

Abstract
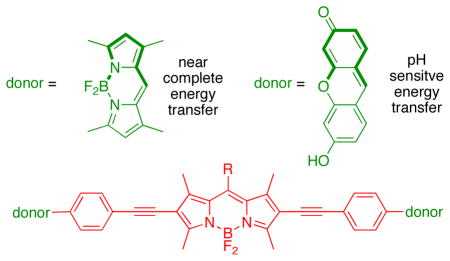
Photophysical data and orbital energy levels (from electrochemistry) were compared for molecules with the same BODIPY acceptor part (red) and perpendicularly oriented xanthene or BODIPY donor fragments (green). Transfer of energy, hence the photophysical properties of the cassettes, including the pH dependant fluorescence in the xanthene containing molecules, correlates with the relative energies of the frontier orbitals in these systems.
Intracellular sensing of protons is often achieved via sensors that switch off completely at certain pH values, but probes of this type are not easy to locate inside cells in their “off-state”. A communication from these laboratories (J. Am. Chem. Soc., 2009, 131, 1642 – 3) described how the energy transfer cassette 1 could be used for intracellular imaging of pH. This probe is fluorescent whatever the pH, but its exact photophysical properties are governed by the protonation-states of the xanthene donors. This work was undertaken to further investigate correlations between structure, photophysical properties, and pH for energy transfer cassettes. To achieve this, three other cassettes 2 – 4 were prepared another one containing pH-sensitive xanthene donors (2), and two “control cassettes” that each have two BODIPY-based donors (3 and 4). Both the cassettes 1 and 2 with xanthene-based donors fluoresce red under slightly acidic conditions (pH < ca 6), and green when the medium is more basic (> ca 7), whereas the corresponding cassettes with BODIPY donors give almost complete energy transfer regardless of pH. The cassettes that have BODIPY donors by contrast, show no significant fluorescence from the donor parts, but the overall quantum yields of the cassettes when excited at the donor (observation of acceptor fluorescence) are high (ca 0.6 and 0.9). Electrochemical measurements were performed to elucidate orbital energy level differences between the pH-fluorescence profiles of cassettes with xanthene donors, relative to the two with BODIPY donors. These studies confirm energy transfer in the cassettes is dramatically altered by analytes that perturb relative orbital levels. Energy transfer cassettes with distinct fluorescent donor and acceptor units provide a new, and potentially useful, approach to sensors for biomedical applications.
Keywords: pH probe, fluorescent sensors, BODIPY, fluorescein, energy transfer
Introduction
Fluorescent sensors are widely used for detection of protons and metals in several applications,1,2 especially intracellular imaging.3 Types of indicators may be divided into three: (i) ones that are insignificantly fluorescent in the absence of analyte, but are much more emissive when it is present; (ii) the inverse, where fluorescence of the probe is quenched by the analyte; and, (iii) sensors which have observable spectroscopic differences when the analyte is present compared to when it is absent. The third type of sensor is “always on”; this is a significant advantage because it is clear that the probe is present even if the analyte is not (Figure 1).
Figure 1.


Fluorescent sensors may be activated (i) or quenched (ii) by analytes; ones that are “always on” (iii) but change wavelength of fluorescence emissions on binding.
Intracellular pH (pHi) is a fundamental property that correlates with many events in cell biology. To measure this, researchers tend to rely on subtle changes in sensors that are “always on”;4–12 for instance, they observe emission intensities as a function of excitation wavelength.12–15 However, these sensors do not change emission wavelength maxima as the pH is varied; if they did, they would be far easier to use. That is why we were excited to realize cassette 1 is always fluorescent at pH ranges around the physiological region, but with emission maxima that varying over a very significant spectral region, ca 530 and 600 nm. Application of probe 1 to monitoring intracellular pH has been communicated.16 It fluoresces red at pH values less than about 6.5, and green at pH values above about 7 (Figure 2). Probe 1 compared favorably with commercial pH sensors such as carboxySNARF-1 (Invitrogen), insofar as its pH response range is complementary, and its quantum yields are higher. Further, probe 1 exhibits a greater wavelength difference (~80 nm) between the two ratiometric emission peaks than carboxySNARF-1. It also has acid functionality to conjugate to proteins while carboxySNARF-1 does not. When BSA labeled with probe 1 was imported inside COS 7 cells and the pH values of the protein and in the cytosol and endosomes were quantified.
Figure 2.


Three forms of the energy transfer cassette that was used as a pH probe for intracellular imaging.
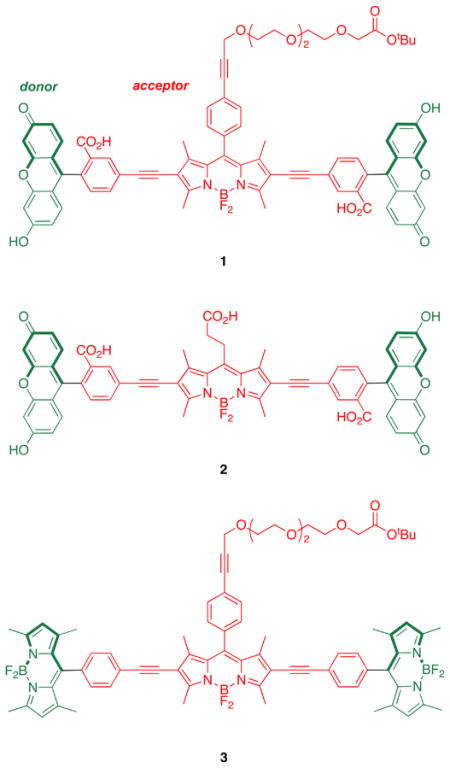
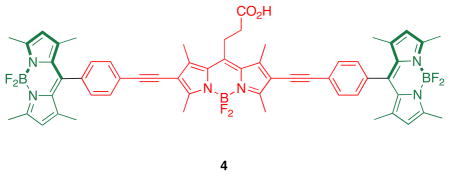
Research described in this paper was undertaken to explore how the photophysical properties of energy transfer cassettes correlate with their structures. Nagano has published on the photoinduced electron transfer (PeT) between meso-phenyl groups and BODIPY cores.17 These PeT processes can negatively impact the fluorescence quantum yields of BODIPYs and we therefore set out to prepare cassette 2, a modification of 1 wherein the meso-phenyl functionality is absent. For comparison, cassettes 3 and 4 were also targeted; these maintain the basic design, two donors perpendicularly aligned to the same central BODIPY18–21-acceptor. However, they are fundamentally different insofar as the donors are also BODIPY-based, and there was no obvious reason to suspect transfer of energy from the donors in these molecules to the acceptors should be pH dependant. These were envisaged as reference compounds for comparison of the extent of energy transfer in these systems.
