


Abstract
The acquisition of genotoxin-induced mutations in the mammalian germline is detrimental to the stable transfer of genomic information. In somatic cells, nucleotide excision repair (NER) is a major pathway to counteract the mutagenic effects of DNA damage. Two NER subpathways have been identified, global genome repair (GGR) and transcription-coupled repair (TCR). In contrast to somatic cells, little is known regarding the expression of these pathways in germ cells. To address this basic question, we have studied NER in rat spermatogenic cells in crude cell suspension, in enriched cell stages and within seminiferous tubules after exposure to UV or N-acetoxy-2-acetylaminofluorene. Surprisingly, repair in spermatogenic cells was inefficient in the genome overall and in transcriptionally active genes indicating non-functional GGR and TCR. In contrast, extracts from early/mid pachytene cells displayed dual incision activity in vitro as high as extracts from somatic cells, demonstrating that the proteins involved in incision are present and functional in premeiotic cells. However, incision activities of extracts from diplotene cells and round spermatids were low, indicating a stage-dependent expression of incision activity. We hypothesize that sequestering of NER proteins by mispaired regions in DNA involved in synapsis and recombination may underlie the lack of NER activity in premeiotic cells.
INTRODUCTION
Unrepaired DNA damage in the mammalian genome results in the induction of mutations during replication. These mutations may interfere with cellular viability or, when present in genes involved in cellular growth control, predispose the cell to develop into malignant cancer. To counteract these threats, cells have evolved various DNA repair pathways. Among these, nucleotide excision repair (NER) is the most versatile DNA repair pathway, capable of removing a wide range of DNA helix distorting lesions. The removal of these DNA lesions by NER occurs via a reaction mechanism that includes DNA damage recognition, lesion demarcation, dual asymmetrical incisions at the 5′ and 3′ sites flanking the lesion, excision of ~30 nt from the single-stranded oligonucleotide, containing the lesion, and gap-filling by DNA synthesis and ligation (1). Two NER sub-pathways, the transcription-coupled repair (TCR) and the global genome repair (GGR) have been identified as acting on the transcribed strand of active genes and on both transcriptionally active and inactive DNA, respectively (2). It is assumed that TCR underlies the recovery of RNA polymerase II-dependent RNA synthesis that is inhibited following exposure to genotoxic agents. Due to its broad substrate specificity, NER plays an important role in protecting the somatic cells of higher eukaryotes against the cytotoxic, mutagenic and carcinogenic effects of a wide variety of DNA damaging agents.
In contrast to somatic cells, mutations occurring in germ cells are transferred to the progeny and may lead to malformations and genetic defects in the offspring. It is conceivable that NER would be of key importance to germ cells to prevent induction of mutations. There is some evidence based on unscheduled DNA synthesis (UDS), suggesting repair activity in certain stages of mammalian spermatogenesis. Cell-stage specific induction of UDS was found following exposure of cultured germ cells in vitro to UV-C irradiation or 2-acetylaminofluorene (2-AAF) (3–5). Additionally, high levels of UDS in spermatogenic cells was reported following in vivo treatment with DNA alkylating agents and X-rays (5–9). More recently, differential UDS responses were observed in spermatogonia and post-spermatogonial stages after ENU and MNU treatment of mice (10). However, with the exception of UV-C and 2-AAF, the genotoxic agents used in these studies induced DNA lesions that were mostly substrates for base excision repair (BER). However, also in the case of UV-C, the response might be due to oxidative DNA damage, considering the high doses used in these studies. Taken together this implies that the UDS responses are related to BER or more likely to other processes such as recombination, as repair by BER results in low levels of UDS due to the small size of the repair patch. In summary, these observations indicate that the ability of germ cells to perform UDS might not be informative of the NER capacity per se.
In a previous study, we showed that isolated rodent germ cells exhibited an unexpectedly low level of incision of UV-C induced DNA lesions (11). In contrast to various somatic cell types, rat germ cells did not accumulate DNA single-strand breaks (SSBs) when incubated in the presence of polymerization inhibitors [cytosine-1-β-d-arabinofuranoside (AraC) (or aphidicolin) plus hydroxyurea (HU)] following UV exposure, a treatment that causes incised DNA lesions to remain open as SSBs. These data suggested that NER in rodent germ cells is non-functional or operates at a very low level. The aim of the present investigation was to elucidate further whether NER is functional in male germ cells. For this purpose we have utilized crude cell suspensions, enriched preparations of spermatocytes and spermatids and segments of seminiferous tubules from rats. These cells were exposed ex vivo to UV light or the chemical carcinogen N-acetoxy-2-acetylaminofluorene (NA-AAF), and the incision events or removal of DNA lesions were measured. UV light-induced photolesions and NA-AAF-induced N-(deoxyguanosine-8-yl)-2-acetylaminofluorene (dG-C8-AAF) and N-(deoxyguanosine-8-yl)-aminofluorene (dG-C8-AF) are DNA helix distorting lesions that are substrates for NER in rodent cells (12–14). Induction and repair of DNA lesions were studied either at the level of the total genome or in single copy genes. In addition, NER-specific incision activity was measured in cell extracts from fractionated germ cell stages. All results indicated that NER appears to be non-functional in intact cells, probably at all germ cell stages. In contrast, cell extracts from fractions containing early/mid pachytene show high levels of incision activity. Taken together, the results suggest that the low level of NER in germ cells is not due to depletion of NER components but rather due to specific features of chromatin in premeiotic cells. Alternatively, NER components are rate limiting in cells, but are present in sufficient quantities in the extracts to allow incision of damaged DNA.
MATERIALS AND METHODS
Isolation, fractionation and characterization of rat germ cells
Media. RPMI culture medium consisted of RPMI 1640 with 25 mM HEPES buffer and l-glutamine supplemented with 10% fetal calf serum (FCS), 0.1 mg/ml pyruvate and antibiotics (100 IU/ml penicillin, 0.1 mg/ml streptomycin). Testis incubation medium (TIM) was prepared as described (15). MEMα culture medium consists of MEMα medium (Gibco BRL, Grand Island, NY) supplemented with 6% FCS, 0.14 mg/ml sodium-lactate, 0.1 mg/ml sodium pyruvate, 0.15 mg/ml l-glutamine, 5 mg/ml HEPES and 0.1 mg/ml gentamicin.
Isolation of testicular cells. In the experiments using single cell gel electrophoresis, rat germ cells from sexually mature Wistar rats (200–300 g) were isolated as described using RPMI 1640 culture medium (16). Viability, as assessed by trypan blue exclusion, was >95%. In all other experiments, testes from 37-day-old male Wistar rats were isolated, decapsulated and minced. Four testes were incubated in 20 ml TIM containing collagenase (1 mg/ml) (type CLS, Worthington Biochemical Corporation, USA) for 1 h at 32°C. The suspension containing fragments of seminiferous tubules was then washed three times with TIM. The suspension (15 ml) was shaken gently for 15 min at 32°C, after the addition of 10 ml TIM containing trypsin (7 mg) (type TRL, Worthington Biochemical Corporation). Then, 5 ml TIM containing 2% bovine serum albumin (BSA) and 20 mg soybean trypsin inhibitor (type II, Sigma, St Louis, MO) was added and the tubules were dissociated by pipetting using a 10 ml pipette for 5 min. This cell suspension was passed through a sterile 40 µm nylon mesh (Falcon, Woerden, The Netherlands). During each incubation a few drops of DNase I (Boehringer Mannheim, Almere, The Netherlands) at a concentration of 0.4 mg/ml were added. Cell viability was checked by erythrosine B dye exclusion.
