

Abstract
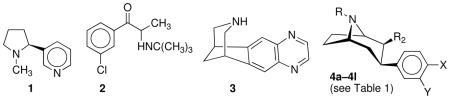
There is a need for different and better aids to tobacco product use cessation. Useful smoking cessation aids, bupropion (2) and varenicline (3), share some chemical features with 3-phenyltropanes (4), which have promise in cocaine dependence therapy. Here we report studies to generate and characterize pharmacodynamic features of 3-phenyltropane analogues. These studies extend our work on the multiple molecular target model for aids to smoking cessation. We identified several new 3-phenyltropane analogues that are superior to 2 in inhibition of dopamine, norepinephrine, and sometimes serotonin reuptake. All of these ligands also act as inhibitors of nicotinic acetylcholine receptor (nAChR) function with a selectivity profile that favors, like 2, inhibition of α3β4*-nAChR. Many of these ligands also block acute effects of nicotine-induced antinociception, locomotor activity, and hypothermia. Importantly, all except one of the analogues tested have better potencies in inhibition of nicotine conditioned place preference than 2. We have identified new compounds that have utility as research tools and possible promise for treatment of nicotine dependence.
Keywords: Nicotine, 3-phenyltropanes, structure activity relationship, dopamine uptake norepinephrine uptake, nAChR antagonism, antinociception, locomotor activity, hypothermia multiple target, conditioned place preference
Introduction
Tobacco product use, principally through cigarette smoking, is the greatest preventable cause of premature mortality, contributing in the United States to over 435,000 deaths annually.1 Tobacco use cessation can halt and reverse the biological damage caused by smoking.2 It is now commonly accepted that smoking behavior is maintained by the reinforcing effects of nicotine (1) and aversive effects of nicotine withdrawal.3–6 Both nonpharmacological and pharmacological interventions have demonstrated efficacy in smoking cessation.7 At present, first-line pharmaceutical treatments include nicotine replacement therapy (NRT),8 bupropion (2) and varenicline (3).9,10 While these treatments are useful in helping 5–20% of smokers abstain long-term, new pharmacotherapies are needed that are either more effective or can impact those individuals not helped by existing treatments.
The effectiveness of varenicline in smoking cessation is thought to be due to its action as a partial agonist at α4β2-containing nicotinic acetylcholine receptors (nAChR).9,10 The mechanism for bupropion’s effectiveness as a smoking-cessation aid appears to be more multi-faceted.11 Its behavioral and neurophysiological effects resemble those of psychomotor stimulants,12 and similar to other psychomotor stimulants, 2 inhibits the reuptake of dopamine (DA).13,14 It also inhibits the reuptake of norepinephrine (NE)13,14 and is a noncompetitive inhibitor of α3β4*-(where the * indicates that nAChR subunits are known or possible assembly partners in addition to those indicated) and α4β2-nAChR.14 In animal behavioral pharmacology studies, 2 induced locomotor activity,15,16 generalized to cocaine and amphetamine in drug discrimination studies,17,18 produced conditioned place preference (CPP),19 and was self-administered by both rats20 and nonhuman primates.21
Over the last several years, we have synthesized a large number of 3-phenyltropane analogues and evaluated them for binding at monoamine transporters.22,23 Similar to 2, some analogues were better at inhibiting the dopamine (DA) transporter (DAT) and the norepinephrine (NE) transporter (NET) than the serotonin (5HT) transporter (SERT). Others were selective for DAT relative to NET and SERT, whereas others showed similar inhibition at all three transporters.22,23 In 1995, Lerner-Marmarosh et al. reported that a number of 3-phenyltropane analogues were effective in blocking nicotine-induced seizures in mice and that a good correlation was observed between pharmacological potencies and abilities to block [3H]mecamylamine binding to brain membranes.24 Based on these results, Lerner-Marmorosh et al. concluded that the 3-phenyltropane analogues are neuronal nicotinic antagonists acting on a similar site to that of mecamylamine, a noncompetitive nicotinic antagonist. In another study, we reported that several 3-phenyltropane analogues blocked nicotine-induced antinociception in the tail-flick test in mice with potencies greater than that of 2.25 These intriguing results suggested that an additional pharmacological study of the 3-phenyltropane class of monoamine uptake inhibitor might provide information about the mechanism of action of noncompetitive nicotinic antagonists like 2 and could provide lead compounds for development as aids to smoking cessation or as drugs for treatment of neurological or psychiatric disorders involving nicotinic mechanisms.
In this study, we report the synthesis and biological evaluation of 3-phenyltropane analogues 4a–4l. All analogues show inhibitory potency at human DAT and NET and functional antagonism of human α3β4*-nAChR. Similar to 2, the compounds antagonize the antinociceptive, hyperlocomotor, and/or hypothermic effects induced by an acute injection of nicotine in mice and blocked nicotine CPP after repeated injection.
Chemistry
The 3-phenyltropane analogues 4a–e, 4g, and 4l were synthesized as previously reported.26–29 Scheme 1 outlines the synthetic route used to prepare 3β-(4-chlorophenyl)-2β-(4′,5′-dimethylbenzimidazol-2′-yl)tropane (4f). 3β-(4-Chlorophenyl)tropane-2β-carboxylic acid (5)28 is converted to the acid chloride and then treated with 4,5-dimethyl-1,2-phenylenediamine to give amide 6. Treatment of 6 with phosphorus oxychloride gave the desired 4f. The nortropane analogue 4h was prepared from 4c as shown in Scheme 2. Treatment of 4c in 1,2-dichloroethane containing excess 1-chloroethyl chloroformate (ACE-Cl) followed by refluxing the intermediate urethane in methanol gave 4h.
Scheme 1a.

aReagents: (a) (COCl)2, CH2Cl2; (b) 4,5-dimethyl-1,2-phenylenediamine, CH2Cl2; (c) POCl3.
Scheme 2a.

aReagents: (a) ACE-Cl, DCE, reflux; (b) CH3OH, reflux; (c) CH3(C=NOH)R, n-C4H9Li, THF, 0 °C; (d) H2SO4, THF.
The synthesis of the 3β-(4-chloro-3-methylphenyl)-2β-(3′-substituted isoxazol-5′-yl)tropanes (4i and 4j) is also outlined in Scheme 2. A solution of 4c was added to the dilithium salt of the appropriate ketone oxime in tetrahydrofuran (THF) at 0 °C under nitrogen, and the reaction mixture was allowed to warm to 25 °C. After a few hours, the reaction mixture was added to a THF solution containing sulfuric acid and refluxed for 1 h to give the desired products 4i and 4j.
The synthesis of the 3α,2β-tropane 4k is outlined in Scheme 3. Addition of a solution of (1R,5S)-2-(3′-methyl-1′,2′,4′-oxadiazol-5′-yl)-8-methyl-8-azabicyclo[3.2.1]oct-2-ene (7)30 in anhydrous THF at −78 °C to a solution of the 3-chloro-4-methylphenyllithium (prepared from the appropriate aryl bromide and butyl lithium) followed by quenching with 1 N hydrochloric acid at −78 °C formed the 3α-(substituted phenyl)tropane-2α-(3′-methyl-1′,2′,4′-oxadiazol-5′-yl)tropanes (8). In addition to the desired isomer, the 2α,3β-isomer was also formed, which was removed by flash chromatography or carried through and removed at the next reaction. Transformation of oxadiazole 8 to the desired methyl ester 4k was accomplished by reduction with nickel boride (generated in situ by reaction of sodium borohydride and nickel tetraacetate) and hydrochloric acid in refluxing methanol. Under such conditions, a complete epimerization of C-2 to form the 3α,2β-stereoisomer was observed.
Scheme 3a.

aReagents: (a) n-C4H9Li, THF, −78 °C, 4 h; (b) TFA; (c) Ni2B, CH3OH, HCl.
