we were also able verify the previously published interactions between S. typhimurium SspH1 and serine/threonine protein kinase N1 (PKN1) as well as S. typhimurium SseJ and small GTPases RhoA and RhoC (29, 30). As both SspH1 and SseJ expressed well in tissue culture, these interactions were confirmed using strategy A (Fig. 4). Nonspecific interactors, such as glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and elongation factor 1-α (eEF1a) resulted in SILAC ratios of about 1 (Fig. 5, A and D). Specific binders RhoA, RhoC, and PKN1, on the other hand, had very high SILAC ratios in immunoprecipitations using SseJ and SspH1 (Fig. 5, B, C, and E, respectively).
FIGURE 5.

Representative mass spectra for immunoprecipitations (IP) with SseJ (A–C) and SspH1 (D and E). Mass/charge (m/z) spectra of individual peptides are shown for GAPDH (A), RhoA (B), RhoC (C), eEF1a (D), and serine/threonine PKN1 (E). Peptide sequences and SILAC ratios (light/heavy) for each peptide are shown.
Most importantly, we were able to identify several new interactions (Table 3). Previously unknown interaction partners were identified for four effectors: SseF, SseG, SseL, and SspH2. Three of these effectors, SseG, SseL, and SspH2, bound more than one host protein with as many as five host interaction partners identified for SspH2. SseF and SseG are thought to act together and to localize to cellular membranes (31, 32). Consistent with this observation, we found SseF and SseG to target junction plakoglobin and desmoplakin, two proteins located at cell-cell junctions (33). SseG furthermore also bound Caprin-1 (cytoplasmic activation/proliferation-associated protein 1), a protein with a suggested role in cell proliferation (34, 35). SseL is a deubiquitinase, whereas SspH2 is a member of the IpaH-like family of E3 ubiquitin ligases (10, 11, 36). Hence, host interactors for these two effectors might be putative cellular substrates for these ubiquitin-modifying enzymes. SseL interacted with Talin-1, a regulator of integrin signaling, as well as with OSBP, a protein implicated in signaling, nonvesicular cholesterol trafficking, and the regulation of lipid metabolism and vesicular transport (37, 38). SspH2 specifically bound Sgt1, AH receptor-interacting protein, Bub3, 14-3-3γ, and BAG (Bcl-2-associated athanogene) regulator. Sgt1 is conserved among eukaryotes, functioning primarily in cell cycle progression, but it also plays an important role as a co-chaperone for nucleotide-binding domain and leucine-rich repeat-containing (NLR)-like proteins during innate immune signaling. AH receptor-interacting protein is involved in aryl hydrocarbon receptor signaling and has been implicated in hepatitis B virus infections; Bub3 is important for kinetochore-microtubule interactions during mitosis; 14-3-3γ is an adaptor molecule mediating protein-protein interactions; and BAG-2 is a co-chaperone that regulates the activity of heat shock complexes (39–44).
Confirmation of Novel Interactions
To further validate the quality of our interaction partner screen, we aimed to confirm individual hits using reciprocal co-immunoprecipitations and Western blot analysis. One particularly interesting interaction is that between SspH2 and Sgt1. Sgt1 regulates innate immune signaling by stabilizing NLR proteins, acting as a co-chaperone in concert with Hsp90 (39). NLR proteins are cytoplasmic receptors that sense the presence of microbe associated molecular patterns. Hence, SspH2 may function by inhibiting the ability of the host to recognize the presence of S. typhimurium. To confirm this interaction, HEK293T cells were transfected either with SspH2 carrying an N-terminal tandem HA tag or with a vector control. Sgt1 was immunoprecipitated from SspH2-transfected lysates using an antibody against SspH2 (i.e. HA) but not in the absence of SspH2. Vice versa, an antibody against Sgt1 immunoprecipitated SspH2 (Fig. 6A).
FIGURE 6.