Purification and characterization of germ cell stages. Cells were separated by centrifugal elutriation by a method based upon that of Meistrich et al. (17). A single cell suspension of four testes was centrifuged at 1000 r.p.m. (Beckman JE6.1 rotor) for 5 min. Cells were resuspended in 7 ml TIM and the suspension was fractionated by centrifugal elutriation at 10°C (Beckman JE 6 rotor) using TIM containing BSA and antibiotics. Three separate cell populations, each in 200 ml, were obtained at flow rates of 22.5 ml/min (fraction E2), 30 ml/min (E4) and 40 ml/min (E5), using a constant rotor speed of 1800 r.p.m. Fractionated cells were centrifuged at 1000 r.p.m. for 5 min (Beckman JE6.1 rotor) and resuspended in 20 ml MEMα culture medium. Cells from fraction E2 were further separated using Percoll density centrifugation. Cells were resuspended in 28 ml Percoll solution (TIM supplemented with 27% Percoll, 0.17 mg/ml trypsin inhibitor and 0.1 mg/ml DNase I) and centrifuged at 10 000 r.p.m. for 20 min (Beckman JA20 rotor). After Percoll centrifugation, the following fractions were isolated: E2.1, E2.2, E2.4 and E2.5 (from top to bottom) and concentrated in MEMα culture medium.
From each fraction a microdrop taken from a viscous concentrated cell suspension using a drawn pasteur pipette and was fixed by touching this drop to a drop of Carnoy’s fixative, as described by Lammers et al. (18). The cells were scored according to the criteria of Lammers et al. (18) after Giemsa staining. Round spermatids could easily be distinguished by their light chromatin and distinct heterochromatin spots. Alternatively, 10 µl of a concentrated cell suspension was spread over a paraformaldehyde coated microscope slide and cells were allowed to sediment for 1–2 h in a humidified box as described by Peters et al. (19). Preparations were immunostained with the rabbit polyclonal serum 175 (1:1500), elicited against whole synaptonemal complexes (SCs) as described by Offenberg et al. (20). Serum 175 predominantly recognizes the Mr 30 000–33 000 SCP3 axial and lateral SC components. FITC-labeled swine anti rabbit (Monosan/Sanbio PS 117F) was used as the second antibody at a final concentration of 1:100.
Isolation and identification of defined stages of seminiferous tubule segments. Testes from sexually mature Wistar rats were decapsulated, and seminiferous tubules were separated with fine forceps in a Petri dish with cold (+4°C) RPMI 1640 medium. Segments (2–3 mm) of seminiferous tubules from defined stages of the epithelial cycle were isolated by a transillumination technique (21) using fibre optic light. Flow cytometric (Bio-Rad Bryte HS, Milano, Italy) and microscopic analyses confirmed the cellular composition of the segments. Segments of the strong spot zone (stages II–IV) were dissected out to be exposed to UV-B light as described above.
Preparation of blood mononuclear cells
Fresh blood was taken from male rats by heart puncturing into Vacutainers containing EDTA. Blood mononuclear cells (MNC) were isolated by isopaque-Ficoll density centrifugation (22,23) and resuspended in RPMI culture medium without pyruvate.
Culturing and treatment of germ cells
Single cell suspensions were cultured in MEMα culture medium or in RPMI culture medium (1–2 × 106 cells/ml) in a humidified atmosphere at 32°C, 5% CO2 for up to 48 h after isolation.
Cells of fractionated germ cell stages were exposed to 10–8 M paraoxon in 10 ml TIM at 32°C for 15 min. Then, 300 µM NA-AAF was added and the cell suspension was incubated at 32°C for another 30 min. The cells were washed twice with MEMα culture medium.
Single cell suspensions of germ cells or MNC were irradiated with UV-C while being continuously shaken (Philips T.U.V. lamp, predominantly 254 nm), whereas three seminiferous tubule segments each of 2.5–3 mm from the strong spot zone (in 2 ml of ice-cold culture medium in 35 mm Petri dishes) were exposed to UV-B at a distance of 15 cm. The solar simulating light source consisted of five tubes (Wolf Helarium B1-01-40W; Germany) giving a total irradiance of 10 W/m2 and a biological efficiency for cyclobutane pyrimidine dimers (CPD) induction of ∼0.05 W/m2, i.e. about one-third of the efficiency of solar UV (24).
Repair assays
Post-irradiation incubations using repair inhibitors. (i) Cell suspensions. Immediately after UV-C irradiation of the cells (2 × 106 in 1.5 ml RPMI culture medium in 50 mm Petri dishes kept on ice), HU and AraC (Sigma) were added to final concentrations 2 and 0.1 mM respectively (11), at 4°C. The cultures were maintained at 32 (germ cells) or 37°C (MNC) in an atmosphere of 5% CO2 in air for periods of up to 24 h. Placing the dishes on ice terminated the incubation, and the cells were analyzed for DNA damage by SCGE.
(ii) Seminiferous tubule segments. Following UV-B exposure the dishes containing seminiferous tubule segments were maintained in RPMI culture medium containing HU and AraC at 32°C for 1 h in an atmosphere of 5% CO2 in air. Repair incubations were performed in RPMI culture medium for periods up to 6 h thereafter, the segments were transferred into fresh, ice-cold culture medium. The contents of the seminiferous tubules were squeezed out, transferred to tubes with 200 µl culture medium, and mechanically resuspended into single cell suspensions. Between 175 000 and 210 000 cells were obtained from three such segments for SCGE, and for flow cytometric and fluorescence microscopic analysis.
Single cell gel electrophoresis. DNA SSBs and alkali labile sites were analyzed with the comet assay (SCGE) as described (25), with modifications (16). When exogenous enzymes were used, one-third of the gel area was removed and 50 µl buffer C (40 mM HEPES, 0.1 M KCl, 0.5 mM EDTA pH 8.0, 0.02% BSA) with or without 47 ng purified T4 endonuclease V (T4Endo V) was added, followed by incubation for 30 min at 32 (germ cells) or 37°C (MNC). This was followed by unwinding and electrophoresis as in a standard SCGE protocol. Comets were analyzed with the Fenestra Comet version 3 system (Kinetic Imaging Ltd, Liverpool, UK). With this system the DNA content of each comet can be recorded to allow SSB analysis in cells of different ploidy. Fifty or 100 comets were scored per slide.