In outline, the research described here investigates: (i) syntheses of the cassettes; (ii) effects of changing of the meso-substituent on the central BODIPY {acceptor} fragment represented by the difference between cassettes 1 and 2 and between 3 and 4; (iii) changes of donor fragment from xanthene to BODIPY represented by the structural differences between 1 or 2, and 3 or 4. Comparison of the cassettes is made on the basis of photophysical and electrochemical data measured at different pH levels. The latter measurements are related to orbital levels in the donor and acceptor fragments.
Results and Discussion
Syntheses of the Cassettes 1 – 4
Two key diiodinated BODIPY intermediates were prepared to make the cassettes featured in this paper. The first, compound 6 (Scheme 1a), was designed to have a triethylene glycol linker that would somewhat separate the dye from the protein as well as increase solubility in polar solvents. Conditions for the diiodination reactions shown in Scheme 1 were based on work from Nagano et al.22 The second, compound 8 (Scheme 1b), was formed via a route that is analogous to one used for a homolog formed from glutaric anhydride.23 Synthons 6 and 8 lead to cassettes with different meso substituents: aryl and alkyl functionalities.
Scheme 1.


Syntheses of pivotal diiodinated synthons: a BODIPY 6; and, b BODIPY 8.
Sonogashira reactions24,25 were used to assemble the cassettes from the acceptor components 6 and 8, and the fluorescein-based and BODIPY-based donor components C26,27 and D28 (Scheme 2). The diacetate intermediate 9 is of some importance because this compound has been shown to be cell permeable, and hydrolyzes in the cytosol to give green fluorescence.16 The fluorescein-based cassettes 1 and 2 are soluble in lower alcohol solvents giving pink solutions.
Scheme 2.


Syntheses of cassettes: a, 1; and, b, 3.
Cassette 2 (Scheme 3) was difficult to purify. Flash chromatography did not give pure material, but the compound was isolated via preparative reverse phase HPLC in 4 % yield. Both cassettes 3 and 4 are soluble in lipophilic solvents like CH2Cl2, and give strongly colored pink or red solutions.
Scheme 3.

Syntheses of cassettes: a, 2; and, b, 4.
The methyl ester of cassette 4 was crystallized for single crystal X-ray diffraction studies (Figure 3). They show the BODIPY donor fragments resting perpendicular to the acceptor part. In part, it is this molecular twist that differentiates cassettes from planar dyes consisting of a single conjugated chromophore. Interestingly, the molecule appears to “sag” around the central BODIPY fragment, because the alkyne parts are not exactly in the same plane; an ideal linear arrangement would give a 180° angle, but the observed angle was 168.2 degrees. This parameter may have some relevance because if the angle were 180° and rigid then the transition dipoles of the BODIPY acceptor and the two donor fragments (which are aligned with their long axes)29 would be exactly perpendicular in any conformation about the alkyne. In that orientation there can be no dipole-dipole coupling hence fluorescence resonance energy transfer (FRET) could not occur. The fact that the molecule is not perfectly linear means that FRET cannot be completely excluded because rotation about the alkyne bond could place the BODIPY donors in conformations in which weak dipole-dipole coupling could occur. However, the “sag-angle” is small, and conformations that allow dipole-dipole coupling also take the phenyl group out of conjugation with the rest of the acceptor; consequently, energy transfer via this mechanism30 is unfavorable.
Figure 3.


Single crystal X-ray structure of 10.
An important set of new acceptor fragments 11 – 14 and known BODIPY E31 and xanthene F32 reference compounds were also generated for this study. Photophysical and electrochemical properties in cassettes tend to be accurately represented by the individual donor and acceptor fragments.33 Consequently, electrochemical studies were performed on these constituents, thus avoiding the need for destructive experiments (electrochemistry) on the valuable cassette samples. Syntheses of the new materials are outlined in the supporting material.
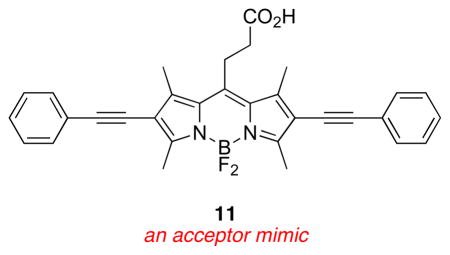
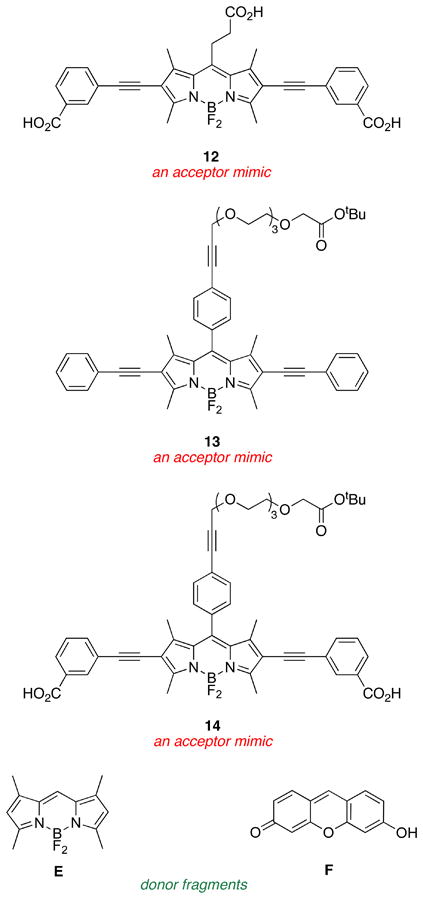
Photophysical Properties
Salient photophysical properties of the cassettes are shown in Table 1. The acceptor fragment is formed by the 2,6-(alkyne-aryl) substituents because these impose a dramatic red-shift on the absorbance and fluorescence properties of the BODIPY core.25 The fluorescein and BODIPY donor parts exhibit characteristically large molar absorptivities, and absorb/fluoresce at wavelengths that are characteristic of the free dye fragments (see Table 2 below).
