

Abstract
Chronic pain associated with inflammation is a common clinical problem, and the underlying mechanisms have only begun to be unraveled. GRK2 regulates cellular signaling by promoting G-protein-coupled receptor (GPCR) desensitization and direct interaction with downstream kinases including p38. The aim of this study was to determine the contribution of GRK2 to regulation of inflammatory pain and to unravel the underlying mechanism. GRK2+/− mice with an ∼50% reduction in GRK2 developed increased and markedly prolonged thermal hyperalgesia and mechanical allodynia after carrageenan-induced paw inflammation or after intraplantar injection of the GPCR-binding chemokine CCL3. The effect of reduced GRK2 in specific cells was investigated using Cre–Lox technology. Carrageenan- or CCL3-induced hyperalgesia was increased but not prolonged in mice with decreased GRK2 only in Nav1.8 nociceptors. In vitro, reduced neuronal GRK2 enhanced CCL3-induced TRPV1 sensitization. In vivo, CCL3-induced acute hyperalgesia in GRK2+/− mice was mediated via TRPV1. Reduced GRK2 in microglia/monocytes only was required and sufficient to transform acute carrageenan- or CCL3-induced hyperalgesia into chronic hyperalgesia. Chronic hyperalgesia in GRK2+/− mice was associated with ongoing microglial activation and increased phospho-p38 and tumor necrosis factor α (TNF-α) in the spinal cord. Inhibition of spinal cord microglial, p38, or TNF-α activity by intrathecal administration of specific inhibitors reversed ongoing hyperalgesia in GRK2+/− mice. Microglia/macrophage GRK2 expression was reduced in the lumbar ipsilateral spinal cord during neuropathic pain, underlining the pathophysiological relevance of microglial GRK2. Thus, we identified completely novel cell-specific roles of GRK2 in regulating acute and chronic inflammatory hyperalgesia.
Introduction
Patients suffering from inflammatory disorders often present with chronic pain that is difficult to treat. Moreover, a significant subgroup of patients continues to experience pain even when inflammation has resolved (Scholz and Woolf, 2007). Pain caused by tissue injury and inflammation is accompanied by sensitization to noxious stimuli (hyperalgesia) (Andrew and Greenspan, 1999; Marchand et al., 2005). Both peripheral mechanisms at the site of inflammation and central processes contribute to the development and maintenance of hyperalgesia. The excitability of peripheral and/or spinal cord sensory neurons increases after exposure to inflammatory mediators including cytokines and chemokines (Oh et al., 2001; Cunha et al., 2005; Hucho and Levine, 2007). There is also evidence that spinal cord microglial and astrocyte activation participates in maintaining a hyperalgesic state (Deleo and Yezierski, 2001; Milligan and Watkins, 2009). However, the mechanisms defining intensity and duration of inflammatory hyperalgesia have only begun to be unraveled.
G-protein-coupled receptor kinase 2 (GRK2) is a ubiquitously expressed member of the family of GRKs. GRK2 restrains cellular activation by phosphorylating specific agonist-occupied G-protein-coupled receptors (GPCRs) leading to receptor desensitization and internalization (Zhang et al., 1997; Ribas et al., 2007). More recently, it has become clear that GRK2 also restrains signaling via direct interaction with downstream intracellular kinases such as Akt, MEK1/2, phosphoinositide-3 kinase, and p38, leading to inhibition of their activity (Reiter and Lefkowitz, 2006; Ribas et al., 2007). The cellular level of GRK2 is regulated via various pathways. Inflammatory mediators decrease cellular levels of GRK2 in vitro (Lombardi et al., 1999; Ramos-Ruiz et al., 2000; Kleibeuker et al., 2007). In vivo, the level of GRK2 is decreased by 40–60% in peripheral blood mononuclear cells of humans with chronic inflammatory diseases including rheumatoid arthritis or multiple sclerosis, or in rodent models of these diseases (Lombardi et al., 1999; Vroon et al., 2005). More recently, we analyzed GRK2 protein levels in spinal cord dorsal horn in two different models of neuropathic pain. Chronic constriction injury of the sciatic nerve in rats significantly reduced GRK2 protein in neurons in the dorsal horn of the lumbar but not the thoracic region of the spinal cord (Kleibeuker et al., 2007). In vitro culture of spinal cord slices with IL-1β reduced GRK2 protein as well. In addition, we showed that the L5 spinal nerve transection (SNT) model of neuropathic pain in mice is associated with decreased spinal cord neuronal GRK2 protein in the relevant areas. Both the reduction in GRK2 and the increase in mechanical allodynia after spinal nerve transection were attenuated in mice deficient in IL-1 signaling (Kleibeuker et al., 2008a). These findings are indicative of a functional relation between the decrease in GRK2 and development of mechanical allodynia in this model.
In the present study, we used GRK2+/− mice and mice with reduced GRK2 in only peripheral sensory neurons, in microglia/macrophages, or in astrocytes to determine the contribution of reduced GRK2 to the course of inflammatory pain and to investigate the underlying mechanism. In addition, we analyzed GRK2 levels specifically in spinal cord microglia/macrophages in the spinal nerve transection model of neuropathic pain.
Materials and Methods
Animals.
Female C57BL/6 mice (12–14 weeks) heterozygous for targeted deletion of the GRK2 gene (GRK2+/−) and their wild-type (WT) littermates were used. GRK2−/− mice die in utero (Jaber et al., 1996). GFAP-GRK2f/+, LysM-GRK2f/+, or Nav1.8-GRK2f/+ and control GFAP-GRK2+/+, LysM-GRK2+/+, or Nav1.8-GRK2+/+ offspring were generated by breeding heterozygous GFAP–Cre (Jackson Laboratory), homozygous lysozyme M (LysM)-Cre (Jackson Laboratory), or homozygous Nav1.8-Cre transgenic mice (Stirling et al., 2005) with heterozygous GRK2-Lox mice (GRK2f/+) (Matkovich et al., 2006). Heterozygous floxed GRK2 mice were used to obtain a similar reduction in GRK2 levels as in GRK2+/− mice. Mice were genotyped by PCR analysis on genomic DNA. Mice were bred and maintained in the animal facility of the University of Utrecht (Utrecht, the Netherlands).
Female Sprague Dawley rats (∼250 g; Charles River Laboratories) were used for L5 spinal nerve transection.
All experimental procedures were performed according to the recommendations of the European Commission, and protocols were approved by the University Medical Center Utrecht Experimental Animal Committee or the Committee on Animal Research of Maastricht University.
Induction of hyperalgesia.
Mice received an intraplantar injection of 5 μl of λ-carrageenan (1%; Sigma-Aldrich) in saline, or 2.5 μl of recombinant murine CCL3 (100 μg/ml; R & D Systems) in PBS, pH 7.5, or 5 μl of recombinant mouse IL-1β (200 ng/ml; Preprotech) in the hindpaw. As a control, the same volume of vehicle was injected. Heat withdrawal latency times were determined using the Hargreaves test (IITC Life Science) as described previously (Hargreaves et al., 1988). Intensity of the light beam was chosen to induce a heat withdrawal latency time of ∼8 s at baseline. Baseline withdrawal latencies were determined on 3 consecutive days before intraplantar injection of the inflammatory agent. Mechanical nociception was tested with a calibrated von Frey hair monofilament (Stoelting). First, mice were acclimated for 15 to 20 min in a transparent box with a metal mesh floor. The von Frey hair monofilament was applied through the mesh floor to the plantar skin of the hindpaw. Mechanical nociception was measured as the total number of paw withdrawals in response to a series of six applications of a 0.02 mg von Frey hair, which does not elicit a response in untreated animals (Alessandri-Haber et al., 2006). Paw thickness was measured in a separate set of experiments using a Digimatic micrometer (Mitutoyo).
Drugs.
The specific p38 inhibitor SB239063 (5 μg in 10% DMSO in saline; Sigma-Aldrich) (Cao et al., 2007) was injected intrathecally following the method described by Hylden and Wilcox (1980) in a volume of 5 μl. Minocycline (Sigma-Aldrich) was administered intrathecally (30 μg/5 μl) twice with a 6 h interval on day 7 after carrageenan administration. Intraperitoneal administrations (50 mg/kg) of minocycline were performed from day −1 to day 4 after intraplantar carrageenan injection (Tikka et al., 2001; Raghavendra et al., 2003). Capsazepine (20% DMSO, 1% Tween-80 in saline) was injected intraplantarly 30 min before measurement of heat withdrawal latency times. Etanercept (Wyeth Pharmaceuticals) was administered intrathecally (100 μg/5 μl) at day 7 after carrageenan (Svensson et al., 2005).
Tissue collection.
For biochemical analyses, mice were killed by CO2 asphyxiation. Paw biopsies were taken using a 4 mm biopsy punch (Stiefel), and thoracic (T1–T8) and lumbar spinal cords (L1–L5) were isolated. All samples were frozen in liquid nitrogen.
Peripheral inflammatory activity.
Myeloperoxidase (MPO) activity in biopsies from the paw was determined as a measure of infiltrated neutrophils.
Frozen paw biopsies were homogenized in 50 mm HEPES buffer, pH 8.0, using a Potter homogenizer. Homogenates were centrifuged for 30 min at 10,000 × g at 4°C, and pellets were used to determine MPO activity (Eijkelkamp et al., 2007).
Frozen paw biopsies were homogenized in 50 mm HEPES and centrifuged for 15 min at 10.000 × g at 4°C. Supernatant was used to determine IL-1β content by ELISA (R & D Systems).
Real-time reverse transcriptase PCR.