Quantification of specific DNA lesions. The quantification of NA-AAF-induced lesions in DNA samples of rat germ cells was performed using HPLC/ECD analysis (26). Gene-specific repair analysis of CPD was performed as described previously (14). Briefly, a suspension of 60 × 106 fractionated germ cells in TIM (12.5 × 106 cells/ml) was exposed to UV-C (30 J/m2). After irradiation, cells were cultured in MEMα culture medium at 32°C, 5% CO2 for 0, 16 or 24 h. More than 90% of the spermatogenic cells remained viable, as detected by erythrosine B dye exclusion. Following culturing, DNA was isolated, restricted and mock-treated or treated with T4Endo V. Each DNA sample was subjected to alkaline agarose gel electrophoresis, transferred to membranes and hybridized with 32P-labeled DNA probes.
To generate a DNA probe recognizing the transcribed strand of the rat SCP1 gene, a 823 bp fragment of rat SCP1 cDNA (clone 536; kindly provided by C.Heyting, Wageningen University, The Netherlands) was amplified in vitro in a standard PCR using oligonucleotide primers SCP13 (5′-GGAAGATTTACAGATAGCAACA-3′) and SCP14 (5′-CTTCATCTGCTTTTCTTTC-3′). After purification, this PCR product was used in a linear PCR using oligonucleotide primer SCP13 and [α-32P]dATP (27). A DNA probe specific for the transcribed strand of the Aprt gene was generated by linear PCR of the Chinese hamster Aprt gene (14).
Cell extracts and incision assay. Cell extracts were prepared from fractionated cell populations E2.1, E2.4, E4 and E5 that were cultured for 16 h at 32°C, 5% CO2 as described (28). Extracts of E2.4, E4 and E5 cells were prepared from >108 cells, whereas the protein extract from E2.1 cells was prepared from >3 x 108 cells. Incision activities of cell extracts were determined according to previously described methods (29–31). Briefly, 25 µg cell extract was incubated for 40 min at 30°C in a total volume of 25 µl with ∼5 pmol of internal 32P-labeled DNA fragments (75 000 d.p.m.) of 147 bp in length containing a dG-C8-AAF or a pivaloyl C4′-deoxyribose modification in a region of three consecutive mispairs (31). Radioactive reaction products of 24–32 nt in length were resolved on 10% polyacrylamide denaturing gels and visualized by autoradiography. Quantification of incision products was performed by laser scanning densitometry.
RNA synthesis
A suspension of 30 × 106 fractionated germ cells in TIM (6 × 106 cells/ml) was irradiated with UV-C (20 J/m2) in a 9 cm Petri dish. Subsequently, the cells were washed with MEMα culture medium. At 0, 16 and 24 h after irradiation, 105 cells were incubated in MEMα culture medium containing [3H]uridine (10 µCi/ml, 39 Ci/mmol) for 30 min at 32°C. The cells were washed with PBS and the amount of radioactivity in trichloroacetic acid precipitated material was determined according to Van Oosterwijk et al. (32).
Western blot analysis
Cell lysates were analyzed by 6% SDS–PAGE and transferred to a nitrocellulose membrane. Aspecific sites were blocked in sterilized skimmed milk in the presence of 0.05% Tween-20 and the membrane was incubated with an appropriate antibody. For analysis of native PARP or the apoptosis-related PARP-cleavage, a suspension of 35 × 106 freshly isolated germ cells in TIM (7 × 106 cells per ml) was irradiated with UV-C (20 J/m2). At 0 and 24 h after irradiation, 3.5 × 106 cells were washed twice with PBS, lysed in 40 µl cell lysis buffer consisting of 62.5 mM Tris–HCl pH 6.8, 5% 2-mercaptoethanol, 2% SDS, 10% glycerol and 0.05% bromophenol blue. Membranes were incubated with anti-PARP monoclonal antibody C-2–10 at a final concentration of 1:1000 (Clontech, Palo Alto, CA) and HRP-conjugated secondary antibody at a final concentration of 1:10 000 (Promega, Madison, WI). Finally, the membrane was incubated for 10 min in Supersignal West Dura solution (Pierce, Rockford, IL) and exposed to X-ray film.
RESULTS
Enrichment and characterization of rat testicular cell fractions
A suspension of spermatogenic cells from testes of 37-day-old Wistar rats was separated into three fractions (E2, E4 and E5) using centrifugal elutriation, which separates cells by size. The E4 and E5 fractions predominantly contain mid to late first meiotic prophase spermatocytes with a small percentage of contaminating spermatogonia and early first meiotic prophase stages (Table 1). The E2 fraction contains a mixture of spermatogonia, early first meiotic prophase stages, secondary spermatocytes and round spermatids. The E2 fraction was further separated using Percoll centrifugation and usually four fractions were isolated: E2.1, E2.2, E2.4 and E2.5 (Table 1). The first two are found in the upper region of the tube and the second two in the lower region (33). Fractions E2.1 and E2.2 contain predominantly round spermatids. Fraction E2.4 contains mostly early meiotic stages, including leptotene/zygotene and early pachytene. In fraction E2.5 spermatogonia can be found, adjacent to leptotene, zygotene and early pachytene stages (Table 1). The isolated fractions contained <1% Sertoli cells as checked by bright field microscopy after Giemsa staining. A detailed account of fraction composition will appear elsewhere (M.Giele and P.de Boer, manuscript in preparation).
Table 1. Purity of fractionated rat testicular cells.
| Fraction |
Germ cell stagea |
| E2.1 |
Round spermatids (>90%) |
| E2.2 |
Round spermatids (∼80%);
spermatogonia (∼15%) |
| E2.4 |
leptotene/zygotene (∼35–55%);
early/mid pachytene (∼25–50%) |
| E2.5 |
spermatogonia (∼18%);
leptotene/zygotene; early pachytene |
| E4 |
mid pachytene (∼75%);
diplotene (∼15%) |
| E5 | mid pachytene (∼35%); diplotene (∼60%) |
aCharacterized by Carnoy fixation, Giemsa staining, Okadaic acid and by immunostaining of SCP proteins.
Induction and removal of UV-C-induced DNA lesions in testicular cells in suspension
To test the NER capacity of rat testicular cells, incision and removal of UV-C-induced photolesions was studied in testicular cell suspensions using the SCGE assay. This assay allows the identification of DNA lesions in individual cells versus their ploidy. Crude Wistar rat testicular cell suspensions (200–300 g) were exposed in vitro to increasing doses of UV-C and incubated in the presence of the polymerization inhibitors (AraC plus HU) to accumulate incision events. Low UV doses were chosen to avoid saturation of repair. When the tail moment for the individual cell was related to its relative amount of DNA, both 4C spermatocytes and 1C round spermatids exhibited very low rates of incision events (Fig. 1A and B). In contrast, extensive DNA incision was observed in MNCs, representing somatic cells from the same species (Fig. 1C). To quantify UV-specific lesions in cellular DNA, the standard SCGE assay was modified by adding the CPD-specific DNA glycosylase T4Endo V to the agarose-embedded cells. The induction was similar in spermatocytes, round spermatids and MNCs, confirming that there were no cell type specific differences in the number of initial CPD per unit dose (Fig. 1D–F). Analysis of CPD following cultivation of UV-C exposed cells for 24 h indicated no significant removal of CPD (Fig. 2A). Cell populations enriched with respect to spermatocytes by centrifugal elutriation and containing >90% SCP3-positive spermatocytes (data not shown), were exposed to UV-C and analyzed similarly. The level of CPD was persistent (4C comets) (Fig. 2B).