In Vitro Assays
The 3-phenyltropane analogues 4a–4l were evaluated for their ability to block reuptake of [3H]dopamine ([3H]DA), [3H]serotonin ([3H]SERT), and [3H]norepinephrine ([3H]NE) using human (h) DAT, hSERT, and hNET stably expressed in HEK293 cells using conditions similar to those previously reported.31,32 The results are given in Table 1.
Table 1.
3-Phenyltropane Analogue Inhibition of Monoamine Uptake and Nicotinic Acetylcholine Receptor (nAChR) Function
 | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Compd | Stereo-chemistry | R1 | R2 | X | Y | Monoamine Uptake Inhibition IC50 (nM)a | nAChR Inhibition IC50, (μM)b | |||||
| [3H]DA | [3H]NE | [3H]5HT | α3β4*- | α4β2- | α4β4- | α1*- | ||||||
| 2 | 658 ± 180 | 1850 ± 300 | IA | 1.8 (1.15) | 12 (1.15) | 15 (1.07) | 7.9 (1.12) | |||||
| 4a | 2β,3β | CH3 | CO2CH(CH3)2 | CH3 | H | 0.83 ± 0.20 | 52 ± 10 | 2830 ± 500 | 1.8 (1.07) | 12 (1.15) | 8.8 (1.13) | 12 (1.07) |
| 4b | 2β,3β | CH3 | CH3 | H | 4.8 ± 0.5 | 122 ± 20 | 2010 ± 800 | 1.3 (1.06) | 11 (1.12) | 7.3 (1.15) | 9.5 (1.05) | |
| 4c | 2β,3β | CH3 | 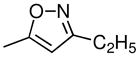 |
CH3 | H | 0.75 ± 0.1 | 11 ± 2 | 2600 ± 1300 | 1.7 (1.05) | 15 (1.10) | 9.9 (1.11) | 9.9 (1.05) |
| 4d | 2β,3β | CH3 | CH3 | H | 11 ± 3 | 310 ± 130 | IA | 20 (1.12) | 942 (1.74) | 57 (4.47) | 95 (1.06) | |
| 4e | 2β,3β | CH3 | Cl | H | 3.9 ± 1 | 26 ± 7 | 2000 ± 300 | 1.8 (1.03) | 15 (1.15) | >100 | 19 (1.03) | |
| 4f | 2β,3β | CH3 | 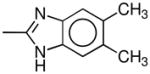 |
Cl | H | 7.3 ± 2 | 19 ± 4 | IA | 0.57 (1.03) | 1.8 (1.15) | 4.3 (1.03) | 4.7 (1.06) |
| 4g | 2β,3β | CH3 | CO2CH3 | Cl | CH3 | 2 ± 1 | 1.1 ± 0.3 | 1.0 ± 0.3 | 3.6 (1.10) | 9.8 (1.18) | 24 (1.16) | 26 (1.07) |
| 4h | 2β,3β | H | CO2CH3 | Cl | CH3 | 2.3 ± 0.4 | 0.9 ± 0.3 | 6.2 ± 2 | 3.1 (1.12) | 16 (1.06) | 12 (1.04) | 11 (1.07) |
| 4i | 2β,3β | CH3 | 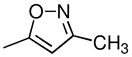 |
Cl | CH3 | 1.5 ± 0.3 | 6.3 ± 0.9 | 115 ± 20 | 0.73 (1.12) | 7.7 (1.26) | 5.8 (1.08) | 13 (1.18) |
| 4j | 2β,3β | CH3 | 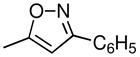 |
Cl | CH3 | 15 ± 5 | 14 ± 3 | 700 ± 200 | 1.4 (1.19) | 14 (1.06) | 4.6 (1.04) | 3.0 (1.09) |
| 4k | 2β,3α | CH3 | CO2CH3 | Cl | CH3 | 3 ± 1 | 6.5 ± 2 | 105 ± 1 | 3.2 (1.08) | 29 (1.04) | 15 (1.04) | 13 (1.05) |
| 4l | 2β,3α | H | CO2CH3 | CH3 | F | 6.4 ± 2 | 0.5 ± 0.1 | 110 ± 7 | 2.6 (1.07) | 11 (1.12) | 25 (1.36) | 7.3 (1.04) |
Values for mean ± standard error of three independent experiments, each conducted with triplicate determination.
Mean micromolar IC50 values (to two significant digits) for bupropion and the indicated 3-phenyltropane analogues from three independent experiments for inhibition of functional responses to an EC80–EC90 concentration of carbamylcholine mediated by nAChR subtypes composed of the indicated subunits (where * indicates that additional subunits are or may be additional assembly partners with the subunits specified; see Methods and Materials). Numbers in parentheses indicate S.E.M. as a multiplication/division factor of the mean micromolar IC50 values shown [i.e., the value 1.8 (1.15) reflects a mean IC50 value of 1.8 μM with an S.E.M. range of 1.8 × 1.15 μM to 1.8/1.15 μM or 1.6–2.1 μM]. IA: IC50 >100 μM.
The 3-phenyltropane analogues 4a–4l were also evaluated for their ability to antagonize functional responses of α3β4*-, α4β2-, α4β4-, and α1*-nAChR and mechanisms involved for functional inhibition using previously reported methods.33 Results are given in Table 1 and in Figures 1–2.32
Fig. 1.

Specific 86Rb+ efflux (ordinate; percentage of control) was determined for functional, human muscle-type α1β1γδ-nAChR (△), ganglionic α3β4*-nAChR (▼), α4β2-nAChR (○), or α4β4-nAChR (▲) naturally or heterologously expressed in human cell lines in the presence of a receptor subtype-specific, EC80–EC90 concentration of the full agonist, carbamylcholine, either alone or in the presence of the indicated concentrations (abscissa, log molar) of 3-phenyltropane analogues 4b, 4c, 4e, or 4f as indicated. Mean micromolar IC50 values and SEM as a multiplication/division factor of the mean micromolar IC50 value are provided in Table 1.
Fig. 2.

Specific 86Rb+ efflux (ordinate; percentage of control) was determined for functional, human ganglionic α3β4*-nAChR naturally expressed by SH-SY5Y human neuroblastoma cells in the presence of the full agonist, carbamylcholine, at the indicated concentrations (abscissa, log molar) either alone (■) or in the presence of 10 μM 3-phenyltropane analogues 4b (△), 4c (□), 4e (▼), or 4f (●) as indicated. The reduction of apparent agonist efficacy without an effect on potency is consistent with a noncompetitive mechanism for nAChR block.
In Vivo Assays
Acute nicotine testing
The 3-phenyltropane analogues 4a–4l were also evaluated for their ability to antagonize behavioral responses to acute nicotine administration as previously described.32 Results are given in Table 2.
Table 2.
Comparison of behavioral potency for 3-phenyltropane analogues in blocking acute effects of nicotine (antinociception, hypomotility, and hypothermia) and development of nicotine CPPa
| Compd | AD50 (mg/kg) |
||||
|---|---|---|---|---|---|
| Tail-flick | Hot-plate | Locomotion | Hypothermia | CPP | |
| 2 | 1.2 (1–1.8) | 15 (6–19) | 4.9 (0.9–46) | 9.2 (4–23) | 0.35 |
| 4a | 0.015 (0.001–0.16) | IA | 1.5 (0.3–7) | 1.6 (0.3–9) | NT |
| 4b | 0.28 (0.09–0.64) | 9.8 (0.7–34) | 1 (0.1–2.1) | 7 (2.5–19) | NT |
| 4c | 0.008 (0.001–0.04) | 1.6 (0.7–3.4) | 0.2 (0.1–2.1) | 6.9 (2.5–19) | 0.045 |
| 4d | IA | IA | 8.7 (6.5–11.2) | IA | 0.013 |
| 4e | 0.018 (0.002–0.16) | IA | IA | IA | NT |
| 4f | 0.11 (0.04–0.3) | 19 (5.2–70) | IA | IA | 0.45 |
| 4g | 0.21 (0.06–2.7) | 0.35 (0.1–1.2) | 0.11 (0.03–0.3) | 0.14 (0.1–0.18) | 0.25 |
| 4h | 0.003 (0.001–0.01) | 0.78 (0.05–12) | 0.41 (0.1–1.7) | 0.23 (0.1–4) | 0.25 |
| 4i | 0.009 (0.002–0.03) | 3.3 (1.4–7.8) | 0.37 (0.06–1.5) | 0.61 (0.24–1.5) | 0.09 |
| 4j | 2.5 (1.3–5.8) | IA | IA | IA | 0.07 |
| 4k | 0.026 (0.004–0.1) | IA | 5.3 (0.5–48) | 7 (3.3–18) | 0.15 |
| 4l | 0.002 (0.001–0.06) | 2.6 (0.6–16) | 1.65 (0.7–3.9) | 7.4 (0.8–14.2) | 0.03 |
Results for nicotine acute effects were expressed as AD50 (mg/kg) ± confidence limits (CL). Dose-response curves were determined using a minimum of four different doses of test compound, and at least eight mice were used per dose group. AD50 (mg/kg) values were estimated in the CPP test using a minimum of three different doses of test compound, and at least eight mice were used per dose group. IA: AD50 > 20 mg/kg; n/a: not studied. NT is not tested.
