

Abstract
BACKGROUND AND PURPOSE
Deletion of the cyclooxygenase-2 (COX-2) gene causes impairment of kidney development, but the effect of selective inhibitors of COX-2 (coxibs) or the non-selective inhibitors of COX (the classical non-steroidal anti-inflammatory drugs; NSAIDs) on kidney development was less well described.
EXPERIMENTAL APPROACH
We assessed the effects of equipotent analgesic doses of celecoxib, rofecoxib, valdecoxib, etoricoxib and lumiracoxib and of the NSAIDs, diclofenac and naproxen, on postpartum kidney development in mice, from postnatal day 1 (P1) to P21.
KEY RESULTS
All the COX inhibitors, at the doses used, blocked COX-2 activity by more than 80% as assayed by PGE2 synthesis in lipopolysaccharide-stimulated mouse blood samples. Rofecoxib, etoricoxib and lumiracoxib exerted the most marked impairment of postpartum kidney development, demonstrated by attenuation of kidney growth, reduction in size of glomeruli, increase in immature superficial glomeruli, thinning of subcapsular cortical mass and reduction in size of juxtamedullary glomeruli. These defects were less severe than those in kidneys from COX-2−/− mice. Administration of diclofenac and naproxen revealed renal defects similar to those after coxib treatment, but both NSAIDs induced greater arrest of immature superficial glomeruli in the outer cortex and increased the number of undifferentiated proliferating cell nuclear antigen-positive cells. Treatment with celecoxib or valdecoxib caused only minimal changes in renal morphology.
CONCLUSIONS AND IMPLICATIONS
Classical NSAIDs cause similar or even stronger nephrodysgenesis than the coxibs. Also, the ranking of coxibs regarding adverse effects on renal development, using equi-analgesic doses, is rofecoxib = etoricoxib = lumiracoxib > valdecoxib > celecoxib.
Keywords: kidney, nephrogenesis, cyclooxygenase, coxib, prostaglandin
Introduction
Cyclooxygenase (COX) inhibitors, also called non-steroidal anti-inflammatory drugs (NSAIDs), are among the most used drugs over the world due to their efficient analgesic, antipyretic and anti-inflammatory effects (Vane et al., 1998). COX is the rate-limiting enzyme in the biosynthesis of prostaglandins responsible for the conversion of arachidonic acid into PGH2, which is then converted to various prostaglandins (Smith et al., 2000), mostly PGE2, which modulates pain, fever and inflammation (Simmons et al., 2004). Apart from pathophysiological roles, prostaglandins are known to contribute to organ physiology (Smith, 1992). The unwanted renal side effects of NSAIDs, such as reduced renal blood flow and glomerular filtration, disturbed electrolyte regulation or impairment of renal development are the consequences of prostaglandin inhibition (Hao and Breyer, 2008). The discovery of two isoforms, COX-1 and COX-2 and the identification of COX-2 as the prominent pharmacological target led to the development of a new subclass of NSAIDs, selective COX-2 inhibitors called coxibs, such as celecoxib or rofecoxib (Flower, 2003). However, a number of studies have shown that the coxibs exert renal side effects similar to those of classical NSAIDs (Harris and Breyer, 2006). As implied by these results, the COX-2 enzyme has been found constitutively expressed in macula densa, cortical thick ascending limb and papillary parenchyma of the kidney (Harris et al., 1994; Guan et al., 1997; Kömhoff et al., 1997). COX-2 expression has also been shown in human fetal kidneys with high levels at gestational age 15–24 weeks, a time of active nephrogenesis (Kömhoff et al., 1997). Treatment with NSAIDs during pregnancy can cause renal dysgenesis in neonates as well as oligohydramnion, due to reduced urine production and, in a few cases, death by renal failure (van der Heijden et al., 1994; Kaplan et al., 1994; Voyer et al., 1994). The assumption that the non-selective inhibition of COX-2 by NSAIDs may be responsible for impaired renal development is supported by the observation that mice with genetic deletion of COX-2 (COX-2−/− mice) exhibited morphological damages to the kidney, similar to those found in human neonates after NSAIDs (Dinchuk et al., 1995; Morham et al., 1995). COX-2−/− mice exhibit renal dysgenesis associated with markedly reduced renal cortical volume but maintaining normal papillary volume, and hypoplastic glomeruli. The small subcapsular glomeruli appear immature, as judged by more densely packed glomerular cells and the presence of cuboidal shaped podocytes (Kömhoff et al., 2000). In addition the kidneys show periglomerular and diffuse interstitial fibrosis (Norwood et al., 2000). Treatment with the experimental COX-2 inhibitor, SC-236, postnatally from the day of birth (P0) to postnatal day 21 (P21) caused comparable renal pathologies, but treatment with the COX-1 inhibitor SC-560 in drinking water or genetic disruption of COX-1 gene did not cause such damage (Kömhoff et al., 2000; Norwood et al., 2000; Koki et al., 2002). Exposure to COX-2 inhibitors limited to the prenatal period (E0.5 to P0) was without effect on glomerular size. These results suggest that COX-2 activity is important at least in postnatal renal development and renal developmental defects are likely to be responsible for reduced GFR or even renal failure observed in adult COX-2−/− mice (Dinchuk et al., 1995; Norwood et al., 2000).
For clinically used coxibs but also for classical NSAIDs, there are still no data available regarding their ability to induce renal dysgenesis. Extrapolation of the observations made in COX-2−/− mice or in SC-236-treated mice, on the effect of clinically used coxibs on renal development may be questionable for two reasons. First, it cannot be ruled out that genetic deletion of the COX-2 gene may result in additional effects, not directly associated with the loss of COX-2 activity. Then; SC-236 is known to exhibit an extremely long half-life (several days) and may accumulate in kidney tissue; this is one reason why SC-236 was rejected for further clinical development during preclinical evaluation (Penning et al., 1997). Of note, COX-2 knockdown mice (COX-2Neo/Neo), expressing low levels of COX-2 and mimicking level of maximal pharmacological suppression obtained with COX-2 inhibitors, show only mild renal dysgenesis and normal GFR was observed (Seta et al., 2009).