Figure 1.

Accumulation of incised photolesions in spermatocytes, round spermatids and MNC. (A–C) Cells incubated with (filled columns) or without (open columns) HU (2 mM) and AraC (0.1 mM) for 1 h at 32°C (SC and RST) or 37°C (MNC), followed by analysis with conventional SCGE. (D–F) Analysis with SCGE in combination with (filled columns) or without (open columns) T4Endo V. In (A), (B), (D) and (E), comets were sorted according to their DNA content. Means ± SD of four experiments (A), (B), (D) and (E) or data from one representative experiment (C) and (F) are shown.
Figure 2.

Persistence of CPD in germ cells exposed to UV-C. Crude germ cell suspension (A) and enriched spermatocytes (B) were cultivated post-exposure at 32°C for 24 h and analyzed by SCGE in combination with T4Endo V. SCGE comets were sorted according to their DNA content. Means ± SD of four different experiments are shown. Open columns, without T4Endo V; filled columns, with T4Endo V.
Induction and repair of photolesions in cells treated in situ in seminiferous tubules
Germ cells in suspension may have a reduced metabolic capacity that in turn could suppress their ability to carry out NER. Experiments were hence carried out with seminiferous tubules. Specific segments of seminiferous tubules from mature rats were isolated and exposed to UV-B (to avoid light absorption by the outer tissue of the seminiferous tubule). The selected spermatogenic stages II–IV of rat seminiferous tubules contain step 2–4 round spermatids (1C), step 16–17 elongated spermatids (1C), mid pachytene spermatocytes (4C), type A spermatogonia (2C) and Sertoli cells (2C) (34). Less than 7% of the cells squeezed out from stages II–IV segments were somatic cells as determined by vimentin staining described previously (35), and the majority of these were Sertoli cells (data not shown). The response was determined in 4C (early pachytene) and 1C (round spermatids) populations in the SCGE assay. Cells omitted from analysis were step 16–17 spermatids (sigmoid shape) and 2C cells. Even when step 16–17 spermatids were heavily irradiated with X-rays, SSB were not measurable with SCGE (R.Wiger, unpublished results). 2C cells were infrequent and represent a mixture of germ and somatic cells.
The seminiferous tubules in ice-cold medium were treated with UV-B (3–300 s, i.e. 0.15–15 J/m2 CIE-weighted irradiance) and incubated in the presence of polymerization inhibitors. Ultimately, cells were squeezed out from the segments and analyzed with SCGE. Little or no accumulation of SSB was observed (Fig. 3). SCGE combined with T4Endo V treatment confirmed that CPD were induced in a dose-dependent manner in germ cells in situ in seminiferous tubules (Fig. 4).
Figure 3.

Incision of photolesions and induction of CPD in germ cells in situ in seminiferous tubules. Tubular segments at stages II–IV were exposed to UV-B irradiation (0–300 s). Cells from such segments were incubated with (squares) or without (diamonds) repair inhibitors HU (2 mM) and AraC (0.1 mM) for 1 h at 32°C and analyzed by conventional SCGE. Alternatively, cells were analyzed post-exposure for CPD by SCGE in combination with T4Endo V (triangles). Data are taken from one representative experiment.
Figure 4.

Persistence of CPD induced by UV-B in germ cells of different ploidy after 0 h (open columns) and 6 h (filled columns) of in situ incubation in seminiferous tubules. Tubular segments at stages II–IV were exposed to UV-B for 15 s, followed by analysis for CPD with SCGE in combination with T4Endo V. Comets were sorted according to their DNA content (ploidy 1C, 2C and 4C). Photolesions are expressed as tail moments.
Measurement of the level of CPD in cells from UV-B-exposed tubules, following incubation for 6 h, revealed no significant repair of CPD either in 1C (round spermatids) or 4C (spermatocytes) (Fig. 4).
RNA synthesis recovery following irradiation with UV-C
Despite poor removal of CPD from the genome overall, rat testicular cells may still efficiently repair CPD in transcriptionally active genes by TCR (12,14,36). Since ongoing RNA synthesis depends on TCR, RNA synthesis recovery was determined in fractions enriched for mid pachytene (E4), diplotene (E5) or round spermatid (E2.1) cell stages following UV-C irradiation at an effective dose of 10 J/m2. RNA synthesis is expressed relative to the level of synthesis in unexposed cells. Following UV irradiation, RNA synthesis was repressed to 40% in the various cell preparations. No RNA synthesis recovery was found 16 and 24 h after UV-irradiation (data not shown).
Gene specific repair of CPD
The lack of RNA synthesis recovery in different rat testicular cell stages suggests that fractionated rat testicular cells are unable to perform TCR. The capacity of fractionated rat testicular cells to perform TCR of CPD was studied in a 7 kb BclI fragment containing the rat SCP1 gene. The transcription activity of the SCP1 gene is probably restricted to leptotene/zygotene and pachytene cell stages based on detectable levels of SCP1 mRNA in these cell stages (37). The initial CPD frequency was determined in the SCP1 gene after irradiation of rat testicular cells with 30 J/m2 of UV-C. This dose of UV-C induced 1.5 ± 0.3 (n = 4), 1.0 ± 0.1 (n = 10), 0.9 ± 0.1 (n = 9) and 1.3 ± 0.1 (n = 2) CPD per 10 kb in zygotene (E2.5), mid pachytene (E4), diplotene (E5) and round spermatid (E2.1) cell stages, respectively. Removal of CPD from the transcribed strand of the SCP1 gene was measured at 16 h after UV-C irradiation. In this period of time, only 16.5 ± 12.4 (n = 4), 16.8 ± 9.2 (n = 10) and 15.5 ± 5.3% (n = 9) CPD was removed from the transcribed strand of the SCP1 gene in fractions with zygotene/leptotene (E2.5), mid pachytene (E4) and diplotene (E5) stages, respectively. These cell stages also showed almost no removal of CPD from the transcribed strand of the housekeeping gene Aprt (data not shown). The results indicate that enriched fractions of pre- and post-meiotic germ cells perform TCR poorly.
Repair of NA-AAF-induced DNA lesions
Rat hepatocytes as well as human diploid fibroblasts efficiently remove NA-AAF-induced dG-C8-AAF by NER, predominantly via the GGR sub-pathway (32,38,39). In most mammalian cell lines treated with NA-AAF, the predominant lesion is dG-C8-AF (38), but a preferential induction of dG-C8-AAF can be achieved by pre-treatment with the deacetylase inhibitor paraoxon (38). Fractionated germ cell stages were pre-treated in vitro with 10–8 µM paraoxon and then exposed to 300 µM NA-AAF and the frequencies of dG-C8-AAF and dG-C8-AF were determined by HPLC/ECD analysis. In two experiments, the fractions containing spermatocytes in early pachytene (E2.4) showed the highest frequencies of dG-C8-AAF (54.8–56.9 adducts/106 nt), whereas fractions containing spermatocytes in mid pachytene (E4) and diplotene (E5) showed somewhat lower frequencies of dG-C8-AAF in their DNA: 32.4–34.0 and 18.7–34.5 lesions/106 nt, respectively. The frequencies of dG-C8-AF were 4–16-fold lower than those found for dG-C8-AAF. HPLC/ECD analysis revealed that only 15% of dG-C8-AAF adducts were removed from the genome overall within 24 h after treatment of fractionated rat testicular cells with 10–8 M paraoxon and 300 µM NA-AAF.
