

Abstract
Defects in mitochondrial DNA (mtDNA) maintenance comprise an expanding repertoire of polymorphic diseases caused, in part, by mutations in the genes encoding the p140 mtDNA polymerase (POLG), its p55 accessory subunit (POLG2) or the mtDNA helicase (C10orf2). In an exploration of nuclear genes for mtDNA maintenance linked to mitochondrial disease, eight heterozygous mutations (six novel) in POLG2 were identified in one control and eight patients with POLG-related mitochondrial disease that lacked POLG mutations. Of these eight mutations, we biochemically characterized seven variants [c.307G>A (G103S); c.457C>G (L153V); c.614C>G (P205R); c.1105A>G (R369G); c.1158T>G (D386E); c.1268C>A (S423Y); c.1423_1424delTT (L475DfsX2)] that were previously uncharacterized along with the wild-type protein and the G451E pathogenic variant. These seven mutations encode amino acid substitutions that map throughout the protein, including the p55 dimer interface and the C-terminal domain that interacts with the catalytic subunit. Recombinant proteins harboring these alterations were assessed for stimulation of processive DNA synthesis, binding to the p140 catalytic subunit, binding to dsDNA and self-dimerization. Whereas the G103S, L153V, D386E and S423Y proteins displayed wild-type behavior, the P205R and R369G p55 variants had reduced stimulation of processivity and decreased affinity for the catalytic subunit. Additionally, the L475DfsX2 variant, which possesses a C-terminal truncation, was unable to bind the p140 catalytic subunit, unable to bind dsDNA and formed aberrant oligomeric complexes. Our biochemical analysis helps explain the pathogenesis of POLG2 mutations in mitochondrial disease and emphasizes the need to quantitatively characterize the biochemical consequences of newly discovered mutations before classifying them as pathogenic.
INTRODUCTION
Mitochondria are ultramicroscopic dynamic organelles required for aerobic life. Each mitochondrion contains 2–10 copies of DNA (mtDNA) and there are hundreds to thousands of mitochondria per mammalian cell (1–3). The large variability of mtDNA copy number in mammals has been linked to different developmental stages (4) and to varying energy demands of different tissues (5). In human cells, DNA polymerase gamma (pol γ) is the only known DNA polymerase responsible for replication and repair of the ∼16.6 kb mtDNA genome (6). Pol γ is a heterotrimer that consists of one 140 kDa catalytic subunit, encoded by the POLG gene, and one ∼110 kDa p55 homodimeric accessory subunit, encoded by the POLG2 gene (7–9). The p55 accessory subunit has been well-characterized biochemically as a DNA-binding protein that confers processivity and physiological salt tolerance on the pol γ catalytic subunit, p140 (10–12). The p55 homodimer binds asymetrically to the catalytic subunit where each monomer of the homodimeric accessory subunit plays a distinct role in conferring processivity on pol γ (9,13). The proximal p55 subunit appears to strengthen the interaction with DNA, while the distal p55 subunit is important for accelerating the nucleotide incorporation step (13). A recent study using human cultured cells by Di Re et al. (14) has also reported that p55 determines the mtDNA content of mitochondrial nucleoids, nucleoprotein complexes found in the mitochondrial matrix, suggesting a new role for p55 in nucleoid organization.
Structurally, the pol γ accessory subunit is distinct when compared with the three other known types of processivity factors. The first type of factors includes the toroidal complexes or ‘donuts’ that encircle and slide along DNA thereby decreasing the dissociation of the holoenzyme from the template. These factors include the Escherichia coli β-sliding clamp, eukaryotic proliferating cell nuclear antigen and the Gp45 trimer of T4 bacteriophage DNA polymerase (15–17). The second type of processivity factor is the ‘partial donut' that include human viral representatives UL44 of the cytomegalovirus DNA polymerase and UL42 of the herpes simplex virus DNA polymerase (18,19). Both of these factors bind to DNA non-specifically and do not require clamp loaders or adenosine triphosphate (ATP) hydrolysis. The third type of processivity factor is exemplified by E. coli thioredoxin of the bacteriophage T7 DNA polymerase, gp5 (20). Thioredoxin causes a conformational change in gp5 that allows basic residues in the polymerase to bind to DNA thereby conferring high processivity on the gp5-thioredoxin holoenzyme (21). The last type of factors includes the unique mitochondrial pol γ accessory subunits. Pol γ accessory subunits from human, mouse, Drosophila and Xenopus all share homology with prokaryotic tRNA synthetases, suggesting a hypothetical model of p55-mediated loading of pol γ onto primers for initiation of mtDNA replication (14,22–24). In vitro, the human minimal mtDNA replisome consists of the heterotrimeric pol γ that works in concert with the mtDNA helicase, encoded by C10orf2, to synthesize thousands of nucleotides of nascent DNA (25). The three genes encoding the minimal replisome components p140, p55 and the helicase have all been implicated in human mitochondrial diseases associated with mtDNA maintenance (3). To date, over 150 mutations in POLG and 40 mutations in C10orf2 have been reported in association with mitochondrial diseases [http://tools.niehs.nih.gov/polg/ (3,26,27)]; however, only two disease-associated mutations have been identified in POLG2 (12,27). A c.1352G>A (G451E) missense mutation in POLG2 was shown to co-segregate with autosomal dominant progressive external opthalmoplegia (adPEO), and biochemical defects in the resulting G451E p55 variant have been described (12). More recently, a heterozygous 24 bp insertion in exon 7 that causes missplicing was found in a patient with late-onset ptosis, myopathy and multiple POLG2 mutations (Table 1) (27). A role for POLG2 in human disease was also suggested by a study of two separate null mutations in the Drosophila melanogaster pol γ accessory subunit gene, which caused lethality in the early pupal stage of fly development (28). Accordingly, we undertook a broad survey for mutations in the POLG2 locus in subjects diagnosed with mitochondrial disease that lacked mutations in POLG.
Table 1.
Mutations identified in the POLG2 gene in mitochondrial disease patients
| Patient | Agea | Gender | cDNA mutation | Amino acid change | Reference and Symptoms |
|---|---|---|---|---|---|
| 1b | 8 years | F | c.307G>A | p.G103S | This study. No symptoms available |
| 2 | 11 years | M | c.457C>G | p.L153V | This study. Headaches/migraines, Peripheral neuropathy, Exercise intolerance/easy fatigue, muscle weakness/DMD-like, easy fatigability, gastrointestinal reflux, delayed gastric emptying |
| 3 | 0.5 year | M | c.614C>G | p.P205R | This study. Irritable, failure to thrive, lethargy, central hypotonia, liver disease, refractory seizures |
| 4 | 19 years | F | c.1105A>G | p.R369G | This study. Exercise intolerance/easy fatigue, muscle weakness/DMD-like, cramps after exercise, easy fatigability, ophthalmoparesis, CPEO, abnormal EMG/NCV, ptosis, gastrointestinal reflux, delayed gastric emptying, apnea/hypoventilation, respiratory deficiency/failure, lactic acidosis, CPK abnormalities, failure to thrive, abnormal histology, abnormal ultrastructure (EM) mitochondria |
| 5 | 11 years | F | c.1158T>G | p.D386E | This study. Developmental delay, dementia/encephalopathy, ataxia, seizures, refractory/intractable seizure, muscle weakness/DMD-like, constipation, high CSF protein, short stature, failure to thrive, microcephaly, abnormal EEG, cerebellar atrophy, abnormal MRI |
| 6 | 1 year | M | c.1158T>G | p.D386E | This study. Hypotonia/floppy baby, hepatic failure, ketosis, lactic acidosis, alopecia |
| 7 | 9 years | M | c.1268C>A | p.S423Y | This study. Developmental delay, hypotonia/floppy baby, seizures, spasticity, gastrointestinal reflux, respiratory deficiency/failure |
| 8c | 2 years | F | c.1247G>C | p.G416A | This study and reference (34). Developmental delay, hypotonia/floppy baby, short stature, dysmorphic features, MRS/lactate peak, abnormal MRI, alopecia. No biochemical defect found and previously published as a possible SNP |
| 9d | 60 years | F | c.1352G>A | p.G451E | Reference (12). adPEO, ptosis, exercise intolerance, muscle pain, mild weakness of facial and limb muscles, impaired glucose intolerance |
| 10 | 8 years | M | c.1423_1424delTT | p.L475DfsX2 | This study. Developmental delay, hypotonia/floppy baby, seizures, constipation, elevated transaminases, ketosis, high CSF protein, abnormal EEG, cerebellar atrophy, abnormal MRI, cortical blindness |
| 11e | 62 years | F | c.1207_1208ins24; c. 505G>A; c.1110+44T>C; c.1191+7_1191+8insT | p.deltaV398-K431 and p.A169T | Reference (27). Bilateral ptosis at 30 years, exercise intolerance, muscle weakness, mild creatine kinase increase and multiple mtDNA deletions |
All mutations were found as heterozygous changes.
aAge at molecular diagnosis.
bSequence determined from a control subject in this study not diagnosed with POLG-related mitochondrial disease.
cPreviously published mutation in POLG2 found in one subject in this study.
dPreviously published mutation in POLG2 not found in this study.
ePreviously published mutations not found in this study with the predicted protein sequence pdeltaV398-K431 based on the observation that POLG2 exon 7 was skipped in cDNA analysis (27). It is unknown whether or not the patient is a complex heterozygote (all mutations on one allele) or whether she is a compound heterozygote (different mutations on different alleles); however, only one of her three unaffected brothers also carried all four mutations suggestive of a complex heterozygote.
POLG-related mitochondrial diseases comprise a continuum of overlapping phenotypes, and the age of onset of these diseases ranges from early childhood to late adulthood. POLG-related disorders are currently defined by six major phenotypes of neurodegenerative disease that include: Alpers–Huttenlocher syndrome, childhood myocerebrohepatopathy spectrum, myoclonic epilepsy myopathy sensory ataxia, the ataxia neuropathy spectrum, autosomal recessive progressive external ophthalmoplegia and adPEO (29,30). Out of 112 samples from patients suspected of POLG-related mitochondrial disorders but with no identifiable mutations in POLG, we explored the possibility of other gene mutations. In this cohort, seven missense mutations and one frameshift mutation in the POLG2 coding region were found in eight subjects and one control. Six of these mutations are novel (Table 1). In the absence of available family genetic history, we adopted a biochemical approach to determine whether these POLG2 variants were contributing to mitochondrial disease. Biochemical characterization of each purified recombinant p55 variant included the ability to associate physically and functionally with p140, determination of intrinsic affinity to double-stranded DNA and analysis of the oligomeric state of the accessory subunits.