The main clinical aim for the use of COX inhibitors, NSAIDs and coxibs, is the treatment of pain. We therefore investigated the impairment of renal development in mice following administration of COX inhibitors, in equipotent analgesic doses, from postnatal day P1 to P21. In our present experiments, the coxibs, celecoxib, rofecoxib, valdecoxib, etoricoxib and lumiracoxib, used in clinical practice currently and in the past, as well as the classical NSAIDs, diclofenac and naproxen, were studied in comparison with methyl-celecoxib (DMC), a derivative of celecoxib, which exhibits no inhibition of COX-2, and the selective COX-1 inhibitor, SC-560.
Methods
Animals and treatment
All animal care and experimental procedures were in accordance with institutional guidelines and approved by the Animal Welfare Committee of the State Agency Darmstadt (Germany). Breeder pairs of C57/BL6J wild-type mice were obtained from Jackson Laboratories (Bar Harbour, ME, USA). Breeder pairs of COX-2−/− mice were kindly provided by Robert Langenbach, Nashville, TN, USA. The animals were maintained in individually ventilated cages in a temperature controlled (21°C) room with 12 h light-12 h dark cycle. Food and water were provided ad libitum.
The day of birth was defined as postnatal day 0 (P0). Mice were treated with the experimental selective COX-2 inhibitor SC-236 (Calbiochem, Darmstadt, Germany), the non-specific COX inhibitors diclofenac (Sigma-Chemie, Deisenhofen, Germany) and naproxen (Sigma-Aldrich, Steinheim, Germany), the selective COX-2 inhibitors celecoxib, rofecoxib, etoricoxib, valdecoxib (all were synthesized by Witega-Laboratory-Adlershofen (Berlin, Germany), and lumiracoxib (Novartis, Germany), COX-1 inhibitor SC-560 (Calbiochem, Darmstadt, Germany) and DMC, which exhibits structural similarity to celecoxib but has minimal COX inhibitory potency (Schiffmann et al., 2008). Drug and molecular target nomenclature follows (Alexander et al., 2009).
All substances were dissolved in dimethylsulphoxide. Doses of coxibs and NSAIDs used were based on given or extrapolated ED80 values for anti-nociceptive effects from the literature (Table 1). As the ED values depended on the model of anti-nociception, we used the highest dose reported. Drugs were injected s.c. between the shoulder blades of the pups from day P1 to P6 and i.p. from day P7 to P21. The volume injected was roughly adjusted to body weight, so that from P1 to P6, substances were dissolved in a volume of 5 µL, from P7 to P14 in a volume of 7.5 µL and from P15 to P21 in a volume of 10 µL. Control groups received dimethylsulphoxide alone.
Table 1.
Cyclooxygenase inhibitors used in the study
| Drug name | Inhibition of COX isoform | Reported analgesic ED50 (mg·kg−1) | Dose used in this study corresponding to ED80 (mg·kg−1) |
|---|---|---|---|
| SC-236 | COX-2 | – | 10 |
| SC-560 | COX-1 | – | 10 |
| Naproxen | COX-1/COX-2 | 11 (Anikwue et al., 2002) | 50 |
| Diclofenac | COX-1/COX-2 | 0.8–8 (Anikwue et al., 2002; Miranda et al., 2006) | 20 |
| Celecoxib | COX-2 | 5–25 (Anikwue et al., 2002; Schmelzer et al., 2006) | 100 |
| Rofecoxib | COX-2 | 6–8 (Schmelzer et al., 2006; Bhat et al., 2008) | 10 |
| Valdecoxib | COX-2 | 0.03–5.9 (Talley et al., 2000; Gierse et al., 2005) | 10 |
| Etoricoxib | COX-2 | 0.58 (Singh et al., 2006) | 20 |
| Lumiracoxib | COX-2 | 0.4–5 (Esser et al., 2005) | 50 |
| Methyl-celecoxib | – | – | 100 |
Tissue preparation and micrography
Following short-term anaesthesia with isoflurane, mice were weighed and killed by cardiac puncture. Kidneys were immediately removed, weighed, transversally hemisected and immersion-fixed overnight in 4% paraformaldehyde/phosphate buffered saline. Thereafter kidney tissue was embedded in paraffin and 4 µm sections were cut, dewaxed in xylene and stained with haematoxylin and eosin. Three kidney sections at least 50–60 µm apart from three individual animals from each group have been selected. Photomicrographs were obtained using a Nikon microscope (Nikon GmbH, Düsseldorf, Germany) and the Kappa image version software and a total of 300 glomeruli chosen at random from the superficial cortex, mid-cortex and juxtamedullary region was digitized for each group. The cross-sectional diameters of glomeruli were measured and the distances from the kidney edge to Bowman's capsule were measured perpendicularly to the renal capsule using NIH ImageJ software. In addition diameters of juxtamedullary glomeruli located close to the medullary border were recorded separately. Glomeruli that appeared to be cut tangentially were excluded from the observation group. To obtain the distribution of glomeruli sizes, values of all diameters obtained were categorized according to their size and arranged in ‘bins’ of 5 µm steps, up to 65–70 µm. Data are presented as the amount in each bin as per cent of the total amount. To count for relative amount of superficial glomeruli, we set a distance limit of 58 µm from the cortical edge, defined as the sum of the distance of glomeruli located nearest to the surface plus one mean diameter of glomeruli in control kidneys. To determine luminal diameter of cortical tubules a total of 150 transversally cut tubules were randomly chosen, digitized and measured perpendicularly to luminal wall.
Ex vivo COX-2 assay
Mice were treated with vehicle or coxibs from day P1 to P21. Four hours following the last injection 100 µL heparinized blood was taken and inhibition of COX-2 activity was measured by ex vivo assay (Patrignani et al., 1994). Briefly, vehicle or lipopolysaccharide (LPS) at a final concentration of 10 µg·mL−1 were added and samples were incubated at 37°C for 20 h. Blood samples were centrifuged and PGE2 concentration was determined in the supernatant by enzyme immunoassay. Data were expressed relative to control blood samples from vehicle-treated mice.
Histochemistry and immunohistochemistry
Sections of fixed renal tissue were deparaffinized, rehydrated and stained with Sirius red (Science Services, Munich, Germany) to evaluate fibrosis by determination of the area of basement of Bowman's capsule. For each group 30 glomeruli were chosen randomly and digitally imaged. Glomeruli that appeared to be cut tangentially were excluded from the observation group. Quantitative analysis of the pictures was performed, without knowledge of the treatments, using Adobe Photoshop CS2 and QuantityOne software (Bio-Rad), which allows counting of the pixels stained specifically. The results are given as stained area of basement of Bowman's capsule, normalized to glomerulus diameter.