Incision activity in cell extracts
The poor repair capacity of rat testicular cells, and particularly the lack of UV-induced incision events, suggests a deficiency in performing dual incisions in the vicinity of the DNA lesion. To test this possibility, cell extracts were prepared from fractionated germ cell populations and tested for incision activity in vitro using the assay devised by Huang et al. (29). A linear 147mer substrate containing a dG-C8-AAF adduct or a C4′ pivaloyl adduct incorporated into a 3mer segment of mispaired bases (31) was incubated with cell extracts prepared from fractions enriched for early pachytene (E2.4), mid pachytene (E4), diplotene (E5) or round spermatids (E2.1 and E2.2). Subsequently, reaction products were resolved by denaturing gel electrophoresis and visualized by autoradiography. Incubation of the substrate containing the dG-C8-AAF adduct or the C4′ pivaloyl adduct with cell extracts from each germ cell stage resulted in the generation of radioactive products of 24–32 nucleotides in length, indicative of dual DNA incision activity of the extracts (Fig. 5A). However, the level of incision activity differed strongly with respect to the germ cell stage. Incision activity of cell extracts from diplotene (fraction E5) and round spermatids (combined fractions E2.1 and E2.2) was reduced at least 6- and 12-fold respectively compared to the levels found for pachytene cell stages (fractions E2.4 and E4). Fractions E2.4 and E4 showed incision activities that were at the same level or even higher than those of extracts prepared from mouse embryonic fibroblasts, mouse erythroleukemia cells or from human HeLa cells (Fig. 5B).
Figure 5.
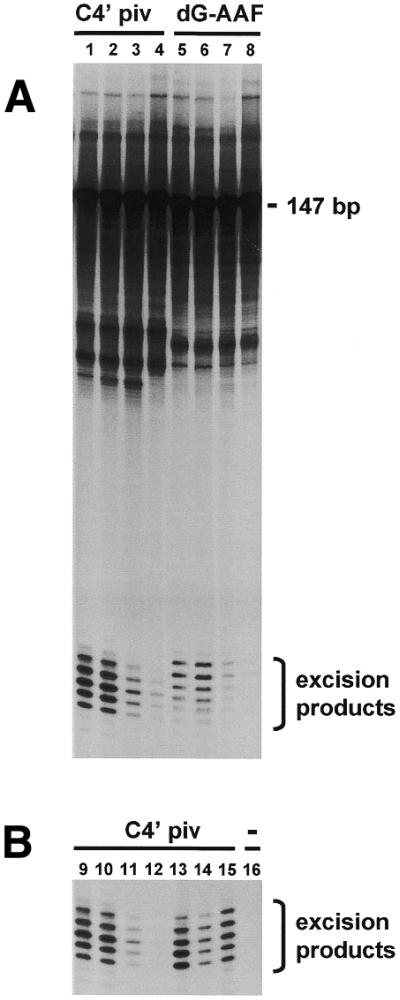
Incision activity in protein extracts from fractions containing different germ cell stages. (A) A 147mer DNA substrate containing a C4′ pivaloyl adduct (lanes 1–4) or a dG-C8-AAF adduct (lanes 5–8) incorporated in a stretch of three consecutive mispairs was incubated with protein extracts from fractionated germ cell populations enriched for early pachytene (E2.4) (lanes 1 and 5), mid pachytene (E4) (lanes 2 and 6), diplotene (E5) (lanes 3 and 7) or round spermatids (E2.1) (lanes 4 and 8). (B) A 147mer DNA substrate containing a C4′ pivaloyl adduct (lanes 9–15) or no adduct (lane 16) incorporated in a stretch of three consecutive mispairs was incubated with protein extracts from early pachytene (E2.4) (lanes 9 and 16), mid pachytene (E4) (lane 10), diplotene (E5) (lane 11), round spermatids (E2.1) (lane 12), HeLa cells (lane 13), mouse erythroleukemia cells (lane 14) or mouse embryonic fibroblasts (lane 15).
Apoptotic activity of enriched germ cell fractions following irradiation with UV-C
Reduced repair of UV-induced photolesions in mouse cells is accompanied by induction of the apoptotic pathway (40,41). To test whether fractionated germ cell stages undergo UV-induced apoptosis, freshly isolated cell populations were irradiated with UV-C (20 J/m2) or mock-treated, cultured for 0 or 24 h and subsequently assayed for PARP cleavage. PARP cleavage was analyzed by immunoblotting using a monoclonal antibody that recognizes 116 kDa native PARP and the 85–89 kDa cleavage fragment indicative of apoptosis. A major 116 kDa band representing native PARP was observed in all cell lysates tested (Fig. 6). In addition to the major 116 kDa band, a minor 89 kDa cleavage product was found in cell lysates prepared from UV-exposed mid pachytene or diplotene cells that were cultured for 24 h. Under these conditions, cleavage of native PARP appeared to be independent of UV exposure, since similar results were found in cell lysates from unirradiated cells. In contrast to cell lysates from pachytene cell stages, all cell lysates from leptotene/zygotene stages showed the major 116 kDa PARP fragment and the minor 89 kDa cleavage product, indicating that PARP cleavage in these cell stages is not related to UV exposure or cell culturing. The lack of PARP cleavage in fractionated rat testicular cells was consistent with a lack of DNA laddering (data not shown).
Figure 6.
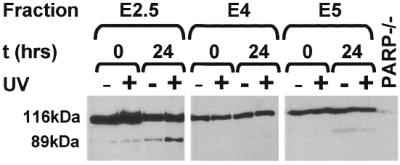
PARP cleavage in extracts from cell populations enriched for leptotene/zygotene (fraction E2.5), mid pachytene (fraction E4) and diplotene (fraction E5) as assayed by immunoblotting.
DISCUSSION
This work and that of others (16,42) showed that rat testicular cells exposed ex vivo to UV light or NA-AAF were viable and metabolically active based on dye exclusion, transcriptional activity, lack of apoptosis and efficient repair of DNA strandbreaks. This indicates that germ cells should be able to perform NER assuming that this repair pathway is functional in these cells.
The salient finding of our work is that rat male germ cells treated ex vivo exhibit no or only low levels of global genome NER activity. In particular, the SCGE analysis showed that UV-exposed rat germ cells are deficient in the incision step of NER. A possible explanation for the absence of NER activity in cultured germ cells might be the loss of tissue architecture as suggested by the finding that methylmethane sulfonate-induced UDS is 4–25 times greater in rat spermatocytes in situ in cultured seminiferous tubules than in spermatocytes cultured in suspension (6). However, cells in seminiferous tubules showed neither accumulation of UV-induced incision events nor removal of CPD.