Table 1.
Photophysical properties of 1 – 4 in 1:1 ethanol:CH2Cl2.
| cmpd | basea | absorptionb |
fluorescencec |
φacceptor excited at acceptord | φacceptor excited at donore,j | ETE (%)f | φdonorg | ||
|---|---|---|---|---|---|---|---|---|---|
| λmax donor (nm)/ε × 10−4 | λmax acceptor (nm)/ε × 10−4 | λmax donor (nm) | λmax acceptor (nm) | ||||||
| 1 | − | 505/5.6 | 575/3.5 | 521 | 600 | 0.30i | 0.15 | 51 | 0.05 |
| 1 | + | 505/11 | 578/3.0 | 529 | - | 0.01i | - | < 5 | 0.09 |
| 2 | − | 506/3.7 | 560/1.9 | 528 | 579 | 0.24j | 0.09 | 38 | 0.07 |
| 2 | + | 505/13 | 561/2.8 | 529 | - | 0.01j | - | < 5 | 0.08 |
| 3 | − | 502/14 | 566/6.6 | 513 | 606 | 0.62i | 0.58 | 93 | -h |
| 3 | + | 502/14 | 566/6.6 | 516 | 604 | 0.66i | 0.60 | 92 | -h |
| 4 | − | 502/11 | 561/4.7 | 521 | 588 | 0.84j | 0.79 | 94 | -h |
| 4 | + | 502/11 | 560/5.2 | 515 | 588 | 0.93j | 0.85 | 91 | -h |
with nBu4NOH at a concentration of 8 × 10−5 M.
At 1 × 10−5 M.
At 1 × 10−6 M.
Quantum yield of acceptor when excited at the acceptor.
Quantum yield of acceptor while excited at the donor.
Energy transfer efficiency calculated with the quantum yield of the acceptor with excitation at donor divided by that with excitation at the acceptor.
Fluorescein (φ = 0.92 in 0.1 M NaOH)34 was used as a standard.
Donors in these cassettes show no significant fluorescence emission.
Rhodamine 101 (φ = 1.00 in EtOH)35 was used as a standard.
Rhodamine B (φ = 0.97 in EtOH)34 was used as a standard.
Table 2.
Photophysical properties of reference compounds in 1:1 ethanol/CH2Cl2
| cmpd | baseb | λabs (nm)/ε × 10−4 | λem (nm) | fwhm (nm) | φa |
|---|---|---|---|---|---|
| 11 | − | 560/2.6 | 587 | 41 | 0.72 |
| 11 | + | 559/2.6 | 587 | 41 | 0.71 |
| 12 | − | 565/0.9 | 593 | 46 | 0.46 |
| 12 | + | 563/1.0 | 592 | 43 | 0.61 |
| 13 | − | 574/3.2 | 606 | 44 | 0.52 |
| 13 | + | 574/3.2 | 605 | 53 | 0.54 |
| 14 | − | 573/2.3 | 612 | 53 | 0.42 |
| 14 | + | 578/2.5 | 613 | 52 | 0.44 |
| E | − | 506/8.7 | 511 | 16 | 0.98 |
| E | + | 506/8.7 | 511 | 17 | 0.90 |
| F | − | 508/7.4 | 517 | 26 | 0.76 |
| F | + | 508/11 | 517 | 25 | 0.95 |
Through bond energy transfer cassettes are usually designed to absorb light at the donor excitation wavelength, relay it to the acceptor part, then emit fluorescence from there. The term “energy transfer efficiency” (ETE %) quantifies this, it is defined as follows:
ETE % is a measure of the quantum yield of the cassette when irradiated at the donor. It reflects the extent of energy transfer including the negative effects of non-radiative loss in the transfer process. The product of the molar absorptivity of the donor in the cassette and the ETE give a measure of the brightness of the acceptor in the system.
Values of the ETE % for the cassettes 1 – 4 are shown in Table 1. Several important observations are clear. First, the fluorescein-based cassettes 1 and 2 have moderate ETE values in the absence of base, but not when Bu4NOH is added. Second, the BODIPY-based cassettes 3 and 4 have excellent ETE values, and these are not influenced by base.
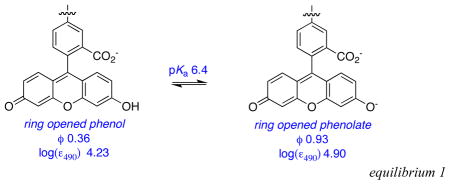
Effects of base on the cassettes are probably best visualized from their absorbance and fluorescence spectra (Figures S1 {supporting information} and 4). Addition of nBu4NOH to cassettes 1 and 2 increases the absorption corresponding to the fluorescein component relative to that from the BODIPY acceptor part. This is logical because addition of base forces the fluorescein donor into its ring-opened phenolate carboxylate form. Absorbance spectra of cassettes 3 and 4 are almost completely insensitive to base, as would be predicted since the electronic spectra of BODIPY dyes are not significantly affected by pH.