Immunohistochemical studies for the detection of macrophages and the proliferating cell nuclear antigen (PCNA) were performed as follows. Deparaffinized sections were incubated in a humidified atmosphere with the following primary antibodies: rat anti-mouse F4/80 (AbD Serotec, Oxford, UK), which reacts with interstitial macrophages, and rabbit anti-PCNA antibody (Santa Cruz Biotechnology, CA). Prior to application of F4/80 antibody sections were treated with target retrieval solution (DAKO, Hamburg, Germany). For visualization of F4/80 DAKO ABC System was used and for PCNA Vectastain Elite System (Vector Laboratories, Burlingame, CA) according to the protocols of the suppliers. Sections were counterstained with haematoxylin. For all samples, negative controls for the immunohistochemical procedures included substitution of the primary antibodies with non-immune sera. Analysis was performed by an operator unaware of the origin of each kidney section. For PCNA and macrophage determination marked cells per index field were counted. Under a light microscope at 200× magnification, five to eight non-overlapping fields (to obtain around 90% of the cortical kidney section) per kidney section were captured with a numeric camera connected to the microscope. Results are presented as positive cells per field.
Statistics
Results were expressed as mean ± SEM. anova with Bonferroni's multiple comparison post hoc analysis using Prism software (Graphpad) was used to determine statistical differences between multiple groups. P < 0.05 was considered statistically significant.
Results
In order to study alterations in postnatal mouse nephrogenesis caused by selective and non-selective inhibitors, targeting the two isoforms of COX, we treated mice with COX inhibitors from postnatal day P1 to P21 and determined the following kidney characteristics at day P21: (i) ratio of kidney weight to body weight, which gives an estimate of the relative organ growth; (ii) glomerular and cortical tubular diameter; (iii) distance of superficial glomeruli to the cortical edge, which gives an estimate of the subcapsular cortical growth; (iv) relative amount of superficial glomeruli within 58 µm of the cortical edge to give a measure for maturational arrest of newly formed nephrons in the outer cortex; (v) the size distribution of glomeruli, disclosing relative hypertrophy of glomeruli; (vi) size of juxtamedullary glomeruli to establish whether these early differentiated glomeruli are also affected by COX inhibition; and (vii) number of interstitial macrophages, proliferating cells and periglomerular fibrosis. Histomorphological observations in mice treated with COX inhibitors were compared with data collected in vehicle-treated control mice.
We studied COX-2−/− mice as positive controls for renal maldevelopment. These mice presented significantly altered kidney characteristics compared with control mice (COX-2+/+) at day P21. The kidney to body weight ratio of COX-2−/− mice was significantly lower (Table 2) but ratio of heart weight to body weight was unaltered (data not shown). Regarding the size of glomeruli, we observed a significant reduction in mean diameter, which gave a reduced mean volume of glomeruli (assuming a spherical form for glomeruli) from 36 679 ± 1762 µm3 to 6835 ± 536 µm3. Cortical thickness was markedly decreased and the number of glomeruli in the outer cortex within 58 µm to the cortical edge was significantly increased (Table 2). Analysis of size distribution of glomeruli revealed an asymmetric shift to the left with a shoulder on the right, indicating the presence of relatively hypertrophic glomeruli (Figure 1), as reported before (Kömhoff et al., 2000; Norwood et al., 2000). No significant difference in any of our renal characteristics was observed, comparing COX-2+/+ mice to C57BL6 mice (data not shown).
Table 2.
Histomorphological analysis of mouse kidneys
| COX-2+/+ | COX-2−/− | C57BL6 + SC-236 | C57BL6 + SC-560 | C57BL6 + DMC | |
|---|---|---|---|---|---|
| Kidney to body weight ratio | 17.53 ± 0.56 | 9.22 ± 0.23* | 11.50 ± 0.41* | 16.50 ± 0.29 | 15.87 ± 0.44 |
| Diameter of glomeruli (µm) | 41.23 ± 0.65 | 23.55 ± 0.60* | 28.87 ± 0.69* | 37.80 ± 1.10 | 39.35 ± 1.32 |
| Subcapsular cortical thickness (µm) | 43.60 ± 1.03 | 10.73 ± 0.32* | 15.59 ± 2.08* | 45.62 ± 3.56 | 45.92 ± 2.23 |
| Relative amount of superficial glomeruli (%) | 11.62 ± 0.89 | 68.51 ± 2.03* | 40.58 ± 1.47* | 12.73 ± 1.48 | 11.40 ± 1.68 |
P < 0.05 versus COX-2+/+.
DMC, methyl-celecoxib.
Figure 1.
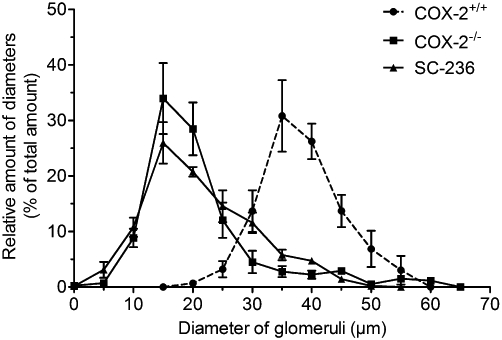
Size distribution of glomeruli from COX-2−/− mice. Mouse kidneys were harvested at P21 and stained with haematoxylin/eosin. Diameters of glomeruli were measured and their relative frequency was determined in 5 µm bins. The peak size shifted in kidneys from COX-2−/− mice to 15 µm, compared with 35 µm in control mice. Please note the presence of hypertrophic glomeruli (>50 µm) in COX-2−/− kidneys.
As in COX-2−/− mice, renal histological alterations were observed following administration of the COX-2 selective inhibitor SC-236 to wild-type C57BL6 mice from day P1 to P21 (Table 2). Interestingly in contrast to kidneys from COX-2−/− mice, hypertrophic glomeruli were not observed (Figure 1). Treatment of wild-type mice with SC-560, a selective inhibitor of COX-1 did not induce any morphological changes in the kidney. We also studied the effect of dimethyl-celecoxib, which is structurally related to the selective COX-2 inhibitor celecoxib but exhibits only minimal COX-2 inhibitory activity and found it did not show any effects on normal renal morphology (Table 2) in mice.