The low NER capacity of rat germ cells also reflects a low level of TCR as repair of the transcriptionally active SCP1 gene in pachytenes is very inefficient. It cannot be excluded, however, that the SCP1 gene is only expressed in leptotene giving stable transcripts during meiotic prophase, and that the poor TCR is actually due to lack of transcription of the SCP1 gene. Nevertheless, the lack of RNA synthesis recovery in UV-exposed cells is consistent with low levels of TCR.
In contrast to the low level of NER in intact rat cells, cell extracts from different germ cell stages display cell-stage-specific incision activities. Dual incision and excision of a damaged DNA strand under in vitro conditions requires the following polypeptides: XPA, ERCC1-XPF, XPG, RPA as well as subunits from basal transcription factor TFIIH (41,43). In addition to these polypeptides, the protein complex XPC–HHR23B is also needed, depending on the DNA lesion (43). XPA is known to be limiting for repair in normal fibroblasts when <25% of normal amount. Low expression levels of XPA have been shown to cause deficiencies in incision of cisplatin-induced DNA damage by extracts of testicular tumor cell lines (44,45). Our current results suggest that cell extracts from early/mid pachytene must contain concentrations of XPA protein comparable to somatic cells such as HeLa cells. Indeed, in preliminary experiments we observed that XPA as well as XPC–HHR23B were expressed in equal amounts in cell extracts of early/mid pachytene and primary somatic cells (J.G.Jansen, A.-K.Olsen, J.Ng and L.H.F.Mullenders, unpublished results).
Remarkably, cell extracts from early spermatocyte stages show high levels of incision activities whereas intact early spermatocyte stages show no or low NER activity. How can this discrepancy be explained? We consider three factors that might influence NER efficiency: (i) apoptosis, (ii) chromatin structure and (iii) subcellular localization of proteins and gene expression.
UV-induced apoptosis can impair NER in intact rodent cells (40,41). Apoptosis has been detected in all stages of spermatogenesis following exposure of rat seminiferous tubules to ionizing radiation (46) or in vivo exposure of rat and mouse spermatocytes to genotoxic chemicals (47,48). The present study, however, indicates that UV light does not induce apoptosis in any of the cell types studied. Therefore, impairment of NER by apoptosis is not a likely explanation for the inability of intact rat spermatocytes to perform NER.
Chromatin structure may have a significant impact on the efficiency of repair of UV-induced photolesions or dG-C8-AAF lesions (32,49,50). Conceivably, the low NER activity in intact rat early/mid pachytene could be caused by inaccessibility of DNA lesions in condensed chromatin looped on the synaptonemal complex (51,52). The observation that TCR is as inefficient as global genome repair makes it unlikely that inaccessibility of chromatin underlies the low NER activity in intact cells.
Subcellular localization of proteins, particularly the single-stranded DNA binding protein complex RPA, could play a crucial role in the observed differences in repair. Firstly, immunolocalization studies have shown that RPA (i) is not detectable in leptotene, (ii) forms foci along the synaptonemal complexes during synapsis of homologous chromosomes in zygotene and early pachytene, and (iii) disappears during mid pachytene (53,54). Secondly, RPA is suggested to be involved in homology search during meiotic recombination in early pachytene (53), possibly via binding to single-stranded regions containing mispaired nucleotides. Recently, it has been reported that XPA has a strong affinity for RPA bound to DNA substrates containing mismatched nucleotides lacking hydrogen bonding (30). Sequestering of XPA by RPA bound to mispaired regions in the DNA of early pachytene could explain the low levels of incision activity in intact early pachytenes following exposure to UV. During the preparation of cell extracts used for the in vitro incision assay, protein complexes involved in NER are extracted from chromatin. Therefore, in contrast to intact cells, cell extracts of early pachytene cell stages may show high levels of incision activity in vitro. Since RPA is essential for generating dual incisions in vitro (43), the absence of RPA in diplotene could explain the low levels of incision activity found in cell extracts prepared from cell populations enriched for this cell stage.
Since the present study indicates that NER is not active in spermatogenic cells, the observed UDS in previous reports may reflect other DNA repair pathways such as recombinational repair, mismatch repair or BER. We have recently shown that a number of enzymes involved in BER are present in high amounts in rat male germ cells and that both spermatocytes and round spermatids are able to perform BER after chemical insult (42). The finding of low levels of NER in rat male germ cells is rather surprising and unexpected as the lack of NER function may lead to high susceptibility to environmental chemicals, which in turn can influence both fertility and the integrity of the genome. A surveillance system alternative to NER would be the elimination of damaged spermatogenic cells by apoptosis similar to that proposed for mouse embryonal stem cells (40).
Acknowledgments
ACKNOWLEDGEMENTS
We are indebted to Dr R.Stephen Lloyd, University of Texas Medical Branch, Galveston, TX, for the gift of purified T4Endo V, Dr Martin Hess for generating the substrates for the in vitro incision assay and technical assistance, and Dr Niels de Wind for critical reading of this manuscript. This work was supported by a grant from the Commission of the European Communities, contract number ENV4-CT95-0204.