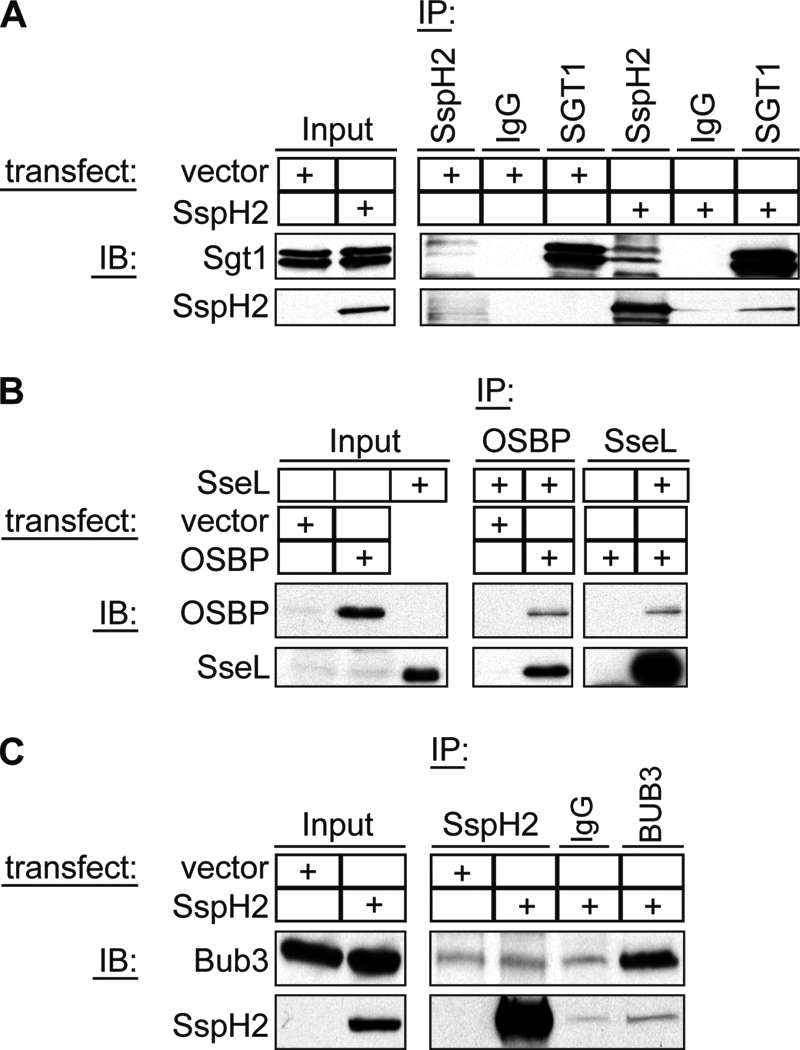
Confirmation of binding of SspH2 to Sgt1 and Bub3 and SseL to OSBP. A, HEK293T lysates transfected with either SspH2 or a vector control were subjected to immunoprecipitation using antibodies directed against SspH2 or Sgt1 or with normal mouse IgG as an antibody control. Samples were analyzed by Western blot (IB) probing for Sgt1 and SspH2. B, HEK293T lysates transfected with either OSBP or a vector control were incubated with recombinant SseL and subjected to immunoprecipitation using antibodies against OSBP or SseL. Samples were analyzed by Western blot probing for SseL and OBSP. C, HEK293T lysates transfected with either SspH2 or a vector control were subjected to immunoprecipitation using antibodies directed against SspH2 or Bub3 or with normal mouse IgG as an antibody control. Samples were analyzed by Western blot probing for Bub3 and SspH2.
Additionally, we chose to confirm the binding of OSBP by SseL. Given the role of OSBP in mitogen-activated protein (MAP) kinase signaling, vesicular trafficking, and lipid metabolism, the role of the SseL-OSBP interaction could be in regulating innate host defenses, as well as nutrient acquisition or trafficking of intracellular S. typhimurium. As intrinsic OSBP levels in HEK293T were too low for detection by Western blot, immunoprecipitations were performed using cells that overexpressed OSBP. Lysates from these cells were incubated with recombinant purified HA3-SseL, and complexes were precipitated using antibodies directed against either OSBP or SseL (HA). As expected, an antibody directed against OSBP was able to precipitate SseL from OSBP-transfected but not vector-transfected cells. Equally, if SseL was present in the sample, an antibody against SseL (HA) precipitated OSBP (Fig. 6B).
We also analyzed Bub3 binding to SspH2 (Fig. 6C). Nonspecific binding of Bub3 to the resin was detected under the experimental conditions used; nevertheless, there was enhanced binding of Bub3 in the presence of SspH2. Similarly, precipitation of SspH2 was stronger in the presence of Bub3 antibody as compared with an antibody control. This suggests a specific interaction between Bub3 and SspH2.