RESULTS
Clinical identification of novel heterozygous POLG2 mutations
In 2006, progressive external ophthalmoplegia was shown to be associated with a heterozygous c.1352G>A mutation in POLG2, and biochemical characterization of the resulting G451E-substituted p55 protein indicated defective binding of the catalytic and accessory subunits within pol γ (12). The newfound role of POLG2 mutation in mitochondrial disease and its interaction with the catalytic POLG subunit prompted us to sequence the eight coding exons and the adjacent intronic regions of POLG2.
Based on clinical, biochemical and imaging findings, patient samples were submitted to the Mitochondrial Diagnostic Laboratory at Baylor College of Medicine for suspected POLG-related disorders. Sequence analysis of 112 samples from patients suspected of dysfunctional mtDNA replication did not show any mutations in the POLG gene, thus POLG2 was considered, sequenced and revealed seven variants in eight subjects, all of which were heterozygous for POLG2 (Table 1). These subjects exhibited heterogeneous clinical symptoms similar to POLG-related diseases typically involving dysfunctions of the central nervous and/or neuromuscular systems. Intragenic large deletions in mitochondrial-related nuclear genes were ruled out in patients 3, 4, 6 and 7 by a comparative genomic hybridization analysis on a custom-targeted oligonucleotide array (31,32). Five of the POLG2 mutations were novel: L153V, P205R, R369G, S423Y and L475DfsX2. The frameshift variant, c.1423_1424delTT, deletes two nucleotides in the last exon, resulting in a non-synonymous change at position 475 of the protein and a premature stop codon that truncated 10 carboxyl-terminal amino acid residues (L475DfsX2). The location of the frameshift within the last exon of POLG2 is predicted to escape nonsense-mediated decay in vivo due to the lack of formation of an exon–junction complex downstream of the premature stop (33). The sixth G416A variant was previously reported by our group as a possible neutral single nucleotide polymorphism (SNP) (34). Two subjects harbor the seventh D386E variant, which has been deposited previously in the NCBI ENTREZ Single Nucleotide Polymorphism database without determination of heterozygosity, allele frequency or clinical associations (http://www.ncbi.nlm.nih.gov/projects/SNP/snp_ref.cgi?locusId=11232). Only three of the eight subjects with mitochondrial disease were older than 10 years of age, two 11 years old and one 19 years old (Table 1). We also identified an eighth mutation (the sixth novel) encoding the G103S p55 variant in a subject who was not diagnosed with POLG-related mitochondrial disorder.
This study expands the list of published p55 clinical variants from 4 (G451E, G416A, deltaV398-K431 and A169T) to a total of 11. Interestingly, 7 of the 11 variants (R369G, D386E, deltaV398-K431, G416A, S423Y, G451E and L475DfsX2) are found in domain 3 of the carboxyl–terminal region (Fig. 1A), which is involved in binding the catalytic subunit (9) and has homology to the anticodon binding domain of type IIa aminoacyl-tRNA synthetases (35). Of the remaining four p55 variants, two (L153V and A169T) are found in domain 2 which contains a four-helix bundle necessary for p55 dimerization, whereas the other two (G103S and P205R) are found in domain 1 (Fig. 1A). Domain 1 has homology to the catalytic domain of aminoacyl-tRNA synthetases and is involved in p55 dimerization as well as binding to p140 (9,23,35,36).
Figure 1.

Location of residues altered by mutation within the p55 crystal structure and conservation of these residues among select vertebrates. (A) Structure of the human p55 homodimer. One monomer is white, while the other monomer is colored yellow, green and red to highlight the previously defined domains 1, 2 and 3, respectively (35). Residue Gly451 and those changed by mutation in the current cohort are emphasized in black in only the colored monomer. The figure was generated using the program PyMOL and the published crystal structure PDB ID 2G4C (36). (B) Alignment of vertebrate POLG2 sequences. Substitutions are indicated above the alignment and invariant residues are bold within the alignment. The alignment was generated with ClustalW2 (http://www.ebi.ac.uk/Tools/clustalw2/index.html) using the Human (NP_009146), Bos Taurus (NP_001068659), Mus musculus (NP_056625) and Xenopus laevis (NP_001081904) sequences. Red, AVFPMILW; blue, DE; magenta, RK; green, STYHCNGQ. An asterisk indicates that the residues in that column are identical in all sequences in the alignment; a colon indicates that a substitution is conserved according to the aforementioned color scheme; a period indicates a semi-conserved substitution.
Excluding the previously studied G416A SNP from our analysis, an alignment of primary sequences of four vertebrates revealed evolutionary conservation of seven residues in our cohort (Fig. 1B), and these accessory subunit variants comprise six novel substitutions (G103S, L153V, P205R, R369G, S423Y and L475DfsX2) as well as D386E p55. The G103S substitution found in an asymptomatic subject may exist in the population as a neutral non-pathogenic SNP, similar to the situation of G416A and therefore, we predict that this protein would behave identically to wild-type p55. Five of the seven amino acids that become substituted by mutation in this study are invariant from human to frog. Leu153 differs only in the mouse sequence which harbors a similar hydrophobic Ile, and Ser423 is replaced with Gly only in the frog sequence. To determine the biochemical consequences of these seven variants in comparison to the wild-type protein and the previously studied G451E p55, the recombinant forms of these proteins were overproduced and purified to homogeneity (Fig. 2).
Figure 2.

Purified recombinant p55 variants. Variant forms of p55 were expressed in E. coli BL21(DE3) followed by FPLC purification as described in Materials and Methods. The purified proteins (1 μg per lane) were analyzed by 4–20% SDS–PAGE and stained with Coomassie Brilliant Blue. Lane 1, wild-type p55; lane 2, G103S; lane 3, L153V; lane 4, S423Y; lane 5, R369G; lane 6, P205R; lane 7, L475DfsX2; lane 8, D386E; lane 9, G451E.
Analysis of variant accessory subunits oligomeric states
It was recently shown that each monomer of the homodimeric accessory subunit plays a distinct role in conferring processivity on pol γ (13); therefore, we wished to determine the oligomeric states of each p55 variant. Homodimerization was investigated by analytical gel filtration using a Superdex 200 10/300 GL column (Fig. 3). With the exception of the L475DfsX2 variant, all other p55 variants eluted with a major UV peak at ∼13.6 ml (corresponding to a native MW of 117 000) in good agreement with previous results for the WT homodimer (13). A representative western blot shown above the peak (fractions 13–16) revealed that the UV signal indeed represents wild-type p55 (Fig. 3). The L475DfsX2 p55 truncation variant eluted early in the void volume with a major UV peak at 8.5 ml which is above the calculable MW range of the standard curve. This peak was also verified as recombinant L475DfsX2 p55 by western analysis of fractions 1–6 (Fig. 3). Although the major peak for the truncation mutant eluted at 8.5 ml, the UV signal and the protein are actually spread out over a region of 2.4ml (fractions 1–6), whereas the wild-type and other variant peaks eluting at ∼13.6 ml were only distributed over a 1.6ml region.
Figure 3.

Analysis of accessory subunit oligomerization. Chromatograms from nine separate gel filtration experiments are superimposed on the graph. Chromatography was performed as described under Materials and Methods using a Superdex 200 10/300 GL column and 2 μm wild-type (WT) or variant p55. Shown above the graph are a western blot of L475DfsX2 p55 fractions 1–6 containing a major peak centered at 8.5 ml and a representative blot of WT fractions 13–16 containing a major peak centered at 13.6 ml.
Oligomerization of L475DfsX2 p55 was also investigated via partially denaturing polyacrylamide gel electrophoresis (PAGE; data not shown). L475DfsX2 p55 contained reducible laddered bands and smears not present in wild-type p55 that could represent protein aggregates containing aberrant disulfide bonds. To determine whether protein oxidation in vitro formed aberrant disulfides, the wild-type and L475DfsX2 p55 were also prepared under conditions that contained the reducing agent 2-mercaptoethanol at 10 mm (Materials and Methods). When the gel filtration analysis was repeated on these proteins, the L475DfsX p55 variant continued to elute early in the void volume and remained distributed over a large 3 ml region in comparison to the wild-type p55 which eluted as a dimer identically to conditions without 2-mercaptoethanol (data not shown). Therefore, partial oxidation of the L475DfsX2 polypeptide likely occurred in vivo.
Functional and physical interactions of p55 variants with the catalytic subunit
Previously, we have shown that the G451E variant failed to enhance processivity of the holoenzyme due to an inability to physically associate with the catalytic subunit (12). Therefore, we used an end-labeled primer-extension assay to screen the seven variants for their ability to stimulate p140 in vitro (Fig. 4). As judged by alkaline gel electrophoresis (Fig. 4A, lane 3, and Fig. 4B, lane 2) and by denaturing PAGE (data not shown), p140 can synthesize DNA ∼150–200 nucleotides past the primer in the absence of the accessory subunit. The ability of p140 to extend the primer beyond 100 nucleotides becomes severely inhibited in reactions supplemented with 100 mm NaCl (Fig. 4A, compare lane 4 and lane 3; Fig. 4B, compare lane 4 and lane 2), and significant stalling and/or dissociation was also evident by denaturing PAGE (data not shown; 10). Including wild-type p55 in the reaction significantly stimulates primer extension at low salt (Fig. 4A, lane 5, and Fig. 4B, lane 8), and the higher salt concentration further enhances both the quantity and the length of primer-extension products to as much as ∼7 kb or the full length of the M13mp18 template (Fig. 4A, compare lanes 6 and 5; Fig. 4B, compare lanes 10 and 8). Further enhancement of primer extension by wild-type p55 is apparent at 150 mm NaCl (Fig. 4B, lane 11). Synthesis of ∼600 nucleotide products indicates partial inhibition at 200 mm NaCl (Fig. 4B, lane 12) and nearly complete inhibition at 250 mm NaCl (Fig. 4B, lane 13). The L475DfsX2 p55 variant was unable to stimulate p140 at either low- or high-salt concentrations (Fig. 4A, lanes 19 and 20), similar to effect seen for G451E p55 (Fig. 4A, lanes 21 and 22), although less stimulation was seen with L475DfsX2 p55 than with G451E p55 by denaturing PAGE (data not shown). Compared with wild-type p55, P205R p55 and R369G p55 were clearly unable to stimulate robust DNA synthesis on the M13mp18 template at either 0 or 100 mm NaCl (Fig. 4A, compare lane 5 with lanes 11 and 13; compare lane 6 with lanes 12 and 14). With the exception of G103S p55, which stimulated primer extension somewhat better than wild-type p55, the other variants displayed the same degree of stimulation as wild-type at both 0 and 100 mm NaCl (Fig. 4A).