It is not known whether coxibs, especially the clinically available coxibs, and non-specific NSAIDs would also interfere with renal development. We studied the effect of equipotent anti-nociceptive doses of the coxibs celecoxib, rofecoxib, valdecoxib, lumiracoxib and etoricoxib and of the NSAIDs diclofenac and naproxen, two widely used analgesic drugs. In order to test for inhibition of COX-2 activity we measured synthesis of PGE2 in LPS-stimulated heparinized blood samples. All the coxibs and NSAIDs, used in our study, strongly suppressed COX-2 activity, as reflected by inhibition of LPS-induced PGE2 synthesis (Figure 2). At the doses used, PGE2 formation was suppressed by more than 80%, and no significant differences in inhibitory potency were observed between the various COX inhibitors.
Figure 2.
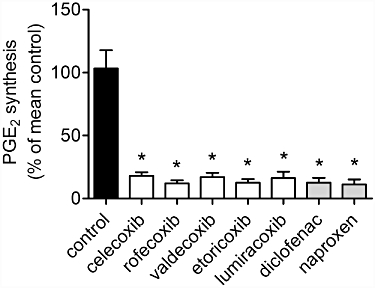
Inhibition of COX-2 activity determined by ex vivo blood assay. Heparinized blood was taken from mice after administration of COX inhibitors or vehicle from day P1 to P21 and incubated with or without LPS (10 µg·mL−1) for 20 h. Following centrifugation of blood cells, PGE2 concentrations in the supernatants were determined by enzyme immunoassay. Data represent mean ± SEM of three independent experiments, *P < 0.05 versus control. No significant difference between the COX inhibitor-treated groups was observed.
All COX inhibitors tested under our experimental conditions affected mouse nephrogenesis, albeit to different extents. For instance, the kidney to body weight ratios for mice treated with celecoxib and valdecoxib were similar to control values (Figure 3). In contrast, administration of rofecoxib, lumiracoxib and etoricoxib and also of diclofenac and naproxen resulted in a significant reduction in kidney mass (Figure 3), expressed by lowered ratio. Ratios of heart weight to body weight were unaltered by administration of COX inhibitors (data not shown).
Figure 3.
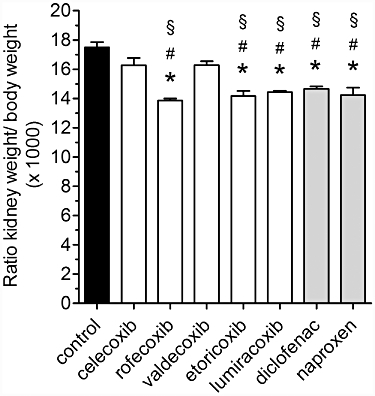
Ratio of kidney weight/body weight in mice treated with COX inhibitors. Treatment of mice with the indicated coxib or NSAID, at doses as shown in Table 1, was from P1 to P21. Following measurement of body weight, blood-free kidneys were removed and weighed. Data represent mean ± SEM of three independent experiments, *P < 0.05 versus control; #P < 0.05 versus celecoxib; §P < 0.05 versus valdecoxib.
In accordance with previous reports on COX-2−/− mice, following COX inhibition in wild-type mice, histologically poorly developed, condensed glomeruli were observed in the subcapsular space (Figure 4). The mean diameters of glomeruli were significantly reduced in all inhibitor groups (Figure 5A) in comparison with the control group values. The most marked effects were observed after treatment with rofecoxib, etoricoxib, diclofenac and naproxen, which lowered glomerulus size significantly compared with the sizes after celecoxib or valdecoxib (Figure 5A). Mean luminal diameters of cortical tubules in COX inhibitor-treated groups were not significantly different from control-treated kidneys (Table 3). We evaluated renal cell proliferation by immunostaining for PCNA as a measure of undifferentiated cells. A very small number of PCNA-positive cells were found in cortical sections of control kidneys with no difference in coxib-treated kidneys. In contrast, treatment with naproxen and diclofenac caused a slight but significant increase in PCNA-positive cells (Table 3).
Figure 4.
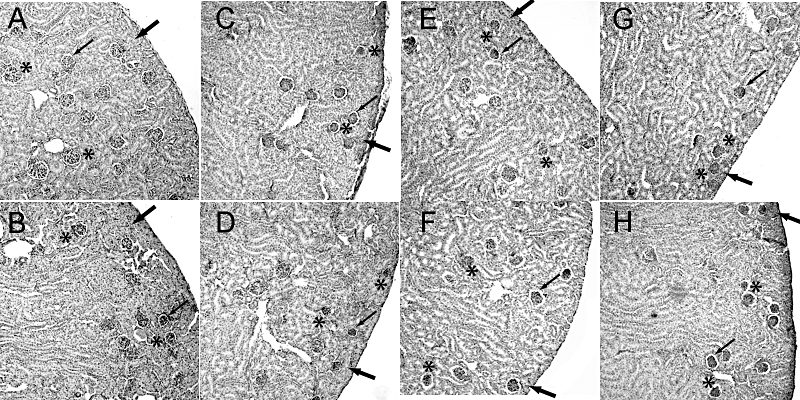
Renal morphological alterations in mice treated with coxibs or NSAIDs. COX inhibitors were given in the doses indicated in Table 1 from P1 to P21. Kidneys were harvested at P21, fixed and stained with haematoxylin/eosin. (A) Control, (B) celecoxib, (C) rofecoxib, (D) etoricoxib, (E) valdecoxib, (F) lumiracoxib, (G) diclofenac, (H) naproxen. A representative cortical section is shown, magnification 200×. In control animals, glomeruli exhibited a mature morphology (indicated by asterisk) with minimal space between the glomerular and Bowman's capsule (indicated by thin arrow) and several layers of tubules occupy the space between the renal capsule and the subcapsular glomeruli (indicated by thick arrow). In COX inhibitor-treated animals (B–H) glomeruli exhibit reduced size (indicated by asterisk), condensed glomerular tuft partially with increased urinary space of Bowman's capsule (indicated by thin arrow), and, except for celecoxib (B), reduced thickness of subcapsular tubules (indicated by thick arrow).
Figure 5.