Nicotine reward using the CPP test
Selected 3-phenyltropane analogues were also evaluated for their ability to antagonize the development of nicotine-induced CPP in mice using an unbiased paradigm.34 Results are given in Table 2.
Results
Effects on Monoamine Uptake
Compound 2 is a relatively weak monoamine uptake inhibitor. In contrast, all of the 3-phenyltropane analogues studied here have higher inhibitory potencies for DA uptake inhibition. 3β-(4-Methylphenyl)tropane-2β-carboxylic acid isopropyl ester (4a) and 3β-(4-methylphenyl)-2β-(3′-ethylisoxoazol-5′-yl)tropane (4c), with IC50 values of 0.83 and 0.75 nM, respectively, were the most potent analogues as inhibitors of DA uptake. However, 3β-(4-chloro-3-methylphenyl)-2β-(3′-methylisoxazol-5′-yl)tropane (4i), 3β-(4-chlorophenyl-3-methyl)tropane-2β-carboxylic acid methyl ester (4g), and its nortropane analogue (4h), with IC50 values of 1.5, 2.0, and 2.3 nM, are almost as potent at inhibition of DA uptake as 4a and 4c. In addition, all of the 3-phenyltropane analogues were potent at NE uptake inhibitors. The most potent NE uptake inhibitors were the nor-tropane analogues 3α-(4-fluoro-3-methylphenyl)nortropane-2β-carboxylic acid methyl ester (4l) and 3β-(4-chloro-3-methylphenyl)nortropane-2β-carboxylic acid methyl ester (4h), with an IC50 values of 0.5 and 0.9 nM, respectively. 3α-(4-Chloro-3-methylphenyl)tropane-2β-carboxylic acid methyl ester (4g), 3β-(4-chloro-3-methylphenyl)-2β-(3′-methylisoxazol-5′-yl)tropane (4i), and 3α-(4-chloro-4-methylphenyl)tropane 2β-carboxylic acid methyl ester (4k), with IC50 values of 1.1, 6.3, and 6.5, respectively, also are very potent NE uptake inhibitors.
Most of the 3-phenyltropane analogues are inactive or have IC50 values greater than 100 nM for 5HT uptake inhibition. However, the 4-chloro-3-methyl carboxylic acid methyl ester 4g and its nortropane analogue 4h have IC50 values of 1 and 6.2 nM for 5HT uptake inhibition. Since these two analogues also have high potency for DA and NE uptake inhibition, they have high potency uptake inhibition at all three monoamine transporters.
Analogue 4g has a slight (~2-fold) preference for inhibition of 5HT and NE over DA uptake inhibition. Compound 4j has comparable inhibitory potencies for DA and NE uptake. Ligand 4h has slight (~2-fold) preference for NE over DA uptake inhibition. Compound 4l has 14-fold preference for inhibition of NE over DA uptake. Otherwise, all the other compounds have preference for inhibition of DA uptake over other monoamine transporters, as low as 2-fold for 4k and 4f to >60-fold for 4a.
Effects on nAChR Function
The effects of 3-phenyltropane analogues 4a–4l on function of diverse human nAChR subtypes naturally or heterologously expressed by human cell lines were assessed using 86Rb+ efflux assays that are specific only for nAChR function in the cells used. None of the analogues has activity as agonists at α1*-, α3β4*-, α4β2-, or α4β4-nAChR, because 86Rb+ efflux in the presence of these ligands alone at concentrations from ~5 nM to 100 μM (data not shown here) was indistinguishable from responses in cells exposed only to efflux buffer.
86Rb+ efflux assays also were used to assess whether ligands had activity as antagonists at human nAChR. Representative concentration-response curves for selected ligands (Fig. 1) illustrate nAChR in vitro inhibitory profiles (see also Table 1).
Compound 2 has IC50 values of 1.8, 12, 15, and 7.9 μM for functional antagonism of α3β4*-, α4β2-, α4β4-, and α1β1*-nAChR, respectively. Analogues 4a, 4b, 4c, 4e, 4f, 4i, and 4j all have IC50 values equal to or lower than that for 2 at α3β4*-nAChR. The most potent inhibitors of α3β4*-nAChR function are 4f and 4i (IC50 = 0.57 and 0.73 μM, respectively). Similar to 2, all the 3-phenyltropane analogues show preference for functional inhibition of α3β4*-nAChR over the other nAChR subtypes tested, with greatest overall preference for 4e (8-, >56-, and 11-fold over α4β2-, α4β4-, or α1*-nAChR), and 4i (11-, 8-, and 18-fold over α4β2-, α4β4-, or α1*-nAChRs). Only 4f had significantly better potency as a functional antagonist of α4β2-nAChR than 2. Analogue 4j had lowest overall preference for α3β4*-nAChR (~2-fold), in part because it (and 4f) had the highest inhibitory potencies at α4β4- and α1*-nAChR. Compounds 4f and 4g had the lowest preference (~3-fold), and compounds 4i and 4j had the highest preference (10–13-fold) for α3β4*- over α4β2-nAChR.
As was the case for 2, all of the 3-phenyltropane analogues tested inhibited nAChR function via an apparently noncompetitive mechanism, lowering apparent agonist efficacy without altering agonist EC50 values [representative data for compounds acting at α3β4*-nAChR are shown (Fig. 2)].
Comparing inhibitory potencies across classes of targets, 4g, 4h, and 4l have 3500–5700-fold selectivity for inhibition of NE uptake over α3β4*-nAChR, and selectivity for inhibition of DA uptake over α3β4*-nAChR ranges from 78–100-fold for 4f and 4j to >2000-fold for 4a and 4c.
In Vivo Effects
Compound 2 blocks nicotine-induced antinociception in the tail-flick and hot-plate tests with AD50 values of 1.2 and 15 mg/kg, respectively. Ten of the 3-phenyltropane analogues were more potent in blocking nicotine’s effects in the tail-flick assay than 2, having AD50 values between 0.002 to 0.28 mg/kg. The most potent analogue in the tail-flick test was 3α-(4-fluoro-3-methylphenyl)-2β-carboxylic acid methyl ester (4l), AD50 = 0.002 mg/kg. Six analogues, 4b, 4c, 4g, 4h, 4i, and 4l, with AD50 values of 0.35 to 9.8 mg/kg were more potent than 2 in blocking nicotine’s acute effects in the hot-plate test, with 4g being most potent (AD50 = 0.35 mg/kg, ~42-fold more potent than 2).
Compound 2 blocks nicotine-induced increases in locomotor activity with an AD50 value of 4.9 mg/kg. Compounds 4a, 4b, 4c, 4g, 4h, 4i, and 4l with AD50 values of 0.11 to 1.65 mg/kg are more potent than 2. The most potent analogue was 4g, which has an AD50 value of 0.11 mg/kg.