DISCUSSION
In this study we applied the versatility of SILAC-based quantitative proteomic technology to gain a better understanding of how the effectors secreted by the SPI-2 T3SS contribute to S. typhimurium virulence. We began by designing an in vitro secretion screen to identify proteins that were secreted in a SPI-2-dependent manner in an effort to catalogue the complete SPI-2 T3SS substrate inventory. Two important considerations emerged from this work. The first was that SILAC is a very powerful tool to use in identifying T3SS substrates. This is best exemplified by the fact that most known SPI-2 effectors identified in this screen had ratios of >3, whereas the bulk of the secreted proteins had ratios ranging from 0 to 2. This argues that SILAC screening is highly specific to the identification of proteins that match the screening criteria. Indeed, our laboratories have recently identified novel T3SS effectors using SILAC-based quantitative proteomics in the mouse pathogen Citrobacter rodentium, which is a model pathogen for enterohemorrhagic E. coli (EHEC) and enteropathogenic E. coli (EPEC) (45).
Nevertheless, we were unable to identify all known SPI-2 effectors in this screen, which underscores the second consideration, that the sensitivity of the screen most likely can be enhanced by a deeper understanding of the T3SS process. A growing paradigm is that T3SS substrates are secreted in a hierarchical manner with a proteinaceous regulator that belongs to the SepL protein family. We speculate that the robustness of this screen could be improved by using additional genetic mutations to enhance secretion, such as the sepD/sepL mutations used to enhance T3SS-dependent effector secretion in C. rodentium (45). Indeed, a recent report identified the SpiC-SsaL-SsaM protein complex and pH as factors that can enhance SPI-2-dependent effector secretion (46). In this context the SpiC-SsaL-SsaM protein complex is analogous to the SepD-SepL complex, responsible for switching from the secretion of translocon components to effectors (46).
During the preparation of this manuscript another study was reported that examined the SPI-2 secretome using spectral counting-based quantitative proteomics (47). That study, which employed a deletion in the ssaL gene, reported the identification of six novel SPI-2 T3SS substrates from S. typhimurium 14028s that were dependent upon the additional ssaL deletion. Our laboratory had previously demonstrated that some SPI-2 effectors are secreted in an SsaL-independent manner, and others have reported that SPI-2-dependent secretion is virtually undetectable in the presence of a functional SpiC-SsaL-SsaM complex (17, 46, 47). Our results unequivocally demonstrate that this is not the case, and in fact, we also identified four of the six (SpvD, SteE, GtgE, and SssA) novel ssaL-biased effectors recently reported (47). Our results suggest that SILAC technology can overcome some limitations imparted by genetic restriction and that this technology augurs well for cataloguing the T3SS proteomes of emerging and poorly studied pathogens.
One interesting SPI-2 T3SS-secreted protein candidate that arose from our study was the large ribosomal subunit protein L21. Four unique peptides were identified from this 100-amino acid protein, and the SILAC ratio was comparable to those of bona fide SPI-2 effectors. Ribosomal components are abundant cytosolic proteins and generally serve as a good indication of cell lysis and contamination of secreted fractions with cellular components. However, this is likely not a significant issue here because we do not anticipate any bias in the lysis rates of ssaR+ and ssaR− strains. As a result, any proteins present in the secreted fraction that originated from cytosolic contamination would be expected to have a ratio of 1. Alternatively, it has been reported that ribosomal components can be secreted inside outer membrane vesicles; these proteins would be identified in our approach, although again with an anticipated ratio of 1 (48). Furthermore, multiple different ribosomal proteins are typically identified from outer membrane vesicle fractions. In contrast, L21 was the lone ribosomal protein identified in this screen with a ratio of >2; all other ribosomal proteins detected had ratios near 1. The solitary identification of L21 and its remarkably high ratio made L21 a candidate for follow-up studies. However, our follow-up study showed that the SPI-2 T3SS did not secrete epitope-tagged L21. We cannot rule out the possibility that the addition of the tandem HA epitope tag interfered with SPI-2 T3SS secretion of this protein. Indeed, the presence of the epitope tag may not be innocuous, as it alters the protein length by 20%. It remains unclear why L21 yielded a high ratio in the SILAC analysis but was not detected in supernatants by Western blot.