Figure 4.

Functional interaction of p55 variants with the catalytic subunit. Reactions were analyzed by alkaline gel electrophoresis (Materials and Methods) and contained 2 nm singly primed M13mp18 DNA, 5 nm p140 and 10.5 nm monomeric p55 or variant unless otherwise indicated. (A) Initial screening for processivity of recombinant variants. L, low salt (none added to the reaction); H, high salt (reactions supplemented with 100 mm NaCl); p140, p140 only added to the reaction; WT, wild-type p55. (B) Reactions containing 0, 50, 100, 150, 200 and 250 mm supplemental NaCl. The gradient from low (L, none added) to high salt (H, 250 mm NaCl) is indicated by black triangles above each variant or the wild-type p55 (WT). The hyphen (-) indicates the absence of p55 in these reactions (p140 only). Primer, no protein added to the reaction. Markers, number of λ-DNA/HindIII fragment nucleotides labeled with [α-32P]dTMP. Log(λ-DNA/HindIII nucleotide fragments) was plotted against band migration distance (millimeters), and linear regression generated a correlation coefficient of 0.97 over the range analyzed (23,130, 9416, 6557, 2322, 2027 and 564 nucleotides). This standard curve was used to estimate the size of polymerase γ products observed on the alkaline gel, whereas shorter products were observed directly on denaturing PAGE (data not shown). Asterisks indicate the position of full-length primer-extension products (7249 nucleotides).
To further investigate the intermediate stimulatory effects of the P205R p55 and R369G p55 accessory subunits, primer-extension reactions were repeated across a range (0–250 mm NaCl) of salt concentrations (Fig. 4B). In the absence of p55, p140 activity is almost undetectable at 100 mm NaCl and is completely abolished at 150 mm NaCl (Fig. 4B, lanes 4 and 5), as was previously shown on poly(rA)•oligo(dT)12–18 and activated salmon sperm DNA templates (10). When wild-type p55 was added to primer-extension reactions, a stimulation of DNA synthesis was seen over a broad range of salt concentrations from 0 to 150 mm NaCl (lanes 8–11). Fewer full-length products were synthesized at 200 mm NaCl (lane 12) and none was detected at 250 mm NaCl (lane 13). P205R p55 and R369G p55 were less able to stimulate extension of primers to maximal length (∼7 kb) at any salt concentration (Fig. 4B, lanes 14–19 and lanes 20–25), and the longest products (∼4.6 kb) were observed at 0–100 mm NaCl for these two variants. P205R p55 was less efficient at 0–50 mm NaCl (lanes 14 and 15) compared with wild-type p55 (lanes 8 and 9) or R369G p55 (lanes 20 and 21). Strikingly, R369G p55 was more sensitive to inhibition by ≥150 mm NaCl when compared with either wild-type p55 or the P205R variant. At 150 mm NaCl, R369G p55 only synthesized products ∼600 bp long compared with ∼7 kb products for the wild-type and ∼4.6 kb long products for R369G p55 (compare lane 23 with lanes 11 and 17). At 200 mm salt, the increased relative stalling and/or dissociation of the R369G p55 containing holoenzyme was also apparent (compare lane 24 with lanes 12 and 18) and at 250 mm NaCl activity is completely inhibited (lane 25).
Whereas primer-extension reactions show the products of multiple DNA-binding events by pol γ, processivity reactions reveal the products of a single DNA-binding event. Thus, we also assessed the processivity of pol γ assembled with each p55 variant by including an excess (∼900-fold) of unlabeled, sonicated, heat-denatured calf-thymus DNA harboring ∼18 pmol of random 3′-ends in primer-extension reactions to ‘trap’ the polymerase following its first disassociation from the labeled substrate (37). In each case, although the total fraction of primer utilized was reduced due to limiting the enzymes to single binding events, the lengths of the resulting DNA products were essentially unchanged. Also, we noted that stimulation by R369G p55 was less efficient than the P205R variant at all salt concentrations tested (data not shown). These altered activity profiles suggest potential defects that are sensitive to ionic strength, such as association of protein subunits or binding to DNA. To determine whether the altered function of pol γ results from reduced physical interaction between the variant accessory subunits and the catalytic subunit, we performed a co-immunoprecipitation (co-IP) experiment (Fig. 5). Protein G-Sepharose beads prepared with polyclonal antibodies specific for p55 were employed to capture interactions between wild-type p55 or variants and p140. Co-IP of p140 was dependent on the presence of p55 (Fig. 5, compare lanes 3 and 4). G451E p55 was unable to immunoprecipitate p140 (lane 12), consistent with previous observations of weakened subunit interactions between p140 and G451E p55 (12). Similarly, L475DfsX2 p55 was also unable to interact with p140 as indicated by the failure of this protein to immunoprecipitate p140 (lane 11). P205R p55 and R369G p55 capture less p140 in comparison to wild-type p55, whereas G103S, L153V, D386E and S423Y p55 show levels of physical interaction similar to the wild-type protein. The P205R and L475DfsX2 reactions (lanes 7 and 11) reproducibly exhibited darker p55 bands than the other lanes. The anti-p55 antiserum used for the co-IP was raised against a fully denatured form of the insoluble 43 kDa carboxyl–terminal portion of p55 (10), and these darker bands could represent enhanced antigenicity or altered conformations for these two variants.
Figure 5.

Western blot analysis of co-IP reactions showing the physical association of p55 variants with the catalytic subunit p140. Co-IP reactions were performed using immobilized anti-p55-specific rabbit antibodies as described under Materials and Methods. Lane 1, 300 ng of purified p140 and 300 ng of purified p55 protein loaded directly; lane 2, wild-type p55 alone; lane 3, wild-type p140 alone (note: minor cross reactivity of rabbit antibody heavy chains are visible at ∼50 kDa in all lanes, including lane 3); lane 4, wild-type p55 and p140; lane 5, G103S p55 and p140; lane 6, L153V p55 and p140; lane 7, P205R p55 and p140; lane 8, R369G p55 and p140; lane 9, D386E p55 and p140; lane 10, S423Y p55 and p140; lane 11, L475DfsX2 p55 and p140; lane 12, G451E p55 and p140.
Binding affinities of p55 variants to p140 and to synthetic double-stranded DNA
To develop a steady-state functional assay to quantify association of p140 with the p55 variants, we searched for reaction conditions under which pol γ only incorporates nucleotides into a homopolymeric substrate in the presence of p55. The salt-dependent stimulation of p140 by p55 has been demonstrated previously with the exonuclease proficient form of p140 on poly(rA)•oligo(dT)12–18 (10), and we saw almost identical stimulation of the exonuclease deficient form of pol γ in this study (Fig. 6A). At 220 mm NaCl, p140 alone incorporates only ∼4–6 pmol dTMP into nascent DNA, while the p140–p552 poly γ complex synthesizes 30–55 pmol dTMP. Accordingly, 220 mm NaCl was included in subsequent polymerase reactions.
Figure 6.

Determination of the affinity of p55 for p140 (Kd(p140)). (A) Salt-dependent stimulation of pol γ by p55. DNA polymerase activity was measured on poly(rA)•oligo(dT)12–18 as described under Materials and Methods. Reactions contained either 50 fmol of p140 alone (open cirles) or in combination with 50 fmol of dimeric p55 (closed circles). Reaction times were 10min at 37° C. On the Y-axis, pmol dTMP represents the amount of dTMP incorporated into the poly(rA)•oligo(dT)12–18 as measured by counting TCA-insoluble radioactivity. Values presented are averages with standard deviations (error bars) of duplicate determinations. (B) Binding isotherm for wild-type p55 dimers binding to p140 as measured by polymerase activity on poly(rA)•oligo(dT)12–18. Reactions (50 μl) contained 50 fmol of p140 and varying amounts of dimeric p55 at the fixed concentration of 220 mm NaCl. Reaction times were 10 min at 37°C. The Kd(p140) was calculated from the quadratic curve fit as described in Materials and Methods. Values presented are averages with standard deviations (error bars) of five determinations.
Next, we obtained binding isotherms by measuring polymerase activity at various concentrations of each p55 variant (calculated as homodimers), and apparent disassociation constants were calculated by nonlinear regression (Fig. 6B and Supplementary Material, Fig. S1). Each of the variants was tested at least in duplicate and representative nonlinear regression graphs are shown in the Supplementary Material, Figure S1. Five of the variants were similar to the wild-type and displayed high affinity for p140 with Kd(p140) values in the range of 0.14 ± 0.03 to 0.25 ± 0.03 nm (Table 2). However, the R369G p55 variant had a 4.5-fold reduced affinity (0.85 ± 0.01 nm) relative to wild-type p55, and the L475DfsX2 and G451E p55 variants displayed affinities for p140 at least two orders of magnitude weaker than wild-type p55.
Table 2.
Dissociation constants of wild-type p55 and variants
| Variant | Kd(p140) (nm) | Kd(DNA) (nm) |
|---|---|---|
| Wild-type | 0.19 ± 0.05 | 49 ± 25 |
| G103S | 0.15 ± 0.05 | 55 ± 4 |
| L153V | 0.14 ± 0.03 | 66 ± 29 |
| P205R | 0.21 ± 0.03 | 214 ± 124 |
| R369G | 0.85 ± 0.01 | 86 ± 50 |
| D386E | 0.25 ± 0.03 | 107 ± 22 |
| S423Y | 0.23 ± 0.10 | 62 ± 21 |
| L475DfsX2 | 48 ± 6 | >1000a |
| G451E | 60 ± 7 | 305 ± 79 |
Kd(p140) values were determined using poly(rA)•oligo(dT)12–18 as a substrate in a functional assay and Kd(DNA) values were determined by EMSAs as described in Materials and Methods. The average values of multiple determinations are shown with errors expressed as standard deviations.
aA Kd(DNA) value could not be accurately determined for L475DfsX2 p55 as DNA binding was below the limits of detection by this assay. At the maximum concentration of protein tested (160 nm), only 10% of the DNA substrate was shifted in comparison to 65% by WT p55 (averages of three independent experiments for each protein).