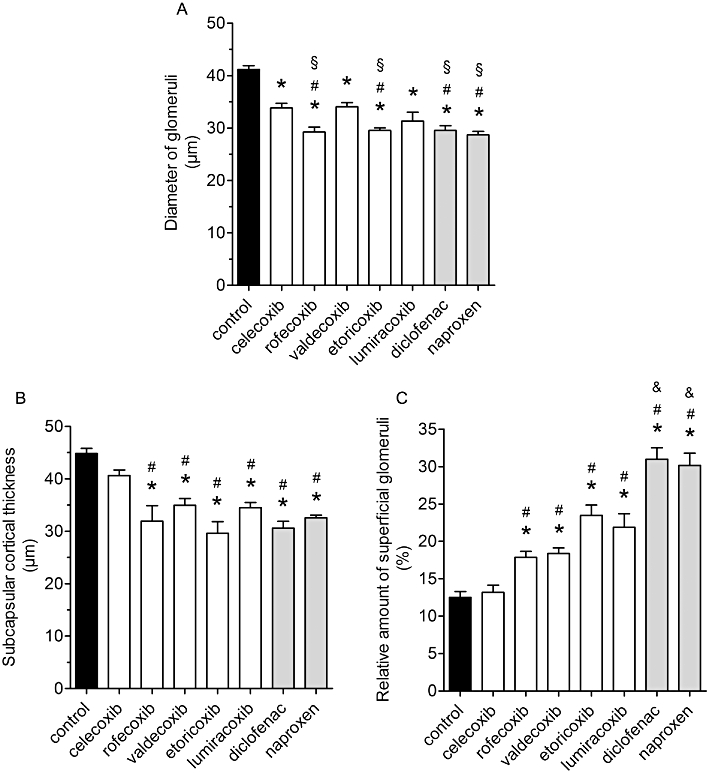
Impairment of postnatal renal development by treatment with coxibs and NSAIDs. COX inhibitors were given in the doses indicated in Table 1 from postnatal day P1 to P21. Thereafter kidneys were removed, fixed in PFA and histomorphometrically analysed. (A) Diameter of glomeruli, (B) subcapsular cortical thickness, (C) relative amount of superficial glomeruli within 58 µm from the renal cortical edge. Data represent mean ± SEM of three independent experiments, *P < 0.05 versus control; #P < 0.05 versus celecoxib; §P < 0.05 versus valdecoxib; &P < 0.05 versus coxibs.
Table 3.
Effect of treatment with COX inhibitors on relative number of cortical macrophages (F4/80-positive), proliferating cells (PCNA-positive), fibrosis (area of basement membrane of Bowman's capsule) and diameter of cortical tubules in mouse kidney sections
| Substance | Number of cortical F4/80-positive cells per index field | Number of PCNA-positive cells per index field | Area of basement membrane (arbitrary units) | Diameter of cortical tubules (µm) |
|---|---|---|---|---|
| Control | 11.2 ± 0.8 | 1.5 ± 0.3 | 0.25 ± 0.03 | 20.67 ± 1.08 |
| SC-236 | 14.5 ± 1.9 | 2.8 ± 0.3 | 0.76 ± 0.11* | 19.15 ± 0.78 |
| DMC | 11.0 ± 1.4 | 0.9 ± 0.2 | 0.23 ± 0.04 | 20.68 ± 1.10 |
| Celecoxib | 12.1 ± 1.1 | 1.4 ± 0.4 | 0.32 ± 0.09 | 20.49 ± 1.33 |
| Rofecoxib | 13.0 ± 1.2 | 2.9 ± 0.5 | 0.85 ± 0.13* | 20.37 ± 1.19 |
| Valdecoxib | 13.5 ± 1.3 | 3.8 ± 0.4 | 0.47 ± 0.11 | 20.28 ± 0.24 |
| Etoricoxib | 13.1 ± 1.2 | 2.0 ± 0.3 | 0.71 ± 0.12 | 19.40 ± 0.70 |
| Lumiracoxib | 12.4 ± 0.9 | 0.9 ± 0.2 | 0.65 ± 0.09 | 19.27 ± 0.23 |
| Naproxen | 9.2 ± 0.9 | 6.7 ± 0.7* | 0.66 ± 0.14 | 19.20 ± 0.86 |
| Diclofenac | 10.2 ± 0.8 | 5.9 ± 0.6* | 0.70 ± 0.13 | 19.14 ± 0.41 |
P < 0.05 versus control.
DMC, methyl-celecoxib; PCNA, proliferating cell nuclear antigen.
Regarding subcapsular cortical growth and relative amount of superficial glomeruli, celecoxib treatment gave values indistinguishable from the control values (Figure 5B, C). All other coxibs significantly reduced cortical thickness of cortical mantle and increased the relative amount of outmost superficial glomeruli (Figure 5B, C). The percentage of superficial glomeruli was significantly higher in kidneys from diclofenac- and naproxen-treated mice than in those from mice treated with the coxibs (Figure 5C).
The size distribution of the glomeruli was affected by all coxibs, resulting in a shift to the left with nearly Gaussian distribution (Figure 6A). The most marked effects were observed following treatment with rofecoxib, etoricoxib, lumiracoxib, diclofenac and naproxen, with intermediate left-wards shift after celecoxib or valdecoxib. Furthermore, all inhibitors reduced the size of juxtamedullary glomeruli, in comparison with vehicle treatment. As shown in Figure 6B, although treatment with celecoxib or valdecoxib decreased the diameter of juxtamedullary glomeruli, the other COX inhibitors were more effective and significantly different from control or celecoxib.
Figure 6.