We also took advantage of quantitative proteomics to probe S. typhimurium effector function through the identification of host binding partners. Interactions that had been published previously for SopB/SigD, SseJ, and SspH1 were confirmed, and several new interactions were identified in the screen. For example, we established junction plakoglobin and desmoplakin as binding partners of SseF and SseG, respectively. SseF and SseG are effectors encoded immediately adjacent to one another within SPI-2 and are translocated by the SPI-2 T3SS (32). They share about 30% sequence similarity and contain predicted transmembrane helices. Indeed, both proteins have been shown to localize to membranes, thus allowing proper intracellular positioning of the SCV, and to influence SCV architecture (32, 49). Interestingly, SseF and SseG interact with one another, and competitive index experiments have revealed that there is no additive virulence defect in a sseFG double mutant as compared with strains carrying deletions of either gene alone (31). This suggests that both proteins act together in a protein complex to fulfill their cellular functions. Consistent with these findings, their interaction partners, plakoglobin and desmoplakin, are also part of the same molecular assembly, the desmosome. Desmosomes are cell junctions in the plasma membrane that ensure the mechanical integrity of organs by connecting cells within the tissue and anchoring cellular membranes to intermediate filaments (33). Hence, the interaction between SseF/Sse and the desmosome might represent a novel pathway by which S. typhimurium influences cytoskeletal rearrangements and cellular membrane dynamics. SseG also bound to Caprin-1. Caprin-1 was identified as a protein that is up-regulated in activated T- and B-lymphocytes and has been shown since to be important for normal cell division (34, 35). Caprin-1 interacts with RNA-binding proteins and has been found in association with mRNAs involved in cellular proliferation (50). Hence, SseG might have an additional role in modulating cell cycle progression through the regulation of Caprin-1 complexes.
Our screen revealed that SseL binds OSBP. This interaction was further confirmed by reciprocal co-immunoprecipitation. SseL is an effector translocated by the SPI-2 T3SS but encoded outside of the pathogenicity island (51). SseL localizes to the SCV membrane during infection and deubiquitinates cellular targets with a preference for Lys63-linked polyubiquitin chains (10). IκBα, an inhibitor of the NF-κB transcription factor, has been identified as a target for deubiquitination by SseL, thus suggesting a role for SseL in modulating proinflammatory signaling (11). OSBP has been implicated in various cellular functions, including vesicular trafficking, lipid biosynthesis, non-vesicular lipid transport, and signaling, all of which are hallmarks of S. typhimurium infection (52–54). Our screen also provided evidence that SseL binds Talin-1, a protein localized at focal adhesions, which links the actin cytoskeleton to the extracellular matrix by directly interacting with the cytoplasmic domains of integrins (38, 55). Talin-1 thereby plays an important role in regulating the affinity of integrin for its ligands. Consistent with our observation, Talin-1 had previously been shown to accumulate around invading S. typhimurium in epithelial cells (56). It is tempting to speculate that SseL, by interacting with Talin-1, influences the integrity of the epithelium, resembling the mechanism employed by the Shigella effector OspE, which has been reported to target focal adhesions to inhibit the detachment of infected cells (57).
A SPI-2 effector with opposing biochemical activity to SseL that also yielded novel hits in the mammalian binding partner screen was the E3 ubiquitin ligase, SspH2. We identified five hits for SspH2 and confirmed a clear, reciprocal interaction with Sgt1. An interaction between SspH2 and Bub3 was also confirmed by reciprocal co-immunoprecipitation; however, these data were more ambiguous, as a fraction of cellular Bub3 was precipitated nonspecifically. Nevertheless, the amount of Bub3 co-immunoprecipitated in the presence of SspH2 was increased over the negative control, suggesting that they interact. Bub3 is part of the SAC (spindle assembly checkpoint) protein complex that ensures the fidelity of chromosome segregation by inhibiting the transition from mitosis to anaphase. This is achieved by SAC inhibition of the anaphase-promoting complex (APC) in the presence of unattached kinetochores (42). Thus, Bub3 plays an important role in cell cycle progression. Interestingly, Sgt1 also plays a role in kinetochore function. Sgt1 binds to Skp1, a component of the SCF ubiquitin ligase complex in yeast, which is required for progression through the G1/S and G2/M transitions by promoting SCF activation and the subsequent degradation of regulatory factors (58). Skp1 has also been implicated in influencing IκB degradation in higher eukaryotes, thereby linking Skp1 to innate immunity (59). Sgt1 has also been shown to influence innate immunity in a Skp1-independent manner via its association with Hsp90 and NLR innate immune molecules. Silencing of sgt1 ablates the ability of Nod1 to elicit cytokine secretion in the presence of its agonist (60). It is tempting to speculate that SspH2 can target Sgt1 for ubiquitin transfer and possible degradation, thereby deactivating the Nod1 signaling pathway, although at present our analysis of Sgt1 peptides identified in this screen did not reveal the presence of a Gly-Gly modification characteristic of ubiquitin transfer. However, it is possible that HEK293T cells do not contain the requisite E1 and E2 enzymes for SspH2. Indeed, it has been demonstrated that SspH2 requires an unconventional E2 substrate, ubiquitin-charged UbcH5, for efficient E3 ligase function (36), and it is unclear whether Ub-UbcH5c is present in HEK293T.