Nicotine-induced hypothermia is blocked by 2 with an AD50 of 9.2 mg/kg. Compounds 4a, 4b, 4c, 4g, 4h, 4i, 4k, and 4l with AD50 values of 0.14 to 7.4 mg/kg are all more potent than 2. Compound 4g and its nortropane analogue 4h with IC50 values of 0.14 and 0.23 mg/kg, respectively, were the two most potent analogues in the hypothermia test.
Pre-treatment with 2 at different doses decreased the development of nicotine-induced CPP in mice conditioned with 0.5 mg/kg nicotine, with an estimated AD50 value of 0.35 mg/kg. Nine of the 3-phenyltropane analogues were tested for their ability to block nicotine’s preference. Six of the compounds, 4c, 4d, 4i, 4j, 4k and 4l, with AD50 values of 0.013 to 0.15 mg/kg are more potent than 2.
Discussion
The results from this study show that the 3-phenyltropane analogues have monoamine uptake inhibition and nAChR antagonism profiles similar to 2. Some of the analogues also have slightly higher potency as antagonists of α3β4*-nAChR, and all analogues retain preference across nAChR subtypes for blockade of α3β4*-nAChR. Moreover, some of these compounds show better potency than 2 as inhibitors of acute effects of nicotine and nicotine-induced CPP, which measures the acute rewarding effect of the drug.
Regardless of the type of substituent at the 2β- and 3β-position, all the 3-phenyltropane analogues (see structures 4a–4l) had high potency in DA uptake inhibition. In addition all the analogues except the 3β-cyclobutyl ester 4b and the 3β-4-chlorophenylisoxazole 4d also had high potency in NE uptake inhibition. From a structural perspective, extensive modifications of the 2β-group (see structures 4a–4l) lowered analogue potency as 5HT uptake inhibitors more than they altered inhibitory potency as DA and NE uptake inhibitors. Only analogues with smaller 2β-substituents (4g and 4h) retained high efficacy for 5HT uptake inhibition. Based on the limited comparisons, stereochemistry seems to influence activity for 5HT and NE uptake inhibition more than for DA uptake inhibition and α3β4*-nAChR antagonist potency (compare 4g and 4k). With the exception of the 3β-4-chlorophenylisoxazole analogue 4d, all the analogues had similar relative potencies at α3β4*- and α4β2-nAChRs.
It is interesting to note that analogue 4c has 150-times more potency than 2 as an inhibitor of nicotine’s analgesic action in the tail-flick assay, 9-fold better activity in the hot-plate assay, 24-fold better activity in hypolocomotion studies, and a marginally more potent effect on body temperature, whereas it has ~8-fold better activity in the CPP assay. Analogue 4c does not differ much from 2 in its antagonistic potency at any of the nAChR subtypes studied, and it has 480-fold better activity as a DA uptake inhibitor, ~170-fold better activity as NE uptake inhibitor and no less than 5-fold better activity as a 5HT uptake inhibitor than 2. Since analogue 4g (AD50 = 0.25 mg/kg) has activity comparable to 2 (AD50 = 0.35 mg/kg) in the CPP assay, is 6-fold more potent than 2 in the tail-flick assay, 40-, 44-, and 65-fold more potent than 2 in the hot-plate locomotor and hypothermia tests and has about the same nAChR inhibition profile as 2 while having ~300/~2000/>10,000-times more potency at DA/NE/5HT uptake inhibition, its CPP effects appears to be more related to antagonism of nAChR. A similar analysis of 4i and 4j shows that the 4- to 5-fold better CPP activity relative to 2 correlates better with their nAChR inhibition profile than with their monoamine transporter and acute nicotinic effects. Compound 4d with an AD50 = 0.013 mg/kg was the most potent analogue in the CPP test and, thus, was 27 times more potent than 2. Somewhat surprisingly, unlike 2, compound 4d was inactive in the acute mouse tail-flick, hot-plate, and hypothermia tests and less potent than 2 in the hypolocomotor test. However, it retained high antagonist potency against α3β4*-nAChR and in DA uptake inhibition. In previous studies, we found that even though 4d was a potent and selective inhibitor of the DAT relative to the NET and SERT, it did not show locomotor activity in rats even though in vivo binding studies showed that appreciable brain levels were achieved.8,26 We also found that 4d was a positive allosteric modulator of the CB-1 cannabinoid receptor and suggested that its interaction with this receptor might in part be the reason for its unusual behavioral pharmacological properties.8 However, the reason for this lack of locomotor activity is still not fully understood. One possibility is that 4d is interacting with some as yet unidentified target, possibly a nAChR subtype other than those studied, that is responsible for its lack of locomotor activity and potent activity in the CPP test.
Although many analogues have much better potency in behavioral assays than 2, it is challenging to determine how those effects relate to ligand actions at molecular targets, in part because improvements over 2 for a given ligand can vary quite widely depending on the behavioral assay. Correlation plots comparing the in vitro and in vivo data did not provide any useful insights. Some of these challenges may be due to metabolism of analogues to more or less active forms. In addition, 2 in mice is converted in humans to hydroxy metabolites, one of which expresses much of the drug’s behavioral activity.35
In summary, ligands have been developed that can serve as useful research tools, having different inhibitory potency profiles across nAChR and monoamine transporter targets. For example, 3β-(4-methylphenyl)-2β-(3′-ethylisoxazol-5′-yl)tropane (4c) has IC50 values of 0.75, 11, and 2600 nM for inhibition of DA, NE, and 5HT uptake, and an IC50 of 1.7 μM for antagonism of α3β4*-nAChR. Like 2, 4c is active in all four acute tests of nicotine action in mice. Importantly, it is 150-, 9-, and 5-times more potent in the tail-flick, hot-plate, and locomotor tests, respectively, than 2. It also is eight times more potent than 2 in blocking nicotine-CPP. Three other compounds (4g, 4h, 4i) have higher potencies than 4c as inhibitors in all four tests of acute nicotine effects, and these compounds as well as 4d, 4j, 4k, and 4l also have potency as antagonists of nicotine-CPP significantly higher than that of 2. Compounds 4d and 4j are inactive or are less active than 2 in the acute test of nicotinic action. However, they retain high antagonist potency against α3β4*-nAChR and DA uptake inhibition. The current studies support the idea that nAChR antagonism, particularly inhibition of α3β4*-nAChR function, and inhibitory actions at monoamine transporters are pharmacodynamic features of 3-phenyltropanes. Perhaps most importantly, and certainly warranting further investigation, is the possible utility of these ligands as aids to smoking cessation, especially given their ability to inhibit acute effects of nicotine and a test of nicotine preference with better potencies than 2, which is a useful pharmacotherapy for nicotine dependence.
Experimental Section
Nuclear magnetic resonance (1H NMR and 13C NMR) spectra were recorded on a 300 MHz (Bruker AVANCE 300) spectrometer. Chemical shift data for the proton resonances were reported in parts per million (δ) relative to internal (CH3)4Si (δ 0.0). Elemental analyses were performed by Atlantic Microlab, Norcross, GA. Purity of compounds (>95%) was established by elemental analysis. Analytical thin-layer chromatography (TLC) was carried out on plates precoated with silica gel GHLF (250 μM thickness). TLC visualization was accomplished with a UV lamp or in an iodine chamber. All moisture-sensitive reactions were performed under a positive pressure of nitrogen maintained by a direct line from a nitrogen source. Anhydrous solvents were purchased from Aldrich Chemical Company or VWR. CMA80 is a mixture of 80% chloroform, 18% methanol, and 2% concentrated ammonium hydroxide.