Our estimation of affinity between wild-type p55 dimers and p140 is significantly higher than that reported in previous studies. Our apparent Kd(p140) value of 0.19 ± 0.05 nm was derived from a functional assay of steady-state reverse transcriptase activity on a homopolymeric template in the presence of p55 at relatively high ionic strength (Table 2). In contrast, the value calculated from pre-steady-state kinetics (34.9 ± 16.2 nm) utilized an oligonucleotide DNA substrate at 100 mm NaCl (38), and the value determined by surface plasmon resonance (27.8 ± 0.03 nm) was measured in the absence of nucleic acids and salt (7). Although the absolute Kd values vary depending on the method of determination, the relative differences in affinity among the p55 variants studied here is compelling (see Discussion).
To screen further for protein dysfunction, we analyzed the DNA-binding affinities of the variants. Electrophoretic mobility shift assay (EMSA) was carried out as described previously (11) with the modifications outlined in Materials and Methods. The fraction of synthetic double-stranded DNA bound was plotted at various concentrations of dimeric p55, and binding isotherms were fit, as in the aforementioned functional subunit-binding assay, with a quadratic equation to estimate Kd(DNA) values (Fig. 7 and Supplementary Materials, Fig. S2). Each of the variants was tested in duplicate and representative nonlinear regression graphs are shown in the Supplemental Materials, Figure S2. Wild-type p55 exhibited a Kd(DNA) of 49 ± 25 nm, and dissociation constants for the mutant collection ranged from 55 to >1000 nm (limit of detection; Table 2). Carrodeguas et al. (11) previously reported a Kd(DNA) of 8.6 ± 1.5 nm for wild-type p55, although their buffer conditions and DNA concentration differed from ours. Among the variant proteins, L475DfsX2, G451E, D386E and P205R p55 displayed statistically significant weaker affinities for DNA than wild-type p55 (Table 2). L475DfsX2 p55 was most deficient at binding DNA with an almost complete inability to shift the double-stranded oligonucleotide, even with a >10-fold excess of protein over DNA substrate. When compared with wild-type p55, G451E p55 showed a ∼6-fold decrease in affinity (6-fold higher Kd) for DNA. P205R p55 was reduced ∼4-fold, while D386E p55 was barely reduced by ∼2-fold. Therefore, although L475DfsX2 and G451E p55 are both unable to bind and stimulate p140, only L475DfsX2 p55 is completely unable to bind DNA.
Figure 7.

A representative determination of the dissociation constant for DNA binding of wild-type p55 (Kd(DNA)). EMSAs were performed as described under Materials and Methods to estimate the Kd(DNA). Lanes 1–8 of the inset gel shows increasing concentrations of the wild-type p55 dimer from 0 to 160 nm.
DISCUSSION
The role of POLG2 in mitochondrial disease was previously established by identification of the first pathogenic mutation, c.1352G>A, associated with adPEO, ptosis, exercise intolerance, muscle pain, mild weakness of facial and limb muscles, and impaired glucose tolerance (12). The G451E p55 variant encoded by this mutation retained the ability to bind DNA but was unable to physically associate with p140 in vitro, which eliminated the ability to stimulate processive DNA synthesis. A second variant, G416A p55, was studied subsequently and shown to behave similar to wild-type p55 in vitro (34). In the current study, we examined the biochemical consequences in the resulting p55 subunit from seven recently identified POLG2 variants (Table 1).
Variants with subtle or undetectable biochemical deficiencies
Patients 2, 5, 6, and 7 encoding L153V, D386E, D386E and S423Y, respectively, all had clinical complications associated with the central nervous system (Table 1), and the subject harboring the G103S change was asymptomatic. Out of the seven proteins examined G103S, L153V, D386E and S423Y p55 had only subtle or undetectable biochemical deficiencies. Similar to the previously characterized G416A p55 (34), the L153V and S423Y variants did not have any detectable biochemical deficiencies. D386E p55 had only a slightly reduced affinity for binding to synthetic double-stranded DNA in comparison to wild-type p55 (Table 2). In all of the our other assays, the D386E p55 was indistinguishable from wild-type p55, so the slight decrease in DNA binding seems inadequate to explain the severity of disease afflicting patients 5 and 6. For the G103S variant, we noticed a greater production of primer-extension products compared with the wild-type (Fig. 4A). Thus, from our biochemical analyses alone, we cannot exclude the possibility that the G103S, L153V, D386E and S423Y variants are non-pathogenic, neutral SNPs.
Our identification of G103S p55 from a subject without POLG-related mitochondrial disease supports the hypothesis that neutral SNPs in POLG2 occur within the population. This idea is also supported by the observation that L153V and S423Y p55 were not absolutely conserved phylogenetically in the sequence alignment (Fig. 1), suggesting these regions of POLG2 may have escaped purifying selection during divergent evolution. Nevertheless, in the absence of detectable biochemical defects and without a large cohort sufficient for a formal segregation study, we suggest these four variants are possible SNPs. Similar to the situation with the G416A p55 variant (34), mutations in other genes may be associated with heritable diseases for these patients. Alternatively, disease-associated mutations in the unsequenced regions of the POLG2 promoter and/or introns may alter the effective POLG2 gene dose and contribute to haplotype insufficiency, as proposed previously (12). Lastly, these p55 variants may indeed disrupt essential protein–protein interactions in vivo, promote proteolytic destruction of the variant proteins or alter intracellular targeting of the variants.
P205R p55 variant encoded by c.614C>G
The P205R substitution encoded by the c.614C>G mutation was found in a half year old male subject with clinical complications involving the central nervous system and the liver (patient 3 in Table 1). The early presentation of these symptoms is similar to the Alpers syndrome, although this patient only harbored one detectable heterozygous mutation. The P205R p55 variant had an ∼4-fold weaker ability to bind DNA compared with the wild-type protein but assembled into a homodimer (Fig. 3) and maintained a wild-type ability to associate with the catalytic subunit as measured in a functional assay (Table 2). The ability of this variant to stimulate primer extension to full-length products was compromised (Fig. 4), and this intermediate stimulatory effect was prominent at both low- and high-salt concentrations (Fig. 4B). Furthermore, the co-IP of p140 by P205R p55 was somewhat reduced compared with wild-type p55 (Fig. 5), which may be predictive of the intermediate stimulation in the primer-extension reactions with higher salt concentrations (Fig. 4), even though the functional assay measurement showed no difference in Kd(p140) (Table 2). Based on the crystal structure of the wild-type pol γ holoenzyme (9), the location of the P205R substitution in p55 would not appear to disrupt the p140–p552 interface. However, the arginine substitution in the P205R variant is located within a sharp bend of the p55 molecule between α-helix F and β-strand 5 of a six-stranded β-sheet in domain 1 (35). The addition of a bulky positively charged side-chain in this region could distort this structural feature, and intramolecular p552 misfolding may sufficiently disrupt the function of p55 homodimer/heterodimers to induce the mild biochemical defects we observed.
R369G p55 variant encoded by c.1105A>G
The R369G substitution encoded by the c.1105A>G mutation was observed in a 19-year-old female subject with neuromuscular and metabolic clinical complications consistent with chronic PEO (Table 1). R369G p55 retains a wild-type ability to bind dsDNA (Table 2). In primer-extension reactions with the addition of a DNA trap, R369G p55 synthesized fewer products over a 0–250 mm range of NaCl in comparison to wild-type p55 (data not shown). In primer-extension reactions without trap DNA, the R369G variant could only synthesize a maximum of ∼4.6 kb similar to P205R; however, a unique feature of this variant was the high degree of inhibition of primer-extension reactions at physiological salt concentration (Fig. 4B, lane 23). Unlike P205R p55, the R369G variant had a 4.5-fold reduced affinity for p140 when compared with wild-type p55 (Kd(p140) of 0.85 mm versus 0.19 nm) (Table 2). This decrease in affinity for the catalytic subunit was further corroborated by the decreased ability of R369G p55 to pull down the catalytic subunit in a co-IP (Fig. 5).
Recently, the structure of the pol γ heterotrimer was solved at 3.2 Å resolution (Fig. 8A; 9), which revealed an asymmetric association of the p55 homodimer with p140 and distinct points of contact with p140 for the proximal and distal p55 monomers. Major regions of contact occur between the accessory-interacting determinant (AID) subdomain of the catalytic subunit's spacer domain and the proximal p55 monomer. Surprisingly, only two interactions between the distal p55 monomer and p140 were observed in the structure (9,39). The Arg369 side-chain of the proximal p55 monomer is located within 4.9 Å of the catalytic subunit (Fig. 8B). The R369G substitution could alter the local van der Waals interactions made between the long side-chain of p55-R369 and the side-chain of the p140-L473 residue (Fig. 8B), thus contributing to the reduced primer-extension activity and affinity for the catalytic subunit. Interestingly, the Arg369 side-chain is also in close proximity to Ala467 within the thumb subdomain of the catalytic subunit (data not shown). Mutation of the Ala467 codon to Thr is the most common POLG disease mutation associated with Alpers, PEO and ataxia neuropathy (40). Biochemical analysis of the A467T pol γ demonstrated a defect in binding the accessory subunit (41). Therefore, we predict that the glycine substitution at position Arg369 decreases the affinity for p140 and causes the apparent salt sensitive stalling of pol γ in vivo due to the modification of a hydrophobic interaction between the thumb subdomain of p140 and the p55 proximal monomer.
Figure 8.