Although it is largely appreciated that SPI-2 effectors play a critical role in maintaining the SCV, thereby providing a replicative niche for this facultative intracellular pathogen, our study has revealed that S. typhimurium may target other host cell processes, some of which are understudied in the context of S. typhimurium infection. These processes include cell adhesion, cell cycle control, and innate immunity, highlighting the importance of these pathways for intracellular survival and replication of S. typhimurium (Fig. 7). This emphasizes that the strength of this proteomic analysis lies not only in the discovery of individual interactions but also in revealing the overall biological processes affected by S. typhimurium infection. Interestingly, several effectors appear to fulfill more than one functional role, influencing several of these processes through interactions with different binding partners (Fig. 7). Also of note is that SPI-2 effectors appear to target multiple points (proteins) in a given cell process, suggesting a concerted subversion of these processes.
FIGURE 7.

Biological processes affected by effectors analyzed in this study. S. typhimurium effectors binding to host cell targets suggest a set of biological contexts to be influenced by infection. SseG (G) and SspH2 (H2) bind, respectively, to Caprin-1 and Bub3/AH receptor-interacting protein (AIP)/Sgt-1, host proteins influencing cell cycle control. SseF (F), SseG, SseL (L), and SseJ (J) target proteins located at desmosomes or focal adhesions, suggesting a role in the regulation of cell attachment. SspH1 (H1), SspH2, and SseL target PKN1, Sgt1, and OSBP, respectively, arguing for a possible role in the regulation of innate immunity.
One major concern in the identification of protein-protein interactions is the high frequency of false positives resulting from common screening methods. For yeast two-hybrid screens, for example, it has been estimated that 25–45% of detected interactions are likely incorrect (61). Binding studies using metabolic labeling through SILAC and simultaneous mass spectrometric analysis of differentially labeled peptides originating from the sample and negative control have been reported to result in a very low rate of false positive results (62). Indeed, among the 14 interactions identified here, four had been found and confirmed elsewhere. We were able to further confirm three novel binding pairs by reciprocal co-immunoprecipitation and Western blot. Thus, the majority of hits in the interaction partner screen described here could be confirmed by others and us, or they agree with known hallmarks of S. typhimurium biology, arguing for a very low false detection rate.
Interestingly, only about one-quarter of the effectors analyzed yielded interaction partners. Using effectors expressed in tissue culture (Fig. 4, strategy A), we found 11 new binding partners, and several interactions could be confirmed that had been published previously or were published during the course of this study. Precipitation with purified recombinant effectors (Fig. 4, strategy B) was able to confirm the interaction between SopB/SigD and Cdc42 and for the first time found that SseL bound to Talin-1. This implies that, although several previously published interactions were missed (63–67), the technology is sensitive. However, using strategy B, false negative results can occur if binding events depend on an intact host cell environment, for example for proper effector processing and localization. It also cannot be ruled out that the N-terminal HA tag used in both strategies may interfere with the formation of certain host protein-effector complexes. Nevertheless, it appears that several effectors do not have host protein interaction partners. These effectors may instead bind and act on lipids, carbohydrates, nucleic acids, or small molecules. There might also be effectors that bind host proteins cooperatively with other S. typhimurium proteins or that bind host proteins only transiently, for example to carry out an enzymatic reaction.
Supplementary Material
Acknowledgments
We thank the members of our group for discussions and valuable advice. Mass spectrometry infrastructure used in this work was supported by the Canada Foundation for Innovation, the British Columbia Knowledge Development Fund, and the BC Proteomics Network.