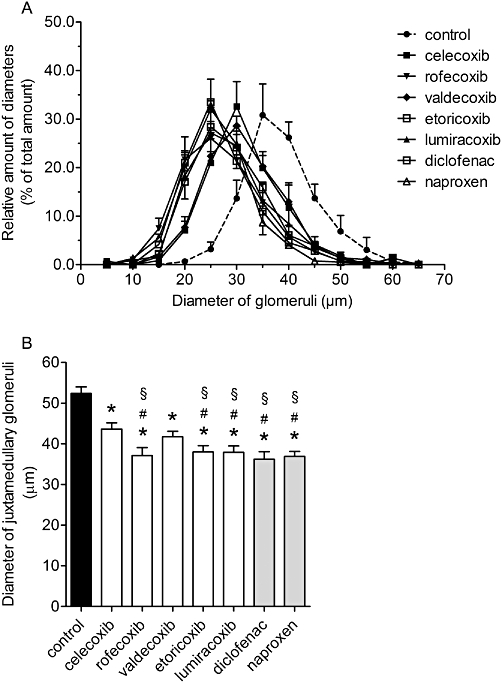
Relative distribution of diameters of glomeruli and impairment of growth of juxtamedullary glomeruli after treatment with coxibs and NSAIDs. COX inhibitors were given in the doses indicated in Table 1 from postnatal day P1 to P21. Thereafter kidneys were removed, fixed in PFA and histomorphometrically analysed. (A) Relative distribution of diameters of glomeruli and (B) diameter of juxtamedullary glomeruli. Data represent mean ± SEM of three independent experiments, *P < 0.05 versus control; #P < 0.05 versus celecoxib; §P < 0.05 versus valdecoxib.
In order to evaluate inflammatory infiltrates we determined the number of macrophages by F4/80 staining. In kidney sections from control mice, low levels of interstitial macrophage infiltration were detected but no difference was found following treatment with coxibs or NSAIDs (Table 3). Periglomerular fibrosis was studied in Sirius red stained kidney sections. Although all COX inhibitors caused a slight increase in the mean area of basement membrane of Bowman's capsules, only the staining in kidneys after SC-236 or rofecoxib treatment reached significance, in comparison with control (Table 3).
Discussion and conclusions
Inhibitors of COX are among the most widely used drugs, but also induce the greatest number of unwanted side effects, often causing hospitalizations (Wolfe et al., 1999). Adverse effects have been reported for non-selective COX, as well as for COX-2 selective, inhibitors. Renal side effects are associated with oedema, kidney hypoperfusion, hypertension and renal maldevelopment (Harirforoosh and Jamali, 2009). Although it has become evident that COX-2 deficiency causes renal dysgenesis in mice (Dinchuk et al., 1995; Morham et al., 1995), less information is available regarding the potency of classical NSAID and of clinically used coxibs to impair renal development, especially at analgesic doses. In the present study we focused on the effect of equipotent analgesic dosages of COX selective and non-selective inhibitors on postnatal kidney development in mice. Our observations let us draw two major conclusions. First, clear differences do exist between different coxibs in causing renal developmental defects with celecoxib being most ‘safe’, and rofecoxib being least. Second, the classical NSAIDs, diclofenac and naproxen, caused the most marked defects, overall, in renal development, among all inhibitors used in this study.
The metanephric kidney undergoes two basic phases of growth, with a considerable overlap among these phases (Kleinman, 1982). The first phase is that of nephrogenesis, the development of new nephrons, which begins in mice at embryonic day 11 and ends 10 days postpartum. The second phase of growth is the anatomical maturation and growth of the nephrons already present, especially an increase in glomerular size and elongation of the tubule system leading to enlargement of subcapsular cortical region and kidney mass. During nephrogenesis, morphological and functional differentiation of nephrons proceeds centrifugally. Thus, in the developing kidney, the first nephrons to form are in the juxtamedullary area and those nephrons located in the outermost region of the cortex are the youngest and least differentiated morphologically (see Horster et al., 1999). All COX inhibitors used in this study affected various aspects of renal development.
As a reference compound for COX-2 inhibition, we used SC-236 as it is known to induce renal dysgenesis in mice (Kömhoff et al., 2000). The effect of SC-236 on kidney growth retardation, glomerular size and cortical area was similar to, but not as strong as, the effect of genetic deletion of the COX-2 gene. Three explanations may be offered. First, SC-236 did not completely block renal COX-2 activity. Second, peripartum COX-2 activity already contributes to renal development. Third, partial compensation for the block of prostaglandins via COX-2 is provided via the remaining COX-1 activity. Support for this last possibility is given by our observation that the non-selective COX inhibitors (which block both COX-1 and COX-2) caused, in some aspects, stronger renal defects than those caused by the coxibs. However, administration of the COX-1 selective inhibitor SC-560 caused no detectable changes in renal morphology, consistent with earlier observations (Kömhoff et al., 2000).
Striking differences in the extent of renal defects were observed between the coxibs and NSAIDs, although equipotent doses, corresponding to the ED80 doses to block hyperalgesia were given and all the COX inhibitors used gave equally extensive (more than 80%) inhibition of the LPS-induced COX-2 activity in blood samples. Compared with control mice, the least detrimental effects on nephrogenesis were recorded for celecoxib, which affected size of glomeruli but not nephron growth or kidney mass. DMC, a structural derivative of celecoxib with no COX-2 inhibitory activity (Kardosh et al., 2005), did not impair renal development, indicating that the effect of celecoxib depends on COX-2 inhibition.
In contrast, treatment with rofecoxib strongly diminished size of glomeruli, subcapsular cortical mass, kidney weight, but induced maturational arrest of superficial glomeruli and periglomerular fibrosis, thereby exhibiting the strongest effects of all the coxibs tested. For valdecoxib, lumiracoxib and etoricoxib, we observed an intermediate impact on renal development. Even though equi-analgesic doses were used, accessibility to renal COX-2 and inhibitory effects on renal COX-2 enzyme activity may differ among the coxibs and provide some explanation for the observed differences in effects on renal development.
The impairment of nephrogenesis by the classical NSAIDs diclofenac and naproxen was comparable to that induced by rofecoxib, in terms of attenuation of glomerular and subcapsular cortical growth. Moreover, both diclofenac and naproxen induced a significantly higher percentage of superficially located glomeruli, indicating a stronger effect on cortical maturation and growth. Compatible with this assumption is our observation that among COX inhibitors used in our study, diclofenac and naproxen held a small number of renal cells in an undifferentiated state, as shown by the increase in PCNA-positive cell number. In general, PCNA expression is restricted to the nephrogenic zone and is down-regulated rapidly as renal epithelial cells differentiate and acquire functional characteristics (Saifudeen et al., 2002).
Apart from the cortical glomeruli, the juxtamedullary glomeruli were also affected by coxibs and NSAIDs, and here growth inhibition was significantly stronger for rofecoxib, etoricoxib, lumiracoxib, diclofenac and naproxen. This indicates that also the maturation program of the earliest glomeruli during nephrogenesis is modulated by COX activity. In contrast to this observation, in COX-2−/− mice, juxtamedullary glomeruli were observed to be relatively hypertrophic (Kömhoff et al., 2000). One explanation may be that embryonic loss of COX activity activates compensatory mechanisms, which do not operate when there is postnatal inhibition of COX.