References
- 1.de Laat W.L., Jaspers,N.G.J. and Hoeijmakers,J.H.J. (1999) Molecular mechanism of nucleotide excision repair. Genes Dev., 13, 768–785. [DOI] [PubMed] [Google Scholar]
- 2.Mullenders L.H.F. (1998) Transcription response and nucleotide excision repair. Mutat. Res., 409, 59–64. [DOI] [PubMed] [Google Scholar]
- 3.Gledhill B.L. and Darzynkiewicz,Z. (1973) Unscheduled synthesis of DNA during mammalian spermatogenesis in response to UV irradiation. J. Exp. Zool., 183, 375–382. [DOI] [PubMed] [Google Scholar]
- 4.Kofman-Alfaro S. and Chandley,A.C. (1971) Radiation-initiated DNA synthesis in spermatogenic cells of the mouse. Exp. Cell Res., 69, 33–44. [DOI] [PubMed] [Google Scholar]
- 5.Working P.E. and Butterworth,B.E. (1984) An assay to detect chemically induced DNA repair in rat spermatocytes. Environ. Mutagen., 6, 273–286. [DOI] [PubMed] [Google Scholar]
- 6.Bentley K.S. and Working,P.K. (1988) Use of seminiferous tubule segments to study stage specificity of unscheduled DNA synthesis in rat spermatogenic cells. Environ. Mol. Mutagen., 12, 285–297. [DOI] [PubMed] [Google Scholar]
- 7.Bentley K.S. and Working,P.K. (1988) Activity of germ-cell mutagens and nonmutagens in the rat spermatocyte UDS assay. Mutat. Res., 203, 135–142. [DOI] [PubMed] [Google Scholar]
- 8.Sotomayor R.E., Sega,G.A. and Cummings,R.B. (1979) An autoradiographic study of unscheduled DNA synthesis in the germ cells of male mice treated with X-rays and methyl methanesulfonate. Mutat. Res., 62, 293–309. [DOI] [PubMed] [Google Scholar]
- 9.Working P.E., Smith-Oliver,T., White R.D. and Butterworth,B.E. (1986) Induction of DNA repair in rat spermatocytes and hepatocytes by 1,2-dibromoethane: the role of glutathione conjugation. Carcinogenesis, 7, 467–472 [DOI] [PubMed] [Google Scholar]
- 10.Sotomayor R.E., Ehling,U.H. and Sega,G.A. (1999) Differential sensitivities of spermatogonial and postspermatogonial cell stages of the mouse to induction of unscheduled DNA synthesis by ethyl- and methyl-nitrosourea. Teratog. Carcinog. Mutagen., 19, 339–351. [PubMed] [Google Scholar]
- 11.Brunborg G., Holme,J.A. and Hongslo,J.K. (1995) Inhibitory effects of paracetamol on DNA repair in mammalian cells. Mutat. Res., 342, 157–170. [DOI] [PubMed] [Google Scholar]
- 12.Ruven H.T.J., Seelen,C.M.J., Lohman,P.H., van Kranen,M.H. van Zeeland,A.A. and Mullenders,L.H.F. (1994) Strand-specific removal of cyclobutane pyrimidine dimers from the p53 gene in the epidermis of UVB-irradiated hairless mice. Oncogene, 9, 3427–3432. [PubMed] [Google Scholar]
- 13.Tang ,M.-S., Bohr,V.A., Zhang,X., Pierce,J. and Hanawalt,P.C. (1989) Quantification of aminofluorene adduct formation and repair in defined DNA sequences in mammalian cells using the UVRABC nuclease. J. Biol. Chem., 264, 14455–14462. [PubMed] [Google Scholar]
- 14.Vreeswijk M.P.G., van Hoffen,A., Westland,B.E., Vrieling,H., van Zeeland,A.A. and Mullenders,L.H.F. (1994) Analysis of repair of cyclobutane pyrimidine dimers and pyrimidine 6-4 pyrimidone photoproducts in transcriptionally active and inactive genes in Chinese hamster cells. J. Biol. Chem., 269, 31858–31863. [PubMed] [Google Scholar]
- 15.Dietrich A.J.J., Scholten,R., Vink,A.C.G. and Oud,J.L. (1983) Testicular cell suspensions of the mouse in vitro. Andrologia, 15, 236–246. [DOI] [PubMed] [Google Scholar]
- 16.Bjorge C., Wiger,R., Holme,J.A., Brunborg,G., Scholz,T., Dybing,E. and Soderlund,E. (1996) DNA strand breaks in testicular cells from humans and rats following in vitro exposure to 1,2-dibromo-3-chloropropane (DBCP). Reprod. Toxicol., 10, 51–59. [DOI] [PubMed] [Google Scholar]
- 17.Meistrich M.L., Longtin,J., Brock,W.A., Grimes,S.R.,Jr and Mace,M.L. (1981) Purification of rat spermatogenic cells and preliminary biochemical analysis of these cells. Biol. Reprod., 25, 1065–1077. [DOI] [PubMed] [Google Scholar]
- 18.Lammers J.H.M., van Aalderen,M., Peters,A.H.F.M., van Pelt,A.A.M., Gaemers,I.C., de Rooij,D.G., de Boer,P. Offenberg,H.H., Dietrich,A.J.J. and Heyting,C. (1995) A change in phosphorylation pattern of the 30000–33000 Mr synaptonemal complex proteins of the rat between early and mid-pachytene. Chromosoma, 104, 154–163. [DOI] [PubMed] [Google Scholar]
- 19.Peters A.H.F.M., Plug,A.W., van Vugt,M.J. and de Boer.,P. (1997) A drying down technique for spreading of mammalian meiocytes from the male and female germline. Chromosome Res., 5, 66–68. [DOI] [PubMed] [Google Scholar]
- 20.Offenberg H.H., Dietrich,A.J.J. and Heyting C. (1991) Tissue distribution of two major components of the synaptonemal complexes of the rat. Chromosoma, 101, 83–91. [DOI] [PubMed] [Google Scholar]
- 21.Parvinen M., Toppari,J. and Lähdetie,J. (1993) Transillumination-phase contrast microscopic techniques for evaluation of male germ cell toxicity and mutagenicity. In Chapin,R.E. and Hendel,J.J. (eds), Male Reproductive Toxicology. Academic Press Ltd, London, pp. 142–165.
- 22.Bøyum A. (1968) Separation of leucocytes from blood and bone marrow. Introduction. Scand. J. Clin. Lab. Invest., 27 (suppl.). [PubMed] [Google Scholar]
- 23.Hongslo J.K., Brunborg,G., Steffensen,I.L. and Holme,J.A. (1993) Paracetamol inhibits UV-induced DNA repair in resting human mononuclear blood cells in vitro. Mutagenesis., 8, 423–429. [DOI] [PubMed] [Google Scholar]
- 24.Kinley J.S., Brunborg,G., Moan,J. and Young,A.R. (1997) Photoprotection by furocoumarin-induced melanogenesis against DNA photodamage in mouse epidermis in vivo. Photochem. Photobiol., 65, 486–491. [DOI] [PubMed] [Google Scholar]
- 25.Singh N.P., McCoy,M.T., Tice,R.R. and Schneider,E.L. (1988) A simple technique for quantitation of low levels of DNA damage in individual cells. Exp. Cell Res., 175, 184–191. [DOI] [PubMed] [Google Scholar]
- 26.Bol S.A.M., de Groot,A.J.L., Tijdens,R.B., Meerman,J.H.N., Mullenders,L.H.F. and van Zeeland,A.A. (1997) Electrochemical detection and quantification of the acetylated and deacetylated C8-deoxyguanosine DNA adducts induced by 2-acetylaminofluorene. Anal. Biochem., 251, 24–31. [DOI] [PubMed] [Google Scholar]
- 27.Ruven H.T.J., Seelen,C.M.J., Lohman,P.H.M., Mullenders,L.H.F. and van Zeeland,A.A. (1994) Efficient synthesis of 32P-labeled single-stranded DNA probes using linear PCR; application of the method for analysis of strand-specific DNA repair. Mutat. Res., 315, 189–195. [DOI] [PubMed] [Google Scholar]
- 28.Manley J.L., Fire,A., Samuels,M. and Sharp,P.A. (1983) In vitro transcription whole cell extract. Methods Enzymol., 101, 568–582. [DOI] [PubMed] [Google Scholar]