In the absence of base, excitation of the fluorescein donor of cassettes 1 and 2 leads to significant fluorescence from the BODIPY acceptor. However, no significant fluorescence is observed from the BODIPY part when base is added to the same solutions (Figure 4a and b). Conversely, addition of base has no significant effect on the extent of energy transfer for the cassettes 3 and 4 that have BODIPY donors. Thus in 1:1 ethanol/CH2Cl2 the fluorescein donor parts of cassettes 1 and 2 are, at least partially, protonated, and energy transfer to the BODIPY acceptor occurs. Energy transfer is quenched when the fluorescein donors are completely deprotonated. Cassettes 3 and 4 have BODIPY, not fluorescein, donor parts, but they are otherwise identical to 1 and 2.
Figure 4.


a – d Fluorescence spectra of cassettes 1 – 4 (1 × 10−6 M in 1:1 ethanol/CH2Cl2); throughout, spectra recorded without added base are shown in blue, and with nBu4NOH (concentration of 1 × 10−4 M) are shown in red.
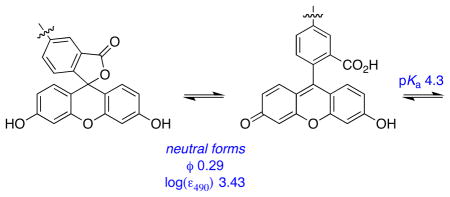
Table 2 shows photophysical properties for the reference compounds 11 – 14, E and F. None of the building blocks that were assembled to give cassettes 1 – 4 have fluorescence characteristics that are significantly changed by added base. The largest change in quantum yield is seen for the dicarboxylate 13 and xanthene F: for these compounds a 25 % increase in the presence of base. There are no appreciable shifts in λabs, λem, or even the peak width values when these fragments are compared without and with base.
None of the reference compounds can undergo changes like that shown in equilibrium 1, but this has been comprehensively studied for fluorescein in aqueous media.14 These data show that the quantum yield of fluorescein is highest in its dianion state. However, Table 1 shows that the quantum yield of the donor part in the fluorescein-based cassettes 1 and 2 are less than 0.1, with or without base. Without base there is significant energy transfer to the acceptor, so some quantum yield reduction is anticipated. With base, however, less of the energy transferred between the donor and acceptor is emitted as acceptor fluorescence. Further, the xanthene quantum yields in the cassettes are much less than for fluorescein in any of the accessible protonation states, or for the xanthene F. Thus, integration of the xanthene donors in cassettes 1 and 2 reduces their quantum yields relative to the parent fragments.
Electrochemical Studies
Oxidation and reduction potentials were measured for the reference fragments E, F, 13 – 14, and for cassette 3 relative to the ferrocene/ferrocinium couple. The electrochemical measurements provide an approximation of the band gap magnitude. The HOMO and LUMO energies were estimated based on the onset of REDOX events as described by Reynolds et al.36 BODIPY 13 shows a reversible reduction wave, while for 14 and E the wave is quasi-reversible for the first reduction events. For F and FNa the first reduction wave is irreversible. The oxidation events for all compounds are irreversible. In a method similar to the one used by Reynolds et al.36 the ferrocene/ferrocenium couple in Volts is estimated to an orbital level of 5.15 eV and 5.16 eV relative to vacuum in DMF and CH2Cl2, respectively.37,38 Thus energy levels of HOMO and LUMOs can be pegged relative to this reference point. The same data was acquired for compounds 11 and 12 (see supporting information).
Figure 5a plots HOMO and LUMO energy levels for the reference BODIPY E and the acceptor mimic 13 that represent cassette 3 (BODIPY donor and acceptor). The LUMO levels of the donor and acceptor parts are close in energy with the latter 0.35 eV lower. This corresponds to the system for which high ETE was observed (93 %), and for which no pH dependence was observed or expected. Figure 5b and c shows data corresponding to the pH dependant cassette 1. Under conditions favoring protonation of the phenolic-O atoms the two LUMO levels are also close in energy (0.1 eV), and again, significant ETE was observed. However, for the same cassette but under conditions wherein the phenol groups would be completely deprotonated, poor energy transfer corresponds with a large energy gap between the donor and acceptor LUMO levels.
Figure 5.

a HOMO and LUMO levels of the reference compounds representing cassettes: a, 3; b, 1 (under neutral conditions); and c, 1 (basic conditions).
Conclusions
Rapid and efficient energy transfer may be possible when a fluorescent donor is joined to an acceptor in such a way that the two fragments would be electronically conjugated if they became planar, but are sterically prevented from doing so.39–42 The fact that they cannot easily achieve planarity means that the absorption spectra of the cassette resembles the sum of the donor and acceptor parts. However, the donor part will not fluoresce when it is excited in an efficient energy transfer cassette; instead the energy will be rapidly transferred to the acceptor that will then fluoresce (Figure 6).43–55
Figure 6.

The concepts of through bond energy transfer cassettes.
Previous studies from these laboratories provided evidence that energy transfer mechanisms besides FRET can be dominant in cassettes like the ones described in this paper. Specifically, energy transfer rates between anthracene (donor) and BODIPY (acceptor) fragments were much faster than would be expected from a FRET mechanism.29,43,45 Further, some of those cassettes have the donor and acceptor parts oriented in ways that should largely exclude the possibility of FRET, yet fast transfer rates were observed. Others in this field had documented similar effects for energy transfer cassettes.33,56–58
In this work, we do not profess to know the mechanisms by which energy is transferred between the donor and the acceptor. It seems highly probable that all possible mechanisms compete; these include dipole-dipole coupling (FRET), electron transfer, and processes in which relaxation of energy from the donor excited state leads to excitation of the acceptor via through-bond mechanisms that rely on orbital overlap. Moreover, the proportions of each energy transfer mechanism operative will be influenced by the protonation state of the donor, at least for the xanthene dyes. Comparisons with the all-BODIPY donor-acceptor systems 3 and 4 reveal that the relative orbital energy levels between the donor and acceptor fragments in these cassettes are comparable to those in the pH-sensitive systems 1 and 2 in protonation states where efficient energy transfer is observed. Moreover, the meso-aromatic substituent of 1 and 3 has no significant effect on the photophysical properties of these cassettes; this can be inferred from the data collected for 2 and 4 which have only aliphatic groups in that position.