Regarding the differences in renal defects caused by coxibs and NSAIDs, we postulate that prostanoids exercise different and subtle control on cortical tubule growth and on maturation of superficial and juxtamedullary glomeruli. From a clinical point of view, the risk of renal maldevelopment may differ between different coxibs and these renal side effects may be more likely to occur following the use of NSAIDs than after the coxibs.
Acknowledgments
This study was supported by the Deutsche Forschungsgemeinschaft (Grant Nu73/10–1).
Glossary
Abbreviations
- COX
cyclooxygenase
- coxib
selective cyclooxygenase-2 inhibitor
- DMC
methyl-celecoxib
- LPS
lipopolysaccharide
- NSAIDs
non-steroidal anti-inflammatory drugs
- PCNA
proliferating cell nuclear antigen
Conflicts of interest
None.
Supporting Information
Teaching Materials; Figs 1–6 as PowerPoint slide.
References
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 4th edn. Br J Pharmacol. 2009;158(Suppl 1):S1–S254. doi: 10.1111/j.1476-5381.2009.00499.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anikwue R, Huffman JW, Martin ZL, Welch SP. Decrease in efficacy and potency of nonsteroidal anti-inflammatory drugs by chronic delta(9)-tetrahydrocannabinol administration. J Pharmacol Exp Ther. 2002;303:340–346. doi: 10.1124/jpet.303.1.340. [DOI] [PubMed] [Google Scholar]
- Bhat AS, Tandan SK, Kumar D, Krishna V, Prakash VR. The interaction between inhibitors of nitric oxide synthase and cyclooxygenase in formalin-induced pain in mice: an isobolographic study. Anesth Analg. 2008;106:978–984. doi: 10.1213/ane.0b013e318163f71b. table of contents. [DOI] [PubMed] [Google Scholar]
- Dinchuk JE, Car BD, Focht RJ, Johnston JJ, Jaffee BD, Covington MB, et al. Renal abnormalities and an altered inflammatory response in mice lacking cyclooxygenase II. Nature. 1995;378:406–409. doi: 10.1038/378406a0. [DOI] [PubMed] [Google Scholar]
- Esser R, Berry C, Du Z, Dawson J, Fox A, Fujimoto RA, et al. Preclinical pharmacology of lumiracoxib: a novel selective inhibitor of cyclooxygenase-2. Br J Pharmacol. 2005;144:538–550. doi: 10.1038/sj.bjp.0706078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flower RJ. The development of COX2 inhibitors. Nat Rev Drug Discov. 2003;2:179–191. doi: 10.1038/nrd1034. [DOI] [PubMed] [Google Scholar]
- Gierse JK, Zhang Y, Hood WF, Walker MC, Trigg JS, Maziasz TJ, et al. Valdecoxib: assessment of cyclooxygenase-2 potency and selectivity. J Pharmacol Exp Ther. 2005;312:1206–1212. doi: 10.1124/jpet.104.076877. [DOI] [PubMed] [Google Scholar]
- Guan Y, Chang M, Cho W, Zhang Y, Redha R, Davis L, et al. Cloning, expression, and regulation of rabbit cyclooxygenase-2 in renal medullary interstitial cells. Am J Physiol. 1997;273:F18–F26. doi: 10.1152/ajprenal.1997.273.1.F18. [DOI] [PubMed] [Google Scholar]
- Hao CM, Breyer MD. Physiological regulation of prostaglandins in the kidney. Annu Rev Physiol. 2008;70:357–377. doi: 10.1146/annurev.physiol.70.113006.100614. [DOI] [PubMed] [Google Scholar]
- Harirforoosh S, Jamali F. Renal adverse effects of nonsteroidal anti-inflammatory drugs. Expert Opin Drug Saf. 2009;8:669–681. doi: 10.1517/14740330903311023. [DOI] [PubMed] [Google Scholar]
- Harris RC, Breyer MD. Update on cyclooxygenase-2 inhibitors. Clin J Am Soc Nephrol. 2006;1:236–245. doi: 10.2215/CJN.00890805. [DOI] [PubMed] [Google Scholar]
- Harris RC, McKanna JA, Akai Y, Jacobson HR, Dubois RN, Breyer MD. Cyclooxygenase-2 is associated with the macula densa of rat kidney and increases with salt restriction. J Clin Invest. 1994;94:2504–2510. doi: 10.1172/JCI117620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Heijden BJ, Carlus C, Narcy F, Bavoux F, Delezoide AL, Gubler MC. Persistent anuria, neonatal death, and renal microcystic lesions after prenatal exposure to indomethacin. Am J Obstet Gynecol. 1994;171:617–623. doi: 10.1016/0002-9378(94)90073-6. [DOI] [PubMed] [Google Scholar]
- Horster MF, Braun GS, Huber SM. Embryonic renal epithelia: induction, nephrogenesis, and cell differentiation. Physiol Rev. 1999;79:1157–1191. doi: 10.1152/physrev.1999.79.4.1157. [DOI] [PubMed] [Google Scholar]
- Kaplan BS, Restaino I, Raval DS, Gottlieb RP, Bernstein J. Renal failure in the neonate associated with in utero exposure to non-steroidal anti-inflammatory agents. Pediatr Nephrol. 1994;8:700–704. doi: 10.1007/BF00869093. [DOI] [PubMed] [Google Scholar]
- Kardosh A, Wang W, Uddin J, Petasis NA, Hofman FM, Chen TC, et al. Dimethyl-celecoxib (DMC), a derivative of celecoxib that lacks cyclooxygenase-2-inhibitory function, potently mimics the anti-tumor effects of celecoxib on Burkitt's lymphoma in vitro and in vivo. Cancer Biol Ther. 2005;4:571–582. doi: 10.4161/cbt.4.5.1699. [DOI] [PubMed] [Google Scholar]
- Kleinman LI. Developmental renal physiology. Physiologist. 1982;25:104–110. [PubMed] [Google Scholar]