Total RNA was extracted using Trizol (Invitrogen). cDNA was synthesized using Superscript RNase H− Reverse Transcriptase (Invitrogen) with 2.5 μm random hexamers (Invitrogen). Quantitative real-time PCR was performed using CyberGreen probe and iCycler IQ5 (Bio-Rad). Data were normalized using average β-actin and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) expression. Sense and antisense sequences are as follows: IL-1β, sense, 5′-CAACCAACAAgTgATATTCTCCATg-3′; antisense, 5′-gATCCACACTCTCCAgCTgCA-3′; tumor necrosis factor α (TNF-α), sense, 5′-gCggTgCCTATgTCTCAg-3′; antisense, 5′-gCCATTTgggAACTTCTCATC-3′; CCL3, sense, 5′-CTgCCCTTgCTgTTCTTCTCTg-3′; antisense, 5′-CgATgAATTggCgTggAATCTTC-3′; GAPDH, sense, 5′-TgAAgCAggCATCTgAggg-3′; antisense, 5′-CGAAggTggAAgAgTgggAg-3′; β-actin, sense, 5′-AgAgggAAATCgTgCgTgAC-3′; antisense 5′-CAATAgTgATgACCTggCCgT-3′.
Western blot analysis.
Tissue samples, F11 cells, primary CD11b+ macrophages, or primary microglia were homogenized in ice-cold cell lysis buffer (20 mm Tris-HCl, pH 7.5, 1% Triton X-100, 2 mm EDTA) containing protease inhibitor mix (Sigma-Aldrich), 100 mm PMSF, and 10 mm β-glycerolphosphate, 1 mm NaVO3, and 20 mm NaF. Proteins were separated by 10% SDS-PAGE and transferred to polyvinylidene difluoride membranes (Millipore). Blots were stained with the following primary antibodies: rabbit anti-p-p38 mitogen-activated protein kinase (MAPK) (Cell Signaling Technology), rabbit anti-p38 MAPK (Cell Signaling Technology), rabbit-anti-GRK2 (Santa Cruz Biotechnology), goat-anti-TNF-α (Cell Signaling Technology), goat-anti-β-actin (Santa Cruz Biotechnology), or mouse-anti-α-tubulin (Santa Cruz Biotechnology). Subsequently, blots were incubated for 1 h with goat anti-mouse peroxidase IgG + IgM (H+L) (The Jackson Laboratory) or donkey anti-rabbit peroxidase IgG (GE Healthcare) and developed by enhanced chemiluminescence (GE Healthcare). Band density was determined using a GS-700 Imaging Densitometer (Bio-Rad).
F11 cell line, overexpression, and small interfering RNA.
The dorsal root ganglion (DRG)-like neuronal F11 cell line was cultured in F12 culture medium supplemented with 20% fetal calf serum, 2 mm/l-glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin (all from Invitrogen). F11 cells did not respond to capsaicin or CCL3 (supplemental Fig. 1, available at www.jneurosci.org as supplemental material) (data not shown), indicating that F11 cells do not express functional TRPV1 or CCR1. Therefore, we prepared F11 cells overexpressing TRPV1, CCR1 (both generous gifts from Dr. N. Zhang, National Cancer Institute, Frederick, MD), and GRK2 using a total of 4 μg of pcDNA3 vector containing TRPV1, CCR1, or GRK2 and Lipofectamine. The small interfering RNA (siRNA) sequence targeting GRK2 (5′-UGACUUCAGUGUGCAUCGA-3′) and the control nonspecific siRNA (5′-GCGCGCUUUGUAGGAUUCG-3′) and Lipofectamine (Invitrogen) were used to reduce GRK2 levels. Cells were grown on coverslips and were used for analysis 24 h after transfection.
Analysis of changes in intracellular calcium.
Transfected F11 cells were grown on coverslips and loaded for 30 min at 37°C with 10 μm fura-2 AM (Invitrogen) in F12 medium. Subsequently, the coverslips were placed in 140 mm NaCl, 5 mm KCl, 2 mm CaCl2, 1 mm MgCl2, 0.33 mm NaH2PO4, 10 mm HEPES, and 10 mm glucose, pH 7.4, for 20 min and mounted in a perfusion chamber on a Zeiss UltraVIEW Live Cell Imager (PerkinElmer). Fluorimetric measurements were made using a CCD camera supported by MetaFluor imaging software. Digital images of the cells were obtained at an emission wavelength of 510 nm using paired exposures to 340 and 380 nm excitation wavelengths sampled approximately every 2 s. Fluorescence values representing spatial averages from a defined pixel area were recorded online. Changes in intracellular [Ca2+] after continuous stimulation with 1 μm capsaicin (Sigma-Aldrich) or 100 ng/ml (Zhang et al., 2005) CCL3 (R & D Systems) are expressed as the ratio of the 510 nm emission at 340/380 nm excitation.
Granulocyte depletion.
For depletion of granulocytes, rabbit anti-mouse polymorphonuclear (PMN) antibody or normal rabbit serum (Accurate Antibodies; prediluted 1:5 in sterile saline 17.5 μl/g body weight) was injected intraperitoneally 24 h before and directly before intraplantar carrageenan or CCL3 injection. Effective depletion of circulating neutrophils was confirmed by a differential count of whole blood.
Primary culture of microglia and astrocytes.
Primary cultures of cortical astrocytes and microglia were obtained from 1-d-old LysM-GRK2+/+ and LysM-GRK2f/+ mice or GFAP-GRK2+/+ and GFAP-GRK2f/+ mice.
Cortices were dissected after removal of meninges and blood vessels, minced, and incubated with 0.25% trypsin for 15 min in Gey's balanced salt solution containing 100 U/ml penicillin, 100 μg/ml streptomycin, and 30 mm d(+)-glucose. Cells were dissociated and cultured in poly-l-ornithine (15 μg/ml) coated culture flasks in DMEM/Ham's F10 (1:1) supplemented with 10% FCS, 2 mm glutamine, and antibiotics as stated above.
After 10–12 divisions, flasks were shaken overnight at 37°C to detach microglia from the astrocyte layer. Microglia were used for Western blot analysis to determine GRK2 levels or cultured for 48 h in poly-l-ornithine-coated 24-well plates at a density of 0.5 × 106 cells/ml before the experimental procedures. Plated microglia were preincubated for 1 h with p38 inhibitors SB203580 (20 μm; Sigma-Aldrich) before stimulation with 10 ng/ml lipopolysaccharide (LPS; Sigma-Aldrich). Three hours after stimulation with LPS, supernatant was collected to determine TNF-α by ELISA (U-Cytech). Astrocytes were used to determine GRK2 levels using Western blot analysis.
Peritoneal macrophages.
Macrophages were collected from the peritoneal cavity after intraperitoneal injection of 3 ml of ice-cold RPMI-1640 (Invitrogen) using CD11b microbeads and a MACS column (Milteny Biotec). Isolated CD11b+ macrophages were then used for Western blot analysis of GRK2 levels.
Immunohistochemistry.
Mice were deeply anesthetized with sodium pentobarbital (50 mg/kg, i.p.), perfused intracardially with 0.9% saline, followed by 4% paraformaldehyde in PBS. Lumbar spinal segments were removed, postfixed for 6 h at 4°C, and then kept overnight in 20% sucrose in PBS at 4°C, followed by 30% sucrose in PBS for 24–48 h. Dissected tissue was mounted in OCT compound and frozen at −20°C. Transverse spinal cord sections (10 μm) of spinal segments L2–L4 were cut using a cryostat. Sections were washed in PBS and blocked in 2% normal goat serum and 0.1% Triton X-100. Sections were then incubated overnight for at 4°C with rabbit-anti-Iba-1 (1:200; Wako Pure Chemical Industries). Tissue samples were then washed and incubated with Alexa-488-conjugated goat-anti-rabbit antibody (1:200; Invitrogen).
L5 transection and measurement of mechanical allodynia.
A total of 16 adult Sprague Dawley rats were randomly divided into two groups of 8 rats: L5 SNT and sham surgery. Anesthesia was induced using 5% isoflurane in air at a flow of 250 ml/min using an Inventor 400 injection system vaporizer (Zevenaar) with an open mask system. Anesthesia was maintained with 2.5% isoflurane in air. Under optical magnification (25×), the L6 transverse process was identified, freed of muscular attachments, and partially removed. For SNT animals, the L5 spinal nerve was isolated, and a 1-mm-long segment was removed using microscissors to prevent regeneration. In sham-operated animals, the L5 spinal nerve was isolated but left intact. Mechanical paw withdrawal thresholds were determined before and at 14 d after SNT or sham surgery using the automatic von-Frey-type testing device (Dynamic Plantar Aesthesiometer; Ugo Basile) with a ramp of 10 s and a maximum force of 50 g. At 14 d, animals were killed by an intraperitoneal injection of pentobarbital (150 mg/kg bodyweight; Schering-Plough), and spinal cords were rapidly removed by hydroextrusion.
Spinal microglia/macrophage isolation and immunohistochemical staining.
The ipsilateral and contralateral spinal cord lumbar enlargements of two to three rats were pooled and grinded briefly using a glass Potter in 4 ml of ice-cold HBSS (Invitrogen) containing 15 mm N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid (Invitrogen) and 0.5% glucose (Sigma). Suspensions were passed through a 70 μm cell strainer (BD Biosciences), and cells were centrifuged at 400 × g for 7 min at 10°C and resuspended in 8 ml of 75% Percoll in HBSS (GE Healthcare). A density gradient consisting of cells in 4 ml of 75% Percoll, 3 ml of 50% Percoll, 3 ml of 35% Percoll, and 2 ml of Dulbecco's PBS (DPBS; Invitrogen) was centrifuged (1000 × g for 20 min at 10°C). Cells at the 50/75 interface collected were washed in ice-cold DPBS and resuspended in DPBS containing 1% bovine serum albumin (BSA). Cells were rapidly centrifuged onto a microscope slide using a cytospin centrifuge.