Communication between the donor and the acceptor in these systems, by whatever mechanisms, does appear to be impacted by the relative energies of the frontier orbitals involved. Specifically, maximal energy transfer was observed when the electrochemical studies showed the donor and acceptor LUMO levels were close, but with the latter being lowest. However, ETE was least in the series when there was an appreciable gap between the LUMO energy levels. This may be a reflection on a larger divergence of the LUMO wavefunction distributions in the latter case.
The work described here may represent the beginning of a new paradigm in which electronically coupled dye pairs can be used to sense analytes in biomedical applications. There is considerable scope for molecular modifications in this series because the donor, acceptor, and linker fragments59 could all be modified to give sensors.
Experimental Section
(1)
16 A mixture of 6 (65 mg, 0.074 mmol), diacetylfluorescein alkyne C26 (82 mg, 0.186mmol), Et3N (0.11 mL, 0.74 mmol), Pd(PPh3)4 (8 mg, 0.007 mmol), CuI (3 mg, 0.014 mmol) were dissolved in THF (2 mL). After the solution was degassed three times via the freeze-thawed method, the mixture was heated up to 45 °C for 16 h. The reaction solvent was removed under reduced pressure and the crude product was purified by flash column eluting with 50% hexane:ethyl acetate to give 9 as a light yellow solid (80 mg, 72%). 1H NMR (500 MHz, CDCl3), δ 8.08 (m, 2H), 7.73 (dd, J =8.0, 1.5 Hz, 2H), 7.65(d, J = 8.0 Hz, 2H), 7.29 (d, J = 8.5 Hz, 2H), 7.15 (d, J = 8.2 Hz, 2H), 7.10 (d, J = 2.0 Hz, 4H), 6.83 (bs, 4H), 6.83 (d, J = 2.0 Hz, 4H), 4.48 (s, 2H), 4.02 (s, 2H), 3.80–3.82 (m, 2H), 3.70–3.75 (m, 10H), 2.75 (s, 6H), 2.32 (s, 12H), 1.58 (s, 6H), 1.47(s, 9H). 13C NMR (125 MHz, CDCl3), δ 169.6, 168.8, 168.2, 159.1, 152.1, 151.8, 151.5, 144.6, 142.1, 137.9, 134.1, 132.8, 131.1, 128.9, 127.9, 127.7, 126.6, 125.8, 124.3, 124.2, 117.8, 116.0, 115.6, 110.5, 94.7, 87.2, 85.3, 84.1, 81.8, 81.5, 70.7, 70.6 (2 C), 70.5, 69.5, 69.0, 59.2, 28.1, 21.1, 13.8, 13.7 MALDI MS calcd for C86H71BF2N2NaO20+ (M+Na)+ 1523.46, found 1523.26. TLC (1:1 EtOAc/Hexane), Rf = 0.20.
To 9 (12 mg, 0.01 mmol) in 5 mL 2:1 methanol/THF in was added Na2CO3 (3.5 mg, 0.03 mmol). The mixture was stirred for 3 h at 25 °C under N2. The reaction was quenched by adding aqueous HCl (0.1M, 10 mL) and the product was extracted out of the solution with 75% CH2Cl2:iPrOH (5 mL × 3). The organic layers were washed with brine solution (10 mL) and dried with magnesium sulfate. 1 was isolated as a purple solid (10 mg, 99 %). 1H NMR (500 MHz, 75% CD3OD:CDCl3), δ 8.00 (s, 2H), 7.74 (dd, J = 8.0 Hz, 1.5 Hz, 2H), 7.65 (d, J = 7.5Hz, 2H), 7.33 (d, J = 8.5 Hz, 2H), 7.15 (d, J = 8.0 Hz, 2H), 6.67 (d, J = 2.5 Hz 4H), 6.59 (d, J = 8.0 Hz, 4H), 6.51 (dd, J = 9.0 Hz, 2.5 Hz, 4H), 4.47 (s, 2H), 4.00 (s, 2H), 3.79–3.81(m, 2H), 3.71–3.73(m, 2H), 3.66–3.69 (m, 8H), 2.71 (s, 6H), 1.58 (s, 6H), 1.44(s, 9H), 13C NMR (125 MHz, 75% CD3OD:CDCl3), δ 170.9, 170.1, 169.8, 159.5, 153.5, 145.2, 142.9, 138.3, 134.8, 133.4, 131.7, 131.2, 129.6, 128.7, 128.5, 128.0, 126.1, 125.9, 124.9, 116.3, 110.3, 108.2, 103.3, 95.6, 87.4, 86.0, 84.1, 82.7, 71.0, 70.9 (2 C), 70.8 (2 C), 69.7, 69.3, 59.5, 30.2, 28.3, 14.0. MS (MALDI) calcd for C78H63BF2N2O16+ (M+H)+ 1333.42, found 1333.44.
(2)
A mixture of 8 (30 mg, 0.05 mmol), C26 (55 mg, 0.13 mmol), Et3N (0.30 mL, 2.1 mmol), PdCl2(PPh3)2 (5 mg, 0.01 mmol), and CuI (2 mg, 0.01 mmol) were dissolved in 1.0 mL DMF under N2. The solution was degassed three times via the freeze-thaw method and then stirred at 40 °C for 4 h and then at 25 °C for 12 h under N2. The solvent was removed under reduced pressure and the crude product partially purified via flash silica column eluting with 7 % methanol:CH2Cl2 to give the acetate protected form of 2 as a purple solid (15 mg).