This work was supported by grants from the Canadian Institutes of Health Research (to L. J. F. and B. B. F.) and from Genome Canada/Genome British Columbia through the PRoteomics for Emerging PAthogen REsponse (PREPARE) project and the VGH & UBC Hospital Foundation.

The on-line version of this article (available at http://www.jbc.org) contains supplemental Fig. S1 and Table S1.
- T3SS
- type three secretion system
- SPI
- Salmonella pathogenicity island
- SCV
- Salmonella-containing vacuole
- SILAC
- stable isotope labeling of amino acids in cell culture
- LTQ
- linear trap quadrupole
- OSBP
- oxysterol-binding protein 1
- NLR
- nucleotide-binding domain and leucine-rich repeat-containing
- PKN1
- protein kinase N1
- LPM
- low phosphate, low magnesium.
REFERENCES
- 1. Galán J. E., Wolf-Watz H. (2006) Nature 444, 567–573 [DOI] [PubMed] [Google Scholar]
- 2. Valdez Y., Ferreira R. B., Finlay B. B. (2009) Curr. Top. Microbiol. Immunol. 337, 93–127 [DOI] [PubMed] [Google Scholar]
- 3. Marcus S. L., Brumell J. H., Pfeifer C. G., Finlay B. B. (2000) Microbes Infect. 2, 145–156 [DOI] [PubMed] [Google Scholar]
- 4. Galán J. E., Curtiss R., 3rd (1989) Proc. Natl. Acad. Sci. U.S.A. 86, 6383–6387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Menendez A., Arena E. T., Guttman J. A., Thorson L., Vallance B. A., Vogl W., Finlay B. B. (2009) J. Infect. Dis. 200, 1703–1713 [DOI] [PubMed] [Google Scholar]
- 6. Zhou D., Galán J. (2001) Microbes Infect. 3, 1293–1298 [DOI] [PubMed] [Google Scholar]
- 7. Shea J. E., Beuzon C. R., Gleeson C., Mundy R., Holden D. W. (1999) Infect. Immun. 67, 213–219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ochman H., Soncini F. C., Solomon F., Groisman E. A. (1996) Proc. Natl. Acad. Sci. U.S.A. 93, 7800–7804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hensel M., Shea J. E., Waterman S. R., Mundy R., Nikolaus T., Banks G., Vazquez-Torres A., Gleeson C., Fang F. C., Holden D. W. (1998) Mol. Microbiol. 30, 163–174 [DOI] [PubMed] [Google Scholar]
- 10. Rytkönen A., Poh J., Garmendia J., Boyle C., Thompson A., Liu M., Freemont P., Hinton J. C., Holden D. W. (2007) Proc. Natl. Acad. Sci. U.S.A. 104, 3502–3507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Le Negrate G., Faustin B., Welsh K., Loeffler M., Krajewska M., Hasegawa P., Mukherjee S., Orth K., Krajewski S., Godzik A., Guiney D. G., Reed J. C. (2008) J. Immunol. 180, 5045–5056 [DOI] [PubMed] [Google Scholar]
- 12. Quezada C. M., Hicks S. W., Galán J. E., Stebbins C. E. (2009) Proc. Natl. Acad. Sci. U.S.A. 106, 4864–4869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Abrahams G. L., Hensel M. (2006) Cell. Microbiol. 8, 728–737 [DOI] [PubMed] [Google Scholar]
- 14. Bhavsar A. P., Auweter S. D., Finlay B. B. (2010) Future Microbiol. 5, 253–265 [DOI] [PubMed] [Google Scholar]
- 15. Blagoev B., Kratchmarova I., Ong S. E., Nielsen M., Foster L. J., Mann M. (2003) Nat. Biotechnol. 21, 315–318 [DOI] [PubMed] [Google Scholar]
- 16. Rogers L. D., Kristensen A. R., Boyle E. C., Robinson D. P., Ly R. T., Finlay B. B., Foster L. J. (2008) J. Proteomics 71, 97–108 [DOI] [PubMed] [Google Scholar]
- 17. Coombes B. K., Brown N. F., Valdez Y., Brumell J. H., Finlay B. B. (2004) J. Biol. Chem. 279, 49804–49815 [DOI] [PubMed] [Google Scholar]
- 18. Coombes B. K., Wickham M. E., Brown N. F., Lemire S., Bossi L., Hsiao W. W., Brinkman F. S., Finlay B. B. (2005) J. Mol. Biol. 348, 817–830 [DOI] [PubMed] [Google Scholar]