Cells were fixed in acetone at −20°C for 5 min. Slides were blocked in PBS containing 0.1% saponin, 2% BSA, and 2% normal goat serum and incubated with rabbit-anti-GRK2 (1:100; Santa Cruz Biotechnology) and mouse-anti-OX-42 (1:200; BD Biosciences). The specificity of GRK2 staining was controlled by using primary anti-GRK2 antibody blocked with a GRK2 blocking peptide (Santa Cruz Biotechnology). Specificity of OX-42 staining was controlled by using isotype control antibody. Staining was visualized using Alexa-488-conjugated goat-anti-rabbit antibody and Alexa-594-conjugated goat-anti-mouse (1:200; Invitrogen), and slides were stained with 4′,6′-diamidino-2-phenylindole (DAPI; Sigma). Using this procedure, >95% of the isolated cells were OX-42 positive. Cells were photographed with a Zeiss Apotome microscope, and GRK2 levels in OX-42+ cells were analyzed with ImageJ software.
Data analysis.
Data are expressed as mean ± SEM. Measurements were compared using Student's t test, one-way ANOVA, or two-way ANOVA followed by Bonferroni's analysis. A p value <0.05 was considered to be statistically significant.
Results
Inflammatory hyperalgesia in GRK2+/− mice
GRK2+/− mice express ∼50% of GRK2 protein in various tissues including spinal cord and DRG compared to WT mice (Fig. 1a) (Kleibeuker et al., 2008a,b; Nijboer et al., 2008). At baseline, thermal sensitivity did not differ between WT and GRK2+/− mice (8.1 ± 0.2 s vs 8.1 ± 0.2 s; n = 40). However, carrageenan-induced thermal hyperalgesia was significantly more pronounced during the first 24 h after injection in GRK2+/− mice than in WT mice (Fig. 1b). Moreover, GRK2+/− mice remained hypersensitive to heat for at least 20 d after carrageenan injection, whereas WT mice recovered from thermal hyperalgesia within 3 d (Fig. 1b). Similarly, mechanical allodynia induced by carrageenan was increased and lasted significantly longer in GRK2+/− mice (Fig. 1c).
Figure 1.

Reduced GRK2 increases and prolongs inflammation-induced thermal hyperalgesia and mechanical allodynia. a, GRK2 protein level in dorsal root ganglia (n = 4) and spinal cord (n = 8) of GRK2+/− and WT animals as determined by Western blot analysis. The inset shows representative Western blots for GRK2 with α-tubulin as a loading control. b, Percentage decrease in heat withdrawal latency after intraplantar carrageenan injection in WT (n = 8) and GRK2+/− mice (n = 8). c, Mechanical allodynia induced by intraplantar carrageenan injection in WT (n = 4) and GRK2+/− (n = 4) mice. Mechanical sensitivity was determined using a calibrated von Frey hair that does not elicit a withdrawal response in naive control mice. Data represent the percentage of withdrawal responses to six applications per paw. d, Percentage decrease in heat withdrawal latency after intraplantar CCL3 injection in WT (n = 11) and GRK2+/− mice (n = 11). e, Mechanical allodynia induced by intraplantar injection in WT (n = 4) and GRK2+/− (n = 4) mice. f, Percentage decrease in heat withdrawal latency induced by intraplantar IL-1β in WT (n = 14) and GRK2+/− mice (n = 14). Data are expressed as mean ± SEM. *p < 0.05; **p < 0.01; ***p < 0.001.
Carrageenan-induced paw swelling and influx of granulocytes into the paw determined as paw MPO content were similar in both genotypes (Fig. 2a,b). Additionally, carrageenan-induced upregulation of the prototypic proinflammatory cytokine IL-1β and of the chemokine CCL3 did not differ between WT and GRK2+/− mice (Fig. 2c,d). These findings indicate that the increased and prolonged carrageenan-induced hyperalgesia in GRK2+/− mice cannot be explained by increased or ongoing inflammation.
Figure 2.

GRK2 does not affect carrageenan- or CCL3-induced inflammatory response. a, Carrageenan-induced increase in paw thickness expressed as percentage of baseline (n = 8). b, MPO content of paw biopsies as a measure of granulocyte infiltration at 6 h, 1 d, and 7 d after carrageenan injection (n = 6 − 12). c, IL-1β mRNA in paw biopsies at 6 h after carrageenan injection. d, CCL3 mRNA in paw biopsies at 6 h after carrageenan injection (n = 7). e, IL-1β mRNA in paw biopsies at 24 h after intraplantar CCL3 injection (n = 4). f, TNF-α mRNA in paw biopsies at 24 h after CCL3 injection (n = 4). g, CCL3-induced increase in paw thickness expressed as a percentage of baseline (n = 7). h, MPO content of paw biopsies at 6 h after intraplantar CCL3 injection (n = 8 − 11). Data are expressed as mean ± SEM. **p < 0.01.
Next, we induced hyperalgesia by a single intraplantar injection of the chemokine CCL3. We used this chemokine because it is known to signal via a GPCR that is regulated by GRK2 (Zhang et al., 2005). In addition, CCL3 is produced in the paw during carrageenan inflammation (Fig. 2d). Acute CCL3-induced thermal hyperalgesia was significantly increased in GRK2+/− mice compared to WT mice (Fig. 1d,e). Moreover, thermal hyperalgesia induced by a single injection of CCL3 persisted in GRK2+/− mice for 10 d, whereas WT mice recovered within 24 h (Fig. 1d). CCL3-induced mechanical allodynia was also more pronounced and prolonged in GRK2+/− mice compared with WT mice (Fig. 1e). Intraplantar injection of CCL3 did not induce detectable inflammation as determined by paw swelling, redness, cytokine expression, or granulocyte influx (MPO content) in both genotypes (Fig. 2e–h).
Since GRK2 not only regulates signaling at the level of GPCR desensitization but also interacts with downstream signaling molecules, we investigated whether hyperalgesia induced by the cytokine IL-1β, a ligand that does not induce signaling via a GPCR, is also affected by GRK2 deficiency. In contrast to what was observed after carrageenan or CCL3, IL-1β-induced acute hyperalgesia (until 6 h after IL-1β) was similar in both genotypes. However, a single intraplantar injection of IL-1β did induce prolonged hyperalgesia in GRK2+/− mice that lasted 8 d, whereas WT mice recovered within 48 h after injection (Fig. 1f).
Role of GRK2 in primary sensory neurons
Cre-Lox technology was applied to investigate whether a reduced level of GRK2 in only Nav1.8-positive peripheral sensory neurons, of which 85% are nociceptors (Stirling et al., 2005), was sufficient to increase and prolong inflammatory hyperalgesia. Homozygous Nav1.8-Cre transgenic mice were crossed with GRK2f/+ mice (Matkovich et al., 2006). GRK2 levels were reduced in DRGs but not in spinal cord of Nav1.8-GRK2f/+ offspring compared with littermate Nav1.8-GRK2+/+ controls (Fig. 3a). Heterozygous Nav1.8-GRK2f/+ mice were used to mimic the 40–60% decrease in GRK2 that was observed in GRK2+/− mice with low GRK2 in all cells. Baseline heat withdrawal latency times were similar in both genotypes (8.6 ± 0.2 s vs 8.6 ± 0.2 s; n = 24). Carrageenan-induced thermal hyperalgesia was significantly enhanced in Nav1.8-GRK2f/+ mice. Carrageenan hyperalgesia was also prolonged in Nav1.8-GRK2f/+ mice (8 d vs 2–4 d in littermate controls) (Fig. 3b), but still lasted a significantly shorter time than in mice with reduced GRK2 in all cells (20 d) (Fig. 1b).
Figure 3.

Reduced GRK2 in Nav1.8+ sensory neurons increases acute hyperalgesia. a, GRK2 protein levels in dorsal root ganglia (n = 4) and spinal cord (n = 4) of Nav1.8-GRK2f/+ and control Nav1.8-GRK2+/+ mice. The inset shows Western blots for GRK2 with α-tubulin as a loading control. b, Percentage decrease in heat withdrawal latency after intraplantar carrageenan injection in Nav1.8-GRK2f/+ (n = 8) mice and in Nav1.8-GRK2+/+ controls (n = 8). c, Percentage decrease in heat paw withdrawal latency after intraplantar CCL3 injection in Nav1.8-GRK2f/+ mice (n = 16) and in Nav1.8-GRK2+/+ controls (n = 16). d, Percentage decrease in heat withdrawal latency after intraplantar IL-1β injection in Nav1.8-GRK2f/+ mice (n = 8) and Nav1.8-GRK2+/+ controls (n = 8). Data are expressed as mean ± SEM. *p < 0.05; **p < 0.01; ***p < 0.001.
Acute CCL3-induced thermal hyperalgesia (0.5 to 3 h after injection) was also significantly increased in Nav1.8-GRK2f/+ mice compared to littermate Nav1.8-GRK2+/+ controls (Fig. 3c). However, we did not observe prolonged hyperalgesia in Nav1.8-GRK2f/+ mice after CCL3 treatment. These data indicate that reduced GRK2 in nociceptors was sufficient to aggravate acute carrageenan- or CCL3-induced hyperalgesia but did not cause the prolonged hyperalgesia that was observed in GRK2+/− mice (Fig. 3c).
Thermal hyperalgesia induced by intraplantar injection of IL-1β did not differ between Nav1.8-GRK2f/+ and Nav1.8-GRK2+/+ control mice (Fig. 3d). This finding is in line with Figure 1f, showing that acute IL-1β-induced hyperalgesia was similar in WT and GRK2+/− mice.