The product from above (15 mg) was treated with sodium carbonate (6 mg, .05 mmol) in 5.0 mL of methanol. The mixture was stirred at 25 °C for 3 h. The solvent was removed under reduced pressure and extraction was performed using CH2Cl2 and 0.1 M HCl aqueous solution. The aqueous layer was washed with CH2Cl2 (2 × 5 mL) and the organic fractions were combined and dried over magnesium sulfate. The solvent was then removed under reduced pressure and purified via C-18 preparative HPLC eluting with a 50 – 95 % MeOH and 0.1 % TFA/water linear gradient over 25 min to give the desired product with a retention time of 18 min as a purple solid (2 mg, 4 %).
1H NMR (500 MHz, CD3OD), δ 8.08 (s, 2H), 7.87 (d, J = 8.0 Hz, 2H), 7.23 (d, J = 8.0 Hz, 2H), 6.70 (s, 4H), 6.67 (d, J = 8.5 Hz, 4H), 6.58 (d, J = 7.5 Hz, 4H), 3.45 (m, 2H), 3.12 (m, 2H), 2.70 (s, 6H), 2.68 (s, 6H) MALDI HRMS calcd for C60H37BF2N2O12 (M-2H/2)−2 513.6243, found 513.6244 TLC (5 % MeOH :CH2Cl2) Rf = 0.20.
(3)
A mixture of 6 (80 mg, 0.09 mmol), D45 (69 mg, 0.20 mmol), Et3N (0.13 mL, 0.91 mmol), PdCl2(PPh3)2 (6 mg, 0.01 mmol), CuI (4 mg, 0.01 mmol) were dissolved in 3.0 mL THF. The solution was degassed three times via the freeze-thaw method and the mixture was heated to 50 °C for 16 h under N2. The reaction solvent was removed under reduced pressure and the crude product was purified via flash silica column eluting with 67% hexane:ethyl acetate to give the desired product as a purple solid (89 mg, 74 %). 1H NMR (500 MHz, CDCl3), δ 7.64 (d, J = 8.0 Hz, 2H), 7.59 (d, J =8.0 Hz, 4H), 7.29 (d, J = 8.5 Hz, 2H), 7.27 (d, J = 8.0 Hz, 4H), 5.99 (s, 4H), 4.48 (s, 2H), 4.02 (s, 2H), 3.80–3.82 (m, 2H), 3.70–3.75 (m, 10H), 2.76 (s, 6H), 2.55 (s, 12H), 1.58 (s, 6H), 1.47(s, 9H), 1.43 (s, 12H), 13C NMR (125 MHz, CDCl3), δ 169.6, 158.9, 155.7, 144.1, 143.0, 141.7, 140.7, 134.8, 134.3, 132.8, 131.9, 131.2, 128.2 (2 C), 128.0, 124.1 (2 C), 121.3, 116.0, 96.0, 87.1, 85.3, 82.9, 81.6, 70.7, 70.6 (3 C), 70.5, 69.5, 69.0, 59.2, 28.1, 14.6 (2 C), 14.6, 13.7. MALDI HRMS calcd for C76H77B3F6N6O6+ (M•+) 1316.6113, found 1316.6172.
(4)
A mixture of 8 (30 mg, 0.05 mmol), D45 (47 mg, 0.14 mmol), Et3N (0.29 mL, 2.1 mmol), Pd(PPh3)4 (8 mg, 0.01 mmol), and CuI (2 mg, 0.01 mmol) were dissolved in 1.5 mL DMF under N2. The solution was degassed three times via the freeze-thaw method and then stirred for 3 d at 25 °C under N2. The solvent was removed under reduced pressure and the crude product was purified via flash silica column eluting with 3 % methanol:CH2Cl2 followed by recrystalization from methanol to give the desired product as a purple solid (28 mg, 53%). 1H NMR (300 MHz, CDCl3), δ 7.65 (d, J = 8.5 Hz, 4H), 7.30 (d, J = 8.0 Hz, 4H), 6.00 (s, 4H), 3.78 (bs, 1H), 3.46 (m, 2H), 2.73 (s, 6H), 2.70 (m, 2H), 2.66 (s, 6H), 2.56 (s, 12H), 1.45 (s, 12H), 13C NMR (125 MHz, CDCl3), δ 158.2, 155.9, 143.1, 141.5, 140.9, 135.1 (2 C), 132.1, 131.3, 128.4, 124.2, 121.5, 116.6, 96.4, 83.1, 29.8, 15.5 (2 C), 14.8 (2 C), 13.9. MALDI HRMS calcd for C58H53B3F6N6O2 (M•−) 1012.4434, found 1012.4472. TLC (1:1 EtOAc:Hexane) Rf = 0.30.
Supplementary Material
Table 3.
Electrochemical data for reference compounds E, F, 13 – 14, and cassette 3.
| cmpd | Eonset,ox (V) | HOMO (eV) | Eonset,red (V) | LUMO (eV) | Eg (eV) |
|---|---|---|---|---|---|
| Ea | +0.68 | 5.84 | −1.32 | 3.84 | 2.00 |
| Fb | +0.33 | 5.48 | −1.03 | 4.12 | 1.36 |
| FNab, d | +0.04 | 5.19 | −1.67 | 3.48 | 1.71 |
| 13a | +0.99 | 6.15 | −0.91 | 4.28 | 1.87 |
| 14b | +0.49 | 5.64 | −0.93 | 4.22 | 1.42 |
| 14c | +0.49 | 5.64 | −0.92 | 4.23 | 1.41 |
| 3a | +0.99 | 6.15 | −0.92 | 4.24 | 1.91 |
| −1.18 | 3.98 | 2.17 |
Cyclic voltammograms were recorded using a glassy carbon working electrode (A = 0.071 cm2) referenced to Fc/Fc+ and a Pt counter electrode at a scan rate of 200 mV/s. All potentials are reported vs. Fc/Fc+ and all HOMO and LUMO energies are derived from electrochemical results based on Fc/Fc+ = 5.15 eV (DMF) and 5.16 eV (CH2Cl2) vs vacuum. All solvents were flushed with Ar(g) before use.
In CH2Cl2.
In DMF.
In DMF and 0.1 M pyridine.