3β-(4-Chlorophenyl)-2β-(4′,5′-dimethylbenzimidozol-2′-yl)tropane (4f) Dihydrochloride
Compound 6 (3.8 g, 0.0095 mol) and POCl3 (50 mL) were mixed and stirred at reflux for 90 min. The reaction mixture was cooled and added to pet. ether (2 L). Most of the solvent was decanted, and concentrated NH4OH-water (1:1) was added until basic to litmus paper then extracted with CH2Cl2. The organic layer was separated, dried (Na2SO4), and concentrated to yield 3.0 g of a tan amorphous solid. This solid was chromatographed on silica gel, eluting with EtOAc then EtOAc-CMA80 (1:1) to afford 1.7 g (47%) of 4f. The free base was dissolved in ether and treated with 1 M ethereal HCl to give 1.65 g of the dihydrochloride salt of 4f as a white solid: mp 224–227 °C; [α]D −166.2 °C (c 0.69, MeOH). 1H NMR (CDCl3, free base) δ 1.73 (bd, 1H), 1.88 (q, J = 9.0 Hz, 2H), 2.22–2.35 (m, 4H), 2.31(s, 3H), 2.33 (s, 3H), 2.35 (s, 3H), 3.20–3.48 (m, 4H), 6.67 (d, J = 9.0 Hz, 2H), 7.00 (d, J = 9.0 Hz, 2H), 7.26 (s, 2H). Anal. (C23H28Cl3N3•1.25 H2O) C, H, N.
(−)-N-Nor-3β-(3-methyl-4-chlorophenyl)tropane-2β-carboxylic Acid Methyl Ester (4h) Tartrate
Compound 4c (0.85 g, 2.76 mmol) was dissolved in anhydrous CH2Cl2 (30 mL), and 1-chloroethyl chloroformate (ACE-Cl, 4.2 mL, 39 mmol) was added. The mixture was refluxed for 8 h. The reaction mixture was concentrated, the residue dissolved in MeOH (30 mL), and the solution refluxed overnight. The MeOH solution was concentrated and the residue partitioned between CH2Cl2 and 25 mL of NH4OH-H2O (1:1). The layers were separated and the aqueous layer extracted twice with CH2Cl2. The combined organic extracts were dried (Na2SO4), filtered, and concentrated to give 0.85 g of yellow solid, which was chromatographed on 50 g of silica gel using 25% CMA80 in CH2Cl2 to obtain 0.54 g (67%) of 4h. This material was converted to the tartrate salt by dissolving 520 mg (1.77 mmol) of 4h in MeOH and adding a MeOH solution of D-tartaric acid 0.286 mg (1.77 mmol). The salt was crystallized from MeOH-ether to give 0.70 g of 4h•tartrate: mp 156–158 °C; [α]25D −100.0 °C (c 1.00, MeOH). C20H25ClNO8: C, 54.12; H, 5.90; N, 3.16; Cl, 7.99. Found: C, 53.88; H, 6.01; N, 3.13; Cl, 7.88. 1H NMR (CD3OD) δ 7.30 (d, 1H), 7.18 (s, 1H), 7.03 (d, 1H), 4.40 (s, 2H), 4.22 (m, 2H), 3.50 (m, 1H), 3.38 (s, 3H), 3.05 (d, 1H), 2.61 (t, 1H), 2.35 (s, 3H), 2.12–2.28 (m, 4H), 1.91 (d, 1H). Anal. (C20H26ClNO8) C, H, N.
(−)-3β-(3-Methyl-4-chlorophenyl)tropane-2β-(3′-methylisoxazol-5′-yl (4i) Hydrochloride
Acetone oxime (262 mg, 359 mmol) was dissolved under N2 in 7.5 mL of THF, and the solution was cooled to 0 °C. Butyl lithium, 25 M in hexanes (2.9 mL, 7.18 mmol), was slowly added, and the solution was stirred in an ice bath for 2–3 h. Compound 4c (85 mg, 2.76 mmol) was dissolved in 4 mL of THF and slowly added to the cold reaction mixture. The mixture was allowed to warm to room temperature and remain for 16 h. A solution of 1.65 g of 36 N H2SO4, 2.1 mL of H2O, and 7.65 mL of THF was slowly added, and the mixture was refluxed for 6 h before being stirred at room temperature for 16 h. The reaction mixture was concentrated and basified with NH4OH-H2O (1:1) to pH 10–11 and extracted with CH2Cl2 (3 × 10 mL). The combined organic extracts were dried (Na2SO4), filtered, and concentrated to give 0.87 g of product, which was purified by flash chromatography on silica gel using 10% Et2O-Et3N (9:1) in 90% hexanes to give 0.26 g of 4c and 0.27 g (44% adjusted) of the desired 4i. The product was converted to the HCl salt by adding 1M HCl in Et2O to an Et2O solution of 4i. Recrystallization from MeOH-Et2O gave 4i•HCl: mp 150 °C (dec.); [α]25D −112.0 °C (c 1.00, MeOH). 1H NMR (CD3OD) δ 7.20 (d, 1H), 7.08 (s, 1H), 6.94 (d, 1H), 5.70, (s, 1H), 4.22 (m, 1H), 4.12 (m, 1H), 3.87 (m, 1H), 3.68 (m, 1H), 2.89 (s, 3H), 2.16–2.66 (m, 16H). Anal. (C19H25Cl2N2O) C, H, N.
(−)-3β-(3-Methyl-4-chlorophenyl)tropane-2β-(3′-phenylisoxazol-5′-yl (4j) Hydrochloride
Acetophenone oxime (617 mg, 4.56 mmol) was dissolved under N2 in 10 mL of THF and cooled to 0 °C. Butyl lithium, 2.5 M in hexanes (3.65 mL, 9.12 mmol), was slowly added, and the solution was stirred in an ice bath for 2 h. Compound 4c (1.08 g, 0.0035 mol) was dissolved in THF (8 mL) and slowly added to the cold reaction mixture. The mixture was allowed to warm to room temperature for 16 h. A solution of 2.00 g of 36 N H2SO4, 2.5 mL of H2O, and 10.5 mL of THF was slowly added, and the mixture was refluxed for 6 h before being stirred at room temperature 16 h. The reaction mixture was concentrated and basified with conc. NH4OH-H2O (1:1) to pH 10–11 and extracted with CH2Cl2 (3 × 25 mL). The combined organic extracts were dried (Na2SO4), filtered, and concentrated to give 1.23 g of product, which was purified by flash chromatography on silica gel using 10% (Et2O-Et3N) (9:1) in 90% hexanes to give 0.39 g of 4c and 0.71 g (80% adjusted yield) of 4j. The 4j was crystallized from petroleum ether to give 0.59 g of fine white crystals: mp 139–141 °C. The product was converted to the HCl salt by adding 1M HCl in Et2O to an Et2O solution of 4j. Recrystallization from MeOH-Et2O gave 0.467 g of 4j•HCl: mp 278 °C (dec.); [α]D −110.2 °C (c 1.00, MeOH). 1H NMR (CD3OD) δ 7.66 (m 2H), 7.43 (m, 3H), 7.21 (d, 1H), 7.12 (s, 1H), 7.00 (d, 1H), 6.20 (s, 1H), 4.30 (m, 1H), 4.16 (m, 1H), 3.98 (m, 1H), 3.75 (m, 1H), 2.93 (s, 3H), 2.04–2.74 (m, 9H). Anal. (C24H27Cl2N2O) C, H, N.
3α-(4-Chloro-3-methylphenyl)tropane-2β-carboxylic acid Methyl Ester (4k) Tosylate
To Ni(OAc)2 (9.21 g, 0.037 mol) in MeOH (50 mL) was added NaBH4 (1.4 g, 0.037 mol) in MeOH (20 mL). Compound 8 (2.20 g, 0.0074 mol) in MeOH (20 mL) was added followed by concentrated HCl (3.1 mL, 0.037 mol) in MeOH (5 mL). The black heterogeneous reaction mixture was stirred at reflux for 17 h. The mixture was filtered through celite, and the filtrate was concentrated in vacuo. The residue was partitioned between Et2O and conc. NH4OH-H2O (1:1). The ether was separated, dried (Na2SO4), and concentrated in vacuo to give an orange oil. This oil was chromatographed on silica gel, eluting with hexane-Et2O-Et3N (50:45:5), to afford 0.92 g (46%) of 4k as a colorless oil. To a solution of 4k in EtOAc (25 mL) was added one equivalent of p-toluene sulfonic acid in a minimal amount of EtOAc. The resulting solids were separated by filtration and dried to give 1.25 g of 4k•CH3C6H4SO3H as a white solid: mp 170–171 °C. 1H NMR (CDCl3, free base) δ 1.32 (t, J = 5.8 Hz, 1H), 1.59 (m, 3H), 2.10 (m, 1H), 2.22 (s, 3H), 2.32 (s, 3H), 2.46 (m, 2H), 3.32 (m, 3H), 3.59 (s, 3H), 6.95 (d, J = 3.0 Hz, 1H), 6.98 (s, 1H), 7.25 (d, J = 5.5 Hz). Anal. (C24H30ClNO5S•0.25 H2O) C, H, N.