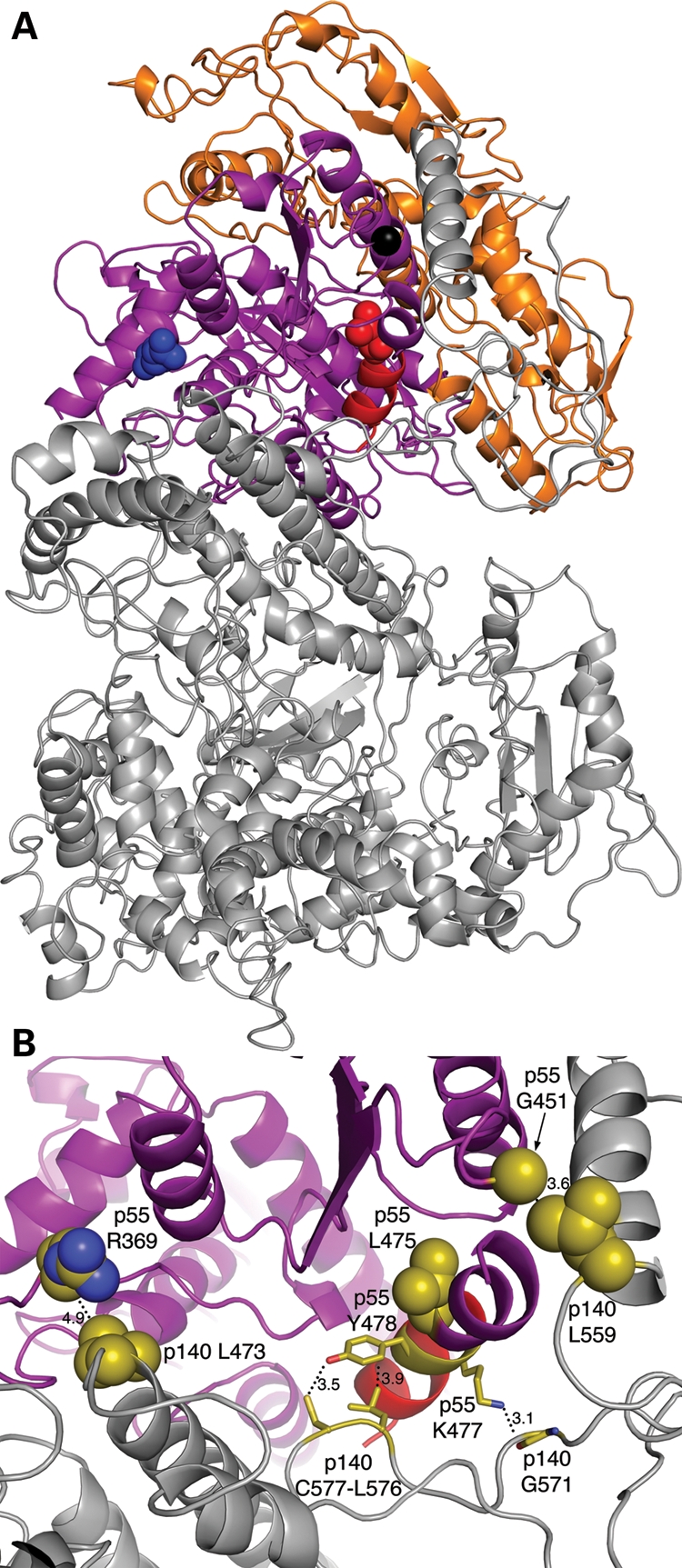
Location of p55 variant residues R369G, G451E and L475DfsX2 in the p140–p552 complex. The p140 catalytic subunit is grey, the proximal p55 monomer is colored purple and the distal monomer is orange. (A) Crystal structure of the p140–p552 complex. Arg369, blue; Leu475 and C-terminal α-helix, red; Gly451, black. The three residues are shown as spheres in the proximal monomer for clarity. (B) Magnified view of the interface between p140 and the proximal p55 monomer. The thumb subdomain of p140 contains the Leu473 residue (olive color), while the AID subdomain contains the Leu559 residue (olive). The carboxyl-terminal α-helix of p55 that becomes truncated in the L475DfsX2 variant is highlighted in red. In the proximal monomer, the carboxyl–terminal α-helix truncated in L475DfsX2 makes three close contacts with the AID subdomain of p140. (i) A potential hydrogen bond is made between the side-chains of the p140-Cys557 thiol group that is 3.5 Å away from the p55-Tyr478 hydroxyl group; (ii) a potential 3.9 Å van der Waals interaction is made between the benzyl ring of the p55-Tyr478 side-chain and the p140-Leu576 side-chain; and (iii) a potential 3.1 Å salt-bridge is made between the p55-Lys477 side-chain and the p140-Gly571 main-chain. Loss of these three intersubunit contacts by carboxyl–terminal truncation of p55 is predicted to disrupt the p140–p552 interface. The figure was generated using the program PyMOL and the published crystal structure PDB ID 3IKM (9).
L475DfsX2 p55 variant encoded by c.1423_1424delTT
To the best of our knowledge, L475DfsX2 is the first case of a clinical variant that is unable to bind to DNA and to have protein folding problems. Previous observations of several p55 truncation mutations identified a 30 residue, carboxyl–terminal region that was essential for interaction with p140 in EMSAs and for stimulating DNA polymerase activity (24). Here we utilized a clinical variant to dissect further this important region, and we conclude that the loss of only 11 carboxyl–terminal residues inactivates DNA binding. The L475DfsX2 p55 variant was found in an 8-year-old male subject with clinical complications involving the central nervous system, metabolism, the gastrointestinal system and the liver (Table 1). Several similar biochemical deficiencies between the G451E and L475DfsX2 p55 were observed: (i) using an end-labeled primer-extension assay, these proteins are defective at stimulating p140 at either low or high salt (Fig. 4A); (ii) when using anti-p55-specific antibodies, they failed to co-immunoprecipitate p140 (Fig. 5); and (iii) in our functional assay for p140–p552 inter-subunit interaction, the affinity for p140 was two orders of magnitude lower than that of wild-type p55 (Table 2).
To gain insight into variant dimerization, we analyzed the proteins via gel filtration chromatography on a Superdex 200 10/300 GL column. Unlike all of the other p55 proteins in this study, the L475DfsX2 variant eluted early in the void volume when prepared in the presence or absence of 2-mercaptoethanol. When analyzed by partially denaturing PAGE, the truncation variant contained reducible crosslinked multimers and the presence of this variant in the void volume is consistent with aggregation in vivo due to the formation of aberrant disulfide bonds (42). Therefore, the inability of L475DfsX2 p55 to stimulate DNA synthesis is 3-fold: (i) propensity to aggregate, (ii) failure to interact with the catalytic subunit, and (iii) failure to bind to DNA.
Further insight into the disease pathogenesis of the patient harboring the c.1352G>A mutation encoding the G451E p55 variant has also been revealed by the recent crystal structure of the holoenzyme (9). A 3.6 Å van der Waals interaction is made between the side-chains of the proximal p55-G451 monomer and the catalytic subunit Leu559 residue in the AID subdomain (Fig. 8B). Replacement of the small, uncharged glycine side-chain at position 451 with the longer carboxyl containing side-chain of glutamate could easily cause a steric clash between the catalytic and accessory subunits. Therefore, we predict that in human disease, the G451E variant or the L475DfsX2 deletion disrupts the pol γ intersubunit interactions at the interface between the catalytic AID subdomain and the proximal p55 subunit (Fig. 8B).
Only seven polymorphisms in POLG2 were previously known and two are associated with mitochondrial disease. This study expands this list to a total of 13 polymorphisms. Of these 13, 9 are missense, 2 induce frameshift mutations (1 deletion and 1 insertion) and 2 reside within introns. Our biochemical findings suggest that the P205R and the R369G p55 variants have moderate defects in processivity, while the L475DfsX2 truncation displays severe defects in p140 binding, dsDNA binding and structural integrity. The consequences of these three variants should be considered damaging to mtDNA synthesis and likely explain the pathogenesis in the subjects. The biochemical defects observed for variants derived from affected heterozygous patients 3, 4 and 10 suggest a dominant mode of inheritance. Further analysis of POLG2 segregation and biochemical analysis of p55 heterodimers will enhance our understanding of POLG2-related mitochondrial diseases.
MATERIALS AND METHODS
Sequencing of the human POLG2 gene
Tissue and blood samples of patients with clinical presentations suggestive of POLG deficiency were submitted to the Mitochondrial Diagnostics Laboratory at Baylor College of Medicine for biochemical and/or molecular evaluation. Clinical history of the patients was provided on a check list at the time of specimen submission. Total DNA was extracted from peripheral blood leukocytes using commercially available DNA isolation kits (Gentra Systems Inc., Minneapolis, MN, USA) according to the manufacturer's protocols. Each coding exon and 50 bp of its flanking intronic regions of the human POLG2 gene were polymerase chain reaction (PCR) amplified using FastStart DNA polymerase (Roche, IN, USA) and sequence-specific oligonucleotide primers linked to the M13 universal primers at the 5′-ends. PCR products were purified using ExcelaPure 96-well UF PCR purification plates (Edge BioSystems, Gaithersburg, MD, USA). Sequencing reactions were performed using the BigDye Terminator Cycle Sequencing kit (version 3.1) and analyzed on an ABI3730XL automated DNA sequencer with the Sequencing Analysis Software version 5.1.1 (Applied Biosystems, CA, USA). The sequencing results were compared with the GenBank POLG2 sequence (NCBI ID: NM_007215.2) using the Mutation Surveyor version 3.24 (SoftGenetics, PA, USA). The POLG2 sequencing primers are listed in the Supplementary Material Table. All analysis on human subjects was done with approval from Baylor's institute review board under IRB protocol #23698.
Construction of p55 and variant proteins
Mutations were generated separately in the codon optimized pET-p55CHIS plasmid (12) using the QuikChange site-directed mutagenesis kit (Stratagene) and therefore all p55 variants harbor C-terminal hexa-histidinyl tags. The oligonucleotides containing the point mutations (lowercase) for introducing the G103S mutation in POLG2 are 5′-ACC CCG GCT TCG GAC CCT TGa GCG TAG AGT TGC GGA AGA ACC TG-3′ and 5′-CAG GTT CTT CCG CAA CTC TAC GCt CAA GGG TCC GAA GCC GGG GT-3′, for the L153V mutation are 5′-CAG GTT AGT TTC TGC AGA AAC TgT ACG CGA AAT CTT GCA AGA C-3′ and 5′-GTC TTG CAA GAT TTC GCG TAc AGT TTC TGC AGA AAC TAA CCT G-3′, for the P205R mutation are 5′-GAT CTG GTA AAC AAG AGG CTA CgT TAT GGC CTT GCT CAG ATT G-3′ and 5′-CAA TCT GAG CAA GGC CAT AAc GTA GCC TCT TGT TTA CCA GAT C-3′, for the R369G mutation are 5′-CAA GAA AGA AAA ATC TTC ATg GAA AGG TAC TTA AAC TTC ACC C-3′ and 5′-GGG TGA AGT TTA AGT ACC TTT CcA TGA AGA TTT TTC TTT CTT G-3′, for the D386E mutation are 5′-AGG TTG CTT TGG AgG TAG GAA GAG GCC-3′ and 5′-GGC CTC TTC CTA CcT CCA AAG CAA CCT-3′ and for the S423Y mutation are 5′-GTT ATT TGG AAA CTA TGC AGT aCT CAT TGG AAC AAC TTT ATT CG-3′ and 5′-CGA ATA AAG TTG TTC CAA TGA GtA CTG CAT AGT TTC CAA ATA AC-3′. For introducing the truncation mutation (lowercase italics, aspartic acid codon) L475DfsX2, the oligonucleotides are 5′-ATA TCC AAA TTA AAA GAC TTT gat AGC CAC CAC CAC CAC CAC CAC-3′ and 5′-GTG GTG GTG GTG GTG GTG GCT atc AAA GTC TTT TAA TTT GGA TAT-3.′ All seven mutations were confirmed by DNA sequencing of the POLG2 insert. Due to decreased expression of P205R p55 and L475DfsX2 p55 relative to the wild-type, and also to rule out second site mutations in these plasmids, the region of the open reading frames harboring these mutations were subcloned separately into pET-p55CHIS. The gel-extracted NdeI–HindIII fragment from the P205R p55 plasmid was cloned into pET-p55CHIS cut with the same enzymes, and the gel-extracted DraIII–HindIII fragment from the L475DfsX2 p55 plasmid was cloned into pET-p55CHIS cut with the same enzymes. The two plasmids were sequenced to confirm addition of mutations.