Xanthene was first reacted with NaOH to obtain the sodium salt.
Acknowledgments
Financial support for this work was provided by The National Institutes of Health (GM72041) and by The Robert A. Welch Foundation (A1121). TAMU/LBMS-Applications Laboratory headed by Dr Shane Tichy provided mass spectrometric support, and the X-ray Diffraction Laboratory (Dr J. Reibenspies) generated the crystallographic data. Dr Yuichiro Ueno, and Mr Juan Castro are thanked for the synthesis of fluorescein precursors, we are grateful to Professor Marcetta Y. Darensbourg for providing equipment and expertise regarding the electrochemical experiments, and to Dr César Pérez-Bolívar and Professor Pavel Anzenbacher (Bowling Green University) for helpful discussions regarding energy transfer in the molecules.
Footnotes
Supporting Information Available. Preparation of compounds 1–14, Cyclic Voltammetry (CV) spectra of 11–14 and E – F, and synthesis of cassette 10 is available free of charge via Internet at http://pubs.acs.org.
References
- 1.DeSilva AP, Gunaratne HGN, Gunnlaugsson T, Huxley AJM, McCoy CP, Rademacher JT, Rice TE. Chem Rev. 1997;97:1515–1566. doi: 10.1021/cr960386p. [DOI] [PubMed] [Google Scholar]
- 2.Callan JF, de Silva AP, Magri DC. Tetrahedron. 2005;61:8551–8588. [Google Scholar]
- 3.Que EL, Domaille DW, Chang CJ. Chem Rev. 2008;108:1517–1549. doi: 10.1021/cr078203u. [DOI] [PubMed] [Google Scholar]
- 4.Thomas JA, Buchsbaum RN, Zimniak A, Racker E. Biochem. 1979;18:2210–2218. doi: 10.1021/bi00578a012. [DOI] [PubMed] [Google Scholar]
- 5.Briggs MS, Burns DD, Cooper ME, Gregory SJ. Chem Commun. 2000:2323–2324. [Google Scholar]
- 6.Galindo F, Burguete MI, Vigara L, Luis SV, Kabir N, Gavrilovic J, Russell DA. Angew Chem Int Ed. 2005;44:6504–6508. doi: 10.1002/anie.200501920. [DOI] [PubMed] [Google Scholar]
- 7.Bizzarri R, Arcangeli C, Arosio D, Ricci F, Faraci P, Cardarelli F, Beltram F. Biophys J. 2006;90:330–3314. doi: 10.1529/biophysj.105.074708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tang B, Liu X, Xu K, Huang H, Yang G, An L. Chem Commun. 2007;36:3726–3728. doi: 10.1039/b707173f. [DOI] [PubMed] [Google Scholar]
- 9.Pal R, Parker D. Chem Commun. 2007;5:474–476. doi: 10.1039/b616665b. [DOI] [PubMed] [Google Scholar]
- 10.Liu Y-S, Sun Y, Vernier PT, Liang C-H, Chong SYC, Gundersen MA. J Phys Chem C. 2007;111:2872–2878. doi: 10.1021/jp0654718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Balut C, vande Ven M, Despa S, Lambrichts I, Ameloot M, Steels P, Smets I. Kidney Int. 2008;73:226–232. doi: 10.1038/sj.ki.5002632. [DOI] [PubMed] [Google Scholar]
- 12.Bradley M, Alexander L, Duncan K, Chennaoui M, Jones AC, Sanchez-Martin RM. Bioorg Med Chem Lett. 2008;18:313–317. doi: 10.1016/j.bmcl.2007.10.075. [DOI] [PubMed] [Google Scholar]
- 13.Diehl H, Horchak-Morris N. Talanta. 1987;34:739–741. doi: 10.1016/0039-9140(87)80232-1. [DOI] [PubMed] [Google Scholar]
- 14.Klonis N, Sawyer WH. J Fluoresc. 1996;6:147–157. doi: 10.1007/BF00732054. [DOI] [PubMed] [Google Scholar]
- 15.Koo MK, Oh CH, Holme AL, Pervaiz S. Cytometry, Part A. 2007;71A:87–93. doi: 10.1002/cyto.a.20361. [DOI] [PubMed] [Google Scholar]
- 16.Han J, Loudet A, Barhoumi R, Burghardt RC, Burgess K. J Am Chem Soc. 2009;131:1642–1643. doi: 10.1021/ja8073374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sunahara H, Urano Y, Kojima H, Nagano T. J Am Chem Soc. 2007;129:5597–5604. doi: 10.1021/ja068551y. [DOI] [PubMed] [Google Scholar]
- 18.Ulrich G, Ziessel R, Harriman A. Angew Chem Int Ed. 2008;47:1184–1201. doi: 10.1002/anie.200702070. [DOI] [PubMed] [Google Scholar]
- 19.Ziessel R, Goze C, Ulrich G. Synthesis. 2007;6:936–949. [Google Scholar]
- 20.Loudet A, Burgess K. In: Handbook of Porphyrin Science: With Applications to Chemistry, Physics, Materials Science, Engineering, Biology and Medicine. Kadish K, Smith K, Guilard R, editors. World Scientific; 2010. p. 203. [Google Scholar]
- 21.Loudet A, Burgess K. Chem Rev. 2007;107:4891–4832. doi: 10.1021/cr078381n. [DOI] [PubMed] [Google Scholar]
- 22.Yogo T, Urano Y, Ishitsuka Y, Maniwa F, Nagano T. J Am Chem Soc. 2005;127:12162–12163. doi: 10.1021/ja0528533. [DOI] [PubMed] [Google Scholar]