- 29.Huang J.-C., Hsu,D.S., Kanantsev,A. and Sancar,A. (1994) Substrate spectrum of human excinuclease repair of abasic sites, methylated bases, mismatches and bulky adducts. Proc. Natl Acad. Sci. USA, 91, 12213–12217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Buschta-Hedayat N., Buterin,T., Hess,M.T., Missura,M. and Naegeli,H. (1999) Recognition of nonhybridizing base pairs during nucleotide excision repair of DNA. Proc. Natl Acad. Sci. USA, 96, 6090–6095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hess M.T., Schwitter,U., Petretta,M., Giese,B. and Naegeli,H. (1997) Bipartite substrate discrimination by human nucleotide excision repair. Proc. Natl Acad. Sci. USA, 94, 6664–6669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.van Oosterwijk M.F., Versteeg,A., Filon,R., van Zeeland,A.A. and Mullenders,L.H.F. (1996) The sensitivity of Cockayne’s syndrome cells to DNA-damaging agents is not due to defective transcription-coupled repair of active genes. Mol. Cell. Biol., 16, 4436–4444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Heyting C. and Dietrich,A.J.J. (1991) Meiotic chromosome preparation and labelling. Methods Cell Biol. 35, 177–202. [DOI] [PubMed] [Google Scholar]
- 34.Toppari J., Eerola,E. and Parvinen,M. (1985) Flow cytometric DNA analysis of defined stages of rat seminiferous epithelium cycle during in vitro differentiation. J. Androl., 6, 325–333. [DOI] [PubMed] [Google Scholar]
- 35.Suter L., Clemann,N., Koch,E., Bobadilla,M. and Bechter,R. (1998) New and traditional approaches for the assessment of testicular toxicity. Reprod. Toxicol., 12, 39–47. [DOI] [PubMed] [Google Scholar]
- 36.Bohr V.A., Smith,C.A., Okumoto,D.S. and Hanawalt,P.C. (1985) DNA repair in an active gene removal of pyrimidine dimers from the DHFR gene of CHO cells is much more efficient than in the genome overall. Cell, 40, 359–369. [DOI] [PubMed] [Google Scholar]
- 37.Meuwissen R.L., Offenberg,H.H., Dietrich,A.J.J., Riesewijk,A., van Iersel,M. and Heyting,C. (1992) A coiled-coil related protein specific for synapsed regions of meiotic prophase chromosomes. EMBO J., 11, 5091–5100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Heflich R.H. and Neft,R.E. (1994) Genetic toxicity of 2-acetylaminofluorene, 2-aminofluorene and some of their metabolites and model metabolites. Mutat. Res., 318, 73–114. [DOI] [PubMed] [Google Scholar]
- 39.van Oosterwijk M.F., Filon,R., de Groot,A.J.L., van Zeeland,A.A. and Mullenders,L.H.F. (1998) Lack of transcription-coupled repair of acetylaminofluorene DNA adducts in human fibroblasts contrasts their efficient inhibition of transcription. J. Biol. Chem., 273, 13599–13604. [DOI] [PubMed] [Google Scholar]
- 40.van Sloun P.P.H., Jansen,J.G., Weeda,G., Mullenders,L.H.F., van Zeeland,A.A., Lohman,P.H.M. and Vrieling,H. (1999) The role of nucleotide excision repair in protecting embryonic stem cells from genotoxic effects of UV-induced DNA damage. Nucleic Acids Res., 27, 3276–3282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vreeswijk M.P.G., Westland,B.E., Hess,M.T., Naegeli,H., Vrieling,H., van Zeeland,A.A. and Mullenders,L.H.F. (1998) Impairment of nucleotide excision repair by apoptosis in UV-irradiated mouse cells. Cancer Res., 58, 1978–1985. [PubMed] [Google Scholar]
- 42.Olsen, A.-K., Bjørtuft,H., Wiger,R., Holme,J.A., Seeberg,E.C., Bjørås,M. and Brunborg,G. (2001) Highly efficient Base Excision Repair (BER) in human and rat male germ cells. Nucleic Acids Res., 29, 1781–1790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mu D., Hsu,D.S. and Sancar,A. (1996) Reaction mechanism of human DNA repair excision nuclease. J. Biol. Chem., 271, 8285–8294. [DOI] [PubMed] [Google Scholar]
- 44.Giannelli F., Pawsey,S.A. and Avery,J.A. (1982) Differences in patterns of complementation of the more common groups of xeroderma pigmentosum possible implications. Cell, 29, 451–458. [DOI] [PubMed] [Google Scholar]
- 45.Köberle B., Masters,J.R.W., Hartley,J.A. and Wood,R.D. (1999) Defective repair of cisplatin-induced DNA damage caused by reduced XPA protein in testicular germ cell tumours. Curr. Biol., 9, 273–276. [DOI] [PubMed] [Google Scholar]
- 46.Henriksén K., Kulmala,J., Toppari,J., Mehrotra,K. and Parvinen,M. (1996) Stage-specific apoptosis in the rat seminiferous epithelium quantification of irradiation effects. J. Androl., 17, 394–402. [PubMed] [Google Scholar]
- 47.Nakagawa S., Nakamura,N., Fujioka,M. and Mori,C. (1997) Spermatogenic cell apoptosis induced by mitomycin C in the mouse testis. Toxicol. Appl. Pharmacol.,147, 204–213. [DOI] [PubMed] [Google Scholar]
- 48.Sjöblom T., West,A. and Lähdetie,J. (1998) Apoptotic response of spermatogenic cells to germ cell mutagens etoposide, adriamycin and diepoxybutane. Environ. Mol. Mutagen., 31, 133–148. [DOI] [PubMed] [Google Scholar]
- 49.van Hoffen A., Venema,J., Meschini,R., van Zeeland,A.A. and Mullenders,L.H.F. (1995) Transcription-coupled repair removes both cyclobutane pyrimidine dimers and 6-4 photoproducts with equal efficiency and in a sequential way from transcribed DNA in xeroderma pigmentosum group C fibroblasts. EMBO J., 14, 360–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Venema J., Bartosova,Z., Natarajan,A.T., van Zeeland,A.A. and Mullenders,L.H.F. (1992) Transcription affects the rate but not the extent of repair of cyclobutane pyrimidine dimers in the human adenosine deaminase gene. J. Biol. Chem., 267, 8852–8856. [PubMed] [Google Scholar]
- 51.Heng H.H.Q., Chamberlain,J.W., Shi,X.-M., Spyropoulos,B., Tsui,L.-C. and Moens,P.B. (1996) Regulation of meiotic chromatin loop size by chromosomal position. Proc. Natl Acad. Sci. USA, 93, 2795–2800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Scherthan H., Eils,R., Trelles-Sticken,E., Dietzel,S., Cremer,T., Walt,H. and Jauch,A. (1998) Aspects of three-dimensional chromosome reorganization during the onset of human male meiotic prophase. J. Cell Sci., 111, 2337–2351. [DOI] [PubMed] [Google Scholar]
- 53.Plug A.W., Peters,A.H.F.M., Xu,Y., Keegan,K.S., Hoekstra,M.F., Baltimore,D., de Boer,P. and Ashley,T. (1997) ATM and RPA in meiotic chromosome synapsis and recombination. Nat. Genet., 17, 457–461. [DOI] [PubMed] [Google Scholar]
- 54.Walpita D., Plug,A.W., Neff,N.F., German,J. and Ashley,T. (1999) Bloom’s syndrome protein, BLM, colocalizes with replication protein A in meiotic prophase nuclei of mammalian spermatocytes. Proc. Natl Acad. Sci. USA, 96, 5622–5627. [DOI] [PMC free article] [PubMed] [Google Scholar]