3β-(4-Chlorophenyl)tropane-2β-N-(3′-amino-4,5-dimethylphenyl Carboxamide) (6)
To compound 5 (5.3 g, 0.0189 mol) in CH2Cl2 (100 mL) was added oxalyl chloride (19.0 mL, 0.0378 mol, 2 M in CH2Cl2). The reaction mixture was stirred at room temperature for 2 h, then concentrated in vacuo. The resulting acid chloride was dissolved in CH2Cl2 (60 mL) and added to 4,5-dimethyl-1,2-phenylenediamine (6.4 g, 0.0473 mol) in CH2Cl2 (50 mL). The reaction mixture was stirred, under nitrogen, for a period of 17 h. The solvent was decanted from a gummy residue, and 10% NaHCO3-CH2Cl2 was added. The organic layer was separated, dried (Na2SO4), and concentrated in vacuo to give 5.4 g of a foam. This material was chromatographed on silica gel, eluting with EtOAc then EtOAc-CMA80 (1:1) to afford 3.8 g (51%) of 6 as a yellow amorphous solid. 1H NMR (CDCl3) δ 1.71–1.80 (m, 3H), 2.12 (s, 3H), 2.14 (s, 3H), 2.13–2.39 (m, 3H), 2.39 (s, 3H), 2.66 (d, J = 3.0 Hz, 1H), 3.19 (p, J = 3.0 Hz, 1H), 3.40–3.60 (bm, 5H), 6.51 (s, 1H), 6.81 (s, 1H), 7.21 (s, 4H).
3α-(4-Chloro-3-methylphenyl)-2β-(3′-methyl-1′,2′,4′-oxadiazol-5-yl)tropane (8)
n-Butyl lithium (12.0 mL, 0.030 mol, 2.5 M) in hexane was added to 5-bromo-2-chlorotoluene (6.0 g, 0.0292 mol) in THF (50 mL) at −78 °C. The resulting creamy white suspension was stirred for 15 min, and anhydroecgonine oxadiazole (7) (3.0 g, 0.0146 mol) in THF (50 mL) was added. The orange reaction mixture was stirred for an additional 3 h, allowing the reaction to come to room temperature. TFA (3.9 mL, 0.050 mol) was added, the mixture was stirred for 15 min and concentrated in vacuo. The resulting residue was treated with conc. NH4OH-H2O (1:1) (100 mL) and CH2Cl2 (100 mL). The organic layer was separated, dried (Na2SO4), and concentrated in vacuo to afford 5.7 g of an orange oil. This oil was chromatographed on silica gel, eluting with ether-Et3N (9:1) to afford 2.0 g (46%) of a mixture that was used without further purification to prepare 4k.
Transporter Assays
The abilities of 2 and its analogues to inhibit uptake of [3H]dopamine ([3H]DA), [3H]serotonin ([3H]5-HT), or [3H]norepinephrine ([3H]NE) by the respective, human transporters were evaluated using the appropriate HEK-293 cell line as previously reported.31
Cell lines and culture
Human embryonic kidney (HEK-293) cells stably expressing human DAT, NET or SERT were maintained as previously described.31
Use was made, as previously described,33 of TE671/RD cells naturally expressing human muscle-type nAChR (α1β1γδ- or α1*-nAChR), SH-SY5Y neuroblastoma cells naturally expressing human autonomic α3β4*-nAChRs (containing α3, β4, probably α5, and sometimes β2 subunits), or transfected SH-EP1 epithelial cells heterologously expressing either human α4β2-nAChR, which are thought to be the most abundant, high affinity nicotine-binding nAChR in mammalian brain, or α4β4-nAChR.36,37
nAChR Functional Assays
Cells were harvested, seeded onto 24-well plates, and subjected to 86Rb+ efflux assays as previously described.33 Specific 86Rb+ efflux was assessed as the response to a fully efficacious concentration of carbamylcholine alone less that in the presence of efflux buffer alone. Any intrinsic agonist activity of test compounds was normalized, after subtraction of non-specific efflux, to specific efflux. Antagonism of carbamylcholine-evoked 86Rb+ efflux was assessed in samples containing the full agonist at a concentration where it stimulates 80–90% of maximal function. For studies of mechanism of antagonism, concentration-response curves were obtained using samples containing the full agonist, carbamylcholine, at the indicated concentrations alone or in the presence of a concentration of the test ligand close to its IC50 value for inhibition of nAChR function. Ion flux assay results were fit using Prism (GraphPad) to the Hill equation, F = Fmax/(1 + (X/Z)n), where F is the test sample specific ion flux as a percentage of control, Fmax is specific ion flux in the absence of test drug (i.e., for control samples), X is the test ligand concentration, Z is the EC50 (n>0 for agonists) or IC50 (n<0 for antagonists), and n is the Hill coefficient. All concentration-ion flux response curves were simple and fit well allowing maximum and minimum ion flux values to be determined by curve fitting, but in cases where antagonists had weak functional potency, minimum ion flux was set at 0% of control. Note that because agonist concentrations used for test ligand antagonism assessments were EC80–EC90 values, not all of the data, even at the lowest concentrations of test antagonist, approaches 100% of specific efflux as separately determined in sister samples exposed to fully efficacious concentrations of agonist. Note also that it has been repeatedly verified that functional parameters for nicotinic ligands and mechanisms of their action as determined using efflux assays are like those determined using whole-cell current recording techniques.38
Behavior
All animal experiments were conducted in accordance with the NIH Guide for the Care and Use of Laboratory Animals and Institutional Animal Care and Use Committee guidelines.
Animals
Male Institute of Cancer Research (ICR) mice (weighing 20–25 g) obtained from Harlan (Indianapolis, IN) were used throughout the study. Animals were housed in an Association for Assessment and Accreditation of Laboratory Animal Care-approved facility, were placed in groups of six, and had free access to food and water. Studies were approved by the Institutional Animal Care and Use Committee of Virginia Commonwealth University.
Tail-Flick Test
Antinociception for pain mediated at the spinal level was assessed by the tail-flick method of D’Amour and Smith.39 In brief, mice were lightly restrained while a radiant heat source was shone onto the upper portion of the tail. To minimize tissue damage, a maximum latency of 10 s was imposed. Latency to remove the tail from the heat source was recorded for each animal. A control response (2–4 s) was determined for each mouse before treatment, and a test latency was determined after drug administration (nicotine as an analgesic 5 min after subcutaneous administration at 2.5 mg/kg; nicotine administration 15 min after exposure to saline of 3-phenytropane analogue to assess the latter drug’s ability to block nicotine-mediated antinociception). Antinociceptive response was calculated as the percentage of maximum possible effect (%MPE), where %MPE = [(test control)/(10 control)] × 100.