Expression and purification of p55 and p140
Human p55 and variants were expressed in BL21(DE3) E. coli without the 25 amino acid N-terminal mitochondrial targeting sequence as described previously (43) with the exception that in some instances, cultures were induced for 17 h at 18°C instead of 30°C to increase the yield of recombinant protein.
We comprehensively re-examined the length of time required for expression and potential oxidation during preparation of L475DfsX2 p55 and wild-type p55 from BL21(DE3). The purification of these proteins was repeated and analyzed at multiple time points under reducing conditions. Briefly, three E. coli background strains (BL21-CodonPlus® (DE3)-RIPL, ArcticExpress™ (DE3)RP, Stratagene; Origami B(DE3), Novagen) were tested to determine the optimal induction time for cultures expressing L475DfsX2 p55. Cultures were grown at 37°C and 220rpm to mid-log followed by chilling on ice for ∼12 min to 10°C. Next, IPTG was added to the culture at 0.9 mm and flasks were incubated at 10°C with shaking at 220 rpm. The optical density of the culture was measured at A600 nm and 0.2 OD600 nm worth of E. coli cells were collected by centrifugation followed by boiling in 100 μl Laemmli's buffer. Samples (20 μl) were analyzed by electrophoresis on a CriterionTM XT 4–12% Bis–Tris gradient gel with XT MES as the running buffer (Bio-Rad) followed by western blotting with anti-p55 rabbit polyclonal antiserum as the primary antibody (see western blotting section below for details). Samples were prepared at 0, 2, 5, 11 and 24 h post-induction to identify ideal p55 expression. Only after 5 h of the time course was a small amount of protein visible on the western blot for BL21-CodonPlus® (DE3)-RIPL and ArcticExpress™ (DE3)RP strains expressing L475DfsX2 p55 while very little was visible for Origami B(DE3) strain (data not shown). After 24 h post-induction, there was a clear band visible on the blot in the BL21-CodonPlus® (DE3)-RIPL and ArcticExpress™ (DE3)RP background strains, while Origami B(DE3) only had minimal expression. Due to the possibility of protein misfolding in vivo, we chose the ArcticExpressTM (DE3)RP expression host (Stratagene) that co-expresses cold-adapted chaperones which may minimize aberrant protein folding.
To minimize oxidation during protein preparation, all buffers were supplemented with reducing agent 2-mercaptoethanol as follows: lysis buffer (50 mm Tris–HCl, pH 7.5, 300 mm NaCl, 25 mm imidazole, 1% Triton X-100, 10 mm 2-mercaptoethanol, 2 mm ATP, 2 mm MgCl2); wash buffer 1 (0.5 m NaCl, 50 mm Tris–HCl, pH 7.5, 25 mm imidazole, 1% Triton X-100, 10 mm 2-mercaptoethanol, 2 mm ATP, 2 mm MgCl2); wash buffer 2 (0.5 m NaCl, 50 mm Tris–HCl, pH 7.5, 25 mm imidazole, 0.01% NP-40, 10 mm 2-mercaptoethanol); elution buffer (50 mm Tris–HCl, pH 9.0, 0.25 m imidazole, 10 mm 2-mercaptoethanol); fast protein liquid chromatography (FPLC) buffer S55A (25 mm KPO4, pH 7.0, 10% glycerol, 1 mm EDTA, 0.005% NP-40, 10 mm 2-mercaptoethanol) and FPLC buffer S55B (25 mm KPO4, pH 7.0, 10% glycerol, 1 mm EDTA, 10 mm 2-mercaptoethanol, 1 m NaCl). Recombinant wild-type and mutant variant p55 proteins were purified as previously described (43).
For this study, the exonuclease-deficient (Exo−) pol γ lacking the N-terminal 25 amino acid residue mitochondrial targeting sequence was denoted as wild-type p140 (37,44). Overexpression of the N-terminal hexa-histidinyl form of this protein, in Spodoptera frugiperda (Sf9) insect cells, and purification was carried out as described (43).
Analytical gel filtration chromatography
The wild-type and p55 variants were analyzed separately on a Superdex 200 10/300 GL column. Samples (300 μl) contained 2 μm monomeric p55 or variant and were analyzed essentially as previously described (13). The column was equilibrated with gel filtration buffer (GFB) that consisted of 25 mm potassium phosphate, pH 7.0, 10% glycerol, 1 mm EDTA, 1 mm 2-mercaptoethanol and 377 mm NaCl. Molecular weight standards used to generate a standard curve were: cytochrome c (12.4 kDa), carbonic anhydrase (29 kDa), bovine serum albumin (66 kDa), alcohol dehydrogenase (150 kDa), β-amylase (200 kDa), apoferritin (443 kDa) and thyroglobulin (669 kDa). Log(MW) was plotted against elution volume, and linear regression generated a correlation coefficient of 0.99 over this MW range. The standards and the samples were eluted isocratically with 1.5 column volumes of GFB at a flow rate of 0.33 ml/min, and eluates were monitored at A280. Fractions were resolved by electrophoresis through a CriterionTM XT 4–12% Bis–Tris gel run in XT MES running buffer (Bio-Rad), and protein peaks were confirmed by western blotting with anti-p55 antiserum as the primary antibody.
Partial denaturing PAGE
Samples of 0.62 μg purified p55 wild-type or variant p55 proteins were individually prepared in a native sample buffer consisting of 31.25 mm Tris–HCl, pH 6.8, 7.5% glycerol and 0.005% bromophenol blue. When supplemented with sodium dodecyl sulfate and/or 2-mercaptoethanol, the final concentrations were 0.03 and 3%, respectively. Samples were heated for 5 min at 95°C or incubated at room temperature for 5 min, followed by analysis on a CriterionTM XT 4–12% Bis–Tris gel in XT MES running buffer (Bio-Rad). Resolved proteins were then electrotransferred to Immobilon-P polyvinylidene fluoride (PVDF) membrane (Millipore) and bands were visualized by western blotting (see below).
Stimulation of activity of the catalytic subunit by the p55 variants
In vitro 5′-32P-end-labeled 35-mer primer-extension assays were carried out as described previously (43) with the exception that 5 nm p140 and 10.5 nm monomeric p55 or variant was utilized and reactions were adjusted to 100 mm NaCl unless otherwise indicated. Samples were resolved by 1% alkaline agarose gel electrophoresis overnight in 30 mm NaOH/1mm EDTA at 24V. Gels were neutralized with one molar equivalent of HCl, dried at 60°C for 3 h and imaged with a phosphor screen on a Typhoon 9400 phosphorimager (GE Healthcare).
Immunoprecipitation assay
1.6 ml of serum containing rabbit polyclonal antibodies raised against recombinant human p55 accessory subunit (10) were immobilized on 1.5 ml Protein G Sepharose beads (GE Healthcare) preincubated in 12 ml phosphate-buffered saline (PBSN; 50 mm KPO4, pH 7.5, 0.05% NP-40, 220 mm NaCl) at 4°C. Binding was carried out for 6 h by rotating end over end followed by three washes with 8 ml PBSN and one wash with 1 ml PBSN/0.25 mg/ml bovine serum albumin (BSA). Centrifugation was done at 2000g. The beads were then mixed for 15 min in another 1 ml PBSN/0.25 mg/ml BSA, centrifuged and stored as a suspension in 1 ml of PBSN/0.25 mg/ml BSA. Prepared bead suspension (15 μl) mixed with 3 μg wild-type p140 and 3 μg wild-type p55 or variant p55 in 0.4 ml PBSN/0.25 mg/ml BSA was rotated end over end for 1 h at 4°C. The beads were collected by microcentrifugation at 250g for 2 min at 4°C, washed twice with 0.8 ml PBSN/0.25 mg/ml BSA and once with 0.8 ml PBSN only and resuspended in 15 μl Laemmli (45). The samples were heated for 10 min at 70°C before analysis on a CriterionTM XT 4–12% Bis–Tris gradient gel with XT MES as the running buffer (Bio-Rad). The proteins were then electrotransferred to an Immobilon-P-charged PVDF membrane (Millipore) and bands were visualized by western blotting.
Western blotting
After western transfer, the membrane was first washed in TNT buffer (50 mm Tris–HCl, pH 7.5, 500 mm NaCl and 0.1% Triton X-100) for 15 min and then blocked with blotto, 5% instant non-fat dry milk in TN buffer (50 mm Tris–HCl, pH 7.5, 500 mm NaCl), at room temperature for 45 min. Next, the blot was incubated with the primary antibody for 1.5 h, washed three times with TN for 8 min and incubated in the secondary antibody for 1 h 25 min. After three 8 min washes in TNT and three 8 min washes in TN, bands were visualized with CDP-Star (Roche) on Kodak Scientific Imaging Film.
Since all p55 variants and p140 harbor hexa-histidinyl tags, the primary antibody for the immunoprecipitation assay was 0.2 μg/ml anti-penta-His monoclonal antibody (Qiagen) in TN buffer also containing 1 mg/ml BSA, and the secondary antibody was a 1/3000 dilution of goat anti-mouse alkaline phosphatase-conjugated antibody (Bio-Rad) also in TN buffer with 1 mg/ml BSA. For the remaining blots, the primary antibody was a 1/100 dilution of anti-p55 rabbit polyclonal antiserum (10) in blotto and the secondary antibody was a 1/3000 dilution of goat anti-rabbit alkaline phosphatase conjugate (Bio-Rad) in TN buffer with 1 mg/ml BSA.