- 19. Shevchenko A., Wilm M., Vorm O., Mann M. (1996) Anal. Chem. 68, 850–858 [DOI] [PubMed] [Google Scholar]
- 20. Brumell J. H., Rosenberger C. M., Gotto G. T., Marcus S. L., Finlay B. B. (2001) Cell. Microbiol. 3, 75–84 [DOI] [PubMed] [Google Scholar]
- 21. Lee A. K., Detweiler C. S., Falkow S. (2000) J. Bacteriol. 182, 771–781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Detweiler C. S., Monack D. M., Brodsky I. E., Mathew H., Falkow S. (2003) Mol. Microbiol. 48, 385–400 [DOI] [PubMed] [Google Scholar]
- 23. Heiland I., Wittmann-Liebold B. (1979) Biochemistry 18, 4605–4612 [DOI] [PubMed] [Google Scholar]
- 24. Petnicki-Ocwieja T., Schneider D. J., Tam V. C., Chancey S. T., Shan L., Jamir Y., Schechter L. M., Janes M. D., Buell C. R., Tang X., Collmer A., Alfano J. R. (2002) Proc. Natl. Acad. Sci. U.S.A. 99, 7652–7657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Woo H. J., Hwang Y. K., Kim Y. J., Kang J. Y., Choi Y. K., Kim C. G., Park Y. S. (2002) FEBS Lett. 523, 234–238 [DOI] [PubMed] [Google Scholar]
- 26. Thöny B., Auerbach G., Blau N. (2000) Biochem. J. 347, 1–16 [PMC free article] [PubMed] [Google Scholar]
- 27. Dittrich S., Mitchell S. L., Blagborough A. M., Wang Q., Wang P., Sims P. F., Hyde J. E. (2008) Mol. Microbiol. 67, 609–618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Rodríguez-Escudero I., Rotger R., Cid V. J., Molina M. (2006) Microbiology 152, 3437–3452 [DOI] [PubMed] [Google Scholar]
- 29. Haraga A., Miller S. I. (2006) Cell. Microbiol. 8, 837–846 [DOI] [PubMed] [Google Scholar]
- 30. Ohlson M. B., Huang Z., Alto N. M., Blanc M. P., Dixon J. E., Chai J., Miller S. I. (2008) Cell Host Microbe 4, 434–446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Deiwick J., Salcedo S. P., Boucrot E., Gilliland S. M., Henry T., Petermann N., Waterman S. R., Gorvel J. P., Holden D. W., Méresse S. (2006) Infect. Immun. 74, 6965–6972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kuhle V., Hensel M. (2002) Cell. Microbiol. 4, 813–824 [DOI] [PubMed] [Google Scholar]
- 33. Stokes D. L. (2007) Curr. Opin. Cell Biol. 19, 565–571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wang B., David M. D., Schrader J. W. (2005) J. Immunol. 175, 4274–4282 [DOI] [PubMed] [Google Scholar]
- 35. Grill B., Wilson G. M., Zhang K. X., Wang B., Doyonnas R., Quadroni M., Schrader J. W. (2004) J. Immunol. 172, 2389–2400 [DOI] [PubMed] [Google Scholar]
- 36. Levin I., Eakin C., Blanc M. P., Klevit R. E., Miller S. I., Brzovic P. S. (2010) Proc. Natl. Acad. Sci. U.S.A. 107, 2848–2853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Raychaudhuri S., Prinz W. A. (2010) Annu. Rev. Cell Dev. Biol. 26, 157–177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Moser M., Legate K. R., Zent R., Fässler R. (2009) Science 324, 895–899 [DOI] [PubMed] [Google Scholar]
- 39. Kadota Y., Shirasu K., Guerois R. (2010) Trends Biochem. Sci. 35, 199–207 [DOI] [PubMed] [Google Scholar]
- 40. Carver L. A., Bradfield C. A. (1997) J. Biol. Chem. 272, 11452–11456 [DOI] [PubMed] [Google Scholar]
- 41. Kuzhandaivelu N., Cong Y. S., Inouye C., Yang W. M., Seto E. (1996) Nucleic Acids Res. 24, 4741–4750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Logarinho E., Bousbaa H. (2008) Cell Cycle 7, 1763–1768 [DOI] [PubMed] [Google Scholar]
- 43. van Hemert M. J., Steensma H. Y., van Heusden G. P. (2001) Bioessays 23, 936–946 [DOI] [PubMed] [Google Scholar]
- 44. Takayama S., Xie Z., Reed J. C. (1999) J. Biol. Chem. 274, 781–786 [DOI] [PubMed] [Google Scholar]