Hot-Plate Test
Mice were placed into a 10-cm wide glass cylinder on a hot plate (Thermojust Apparatus) maintained at 55 °C for assessment of pain responses mediated at supraspinal levels. To minimize tissue damage, a maximum exposure to the hot plate 40 s was imposed. Measures of control latencies (time until the animal jumped or licked its paws; typically 8–12 s) were done twice for stimuli applied at least 10 min apart for each mouse. Antinociceptive responses after test drug administrations were determined and calculated as the %MPE, where %MPE = [(test latency in s − control latency in s)/(40 s − control latency in s) × 100]. Groups of 8 to 12 animals were used for each drug condition. Antagonism studies were carried in mice pretreated with either saline or 3-phenytropane analogues 15 min before nicotine. The animals were then tested 5 min after administration of a subcutaneous dose of 2.5 mg/kg nicotine.
Locomotor Activity
Mice were placed into individual Omnitech photocell activity cages (28 × 16.5 cm; Omnitech Electronics, Columbus, OH) 5 min after subcutaneous administration of either 0.9% saline or nicotine (1.5 mg/kg). Interruptions of the photocell beams (two banks of eight cells each) were then recorded for the next 10 min. Data were expressed as the number of photocell interruptions. Antagonism studies were carried out by pretreating the mice with either saline or 3-phenytropane analogues 15 min before nicotine.
Body Temperature
Rectal temperature was measured by a thermistor probe (inserted 24 mm) and digital thermometer (YSI Inc., Yellow Springs, OH). Readings were taken just before and 30 min after subcutaneous injection of either saline or 2.5 mg/kg nicotine. The difference in rectal temperature before and after treatment was calculated for each mouse. The ambient temperature of the laboratory varied from 21 to 24 °C from day to day. Antagonism studies were carried out by pretreating the mice with either saline or 3-phenytropane analogues 15 min before nicotine. The animals were then tested 30 min after administration of a subcutaneous dose of 2.5 mg/kg nicotine.
Nicotine CPP Assessment
An unbiased CPP paradigm was utilized in this study as described in Kota et al.33 Briefly, place-conditioning chambers consisted of two distinct compartments separated by a smaller intermediate compartment with openings that allowed access to either side of the chamber. On day 1, adult male ICR mice were confined to the intermediate compartment for a 5 min habituation period, and then allowed to move freely between compartments for 15 min. Time spent in each compartment was recorded. These data were used to separate the animals into groups of approximately equal bias. Days 2–4 were the conditioning days during which the saline group received saline in both compartments and drug groups received s.c. vehicle or 3-phenyltropane analogues 15 min before nicotine (0.5 mg/kg, s.c.) in one compartment and saline in the opposite compartment for 20 min. Drug-paired compartments were randomized among all groups. Day 5 was the drug free test day and the procedure was the same as day 1. Activity counts and time spent on each side were recorded via photosensors using Med Associates interface and software. Separate groups of mice were conditioned with saline or 3-phenyltropane analogues alone to investigate if they induce CPP using the same procedure described above. Data were expressed as time spent on drug-paired side minus time spent on saline-paired side. A positive number indicated a preference for the drug-paired side, whereas a negative number indicated an aversion to the drug-paired side. A number at or near zero indicated no preference for either side.
Supplementary Material
Acknowledgments
This work was supported by National Institutes of Health National Cooperative Drug Discovery Group grant U19 DA019377. Other effort was supported by grants (to RJL) from the National Institutes of Health (DA015389) and the Barrow Neurological Foundation. We thank Lawrence E. Brieaddy for the synthesis of 4f and 4k and Fluvanna Josephson for the synthesis of 4h, 4i, and 4j.
Abbreviations
- NRT
nicotine replacement therapy
- DA
dopamine
- 5HT
serotonin
- NE
norepinephrine
- HEK
human embryonic kidney
- DAT
dopamine transporter
- SERT
serotonin transporter
- NET
norepinephrine transporter
- nAChR
nicotine acetylcholine receptor
- VTA
ventral tegmental area
- MPE
maximum possible effect
- CPP
conditioned place preference
Footnotes
Supporting Information Available: Elemental analysis. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Centers for Disease Control and Prevention. Smoking and Tobacco Use-Fact Sheet: Health Effects of Cigarette Smoking. (updated January 2008) http://www.cdc.gov/tobacco/data_statistics/fact_sheets/health_effects/effects_cig_smoking/
- 2.Centers for Disease Control and Prevention. Annual smoking-attributable mortality, years of potential life lost, and productivity losses--United States. MMWR Morb Mortal Wkly Rep. 2005;54:625–628. [PubMed] [Google Scholar]
- 3.Benowitz NL. Nicotine addiction. Prim Care. 1999;26:611–631. doi: 10.1016/s0095-4543(05)70120-2. [DOI] [PubMed] [Google Scholar]
- 4.Benowitz NL. Clinical pharmacology of nicotine: implications for understanding, preventing, and treating tobacco addiction. Clin Pharmacol Ther. 2008;83:531–541. doi: 10.1038/clpt.2008.3. [DOI] [PubMed] [Google Scholar]
- 5.Hughes JR, Higgins ST, Bickel WK. Nicotine withdrawal versus other drug withdrawal syndromes: similarities and dissimilarities. Addiction. 1994;89:1461–1470. doi: 10.1111/j.1360-0443.1994.tb03744.x. [DOI] [PubMed] [Google Scholar]
- 6.Tutka P, Mosiewicz J, Wielosz M. Pharmacokinetics and metabolism of nicotine. Pharmacol Rep. 2005;57:143–153. [PubMed] [Google Scholar]
- 7.West R, McNeill A, Raw M. Smoking cessation guidelines for health professionals: an update. Health Education Authority Thorax. 2000;55:987–999. doi: 10.1136/thorax.55.12.987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Navarro HA, Howard JL, Pollard GT, Carroll FI. Positive allosteric modulation of the human cannabinoid (CB) receptor by RTI-371, a selective inhibitor of the dopamine transporter. Br J Pharmacol. 2009;156:1178–1184. doi: 10.1111/j.1476-5381.2009.00124.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tutka P. Nicotinic receptor partial agonists as novel compounds for the treatment of smoking cessation. Expert Opin Investig Drugs. 2008;17:1473–1485. doi: 10.1517/13543784.17.10.1473. [DOI] [PubMed] [Google Scholar]
- 10.Rollema H, Coe JW, Chambers LK, Hurst RS, Stahl SM, Williams KE. Rationale, pharmacology and clinical efficacy of partial agonists of alpha4beta2 nACh receptors for smoking cessation. Trends Pharmacol Sci. 2007;28:316–325. doi: 10.1016/j.tips.2007.05.003. [DOI] [PubMed] [Google Scholar]
- 11.Dwoskin LP, Smith AM, Wooters TE, Zhang Z, Crooks PA, Bardo MT. Nicotinic receptor-based therapeutics and candidates for smoking cessation. Biochem Pharmacol. 2009;78:732–743. doi: 10.1016/j.bcp.2009.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Carroll FI, Blough B, Abraham P, Mills AC, Holleman JA, Wolckenhauer SA, Decker AM, Landavazo A, McElroy KT, Navarro HA, Gatch MB, Foster MJ. Synthesis and Biological Evaluation of Bupropion Analogues as Potential Pharmacotherapies for Cocaine Addiction. J Med Chem. 2009;52:6768–6781. doi: 10.1021/jm901189z. [DOI] [PubMed] [Google Scholar]