Effect of GRK2 on CCL3-induced TRPV1 sensitization
A study by Zhang et al. (2005) showed that the GPCR ligand CCL3 is capable of sensitizing TRPV1, a heat- and capsaicin-gated ion channel present on a subset on primary nociceptor afferents. We showed previously that the calcium response of GRK2+/− lymphocytes to CCL3 is increased (Vroon et al., 2004). Therefore, we hypothesized that reduced GRK2 will increase signaling via the CCL3 receptor CCR1, thereby potentiating CCL3-induced TRPV1 sensitization and acute hyperalgesia. To address this issue, we used siRNA to reduce GRK2 in DRG-like neuronal F11 cells (Goswami and Hucho, 2007) overexpressing CCR1 and TRPV1. Reduction of GRK2 significantly increased the CCL3-induced calcium signaling in F11 neuronal cells (Fig. 4a). Consistent with data in the literature, preincubation of neuronal cells with CCL3 enhanced their calcium response to subsequent TRPV1 activation (Fig. 4b,c). Moreover, CCL3-induced TRPV1 sensitization was significantly more pronounced in F11 neuronal cells with reduced GRK2 (Fig. 4b,c). Reduced GRK2 did not affect TRPV1 signaling in the absence of the chemokine (Fig. 4b,c). Conversely, overexpression of GRK2 diminished CCL3 signaling and CCL3-induced sensitization of TRPV1 as determined by measuring capsaicin-induced increases in intracellular calcium (Fig. 4d–f). Overall, these results indicate that the intracellular level of GRK2 may regulate acute CCL3-induced hyperalgesia by determining the strength of the response to the GPCR ligand CCL3, thereby facilitating the downstream CCL3-induced TRPV1 sensitization.
Figure 4.

Regulation of CCL3-induced TRPV1 sensitization by GRK. a, CCL3-induced (100 ng/ml) Ca2+ response in F11 cells expressing CCR1 treated with GRK2 siRNA to reduce GRK2 expression or control nonspecific siRNA (scr). The inset shows a reduction in GRK2 protein. b, Capsaicin-induced Ca2+ response in F11 cells overexpressing TRPV1 and CCR1 treated with GRK2 siRNA or control nonspecific siRNA (scr). Cells were pretreated with or without CCL3 (100 ng/ml) for 20 min to induce TRPV1 sensitization. Capsaicin was added and remained in the medium during the entire period of measurement. c, Quantitative analysis of CCL3-induced sensitization of TRPV1 to capsaicin in cells treated with GRK2 siRNA or control nonspecific siRNA (scr). Each bar represents the mean Fura-2 ratio of 15 responding cells as determined during a 100 s recording period in two independent experiments. d, CCL3-induced (100 ng/ml) Ca2+ response in F11 cells expressing CCR1 with or without [empty vector (EV)] GRK2 overexpression. The inset shows GRK2 expression. e, Capsaicin-induced Ca2+ response in F11 cells expressing TRPV1 and CCR1 and overexpressing GRK2 or transfected with control vector (EV). Cells were pretreated with or without CCL3 for 20 min. f, Quantitative analysis capsaicin-induced Ca2+ response in F11 cells expressing TRPV1 and CCR1 overexpressing GRK2 or transfected with empty vector (EV). Cells were pretreated with or without CCL3 for 20 min. Bars represent the mean Fura-2 ratio as determined during a 100 s recording period of 40 responding cells measured in three independent experiments. g, The TRPV1 antagonist capsazepine was injected intraplantarly 30 min before measurement of heat withdrawal latency 2 h after CCL3 injection. Thermal hyperalgesia is depicted as the percentage of decrease in heat withdrawal latency (n = 8 − 12). Data are expressed as mean ± SEM. *p < 0.05; **p < 0.01; ***p < 0.001.
To determine the contribution of TRPV1 to CCL3-induced hyperalgesia in vivo, we applied the TRPV1 antagonist capsazepine. Acute CCL3-induced thermal hyperalgesia was completely prevented by pretreatment with capsazepine as determined 2 h after CCL3 injection in both WT and GRK2+/− mice (Fig. 4g). These findings indicate that the CCL3-induced hyperalgesia is TRPV1 mediated in both GRK2+/− and WT mice.
Role of GRK2 in microglia/macrophages
In Figure 3b we described that carrageenan-induced thermal hyperalgesia in Nav1.8-GRK2f/+ mice was considerably shorter (8 d) than in GRK2+/− mice (20 d) (Fig. 1b). Moreover, reduction of GRK2 in Nav1.8+ nociceptors only increased acute CCL3-induced thermal hyperalgesia but did not lead to the chronic hyperalgesia that we observed in GRK2+/− mice. These data indicate that GRK2 reduction in other cell types contributes to development of chronic hyperalgesia after intraplantar injection of inflammatory stimuli.
Therefore, we determined the contribution of reduced GRK2 in microglia/macrophages and astrocytes to carrageenan-induced hyperalgesia. Transgenic mice expressing Cre under control of the microglia/macrophage/granulocyte-specific promoter LysM (Clausen et al., 1999) were crossed with GRK2f/+ mice. GRK2 levels were decreased in microglia and in macrophages but not in astrocytes of LysM-GRK2f/+ offspring (Fig. 5a), confirming cellular specificity.
Figure 5.

GRK2 in microglia/macrophages regulates chronic hyperalgesia. a, GRK2 protein levels in microglia (n = 4), macrophages (n = 3), and astrocytes (n = 4) of LysM-GRK2f/+ and control LysM-GRK2+/+ mice. The inset depicts Western blots for GRK2 protein and β-actin as a loading control. b, Percentage decrease in heat withdrawal latency after intraplantar carrageenan injection in LysM-GRK2f/+ (n = 8) and control LysM-GRK2+/+ (n = 8) mice. c, Percentage decrease in heat withdrawal latency after intraplantar CCL3 injection in control LysM-GRK2+/+ (n = 8) and in LysM-GRK2f/+ mice (n = 8). d, Percentage decrease in heat withdrawal latency after intraplantar IL-1β injection in control LysM-GRK2+/+ (n = 8) and LysM-GRK2f/+ mice (n = 8). e, No effect of PMN depletion on CCL3-induced thermal hyperalgesia in LysM-GRK2f/+ (n = 8) and control LysM-GRK2+/+ (n = 8) mice. f, No effect of PMN depletion on carrageenan-induced thermal hyperalgesia in LysM-GRK2f/+ (n = 8) and control LysM-GRK2+/+ (n = 8) mice. Data are expressed as mean ± SEM. *p < 0.05; **p < 0.01; ***p < 0.001.
Baseline sensitivity to heat was similar in LysM-GRK2f/+ and control LysM-GRK2+/+ littermates (8.1 ± 0.1 s vs 8.2 ± 0.2 s; n = 22). In LysM-GRK2f/+ mice, hyperalgesia induced by carrageenan lasted for at least 20 d, whereas control LysM-GRK2+/+ littermates recovered within 2–4 d (Fig. 5b). In addition, hyperalgesia induced by a single injection of CCL3 persisted for 10 d in LysM-GRK2f/+ mice, whereas control LysM-GRK2+/+ littermates recovered within 1 d (Fig. 5c). In contrast, acute carrageenan- or CCL3-induced hyperalgesia in mice with reduced GRK2 in microglia/macrophages/granulocytes (LysM-GRK2f/+) did not differ from control littermates (Fig. 5c). IL-1β-induced hyperalgesia lasted at least 8 d in LysM-GRK2f/+ mice and <1 d in control mice (Fig. 5d). In fact, the duration of carrageenan-, CCL3-, and IL-1β-induced hyperalgesia was similar in GRK2+/− mice and in LysM-GRK2+/− mice.
The LysM promoter that we used to drive Cre expression is not only active in microglia/macrophages but also in granulocytes. There is some evidence for a role of granulocytes in inflammatory hyperalgesia (Rittner et al., 2006; Cunha et al., 2008). We showed, however, that the carrageenan-induced influx of granulocytes into the paw was not affected by genotype (Fig. 2b). Moreover, depleting LysM-GRK2+/+ and LysM-GRK2f/+ mice of granulocytes using a specific antiserum did not affect hyperalgesia induced by carrageenan or CCL3 in both genotypes (Fig. 5e,f). Successful depletion was confirmed by differential blood cell counts. Granulocyte depletion did not have any effect on basal pain responses (change in latency time, −4.8 ± 2.8% in granulocyte-depleted LysM-GRK2+/+, 3.6 ± 4.1% in granulocyte-depleted LysM-GRK2f/+ mice). These data indicate that the prolongation of hyperalgesia in LysM-GRK2f/+ mice was mediated via microglia/macrophages and not via granulocytes.
Role of GRK2 in astrocytes
Transgenic mice expressing Cre under control of the astrocyte-specific promoter GFAP were crossed with GRK2f/+ mice. GRK2 levels were decreased in astrocytes but not in microglia of GFAP-GRK2f/+ offspring (Fig. 6a). Mice with reduced GRK2 expression specifically in astrocytes (GFAP-GRK2f/+) showed similar baseline heat sensitivity (8.4 ± 0.1 s vs 8.3 ± 0.1 s; n = 24). Moreover, carrageenan- and CCL3-induced thermal hyperalgesia were similar in GFAP-GRK2f/+ mice compared with control littermates (GFAP-GRK2+/+) (Fig. 6b,c).
Figure 6.