- 23.Li Z, Mintzer E, Bittman R. J Org Chem. 2006;71:1718–1721. doi: 10.1021/jo052029x. [DOI] [PubMed] [Google Scholar]
- 24.Sonogashira K, Tohda Y, Hagihara N. Tetrahedron Lett. 1975:4467–4470. [Google Scholar]
- 25.Bonardi L, Ulrich G, Ziessel R. Org Lett. 2008;10:2183–2186. doi: 10.1021/ol800560b. [DOI] [PubMed] [Google Scholar]
- 26.Thoresen LH, Jiao GS, Haaland WC, Metzker ML, Burgess K. Chem Eur J. 2003;9:4603–4610. doi: 10.1002/chem.200304944. [DOI] [PubMed] [Google Scholar]
- 27.Han J, Jose J, Mei E, Burgess K. Angew Chem Int Ed. 2007;46:1684–1687. doi: 10.1002/anie.200603307. [DOI] [PubMed] [Google Scholar]
- 28.Burghart A, Thoresen LH, Chen J, Burgess K, Bergstrom F, Johansson LB-A. Chem Commun. 2000:2203–2204. [Google Scholar]
- 29.Kim TG, Castro JC, Loudet A, Jiao JG-S, Hochstrasser RM, Burgess K, Topp MR. J Phys Chem. 2006;110:20–27. doi: 10.1021/jp053388z. [DOI] [PubMed] [Google Scholar]
- 30.Lakowicz JR. Principles of Fluorescence Spectroscopy. 3. Springer; New York: 2006. [Google Scholar]
- 31.Treibs A, Kreuzer FH. Liebigs Ann Chem. 1968;718:208–223. doi: 10.1002/jlac.19687180118. [DOI] [PubMed] [Google Scholar]
- 32.Shi J, Zhang X, Neckers DC. J Org Chem. 1992;57:4418–4421. [Google Scholar]
- 33.Holten D, Bocian D, Lindsey JS. Acc Chem Res. 2002;35:57–69. doi: 10.1021/ar970264z. [DOI] [PubMed] [Google Scholar]
- 34.Weber G, Teale FWJ. Trans Faraday Soc. 1957;53:646–655. [Google Scholar]
- 35.Karstens T, Kobs K. J Phys Chem. 1980;84:1871–1872. [Google Scholar]
- 36.Thompson BC, Kim Y-G, McCarley TD, Reynolds JR. J Am Chem Soc. 2006;128:12714–12725. doi: 10.1021/ja061274a. [DOI] [PubMed] [Google Scholar]
- 37.Connelly NG, Geiger WE. Chem Rev. 1996;96:877–910. doi: 10.1021/cr940053x. [DOI] [PubMed] [Google Scholar]
- 38.Hansen WN, Hansen GJ. Phys Rev A: At, Mol, Opt Phys. 1987;36:1396–1402. doi: 10.1103/physreva.36.1396. [DOI] [PubMed] [Google Scholar]
- 39.Burgess K, Gibbs R. 6340750 B1. US Patent. 2002 January 22;
- 40.Jiao G-S, Thoresen LH, Burgess K. J Am Chem Soc. 2003;125:14668–14669. doi: 10.1021/ja037193l. [DOI] [PubMed] [Google Scholar]
- 41.Burgess K. 2005/0032120 A1. US Patent. 2005 February 10;
- 42.Azov VA, Diederich F, Lill Y, Hecht B. Helv Chim Acta. 2003;86:2149–2155. [Google Scholar]
- 43.Wan C-W, Burghart A, Chen J, Bergstroem F, Johansson LBA, Wolford MF, Kim TG, Topp MR, Hochstrasser RM, Burgess K. Chem Eur J. 2003;9:4430–4441. doi: 10.1002/chem.200304754. [DOI] [PubMed] [Google Scholar]
- 44.Kim TG, Castro JC, Loudet A, Jiao JGS, Hochstrasser RM, Burgess K, Topp MR. J Phys Chem A. 2006;110:20–27. doi: 10.1021/jp053388z. [DOI] [PubMed] [Google Scholar]
- 45.Jiao G-S, Thoresen LH, Kim TG, Haaland WC, Gao F, Topp MR, Hochstrasser RM, Metzker ML, Burgess K. Chem Eur J. 2006;12:7816–7826. doi: 10.1002/chem.200600197. [DOI] [PubMed] [Google Scholar]
- 46.Scholes GD, Ghiggino KP, Oliver AM, Paddon-Row MN. J Am Chem Soc. 1993;115:4345–4349. [Google Scholar]
- 47.Scholes GD, Clayton AHA, Ghiggino KP. J Chem Phys. 1992;97:7405–7413. [Google Scholar]
- 48.Scholes GD, Ghiggino KP, Oliver AM, Paddon-Row MN. J Phys Chem. 1993;97:11871–11876. [Google Scholar]
- 49.Scholes GD, Ghiggino KP. Photochem Photobiol. 1994;80:355–362. [Google Scholar]
- 50.Scholes GD, Ghiggino KP. J Phys Chem. 1994;98:4580–4590. [Google Scholar]
- 51.Scholes GD, Ghiggino KP. J Chem Phys. 1994;101:1251–1261. [Google Scholar]
- 52.Harcourt RD, Scholes GD, Ghiggino KP. J Chem Phys. 1994;101:10521–10525. [Google Scholar]
- 53.Scholes GD, Harcourt RD, Ghiggino KP. J Chem Phys. 1995;102:9574–9581. [Google Scholar]
- 54.Scholes GD, Ghiggino KP. J Chem Phys. 1995;103:8873–8883. [Google Scholar]
- 55.Scholes GD, Harcourt RD. J Chem Phys. 1996;104:5054–5061. [Google Scholar]
- 56.Ghiggino KP, Yeow EKL, Haines DJ, Scholes GD, Smith TA. Photochem Photobiol. 1996;102:81–86. [Google Scholar]
- 57.Vollmer MS, Würthner F, Effenberger F, Emele P, Meyer DU, Stümpfig T, Port H, Wolf HC. Chem Eur J. 1998;4:260–269. [Google Scholar]
- 58.Dexter DL. J Chem Phys. 1953;21:836–850. [Google Scholar]
- 59.Coskun A, Baytekin BT, Akkaya EU. Tetrahedron Lett. 2003;44:5649–5651. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.