Affinity of p55 to p140
All steps were carried out on ice. Functional assay enzyme dilution buffer (FEDB) consisted of 50 mm Tris–HCl, pH 7.5, 10% glycerol, 1 mm EDTA, 0.01% NP-40, 75 mm NaCl and 50 μg/ml BSA. Samples (20 μl) of purified p140 (12.5 nm) and p55 variant (varying concentrations) were premixed in FEDB, and 4 μl was added to a cocktail to make reactions (50 μl) containing 25 mm HEPES-KOH, pH 7.5, 2.5 mm 2-mercaptoethanol, 0.5 mm MnCl2, 200 μg/ml BSA, 25 μm dTTP, 13.32 nm [α-32P] dTTP (3000 Ci/mmol, PerkinElmer Life Sciences), 220 mm NaCl, 50 μg/ml poly(rA)•oligo(dT)12–18 (Amersham Biosciences), 1 nm p140 and 0–20 nm p55 or variant protein (calculated as dimers). Reactions were incubated at 37°C for 10 min. Reactions were stopped by placing in an ice bath followed by the immediate addition of 0.2 ml stop solution (1 mg/ml BSA, 0.1 m sodium pyrophosphate tetrabasic decahydrate) and 1 ml of 10% trichloroacetic acid (TCA). TCA-insoluble radioactivity was determined by liquid scintillation counting. Reverse transcriptase activity at 220 mm NaCl indicated functional association of p140 with the p55 accessory subunit in the presence of nucleic acid. Incorporation of dTMP was plotted against the variable concentration of p55 dimers, and binding isotherms were fit to a quadratic equation by nonlinear regression analysis using the KaleidaGraph program to calculate apparent Kd(p140) values (46).
Determination of p55 affinity to DNA
The Kd(DNA) values for wild-type p55 and variants were determined by EMSA. Polyacrylamide gel purified oligonucleotides 47F, 5′-TAT ATC CAA ATT AAA AGC ATT TTT GAT TGC ATA TAT ATC ATC AGC TA-3′ and 47R, 5′-TAG CTG ATG ATA TAT ATG CAA TCA AAA ATG CTT TTA ATT TGG ATA TA-3′ were obtained from Integrated DNA Technologies and were chosen based on a detailed analysis of p55 binding to DNA (11). The 47F oligonucleotide (0.5 pmol/μl) was phosphorylated with T4 polynucleotide kinase (PNK) in a 20 μl reaction for 1 h at 37°C followed by terminating the reaction at 90°C for 5 min. The entire PNK reaction was then mixed with an additional 20 μl containing 40 pmol cold 47F and 55 pmol 47R in 10 mm Tris–HCl, pH 7.5. To anneal the two oligonucleotides at a ratio of 1:1.1 (47F:47R), the mixture was heated to 90°C followed by slowly cooling to room temperature giving a final concentration of 1.25 μm double-stranded oligonucleotides. Reactions for EMSA were assembled essentially as previously described (11). All proteins were diluted in EMSA dilution buffer (EDB) containing 10 mm Tris–HCl, pH 8.0, 0.2 mg/ml BSA, 2 mm DTT, 232 mm NaCl and 6% glycerol and the EDB was diluted one in four when added to the EMSA-binding reaction. Various concentrations of purified p55 or variant protein (0–160 nm p55 dimers) were incubated separately with 15 nm of double-stranded oligonucleotides in 20 μl binding reactions. Reactions also contained 12 mm NaCl, 10 mm Tris–HCl, pH 8.0, 0.2 mg/ml acetylated BSA and 2 mm DTT and were incubated at 30°C for 10 min followed by the addition of 5 μl of 5× loading buffer (10 mm Tris–HCl, pH 8.0, 50% glycerol, 0.1% bromophenol blue). Electrophoresis and imaging were carried out as described (43). The fraction of DNA bound was plotted against the concentration of p55 dimers, and binding isotherms were fit to a quadratic equation by nonlinear regression analysis using the KaleidaGraph program to calculate apparent Kd(DNA) values (46).
SUPPLEMENTARY MATERIAL
FUNDING
This research was supported by the Intramural Research Program of the NIH, National Institute of Environmental Health Sciences (ES 065078).
Supplementary Material
ACKNOWLEDGEMENTS
We thank Dr Robert Petrovich of the NIEHS Protein Expression Core Facility for numerous discussions about protein purification and his help and expertise with circular dichroism studies and gel filtration chromatography. We also thank Dr Katarzyna Bebenek and Alan Clark for critical reading of this manuscript.
Conflict of Interest statement. None declared.
REFERENCES
- 1.Twig G., Hyde B., Shirihai O.S. Mitochondrial fusion, fission and autophagy as a quality control axis: the bioenergetic view. Biochim. Biophys. Acta. 2008;1777:1092–1097. doi: 10.1016/j.bbabio.2008.05.001. doi:10.1016/j.bbabio.2008.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chen H., Chan D.C. Mitochondrial dynamics—fusion, fission, movement, and mitophagy—in neurodegenerative diseases. Hum. Mol. Genet. 2009;18:R169–R176. doi: 10.1093/hmg/ddp326. doi:10.1093/hmg/ddp326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Copeland W.C. Inherited mitochondrial diseases of DNA replication. Annu. Rev. Med. 2008;59:131–146. doi: 10.1146/annurev.med.59.053006.104646. doi:10.1146/annurev.med.59.053006.104646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shoubridge E.A., Wai T. Mitochondrial DNA and the mammalian oocyte. Curr. Top. Dev. Biol. 2007;77:87–111. doi: 10.1016/S0070-2153(06)77004-1. doi:10.1016/S0070-2153(06)77004-1. [DOI] [PubMed] [Google Scholar]
- 5.Masuyama M., Iida R., Takatsuka H., Yasuda T., Matsuki T. Quantitative change in mitochondrial DNA content in various mouse tissues during aging. Biochim. Biophys. Acta. 2005;1723:302–308. doi: 10.1016/j.bbagen.2005.03.001. [DOI] [PubMed] [Google Scholar]
- 6.Kasiviswanathan R., Longley M.J., Chan S.S., Copeland W.C. Disease mutations in the human mitochondrial DNA polymerase thumb subdomain impart severe defects in MtDNA replication. J. Biol. Chem. 2009;284:19501–19510. doi: 10.1074/jbc.M109.011940. doi:10.1074/jbc.M109.011940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yakubovshaya E., Chen Z., Carrodeguas J.A., Kisker C., Bogenhagen D.F. Functional human mitochondrial DNA polymerase gamma forms a heterotrimer. J. Biol. Chem. 2006;281:374–382. doi: 10.1074/jbc.M509730200. doi:10.1074/jbc.M509730200. [DOI] [PubMed] [Google Scholar]
- 8.Yakubovskaya E., Lukin M., Chen Z., Berriman J., Wall J.S., Kobayashi R., Kisker C., Bogenhagen D.F. The EM structure of human DNA polymerase gamma reveals a localized contact between the catalytic and accessory subunits. EMBO J. 2007;26:4283–4291. doi: 10.1038/sj.emboj.7601843. doi:10.1038/sj.emboj.7601843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lee Y.S., Kennedy W.D., Yin Y.W. Structural insight into processive human mitochondrial DNA synthesis and disease-related polymerase mutations. Cell. 2009;139:312–324. doi: 10.1016/j.cell.2009.07.050. doi:10.1016/j.cell.2009.07.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lim S.E., Longley M.J., Copeland W.C. The mitochondrial p55 accessory subunit of human DNA polymerase gamma enhances DNA binding, promotes processive DNA synthesis, and confers N-ethylmaleimide resistance. J. Biol. Chem. 1999;274:38197–38203. doi: 10.1074/jbc.274.53.38197. doi:10.1074/jbc.274.53.38197. [DOI] [PubMed] [Google Scholar]
- 11.Carrodeguas J.A., Pinz K.G., Bogenhagen D.F. DNA Binding Properties of Human pol gamma B. J. Biol. Chem. 2002;277:50008–50014. doi: 10.1074/jbc.M207030200. doi:10.1074/jbc.M207030200. [DOI] [PubMed] [Google Scholar]
- 12.Longley M.J., Clark S., Yu Wai Man C., Hudson G., Durham S.E., Taylor R.W., Nightingale S., Turnbull D.M., Copeland W.C., Chinnery P.F. Mutant POLG2 disrupts DNA polymerase gamma subunits and causes progressive external ophthalmoplegia. Am. J. Hum. Genet. 2006;78:1026–1034. doi: 10.1086/504303. doi:10.1086/504303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lee Y.S., Lee S., Demeler B., Molineux I.J., Johnson K.A., Yin Y.W. Each monomer of the dimeric accessory protein for human mitochondrial DNA polymerase has a distinct role in conferring processivity. J. Biol. Chem. 2010;285:1490–1499. doi: 10.1074/jbc.M109.062752. doi:10.1074/jbc.M109.062752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Di Re M., Sembongi H., He J., Reyes A., Yasukawa T., Martinsson P., Bailey L.J., Goffart S., Boyd-Kirkup J.D., Wong T.S., et al. The accessory subunit of mitochondrial DNA polymerase gamma determines the DNA content of mitochondrial nucleoids in human cultured cells. Nucleic Acids Res. 2009;37:5701–5713. doi: 10.1093/nar/gkp614. doi:10.1093/nar/gkp614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kong X.P., Onrust R., O'Donnell M., Kuriyan J. Three-dimensional structure of the beta subunit of E. coli DNA polymerase III holoenzyme: a sliding DNA clamp. Cell. 1992;69:425–437. doi: 10.1016/0092-8674(92)90445-i. doi:10.1016/0092-8674(92)90445-I. [DOI] [PubMed] [Google Scholar]
- 16.Krishna T.S., Kong X.P., Gary S., Burgers P.M., Kuriyan J. Crystal structure of the eukaryotic DNA polymerase processivity factor PCNA. Cell. 1994;79:1233–1243. doi: 10.1016/0092-8674(94)90014-0. doi:10.1016/0092-8674(94)90014-0. [DOI] [PubMed] [Google Scholar]
- 17.Moarefi I., Jeruzalmi D., Turner J., O'Donnell M., Kuriyan J. Crystal structure of the DNA polymerase processivity factor of T4 bacteriophage. J. Mol. Biol. 2000;296:1215–1223. doi: 10.1006/jmbi.1999.3511. doi:10.1006/jmbi.1999.3511. [DOI] [PubMed] [Google Scholar]