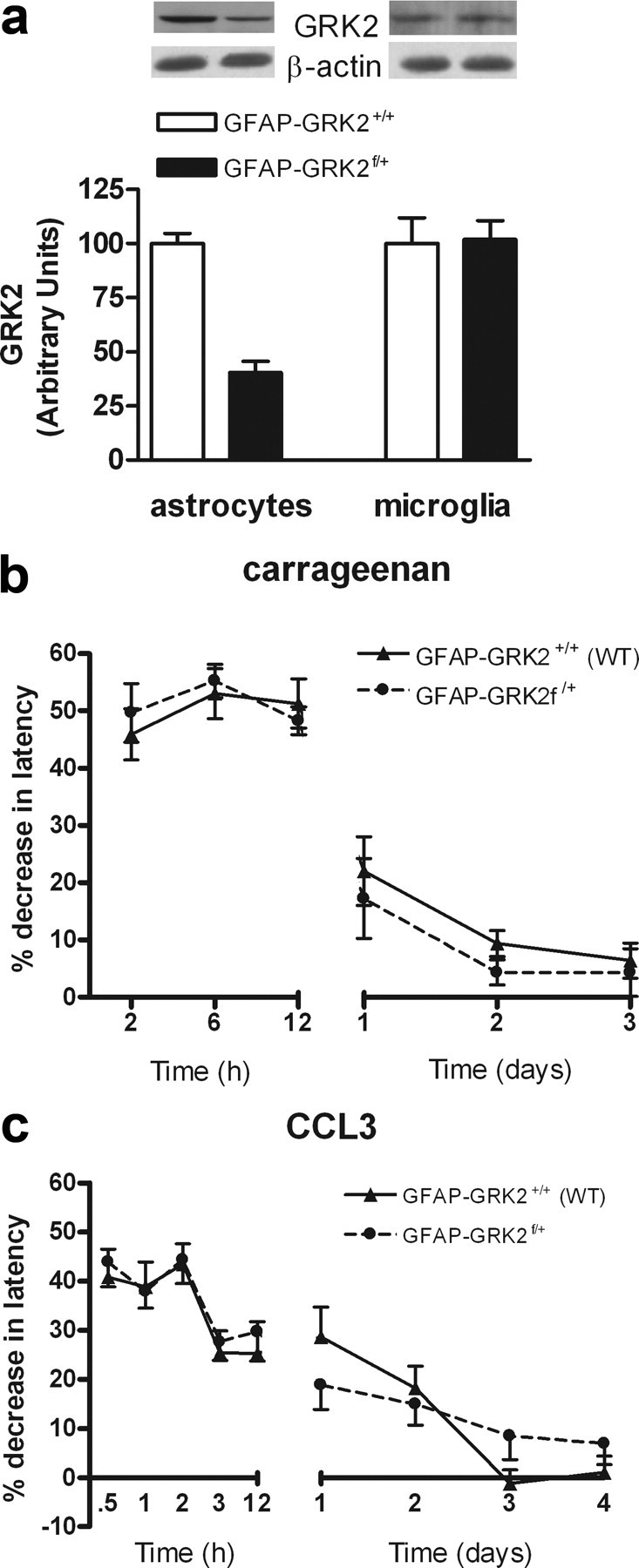
Effect of reduction of GRK2 in GFAP-positive astrocytes on carrageenan- and CCL3-induced hyperalgesia. a, GRK2 protein levels in microglia (n = 4) and astrocytes (n = 4) of GFAP-GRK2f/+ and control GFAP-GRK2+/+ mice. The inset depicts Western blots for GRK2 protein and β-actin as a loading control. b, Percentage decrease in heat withdrawal latency after intraplantar carrageenan injection in GFAP-GRK2+/+ (n = 8) and GFAP-GRK2f/+ mice (n = 8). c, Percentage decrease in heat withdrawal latency after intraplantar CCL3 injection in GFAP-GRK2+/+ (n = 16) and GFAP-GRK2f/+ (n = 16). Data are expressed as mean ± SEM.
Contribution of microglial activity to ongoing hyperalgesia
The data in Figure 5 indicate that reduced GRK2 in microglia/macrophages is sufficient to markedly prolong inflammatory hyperalgesia. Next, we investigated whether carrageenan-induced chronic hyperalgesia in GRK2+/− mice was associated with increased/ongoing spinal cord microglia activation. Iba-1+ microglia in spinal segment L2 had a more activated phenotype in GRK2+/− mice compared to WT mice at 2 d after intraplantar carrageenan (Fig. 7a). To determine whether spinal cord microglial/macrophage activity is required for the development and/or maintenance of chronic hyperalgesia in GRK2 deficient animals, we used the microglial/macrophage inhibitor minocycline (Tikka et al., 2001; Raghavendra et al., 2003). Pretreatment with minocycline completely prevented the development of chronic hyperalgesia in GRK2+/− mice after intraplantar injection of carrageenan but did not have any effect on acute hyperalgesia (<12 h after carrageenan) (Fig. 7b). Moreover, consistent with an important role for ongoing spinal cord microglial activity in prolonged hyperalgesia in GRK2+/− mice, intrathecal administration of minocycline at day 7 after intraplantar carrageenan normalized thermal sensitivity in these mice (Fig. 7c). WT mice did not show hyperalgesia at this time point, and minocycline treatment did not affect thermal sensitivity in these mice.
Figure 7.
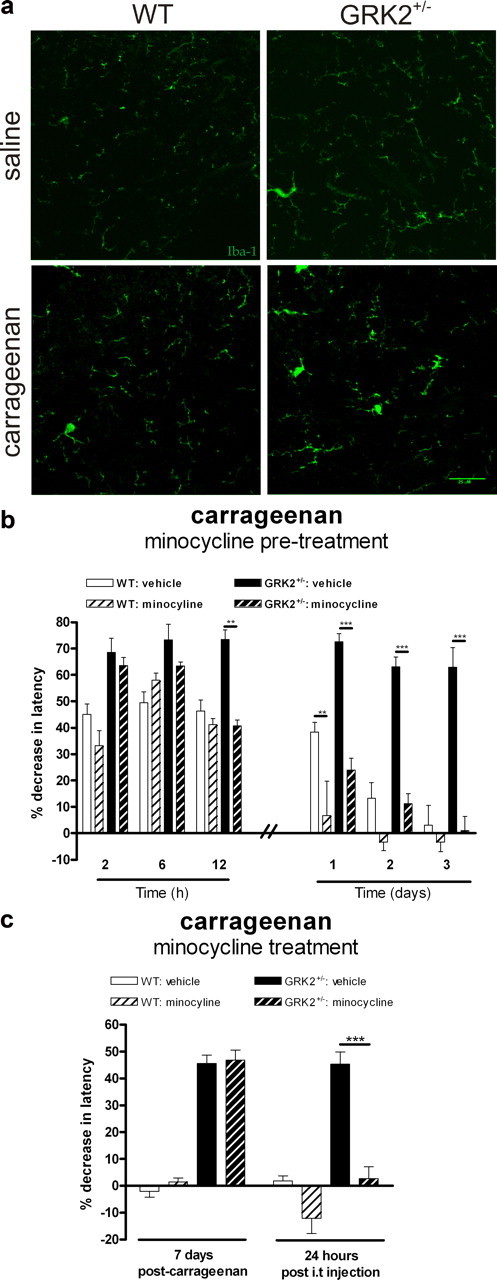
Microglial/macrophage activity is required for chronic carrageenan-induced hyperalgesia. a, WT and GRK2+/− mice received an intraplantar injection of carrageenan or saline. At 2 d after injection, spinal cord was collected, and frozen sections were stained with Iba-1 to visualize microglia. Representative example of lumbar segment L2 of one of three mice per group is displayed. b, WT and GRK2+/− mice were treated with minocycline or PBS starting 1 d before intraplantar injection of carrageenan (n = 8 per group), and heat withdrawal latencies were determined. c, WT and GRK2+/− mice were treated with two intrathecal injections (6 h interval) of minocycline or PBS at day 7 after carrageenan injection. Heat withdrawal latencies were determined 1 h before and 24 h after administration of minocycline (n = 6). Data are expressed as mean ± SEM. **p < 0.01; ***p < 0.001.
Role of p38 MAP kinase and TNF-α in chronic hyperalgesia in GRK2+/− animals
Apart from regulating GPCR desensitization, GRK2 can also inhibit the activity of several downstream signaling molecules, including the MAP kinase p38 (Peregrin et al., 2006). Activation of p38 enhances proinflammatory cytokine production, and there is evidence that p38 contributes to chronic hyperalgesia in rodent models of neuropathic pain (Ji and Suter, 2007; Ji et al., 2009). Moreover, recent evidence suggests that GRK2 directly inhibits p38 activation and that in vitro GRK2+/− macrophages produce more TNF-α than WT macrophages (Peregrin et al., 2006). The data in Figure 8a show that LysM-GRK2f/+ microglia produced significantly more TNF-α after stimulation with LPS compared with WT microglia. Addition of the p38 inhibitor SB203580 in vitro completely abolished the effect of low GRK2 on LPS-induced TNF-α production by microglia. LPS-induced IL-1β production by microglia was not affected by reduced GRK2 (data not shown).
Figure 8.

Role of spinal p38 activation in carrageenan-induced chronic hyperalgesia. a, LPS-induced TNF-α production by LysM-GRK2+/+ WT and LysM-GRK2f/+ microglia. Primary cultures of WT and GRK2+/− microglia were stimulated for 3 h with LPS in presence or absence of the p38 inhibitor SB203580 (20 μm). TNF-α in the supernatant was determined by ELISA. TNF-α levels were undetectable in unstimulated microglia. Data are from two independent experiments performed in triplicate. b, Western blot analysis of p-p38 levels in lumbar spinal cord of saline- or carrageenan-treated WT and GRK2+/− mice at 7 d after intraplantar injection. c, WT and GRK2+/− mice were treated with an intrathecal injection of the p38 inhibitor SB239063 (5 μg/mouse) at day 7 after intraplantar carrageenan injection. Heat withdrawal latencies were determined 1 h before and 2 h after administration of SB239063 (n = 4). d, WT and GRK2+/− were treated with an intrathecal injection of etanercept (100 μg/mouse) at day 7 after intraplantar carrageenan injection and heat withdrawal latencies were determined (n = 6). e, TNF-α levels in spinal segments L1–L5 (lumbar) or T1–T8 (thoracic) of GRK2+/− mice were determined 7 d after intraplantar vehicle or carrageenan injection. The effect of p38 inhibition on carrageenan-induced TNF-α was tested 7 d after intraplantar carrageenan by treating mice intrathecally with SB239063 24 and 2 h before collection of spinal cords. Data represent the level of TNF-α expressed as percentage of vehicle-treated controls. Data are expressed as mean ± SEM. *p < 0.05; **p < 0.01; ***p < 0.001.