- Koki A, Khan NK, Woerner BM, Dannenberg AJ, Olson L, Seibert K, et al. Cyclooxygenase-2 in human pathological disease. Adv Exp Med Biol. 2002;507:177–184. doi: 10.1007/978-1-4615-0193-0_28. [DOI] [PubMed] [Google Scholar]
- Kömhoff M, Gröne HJ, Klein T, Seyberth HW, Nüsing RM. Localization of cyclooxygenase-1 and -2 in adult and fetal human kidney: implication for renal function. Am J Physiol. 1997;272:F460–F468. doi: 10.1152/ajprenal.1997.272.4.F460. [DOI] [PubMed] [Google Scholar]
- Kömhoff M, Wang JL, Cheng HF, Langenbach R, McKanna JA, Harris RC, et al. Cyclooxygenase-2-selective inhibitors impair glomerulogenesis and renal cortical development. Kidney Int. 2000;57:414–422. doi: 10.1046/j.1523-1755.2000.00861.x. [DOI] [PubMed] [Google Scholar]
- Miranda HF, Puig MM, Prieto JC, Pinardi G. Synergism between paracetamol and nonsteroidal anti-inflammatory drugs in experimental acute pain. Pain. 2006;121:22–28. doi: 10.1016/j.pain.2005.11.012. [DOI] [PubMed] [Google Scholar]
- Morham SG, Langenbach R, Loftin CD, Tiano HF, Vouloumanos N, Jennette JC, et al. Prostaglandin synthase 2 gene disruption causes severe renal pathology in the mouse. Cell. 1995;83:473–482. doi: 10.1016/0092-8674(95)90125-6. [DOI] [PubMed] [Google Scholar]
- Norwood VF, Morham SG, Smithies O. Postnatal development and progression of renal dysplasia in cyclooxygenase-2 null mice. Kidney Int. 2000;58:2291–2300. doi: 10.1046/j.1523-1755.2000.00413.x. [DOI] [PubMed] [Google Scholar]
- Patrignani P, Panara MR, Greco A, Fusco O, Natoli C, Iacobelli S, et al. Biochemical and pharmacological characterization of the cyclooxygenase activity of human blood prostaglandin endoperoxide synthases. J Pharmacol Exp Ther. 1994;271:1705–1712. [PubMed] [Google Scholar]
- Penning TD, Talley JJ, Bertenshaw SR, Carter JS, Collins PW, Docter S, et al. Synthesis and biological evaluation of the 1,5-diarylpyrazole class of cyclooxygenase-2 inhibitors: identification of 4-[5-(4-methylphenyl)-3-(trifluoromethyl)-1H-pyrazol-1-yl]benze nesulfonamide (SC-58635, celecoxib) J Med Chem. 1997;40:1347–1365. doi: 10.1021/jm960803q. [DOI] [PubMed] [Google Scholar]
- Saifudeen Z, Marks J, Du H, El-Dahr SS. Spatial repression of PCNA by p53 during kidney development. Am J Physiol Renal Physiol. 2002;283:F727–F733. doi: 10.1152/ajprenal.00114.2002. [DOI] [PubMed] [Google Scholar]
- Schiffmann S, Maier TJ, Wobst I, Janssen A, Corban-Wilhelm H, Angioni C, et al. The anti-proliferative potency of celecoxib is not a class effect of coxibs. Biochem Pharmacol. 2008;76:179–187. doi: 10.1016/j.bcp.2008.04.017. [DOI] [PubMed] [Google Scholar]
- Schmelzer KR, Inceoglu B, Kubala L, Kim IH, Jinks SL, Eiserich JP, et al. Enhancement of antinociception by coadministration of nonsteroidal anti-inflammatory drugs and soluble epoxide hydrolase inhibitors. Proc Natl Acad Sci USA. 2006;103:13646–13651. doi: 10.1073/pnas.0605908103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seta F, Chung AD, Turner PV, Mewburn JD, Yu Y, Funk CD. Renal and cardiovascular characterization of COX-2 knockdown mice. Am J Physiol Regul Integr Comp Physiol. 2009;296:R1751–R1760. doi: 10.1152/ajpregu.90985.2008. [DOI] [PubMed] [Google Scholar]
- Simmons DL, Botting RM, Hla T. Cyclooxygenase isozymes: the biology of prostaglandin synthesis and inhibition. Pharmacol Rev. 2004;56:387–437. doi: 10.1124/pr.56.3.3. [DOI] [PubMed] [Google Scholar]
- Singh VP, Patil CS, Kulkarni SK. Analysis of interaction between etoricoxib and tramadol against mechanical hyperalgesia of spinal cord injury in rats. Life Sci. 2006;78:1168–1174. doi: 10.1016/j.lfs.2005.06.024. [DOI] [PubMed] [Google Scholar]
- Smith W. Prostanoid biosynthesis and mechanism of action. Am J Physiol. 1992;263:F181–F191. doi: 10.1152/ajprenal.1992.263.2.F181. [DOI] [PubMed] [Google Scholar]
- Smith WL, DeWitt DL, Garavito RM. Cyclooxygenases: structural, cellular, and molecular biology. Annu Rev Biochem. 2000;69:145–182. doi: 10.1146/annurev.biochem.69.1.145. [DOI] [PubMed] [Google Scholar]
- Talley JJ, Brown DL, Carter JS, Graneto MJ, Koboldt CM, Masferrer JL, et al. 4-[5-Methyl-3-phenylisoxazol-4-yl]- benzenesulfonamide, valdecoxib: a potent and selective inhibitor of COX-2. J Med Chem. 2000;43:775–777. doi: 10.1021/jm990577v. [DOI] [PubMed] [Google Scholar]
- Vane JR, Bakhle YS, Botting RM. Cyclooxygenases 1 and 2. Annu Rev Pharmacol Toxicol. 1998;38:97–120. doi: 10.1146/annurev.pharmtox.38.1.97. [DOI] [PubMed] [Google Scholar]
- Voyer LE, Drut R, Mendez JH. Fetal renal maldevelopment with oligohydramnios following maternal use of piroxicam. Pediatr Nephrol. 1994;8:592–594. doi: 10.1007/BF00858136. [DOI] [PubMed] [Google Scholar]
- Wolfe MM, Lichtenstein DR, Singh G. Gastrointestinal toxicity of nonsteroidal antiinflammatory drugs. N Engl J Med. 1999;340:1888–1899. doi: 10.1056/NEJM199906173402407. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.