- 18.Zuccola H.J., Filman D.J., Coen D.M., Hogle J.M. The crystal structure of an unusual processivity factor, herpes simplex virus UL42, bound to the C terminus of its cognate polymerase. Mol. Cell. 2000;5:267–278. doi: 10.1016/s1097-2765(00)80422-0. doi:10.1016/S1097-2765(00)80422-0. [DOI] [PubMed] [Google Scholar]
- 19.Appleton B.A., Loregian A., Filman D.J., Coen D.M., Hogle J.M. The cytomegalovirus DNA polymerase subunit UL44 forms a C clamp-shaped dimer. Mol. Cell. 2004;15:233–244. doi: 10.1016/j.molcel.2004.06.018. doi:10.1016/j.molcel.2004.06.018. [DOI] [PubMed] [Google Scholar]
- 20.Doublie S., Tabor S., Long A.M., Richardson C.C., Ellenberger T. Crystal structure of a bacteriophage T7 DNA replication complex at 2.2 A resolution. Nature. 1998;391:251–258. doi: 10.1038/34593. doi:10.1038/34593. [DOI] [PubMed] [Google Scholar]
- 21.Hamdan S.M., Marintcheva B., Cook T., Lee S.J., Tabor S., Richardson C.C. A unique loop in T7 DNA polymerase mediates the binding of helicase-primase, DNA binding protein, and processivity factor. Proc. Natl Acad. Sci. USA. 2005;102:5096–5101. doi: 10.1073/pnas.0501637102. doi:10.1073/pnas.0501637102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Carrodeguas J.A., Kobayashi R., Lim S.E., Copeland W.C., Bogenhagen D.F. The accessory subunit of Xenopus laevis mitochondrial DNA polymerase gamma increases processivity of the catalytic subunit of human DNA polymerase gamma and is related to class II aminoacyl-tRNA synthetases. Mol. Cell Biol. 1999;19:4039–4046. doi: 10.1128/mcb.19.6.4039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fan L., Sanschagrin P.C., Kaguni L.S., Kuhn L.A. The accessory subunit of mtDNA polymerase shares structural homology with aminoacyl-tRNA synthetases: implications for a dual role as a primer recognition factor and processivity clamp. Proc. Natl Acad. Sci. USA. 1999;96:9527–9532. doi: 10.1073/pnas.96.17.9527. doi:10.1073/pnas.96.17.9527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Carrodeguas J.A., Bogenhagen D.F. Protein sequences conserved in prokaryotic aminoacyl-tRNA synthetases are important for the activity of the processivity factor of human mitochondrial DNA polymerase. Nucleic Acids Res. 2000;28:1237–1244. doi: 10.1093/nar/28.5.1237. doi:10.1093/nar/28.5.1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Farge G., Pham X.H., Holmlund T., Khorostov I., Falkenberg M. The accessory subunit B of DNA polymerase {gamma} is required for mitochondrial replisome function. Nucleic Acids Res. 2007;35:902–911. doi: 10.1093/nar/gkl1116. doi:10.1093/nar/gkl1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Longley M.J., Humble M.M., Sharief F.S., Copeland W.C. Disease variants of the human mitochondrial DNA helicase encoded by C10orf2 differentially alter protein stability, nucleotide hydrolysis and helicase activity. J. Biol. Chem. 2010;285:29690–29702. doi: 10.1074/jbc.M110.151795. doi:10.1074/jbc.M110.151795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Walter M.C., Czermin B., Muller-Ziermann S., Bulst S., Stewart J.D., Hudson G., Schneiderat P., Abicht A., Holinski-Feder E., Lochmuller H., et al. Late-onset ptosis and myopathy in a patient with a heterozygous insertion in POLG2. J. Neurol. 2010;257:1517–1523. doi: 10.1007/s00415-010-5565-9. doi:10.1007/s00415-010-5565-9. [DOI] [PubMed] [Google Scholar]
- 28.Iyengar B., Luo N., Farr C.L., Kaguni L.S., Campos A.R. The accessory subunit of DNA polymerase gamma is essential for mitochondrial DNA maintenance and development in Drosophila melanogaster. Proc. Natl Acad. Sci. USA. 2002;99:4483–4488. doi: 10.1073/pnas.072664899. doi:10.1073/pnas.072664899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wong L.J., Naviaux R.K., Brunetti-Pierri N., Zhang Q., Schmitt E.S., Truong C., Milone M., Cohen B.H., Wical B., Ganesh J., et al. Molecular and clinical genetics of mitochondrial diseases due to POLG mutations. Hum. Mutat. 2008;29:E150–E172. doi: 10.1002/humu.20824. doi:10.1002/humu.20824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cohen B.H., Chinnery P.F., Copeland W.C. POLG-Related Disorders. GeneReviews at GeneTests: Medical Genetics Information Resource [database online]. Copyright. Seattle: University of Washington; 2010. 1997–2010. Available at http://www.genetests.org . [Google Scholar]
- 31.Chinault A.C., Shaw C.A., Brundage E.K., Tang L.Y., Wong L.J. Application of dual-genome oligonucleotide array-based comparative genomic hybridization to the molecular diagnosis of mitochondrial DNA deletion and depletion syndromes. Genet. Med. 2009;11:518–526. doi: 10.1097/GIM.0b013e3181abd83c. doi:10.1097/GIM.0b013e3181abd83c. [DOI] [PubMed] [Google Scholar]
- 32.Wong L.J., Dimmock D., Geraghty M.T., Quan R., Lichter-Konecki U., Wang J., Brundage E.K., Scaglia F., Chinault A.C. Utility of oligonucleotide array-based comparative genomic hybridization for detection of target gene deletions. Clin. Chem. 2008;54:1141–1148. doi: 10.1373/clinchem.2008.103721. doi:10.1373/clinchem.2008.103721. [DOI] [PubMed] [Google Scholar]
- 33.Chang Y.F., Imam J.S., Wilkinson M.F. The nonsense-mediated decay RNA surveillance pathway. Annu. Rev. Biochem. 2007;76:51–74. doi: 10.1146/annurev.biochem.76.050106.093909. doi:10.1146/annurev.biochem.76.050106.093909. [DOI] [PubMed] [Google Scholar]
- 34.Ferraris S., Clark S., Garelli E., Davidzon G., Moore S.A., Kardon R.H., Bienstock R.J., Longley M.J., Mancuso M., Gutierrez Rios P., et al. Progressive external ophthalmoplegia and vision and hearing loss in a patient with mutations in POLG2 and OPA1. Arch. Neurol. 2008;65:125–131. doi: 10.1001/archneurol.2007.9. doi:10.1001/archneurol.2007.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Carrodeguas J.A., Theis K., Bogenhagen D.F., Kisker C. Crystal structure and deletion analysis show that the accessory subunit of mammalian DNA polymerase gamma, Pol gamma B, functions as a homodimer. Mol. Cell. 2001;7:43–54. doi: 10.1016/s1097-2765(01)00153-8. doi:10.1016/S1097-2765(01)00153-8. [DOI] [PubMed] [Google Scholar]
- 36.Fan L., Kim S., Farr C.L., Schaefer K.T., Randolph K.M., Tainer J.A., Kaguni L.S. A novel processive mechanism for DNA synthesis revealed by structure, modeling and mutagenesis of the accessory subunit of human mitochondrial DNA polymerase. J. Mol. Biol. 2006;358:1229–1243. doi: 10.1016/j.jmb.2006.02.073. doi:10.1016/j.jmb.2006.02.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Longley M.J., Ropp P.A., Lim S.E., Copeland W.C. Characterization of the native and recombinant catalytic subunit of human DNA polymerase gamma: identification of residues critical for exonuclease activity and dideoxynucleotide sensitivity. Biochemistry. 1998;37:10529–10539. doi: 10.1021/bi980772w. doi:10.1021/bi980772w. [DOI] [PubMed] [Google Scholar]
- 38.Johnson A.A., Tsai Y., Graves S.W., Johnson K.A. Human mitochondrial DNA polymerase holoenzyme: reconstitution and characterization. Biochemistry. 2000;39:1702–1708. doi: 10.1021/bi992104w. doi:10.1021/bi992104w. [DOI] [PubMed] [Google Scholar]
- 39.Lee Y.S., Molineux I.J., Yin Y.W. A single mutation in human mitochondrial DNA polymerase pol gammaA affects both polymerization and proofreading activities, but only as a holoenzyme. J. Biol. Chem. 2010;285:28105–28116. doi: 10.1074/jbc.M110.122283. doi:10.1074/jbc.M110.122283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chan S.S., Copeland W.C. DNA polymerase gamma and mitochondrial disease: understanding the consequence of POLG mutations. Biochim. Biophys. Acta. 2009;1787:312–319. doi: 10.1016/j.bbabio.2008.10.007. doi:10.1016/j.bbabio.2008.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chan S.S.L., Longley M.J., Copeland W.C. The common A467T mutation in the human mitochondrial DNA polymerase (POLG) compromises catalytic efficiency and interaction with the accessory subunit. J. Biol. Chem. 2005;280:31341–31346. doi: 10.1074/jbc.M506762200. doi:10.1074/jbc.M506762200. [DOI] [PubMed] [Google Scholar]
- 42.Baneyx F., Mujacic M. Recombinant protein folding and misfolding in Escherichia coli. Nat. Biotechnol. 2004;22:1399–1408. doi: 10.1038/nbt1029. doi:10.1038/nbt1029. [DOI] [PubMed] [Google Scholar]
- 43.Kasiviswanathan R., Longley M.J., Young M.J., Copeland W.C. Purification and functional characterization of human mitochondrial DNA polymerase gamma harboring disease mutations. Methods. 2010;51:379–384. doi: 10.1016/j.ymeth.2010.02.015. doi:10.1016/j.ymeth.2010.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lim S.E., Copeland W.C. Differential incorporation and removal of antiviral deoxynucleotides by human DNA polymerase gamma. J. Biol. Chem. 2001;276:23616–23623. doi: 10.1074/jbc.M101114200. doi:10.1074/jbc.M101114200. [DOI] [PubMed] [Google Scholar]
- 45.Laemmli U.K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. doi:10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 46.Heyduk T., Lee J.C. Application of fluorescence energy transfer and polarization to monitor Escherichia coli cAMP receptor protein and lac promoter interaction. Proc. Natl Acad. Sci. USA. 1990;87:1744–1748. doi: 10.1073/pnas.87.5.1744. doi:10.1073/pnas.87.5.1744. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.