Analysis of spinal cord p-p38 revealed that at 7 d after intraplantar carrageenan, when GRK2+/− mice still show hyperalgesia, the level of spinal cord phospho-p38 was increased in GRK2+/− mice. At this time point, spinal cord p-p38 levels in carrageenan-treated WT mice were similar to that of saline-treated mice (Fig. 8b).
To determine whether ongoing spinal cord p38 activity contributed to the carrageenan-induced chronic hyperalgesia that develops in GRK2+/− mice, we administered the p38 inhibitor SB239063 intrathecally at 7 d after intraplantar carrageenan. This treatment attenuated the chronic hyperalgesia that had developed in GRK2+/− mice (Fig. 8c). In WT mice, SB239063 administration at this time point did not affect thermal sensitivity (Fig. 8c). Our in vitro data showed that reduced GRK2 in microglia potentiated TNF-α secretion via a p38-dependent pathway. Therefore, we examined whether ongoing spinal TNF-α signaling was involved in the maintenance of carrageenan-induced chronic hyperalgesia in GRK2+/− mice. Mice were treated mice intrathecally with the TNF-α inhibitor etanercept. A single intrathecal injection of etanercept at day 7 after intraplantar carrageenan reversed ongoing carrageenan-induced hyperalgesia in GRK2+/− mice (Fig. 8d). In WT mice, thermal sensitivity had already returned to base line levels at this time point, and etanercept did not have any effect on thermal sensitivity (Fig. 8d).
In line with the effect of etanercept treatment, the level of TNF-α in lumbar segments (L1–L5) of the spinal cord of GRK2+/− mice was increased at 7 d after intraplantar carrageenan injection compared with saline-injected GRK2+/− mice (Fig. 8e). Intrathecal administration of the p38 inhibitor SB239063 slightly but not significantly attenuated the carrageenan-induced increase in TNF-α that we observed in GRK2+/− mice (Fig. 8e). Intraplantar carrageenan with or without intrathecal treatment with the p38 inhibitor did not affect TNF-α levels in thoracic (T1–T8) spinal cord of GRK2+/− mice.
Microglial GRK2 levels in a model of chronic pain
To investigate the pathophysiological relevance of changes in microglial GRK2, we determined whether changes in spinal microglial/macrophage GRK2 levels actually occur during chronic pain. We analyzed GRK2 protein levels in microglia/macrophages isolated from the ipsilateral and contralateral lumbar spinal cord at day 14 after unilateral L5 SNT or sham surgery. Two weeks after SNT, rats were more sensitive to mechanical stimulation of the ipsilateral paw compared with sham-operated animals. Mechanical thresholds were unaffected in the contralateral paws of SNT rats (Fig. 9a). Importantly, at the same time point, GRK2 levels were significantly reduced by ∼35% in OX-42+ cells (microglia/macrophages) isolated from the ipsilateral compared to contralateral lumbar spinal cord of SNT rats (Fig. 9b,c). GRK2 levels in microglia/macrophages isolated from the contralateral lumbar spinal cord of SNT rats were similar to GRK2 levels in microglia/macrophages from ipsilateral and contralateral lumbar spinal cords of sham-operated control rats (Fig. 9b,c).
Figure 9.

Spinal cord microglial/macrophage GRK2 levels after SNT. a, The sensitivity to mechanical stimulation was determined in sham-operated rats or rats subjected to a unilateral L5 SNT two weeks after surgery (n = 8 per group). b, Scatter plot and bar graph of GRK2 levels in OX-42+ microglia/macrophages isolated from ipsilateral and contralateral lumbar spinal cord of sham-operated and SNT rats at day 14 after surgery. GRK2 expression was quantified in 50–100 cells on three separate slides containing cells isolated from two to three rats per slide. c, Representative pictures of GRK2, OX-42, and DAPI staining of isolated spinal cord microglia/macrophages from ipsilateral and contralateral lumbar spinal cord of sham-operated and SNT rats (first 4 columns). Specificity of GRK2 staining was determined by administration of a specific blocking peptide to the primary antibody and for OX-42 by using an isotype control antibody and is shown in the two columns at the right. Data are expressed as mean ± SEM. ***p < 0.001.
Discussion
We describe here novel cell-specific functions of the endogenous kinase GRK2 as a crucial regulator of severity and duration of inflammatory hyperalgesia. GRK2+/− mice, with an ∼50% reduction in GRK2 in all tissues, developed increased and markedly prolonged hyperalgesia after intraplantar injection of carrageenan or the chemokine CCL3.
The ultimate effect of reduced GRK2 on carrageenan- or CCL3-induced hyperalgesia depended on the cell type in which GRK2 was reduced. Low GRK2 in only NaV1.8+ peripheral nociceptors was sufficient to exacerbate acute carrageenan-induced inflammatory hyperalgesia as well as acute hyperalgesia induced by the GPCR-binding chemokine CCL3. Low GRK2 levels in microglia/macrophages transformed transient hyperalgesia into chronic hyperalgesia that lasted for 1 to 3 weeks depending on the inflammatory stimulus used.
The importance of microglial/macrophage GRK2 for the biology of chronic pain was further substantiated by our findings that ipsilateral lumbar spinal cord microglial/macrophage GRK2 was decreased in a rat model of chronic neuropathic pain.
GRK2 can regulate cellular sensitivity at various levels. Classically, GRK2 is thought to fine tune signaling by desensitizing specific GPCRs including the receptor for the chemokine CCL3 (Vroon et al., 2004). In line with the classical model of fine tuning of GPCR activity by GRK2, we show here that CCL3-induced calcium signaling is regulated by the level of GRK2 in neuronal cells. Similar regulation of CCL3 signaling by changes in the level of GRK2 has been described previously. In vitro, reduced GRK2 in T lymphocytes increases CCL3-induced signaling (Vroon et al., 2004). Conversely, overexpression of GRK2 increases the desensitization of CCL3 receptors (Oppermann et al., 1999). Zhang et al. (2005) showed that one of the consequences of CCR1 activation on nociceptors is sensitization of the heat sensing ion channel TRPV1. Here, we confirm in vitro that CCL3 stimulation sensitizes TRPV1. In addition, we show in vitro that reduced GRK2 in a neuronal cell line potentiated CCL3-induced TRPV1 sensitization, whereas overexpression of GRK2 reduced CCL3-induced TRPV1 sensitization. Therefore, we suggest that in GRK2+/− mice, increased CCR1 signaling and subsequent TRPV1 sensitization caused by low GRK2 in nociceptors contributes to the increased acute CCL3-induced hyperalgesia we report here. This hypothesis is further substantiated by the following findings. In vivo, reduction of GRK2 only in Nav1.8-positive peripheral sensory neurons that are responsive to heat in inflammatory conditions (Abrahamsen et al., 2008) and express TRPV1 (Akopian et al., 1996) was sufficient to increase acute CCL3-induced hyperalgesia. Blockade of TRPV1 using a specific antagonist completely prevented acute CCL3-induced hyperalgesia in both genotypes. These observations are compatible with the hypothesis that increased CCL3-induced acute hyperalgesia in mice with low sensory neuron GRK2 is mediated via increased sensitivity of the GPCR CCR1 leading to enhanced TRPV1 sensitization. The latter hypothesis is supported by our finding that acute hyperalgesia induced by IL-1β was not affected by reduced GRK2 in peripheral nociceptors since IL-1 does not signal via a GPCR.
The most striking observation in our study was the marked prolongation of carrageenan-, CCL3-, or IL-1β-induced hyperalgesia in GRK2+/− mice and in mice with reduced GRK2 only in microglia/macrophages. Spinal cord microglial/macrophage activity was required for development of chronic hyperalgesia in our model because GRK2-deficient mice pretreated intrathecally with minocycline did not develop chronic hyperalgesia. The dose of minocycline administered intrathecally is much lower than the dose needed to inhibit peripheral macrophage activity. Therefore, it is unlikely that peripheral macrophage activity is required for development of chronic hyperalgesia in GRK2-deficient mice. We cannot exclude, however, that peripheral macrophage activity and/or macrophages infiltrating into the spinal cord will contribute to the prolongation of hyperalgesia in GRK2-deficient mice. Importantly, minocycline could also alleviate already existing ongoing inflammatory hyperalgesia. Moreover, prolonged hyperalgesia in GRK2+/− mice was associated with signs of ongoing microglial/macrophage activation in the spinal cord. Whether these microglia/macrophages with an activated phenotype originate from resident spinal microglia or peripheral macrophages remains to be determined. Overall, these data indicate that GRK2 is a crucial regulator of microglia/macrophage activation, and that ongoing activity of these cells is necessary for maintaining hyperalgesia in GRK2+/− mice.
There is increasing evidence suggesting that spinal cord microglia/macrophages can contribute to regulation of hypersensitivity to painful stimulation in models of neuropathic pain or chronic inflammation. In these models, signs of microglial/macrophage activation are detected in the spinal cord, and prevention of microglial/macrophage activation by pretreatment with minocycline administration reduced hyperalgesia (Watkins and Maier, 2003; Watkins et al., 2003; Moalem and Tracey, 2006). Notably, our data shows for the first time that partial deletion of one kinase, i.e., GRK2 in microglia/macrophages, is sufficient to prolong spinal cord microglial/macrophage activation and hyperalgesia even in the absence of ongoing peripheral inflammatory activity or nerve damage.