- 13.Dhillon S, Yang LP, Curran MP. Bupropion: a review of its use in the management of major depressive disorder. Drugs. 2008;68:653–689. doi: 10.2165/00003495-200868050-00011. [DOI] [PubMed] [Google Scholar]
- 14.Ferris RM, Cooper BR. Mechanism of antidepressant activity of bupropion. J Clin Psychiatry Monog. 1993;11:2–14. [PubMed] [Google Scholar]
- 15.Nielsen JA, Shannon NJ, Bero L, Moore KE. Effects of acute and chronic bupropion on locomotor activity and dopaminergic neurons. Pharmacol Biochem Behav. 1986;24:795–799. doi: 10.1016/0091-3057(86)90413-2. [DOI] [PubMed] [Google Scholar]
- 16.Nomikos GG, Damsma G, Wenkstern D, Fibiger HC. Effects of chronic bupropion on interstitial concentrations of dopamine in rat nucleus accumbens and striatum. Neuropsychopharmacology. 1992;7:7–14. [PubMed] [Google Scholar]
- 17.Jones CN, Howard JL, McBennett ST. Stimulus properties of antidepressants in the rat. Psychopharmacology (Berl) 1980;67:111–118. doi: 10.1007/BF00431964. [DOI] [PubMed] [Google Scholar]
- 18.Lamb RJ, Griffiths RR. Self-administration in baboons and the discriminative stimulus effects in rats of bupropion, nomifensine, diclofensine and imipramine. Psychopharmacology (Berl) 1990;102:183–190. doi: 10.1007/BF02245920. [DOI] [PubMed] [Google Scholar]
- 19.Ortmann R. The conditioned place preference paradigm in rats: effect of bupropion. Life Sci. 1985;37:2021–2027. doi: 10.1016/0024-3205(85)90033-5. [DOI] [PubMed] [Google Scholar]
- 20.Tella SR, Ladenheim B, Cadet JL. Differential regulation of dopamine transporter after chronic self-administration of bupropion and nomifensine. J Pharmacol Exp Ther. 1997;281:508–513. [PubMed] [Google Scholar]
- 21.Spealman RD, Madras BK, Bergman J. Effects of cocaine and related drugs in nonhuman primates. II. Stimulant effects on schedule-controlled behavior. J Pharmacol Exp Ther. 1989;251:142–149. [PubMed] [Google Scholar]
- 22.Runyon SP, Carroll FI. Dopamine transporter ligands: recent developments and therapeutic potential. Curr Top Med Chem. 2006;6:1825–1843. doi: 10.2174/156802606778249775. [DOI] [PubMed] [Google Scholar]
- 23.Runyon SP, Carroll FI. Tropane-based Dopamine Transporter-Uptake Inhibitors. In: Trudell ML, Izenwasser S, editors. Dopamine Transporters: Chemistry, Biology, and Pharmacology. John Wiley & Sons; 2008. pp. 125–169. [Google Scholar]
- 24.Lerner-Marmarosh N, Carroll FI, Abood LG. Antagonism of nicotine’s action by cocaine analogs. Life Sci. 1995;56:PL67–PL70. doi: 10.1016/0024-3205(94)00438-x. [DOI] [PubMed] [Google Scholar]
- 25.Damaj MI, Slemmer JE, Carroll FI, Martin BR. Pharmacological characterization of nicotine’s interaction with cocaine and cocaine analogs. J Pharmacol Exp Ther. 1999;289:1229–1236. [PubMed] [Google Scholar]
- 26.Carroll FI, Pawlush N, Kuhar MJ, Pollard GT, Howard JL. Synthesis, monoamine transporter binding properties, and behavioral pharmacology of a series of 3beta-(substituted phenyl)-2beta-(3′-substituted isoxazol-5-yl)tropanes. J Med Chem. 2004;47:296–302. doi: 10.1021/jm030453p. [DOI] [PubMed] [Google Scholar]
- 27.Kotian P, Mascarella SW, Abraham P, Lewin AH, Boja JW, Kuhar MJ, Carroll FI. Synthesis, ligand binding, and quantitative structure-activity relationship study of 3β-(4′-substituted phenyl)-2β-heterocyclic tropanes: evidence for an electrostatic interaction at the 2β-position. J Med Chem. 1996;39:2753–2763. doi: 10.1021/jm960160e. [DOI] [PubMed] [Google Scholar]
- 28.Carroll FI, Kotian P, Dehghani A, Gray JL, Kuzemko MA, Parham KA, Abraham P, Lewin AH, Boja JW, Kuhar MJ. Cocaine and 3β-(4′-substituted phenyl)tropane-2β-carboxylic acid ester and amide analogues. New high-affinity and selective compounds for the dopamine transporter. J Med Chem. 1995;38:379–388. doi: 10.1021/jm00002a020. [DOI] [PubMed] [Google Scholar]
- 29.Carroll FI, Mascarella SW, Kuzemko MA, Gao Y, Abraham P, Lewin AH, Boja JW, Kuhar MJ. Synthesis, ligand binding, and QSAR (CoMFA and classical) study of 3β-(3′-substituted phenyl)-, 3β-(4′-substituted phenyl)-, and 3β-(3′,4′-disubstituted phenyl)tropane-2β-carboxylic acid methyl esters. J Med Chem. 1994;37:2865–2873. doi: 10.1021/jm00044a007. [DOI] [PubMed] [Google Scholar]
- 30.Triggle DJ, Kwon YW, Abraham P, Rahman MA, Carroll FI. Synthesis of 2-(3-substituted-1,2,4-oxadiazol-5-yl)-8-methyl-8-azabicyclo [3.2.1]octanes and 2 alpha-(3-substituted-1,2,4-oxadiazol-5-yl)-8-methyl-8-azabicyclo[3.2.1]oct-2-enes as potential muscarinic agonists. Pharm Res. 1992;9:1474–1479. doi: 10.1023/a:1015871131913. [DOI] [PubMed] [Google Scholar]
- 31.Eshleman AJ, Carmolli M, Cumbay M, Martens CR, Neve KA, Janowsky A. Characteristics of drug interactions with recombinant biogenic amine transporters expressed in the same cell type. J Pharmacol Exp Ther. 1999;289:877–885. [PubMed] [Google Scholar]
- 32.Damaj MI, Carroll FI, Eaton JB, Navarro HA, Blough BE, Mirza S, Lukas RJ, Martin BR. Enantioselective effects of hydroxy metabolites of bupropion on behavior and on function of monoamine transporters and nicotinic receptors. Mol Pharmacol. 2004;66:675–682. doi: 10.1124/mol.104.001313. [DOI] [PubMed] [Google Scholar]
- 33.Carroll FI, Blough BE, Mascarella SW, Navarro HA, Eaton JB, Lukas RJ, Damaj MI. Synthesis and Biological Evaluation of Bupropion Analogues as Potential Pharmacotherapies for Smoking Cessation. J Med Chem. 2010;53:2204–2214. doi: 10.1021/jm9017465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kota D, Martin BR, Robinson SE, Damaj MI. Nicotine dependence and reward differ between adolescent and adult male mice. J Pharmacol Exp Ther. 2007;322:399–407. doi: 10.1124/jpet.107.121616. [DOI] [PubMed] [Google Scholar]
- 35.Lukas RJ, Muresan AZ, Damaj MI, Blough BE, Huang X, Navarro HA, Mascarella SW, Eaton JB, Marxer-Miller SK, Carroll FI. Synthesis and Characterization of In Vitro and In Vivo Profiles of Hydroxybupropion Analogues: Aids to Smoking Cessation. J Med Chem. 2010;53:4731–4748. doi: 10.1021/jm1003232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Eaton JB, Peng JH, Schroeder KM, George AA, Fryer JD, Krishnan C, Buhlman L, Kuo YP, Steinlein O, Lukas RJ. Characterization of human alpha 4 beta 2-nicotinic acetylcholine receptors stably and heterologously expressed in native nicotinic receptor-null SH-EP1 human epithelial cells. Mol Pharmacol. 2003;64:1283–1294. doi: 10.1124/mol.64.6.1283. [DOI] [PubMed] [Google Scholar]
- 37.Gentry CL, Lukas RJ. Local anesthetics noncompetitively inhibit function of four distinct nicotinic acetylcholine receptor subtypes. J Pharmacol Exp Ther. 2001;299:1038–1048. [PubMed] [Google Scholar]
- 38.Lukas RJ, Fryer JD, Eaton JB, Gentry CL. Some methods for studies of nicotinic acetylcholine receptor pharmacology. In: Levin ED, editor. Nicotinic Receptors and the Nervous System. CRC Press; Boca Raton: 2002. pp. 3–27. [Google Scholar]
- 39.D’Amour FE, Smith DL. A method for determining loss of pain sensation. J Pharmacol Exp Ther. 1941;72:74–79. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.