- 45. Deng W., de Hoog C. L., Yu H. B., Li Y., Croxen M. A., Thomas N. A., Puente J. L., Foster L. J., Finlay B. B. (2010) J. Biol. Chem. 285, 6790–6800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Yu X. J., McGourty K., Liu M., Unsworth K. E., Holden D. W. (2010) Science 328, 1040–1043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Niemann G. S., Brown R. N., Gustin J. K., Stufkens A., Shaikh-Kidwai A. S., Li J., McDermott J. E., Brewer H. M., Schepmoes A., Smith R. D., Adkins J. N., Heffron F. (2011) Infect. Immun. 79, 33–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Lee E. Y., Bang J. Y., Park G. W., Choi D. S., Kang J. S., Kim H. J., Park K. S., Lee J. O., Kim Y. K., Kwon K. H., Kim K. P., Gho Y. S. (2007) Proteomics 7, 3143–3153 [DOI] [PubMed] [Google Scholar]
- 49. Salcedo S. P., Holden D. W. (2003) EMBO J. 22, 5003–5014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Solomon S., Xu Y., Wang B., David M. D., Schubert P., Kennedy D., Schrader J. W. (2007) Mol. Cell. Biol. 27, 2324–2342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Coombes B. K., Lowden M. J., Bishop J. L., Wickham M. E., Brown N. F., Duong N., Osborne S., Gal-Mor O., Finlay B. B. (2007) Infect. Immun. 75, 574–580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Fairn G. D., McMaster C. R. (2008) Cell. Mol. Life Sci. 65, 228–236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Lehto M., Olkkonen V. M. (2003) Biochim. Biophys. Acta 1631, 1–11 [DOI] [PubMed] [Google Scholar]
- 54. Lehto M., Laitinen S., Chinetti G., Johansson M., Ehnholm C., Staels B., Ikonen E., Olkkonen V. M. (2001) J. Lipid Res. 42, 1203–1213 [PubMed] [Google Scholar]
- 55. Shattil S. J., Kim C., Ginsberg M. H. (2010) Nat. Rev. Mol. Cell Biol. 11, 288–300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Finlay B. B., Ruschkowski S., Dedhar S. (1991) J. Cell Sci. 99, 283–296 [DOI] [PubMed] [Google Scholar]
- 57. Kim M., Ogawa M., Fujita Y., Yoshikawa Y., Nagai T., Koyama T., Nagai S., Lange A., Fässler R., Sasakawa C. (2009) Nature 459, 578–582 [DOI] [PubMed] [Google Scholar]
- 58. Kitagawa K., Skowyra D., Elledge S. J., Harper J. W., Hieter P. (1999) Mol. Cell 4, 21–33 [DOI] [PubMed] [Google Scholar]
- 59. Maniatis T. (1999) Genes Dev. 13, 505–510 [DOI] [PubMed] [Google Scholar]
- 60. da Silva Correia J., Miranda Y., Leonard N., Ulevitch R. (2007) Proc. Natl. Acad. Sci. U.S.A. 104, 6764–6769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Huang H., Jedynak B. M., Bader J. S. (2007) PLoS Comput. Biol. 3, e214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Mittler G., Butter F., Mann M. (2009) Genome Res. 19, 284–293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Bernal-Bayard J., Cardenal-Muñoz E., Ramos-Morales F. (2010) J. Biol. Chem. 285, 16360–16368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Bernal-Bayard J., Ramos-Morales F. (2009) J. Biol. Chem. 284, 27587–27595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Boucrot E., Henry T., Borg J. P., Gorvel J. P., Méresse S. (2005) Science 308, 1174–1178 [DOI] [PubMed] [Google Scholar]
- 66. Miao E. A., Brittnacher M., Haraga A., Jeng R. L., Welch M. D., Miller S. I. (2003) Mol. Microbiol. 48, 401–415 [DOI] [PubMed] [Google Scholar]
- 67. Harrison R. E., Brumell J. H., Khandani A., Bucci C., Scott C. C., Jiang X., Finlay B. B., Grinstein S. (2004) Mol. Biol. Cell 15, 3146–3154 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.