Recent in vitro evidence indicates that GRK2 directly binds p38 and phosphorylates this kinase at a site in the docking groove that prevents activation of p38 by upstream kinases (Peregrin et al., 2006). Our present data show that GRK2+/− microglia with reduced GRK2 produced more TNF-α via a p38-dependent pathway, which is in line with the observation that reduced GRK2 in macrophages facilitates inflammatory cytokine production (Peregrin et al., 2006). However, to the best of our knowledge, we are the first to show that this GRK2/p38 interaction may also be operative in vivo to control microglial/macrophage activation, TNF-α secretion, and duration of inflammatory hyperalgesia. Indeed, we showed that carrageenan-induced chronic hyperalgesia in GRK2+/− mice was associated with ongoing p38 activation in the spinal cord of GRK2+/− mice. Moreover, we demonstrated that intrathecal treatment with the p38 inhibitor SB239063 during the chronic phase alleviated hyperalgesia in GRK2+/− mice after intraplantar carrageenan. Finally, spinal cord TNF-α levels were increased at 7 d after intraplantar carrageenan in GRK2+/− mice, and blocking spinal TNF-α activity by intrathecal etanercept completely reversed ongoing hyperalgesia in GRK2+/− mice. Based on these data, we conclude that low GRK2 leads to increased activation of spinal cord p38 in response to peripheral inflammation, and that spinal cord p38 activity and concomitant TNF-α production is required for maintaining of chronic hyperalgesia in GRK2+/− mice.
P38 inhibition only slightly reduced the carrageen-induced increase in spinal cord TNF-α in GRK2+/− mice. We cannot conclude from our data whether this small reduction in total TNF-α in the spinal cord was sufficient to explain the reversal of ongoing hyperalgesia after p38 inhibition. One of the limiting factors is that we could only determine TNF-α in total spinal cord homogenate, which will reflect the total amount of intracellular and extracellular TNF-α. It is possible that the p38 inhibitor reduced TNF-α secretion (McMahon and Malcangio, 2009) without a major effect on TNF-α production at the time point examined. It is also possible that ongoing hyperalgesia in GRK2-deficient mice is maintained by TNF-α in concert with other downstream effectors and that either a small reduction in multiple mediators after p38 inhibition or the complete inhibition of TNF-α activity by etanercept treatment are both sufficient to block hyperalgesia. Alternatively, it is possible that although p38 activation is indispensable for induction of chronic hyperalgesia, microglial/macrophage GRK2 determines the chronicity of hyperalgesia via another yet unidentified pathway operative in these cells.
The clinical relevance of our findings that mice with reduced GRK2 develop prolonged inflammatory hyperalgesia is substantiated by our studies on GRK2 expression in chronic neuropathic pain. We show here that spinal microglia/macrophage GRK2 is unilaterally reduced in an SNT rat model of chronic pain. Importantly, in this model, mechanical allodynia is unilateral, and the reduction in microglial/macrophage GRK2 was observed only in the ipsilateral spinal cord, indicating that the ongoing unilateral mechanical allodynia is associated with a unilateral reduction of spinal microglial/macrophage GRK2.
We have shown previously that GRK2 levels in neurons of the dorsal horn of the spinal cord are also reduced in two different models of neuropathic pain (Kleibeuker et al., 2007, 2008a). In addition, we described a ∼50% reduction in GRK2 protein in peripheral blood mononuclear cells from patients with the chronic inflammatory diseases rheumatoid arthritis or multiple sclerosis (Lombardi et al., 2001; Vroon et al., 2005). These findings indicate that also in humans the intracellular level of GRK2 is downregulated in the context of chronic inflammatory activity. Whether GRK2 levels are also reduced in spinal microglia/macrophages of patients suffering from chronic pain remains to be determined.
In conclusion, we have presented evidence for a novel role of the kinase GRK2 in the neurobiology of hyperalgesia induced by local peripheral administration of inflammatory stimuli. Low GRK2 in peripheral sensory neurons enhanced severity of acute hyperalgesia. Low GRK2 in microglia/macrophages was sufficient for the transition of an acute to markedly prolonged hyperalgesia. We propose that changes in GRK2 levels contribute to severity and duration of pain in pathological conditions.
Footnotes
J.N.W. was supported by the Biotechnology and Biological Sciences Research Council and by World Class University Grant R31-2008-000-10103-0. We are greatly indebted to Mrs. J. Zijlstra for excellent technical assistance. We thank Dr. T. Hucho and Dr. C. Goswami (Max Planck Institute, Berlin, Germany) for providing the F11 cell line, Dr. N. Zhang (National Cancer Institute, Frederick, MA) for providing the TRPV1 and CCR1 construct, and Dr. T. Christoph (Grünenthal, Aachen, Germany) for help with learning the technique of the intrathecal injection.
The authors declare no competing financial interests.
References
- Abrahamsen B, Zhao J, Asante CO, Cendan CM, Marsh S, Martinez-Barbera JP, Nassar MA, Dickenson AH, Wood JN. The cell and molecular basis of mechanical, cold, and inflammatory pain. Science. 2008;321:702–705. doi: 10.1126/science.1156916. [DOI] [PubMed] [Google Scholar]
- Akopian AN, Sivilotti L, Wood JN. A tetrodotoxin-resistant voltage-gated sodium channel expressed by sensory neurons. Nature. 1996;379:257–262. doi: 10.1038/379257a0. [DOI] [PubMed] [Google Scholar]
- Alessandri-Haber N, Dina OA, Joseph EK, Reichling D, Levine JD. A transient receptor potential vanilloid 4-dependent mechanism of hyperalgesia is engaged by concerted action of inflammatory mediators. J Neurosci. 2006;26:3864–3874. doi: 10.1523/JNEUROSCI.5385-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrew D, Greenspan JD. Mechanical and heat sensitization of cutaneous nociceptors after peripheral inflammation in the rat. J Neurophysiol. 1999;82:2649–2656. doi: 10.1152/jn.1999.82.5.2649. [DOI] [PubMed] [Google Scholar]
- Cao FL, Liu MG, Hao J, Li Z, Lu ZM, Chen J. Different roles of spinal p38 and c-Jun N-terminal kinase pathways in bee venom-induced multiple pain-related behaviors. Neurosci Lett. 2007;427:50–54. doi: 10.1016/j.neulet.2007.09.005. [DOI] [PubMed] [Google Scholar]
- Clausen BE, Burkhardt C, Reith W, Renkawitz R, Forster I. Conditional gene targeting in macrophages and granulocytes using LysMcre mice. Transgenic Res. 1999;8:265–277. doi: 10.1023/a:1008942828960. [DOI] [PubMed] [Google Scholar]
- Cunha TM, Verri WA, Jr, Silva JS, Poole S, Cunha FQ, Ferreira SH. A cascade of cytokines mediates mechanical inflammatory hypernociception in mice. Proc Natl Acad Sci U S A. 2005;102:1755–1760. doi: 10.1073/pnas.0409225102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunha TM, Verri WA, Jr, Schivo IR, Napimoga MH, Parada CA, Poole S, Teixeira MM, Ferreira SH, Cunha FQ. Crucial role of neutrophils in the development of mechanical inflammatory hypernociception. J Leukoc Biol. 2008;83:824–832. doi: 10.1189/jlb.0907654. [DOI] [PubMed] [Google Scholar]
- Deleo JA, Yezierski RP. The role of neuroinflammation and neuroimmune activation in persistent pain. Pain. 2001;90:1–6. doi: 10.1016/s0304-3959(00)00490-5. [DOI] [PubMed] [Google Scholar]
- Eijkelkamp N, Heijnen CJ, Lucas A, Premont RT, Elsenbruch S, Schedlowski M, Kavelaars A. G protein-coupled receptor kinase 6 controls chronicity and severity of dextran sodium sulphate-induced colitis in mice. Gut. 2007;56:847–854. doi: 10.1136/gut.2006.107094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goswami C, Hucho T. TRPV1 expression-dependent initiation and regulation of filopodia. J Neurochem. 2007;103:1319–1333. doi: 10.1111/j.1471-4159.2007.04846.x. [DOI] [PubMed] [Google Scholar]
- Hargreaves K, Dubner R, Brown F, Flores C, Joris J. A new and sensitive method for measuring thermal nociception in cutaneous hyperalgesia. Pain. 1988;32:77–88. doi: 10.1016/0304-3959(88)90026-7. [DOI] [PubMed] [Google Scholar]
- Hucho T, Levine JD. Signaling pathways in sensitization: toward a nociceptor cell biology. Neuron. 2007;55:365–376. doi: 10.1016/j.neuron.2007.07.008. [DOI] [PubMed] [Google Scholar]
- Hylden JL, Wilcox GL. Intrathecal morphine in mice: a new technique. Eur J Pharmacol. 1980;67:313–316. doi: 10.1016/0014-2999(80)90515-4. [DOI] [PubMed] [Google Scholar]
- Jaber M, Koch WJ, Rockman H, Smith B, Bond RA, Sulik KK, Ross J, Jr, Lefkowitz RJ, Caron MG, Giros B. Essential role of beta-adrenergic receptor kinase 1 in cardiac development and function. Proc Natl Acad Sci U S A. 1996;93:12974–12979. doi: 10.1073/pnas.93.23.12974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji RR, Suter MR. p38 MAPK, microglial signaling, and neuropathic pain. Mol Pain. 2007;3:33–42. doi: 10.1186/1744-8069-3-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji RR, Gereau RW, Malcangio M, Strichartz GR. MAP kinase and pain. Brain Res Rev. 2009;60:135–148. doi: 10.1016/j.brainresrev.2008.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleibeuker W, Ledeboer A, Eijkelkamp N, Watkins LR, Maier SF, Zijlstra J, Heijnen CJ, Kavelaars A. A role for G protein-coupled receptor kinase 2 in mechanical allodynia. Eur J Neurosci. 2007;25:1696–1704. doi: 10.1111/j.1460-9568.2007.05423.x. [DOI] [PubMed] [Google Scholar]
- Kleibeuker W, Gabay E, Kavelaars A, Zijlstra J, Wolf G, Ziv N, Yirmiya R, Shavit Y, Tal M, Heijnen CJ. IL-1 beta signaling is required for mechanical allodynia induced by nerve injury and for the ensuing reduction in spinal cord neuronal GRK2. Brain Behav Immun. 2008a;22:200–208. doi: 10.1016/j.bbi.2007.07.009. [DOI] [PubMed] [Google Scholar]
- Kleibeuker W, Jurado-Pueyo M, Murga C, Eijkelkamp N, Mayor F, Jr, Heijnen CJ, Kavelaars A. Physiological changes in GRK2 regulate CCL2-induced signaling to ERK1/2 and Akt but not to MEK1/2 and calcium. J Neurochem. 2008b;104:979–992. doi: 10.1111/j.1471-4159.2007.05023.x. [DOI] [PubMed] [Google Scholar]
- Lombardi MS, Kavelaars A, Schedlowski M, Bijlsma JW, Okihara KL, Van de Pol M, Ochsmann S, Pawlak C, Schmidt RE, Heijnen CJ. Decreased expression and activity of G-protein-coupled receptor kinases in peripheral blood mononuclear cells of patients with rheumatoid arthritis. FASEB J. 1999;13:715–725. doi: 10.1096/fasebj.13.6.715. [DOI] [PubMed] [Google Scholar]
- Lombardi MS, Kavelaars A, Cobelens PM, Schmidt RE, Schedlowski M, Heijnen CJ. Adjuvant arthritis induces down-regulation of G protein-coupled receptor kinases in the immune system. J Immunol. 2001;166:1635–1640. doi: 10.4049/jimmunol.166.3.1635. [DOI] [PubMed] [Google Scholar]
- Marchand F, Perretti M, McMahon SB. Role of the immune system in chronic pain. Nat Rev Neurosci. 2005;6:521–532. doi: 10.1038/nrn1700. [DOI] [PubMed] [Google Scholar]
- Matkovich SJ, Diwan A, Klanke JL, Hammer DJ, Marreez Y, Odley AM, Brunskill EW, Koch WJ, Schwartz RJ, Dorn GW. Cardiac-specific ablation of G-protein receptor kinase 2 redefines its roles in heart development and beta-adrenergic signaling. Circ Res. 2006;99:996–1003. doi: 10.1161/01.RES.0000247932.71270.2c. [DOI] [PubMed] [Google Scholar]
- McMahon SB, Malcangio M. Current challenges in glia-pain biology. Neuron. 2009;64:46–54. doi: 10.1016/j.neuron.2009.09.033. [DOI] [PubMed] [Google Scholar]
- Milligan ED, Watkins LR. Pathological and protective roles of glia in chronic pain. Nat Rev Neurosci. 2009;10:23–36. doi: 10.1038/nrn2533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moalem G, Tracey DJ. Immune and inflammatory mechanisms in neuropathic pain. Brain Res Rev. 2006;51:240–264. doi: 10.1016/j.brainresrev.2005.11.004. [DOI] [PubMed] [Google Scholar]
- Nijboer CH, Kavelaars A, Vroon A, Groenendaal F, van Bel F, Heijnen CJ. Low endogenous G-protein-coupled receptor kinase 2 sensitizes the immature brain to hypoxia-ischemia-induced gray and white matter damage. J Neurosci. 2008;28:3324–3332. doi: 10.1523/JNEUROSCI.4769-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh SB, Tran PB, Gillard SE, Hurley RW, Hammond DL, Miller RJ. Chemokines and glycoprotein120 produce pain hypersensitivity by directly exciting primary nociceptive neurons. J Neurosci. 2001;21:5027–5035. doi: 10.1523/JNEUROSCI.21-14-05027.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oppermann M, Mack M, Proudfoot AE, Olbrich H. Differential effects of CC chemokines on CC chemokine receptor 5 (CCR5) phosphorylation and identification of phosphorylation sites on the CCR5 carboxyl terminus. J Biol Chem. 1999;274:8875–8885. doi: 10.1074/jbc.274.13.8875. [DOI] [PubMed] [Google Scholar]
- Peregrin S, Jurado-Pueyo M, Campos PM, Sanz-Moreno V, Ruiz-Gomez A, Crespo P, Mayor F, Jr, Murga C. Phosphorylation of p38 by GRK2 at the docking groove unveils a novel mechanism for inactivating p38MAPK. Curr Biol. 2006;16:2042–2047. doi: 10.1016/j.cub.2006.08.083. [DOI] [PubMed] [Google Scholar]
- Raghavendra V, Tanga F, Deleo JA. Inhibition of microglial activation attenuates the development but not existing hypersensitivity in a rat model of neuropathy. J Pharmacol Exp Ther. 2003;306:624–630. doi: 10.1124/jpet.103.052407. [DOI] [PubMed] [Google Scholar]
- Ramos-Ruiz R, Penela P, Penn RB, Mayor F., Jr Analysis of the human G protein-coupled receptor kinase 2 (GRK2) gene promoter: regulation by signal transduction systems in aortic smooth muscle cells. Circulation. 2000;101:2083–2089. doi: 10.1161/01.cir.101.17.2083. [DOI] [PubMed] [Google Scholar]
- Reiter E, Lefkowitz RJ. GRKs and beta-arrestins: roles in receptor silencing, trafficking and signaling. Trends Endocrinol Metab. 2006;17:159–165. doi: 10.1016/j.tem.2006.03.008. [DOI] [PubMed] [Google Scholar]
- Ribas C, Penela P, Murga C, Salcedo A, Garcia-Hoz C, Jurado-Pueyo M, Aymerich I, Mayor F., Jr The G protein-coupled receptor kinase (GRK) interactome: role of GRKs in GPCR regulation and signaling. Biochim Biophys Acta. 2007;1768:913–922. doi: 10.1016/j.bbamem.2006.09.019. [DOI] [PubMed] [Google Scholar]
- Rittner HL, Mousa SA, Labuz D, Beschmann K, Schafer M, Stein C, Brack A. Selective local PMN recruitment by CXCL1 or CXCL2/3 injection does not cause inflammatory pain. J Leukoc Biol. 2006;79:1022–1032. doi: 10.1189/jlb.0805452. [DOI] [PubMed] [Google Scholar]
- Scholz J, Woolf CJ. The neuropathic pain triad: neurons, immune cells and glia. Nat Neurosci. 2007;10:1361–1368. doi: 10.1038/nn1992. [DOI] [PubMed] [Google Scholar]
- Stirling LC, Forlani G, Baker MD, Wood JN, Matthews EA, Dickenson AH, Nassar MA. Nociceptor-specific gene deletion using heterozygous NaV1.8-Cre recombinase mice. Pain. 2005;113:27–36. doi: 10.1016/j.pain.2004.08.015. [DOI] [PubMed] [Google Scholar]
- Svensson CI, Schafers M, Jones TL, Powell H, Sorkin LS. Spinal blockade of TNF blocks spinal nerve ligation-induced increases in spinal P-p38. Neurosci Lett. 2005;379:209–213. doi: 10.1016/j.neulet.2004.12.064. [DOI] [PubMed] [Google Scholar]
- Tikka T, Fiebich BL, Goldsteins G, Keinanen R, Koistinaho J. Minocycline, a tetracycline derivative, is neuroprotective against excitotoxicity by inhibiting activation and proliferation of microglia. J Neurosci. 2001;21:2580–2588. doi: 10.1523/JNEUROSCI.21-08-02580.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vroon A, Lombardi MS, Kavelaars A, Heijnen CJ. Changes in the G-protein-coupled receptor desensitization machinery during relapsing-progressive experimental allergic encephalomyelitis. J Neuroimmunol. 2003;137:79–86. doi: 10.1016/s0165-5728(03)00050-x. [DOI] [PubMed] [Google Scholar]
- Vroon A, Heijnen CJ, Lombardi MS, Cobelens PM, Mayor F, Jr, Caron MG, Kavelaars A. Reduced GRK2 level in T cells potentiates chemotaxis and signaling in response to CCL4. J Leukoc Biol. 2004;75:901–909. doi: 10.1189/jlb.0403136. [DOI] [PubMed] [Google Scholar]
- Vroon A, Kavelaars A, Limmroth V, Lombardi MS, Goebel MU, Van Dam AM, Caron MG, Schedlowski M, Heijnen CJ. G protein-coupled receptor kinase 2 in multiple sclerosis and experimental autoimmune encephalomyelitis. J Immunol. 2005;174:4400–4406. doi: 10.4049/jimmunol.174.7.4400. [DOI] [PubMed] [Google Scholar]
- Watkins LR, Maier SF. Glia: a novel drug discovery target for clinical pain. Nat Rev Drug Discov. 2003;2:973–985. doi: 10.1038/nrd1251. [DOI] [PubMed] [Google Scholar]
- Watkins LR, Milligan ED, Maier SF. Glial proinflammatory cytokines mediate exaggerated pain states: implications for clinical pain. Adv Exp Med Biol. 2003;521:1–21. [PubMed] [Google Scholar]
- Zhang J, Ferguson SS, Barak LS, Aber MJ, Giros B, Lefkowitz RJ, Caron MG. Molecular mechanisms of G protein-coupled receptor signaling: role of G protein-coupled receptor kinases and arrestins in receptor desensitization and resensitization. Receptors Channels. 1997;5:193–199. [PubMed] [Google Scholar]
- Zhang N, Inan S, Cowan A, Sun R, Wang JM, Rogers TJ, Caterina M, Oppenheim JJ. A proinflammatory chemokine, CCL3, sensitizes the heat- and capsaicin-gated ion channel TRPV1. Proc Natl Acad Sci U S A. 2005;102:4536–4541. doi: 10.1073/pnas.0406030102. [DOI] [PMC free article] [PubMed] [Google Scholar]