

Abstract
Therapeutic RNA interference has emerged as a promising approach for the treatment of many incurable diseases, including cancer, infectious disease or neurodegenerative disorders. Demonstration of efficacy and safety in animal models is necessary before planning human application. Our group and others have previously shown the potential of this approach for the dominantly-inherited neurological disease DYT1 dystonia by achieving potent shRNA-mediated silencing of the disease protein, torsinA, in cultured cells. To establish the feasibility of this approach in vivo, we pursued viral delivery of shRNA in two different mouse models. Surprisingly, intrastriatal injections of AAV2/1 vectors expressing different shRNAs, whether targeting torsinA expression or mismatched controls, resulted in significant toxicity with progressive weight loss, motor dysfunction and animal demise. Histological analysis showed shRNA-induced neurodegeneration. Toxicity was not observed in animals that received control AAV2/1 encoding no shRNA, and was independent of genotype, occurring in both DYT1 and wild type animals. Interestingly, the different genetic background of both mouse models influenced toxicity, being earlier and more severe in 129/SvEv than C57BL/6 mice. In conclusion, our studies demonstrate that expression of shRNA in the mammalian brain can lead to lethal toxicity. Furthermore, the genetic background of rodents modifies their sensitivity to this form of toxicity, a factor that should be taken into consideration in the design of preclinical therapeutic RNAi trials.
Keywords: RNAi, AAV, DYT1 dystonia, torsinA, TOR1A
Introduction
RNA interference (RNAi) therapy is emerging as a powerful strategy to silence disease-causing alleles, providing potential treatments for previously incurable diseases. Several laboratories have applied therapeutic RNAi in rodent models of neurodegenerative disease, such as Huntington disease (HD) or Alzheimer’s disease, with successful silencing of the disease-linked gene, leading to improvements in disease phenotypes1. However, despite the promise of these early preclinical studies, recent reports raised important concerns on the safety of this therapeutic modality. Adverse effects observed in vivo derive from the saturation of the endogenous microRNA (miRNA) pathway, mostly when highly overexpressing short hairpin RNAs (shRNAs). Animal demise has been observed when applying shRNAs to non-neural tissues, whereas less dramatic toxicity, and likely due to off-targeting effects was described in mammalian brain2–5.
DYT1, a dominantly inherited neurological disorder with no cure, is the most common form of early onset inherited dystonia6. Nearly all patients with DYT1 have a mutation in the gene TOR1A, which results in a glutamic acid deletion in the protein torsinA (torA(ΔE))7. The current hypothesis of DYT1 pathogenesis posits that torA(ΔE) acts through a dominant negative effect over torA(wt), leading to a total loss of torA function8–10. Studies completed in cultured cells by our group and others have shown that RNAi-mediated allele-specific silencing of torA(ΔE) prevents and reverses DYT1-associated phenotypes, thus providing potential therapeutic efficacy8,10,11. Furthermore, non-specific silencing of both torA alleles is detrimental10 and, as a result, could be employed to trigger or worsen the phenotype of DYT1 mice. Here, we designed experiments to establish the therapeutic efficacy and safety of RNAi-mediated allele-specific silencing of torA(ΔE) in a transgenic mouse model of DYT1 dystonia. In parallel, we aimed to trigger a motor phenotype in DYT1 knockin (KI) mice through nonspecific silencing of torA.
Unexpectedly, we detected lethal toxicity caused by striatal expression of U6shRNA in both DYT1 mouse models, an outcome independent of the DYT1 mutation as it was also observed in wild type mice. Interestingly, the genetic background influenced the sensitivity of mice to this toxicity, underscoring the relevance of model selection for therapeutic studies.
Results
Striatal injection of AAV2/1 shRNAs causes lethal toxicity in DYT1 mouse models
We employed two different DYT1 mouse models for our studies. The first is a recently developed transgenic model expressing either a human TorA(ΔE) (D9.hTorA(ΔE) mice) or TorA(WT) (D9.hTorA(WT) mice) under the control of a striatal-specific DARPP-32 (D9) promoter12 and maintained on a C57BL/6 background. Using these mice, we aimed to demonstrate potent allele-specific silencing of human torA(ΔE). To trigger a motor phenotype through non-specific silencing of torA, we selected mice in which the human DYT1 mutation has been knocked into the murine tor1A gene (DYT1 KI mice). This model was maintained on a 129/SvEv background. As delivery vehicle, we employed recombinant adeno-associated virus serotype 2/1 (AAV2/1) with a GFP reporter gene to deliver different U6shRNAs to the striatum of both DYT1 KI mice, D9.hTorA(WT or ΔE) mice and their wildtype littermates (figure 1A–B).
Figure 1. Lethal consequences of U6shRNA expression in mouse striatum.

(A) Diagram of AAV2/1 vector expressing CMV-driven GFP cassette and previously published U6 driven shRNA sequences8. (B) Representative brightfield and GFP autofluorescence images of a DYT1 knockin brain section displaying striatal transduction of AAV2/1.eGFP (no shRNA) (scale = 100µm). (C and D) Postinjection Kaplan Meier survival curves of the DYT1 knockin (C) and D9.hTorA transgenic mice (D) demonstrating U6shRNA-dependent mortality, including significant strain-dependent differences (see controls for both models).
To investigate whether non-selective suppression of torsinA in the striatum would cause motor dysfunction in adult mice we performed intrastriatal injections of a U6shRNA that efficiently suppresses expression of both wild type and mutant murine torsinA (AAV2/1.GFP.U6shTorA) in 2.5–6 month old DYT1 KI mice and their wild type littermates. Control vectors included AAV2/1.GFP with no shRNA, a missense hairpin (U6shMis), or U6shHtorA(ΔE) which targets human but not murine torA(ΔE), thus acting as a second missense control. Unexpectedly, 2–5 weeks post injection we found progressive weight loss and significant lethality of both experimental and control shRNAs, whereas no death was observed in mice receiving virus expressing no shRNA (figure 1C). The shRNAs showed varying levels of toxicity, with mice injected with the U6shTorA hairpin showing markedly lower mortality rates than either U6shHtorA(ΔE) or U6shMis. The presence of the DYT1 mutation did not influence this effect.
In a parallel set of experiments, D9.hTorA(WT) transgenic mice were used to test the efficacy of the therapeutic shRNA, U6shHtorA(ΔE), to achieve allele-specific silencing of human torA(ΔE) in vivo. While the full characterization of these mice will be published elsewhere (Ehrlich et al, manuscript in preparation), we detected striatal expression of the transgene by western blot analysis and immunostaining that would allow us to measure silencing (data not shown). Mice were injected in the striatum with AAV2/1.GFP encoding either the therapeutic construct (U6shHtorA(ΔE)), a missense control shRNA (U6shMis), or a virus expressing no shRNA. Mortality in the first 5 weeks post-injection was significantly lower than in DYT1 KI mice, even though they received the same viral preps. We observed delayed mortality in D9.hTorA(WT or ΔE) and non-transgenic littermates injected with U6shhTorA(ΔE) or U6shMis, with no toxicity in mice injected with AAV2/1.GFP (figure 1D). The differences in the magnitude and timing of mortality between the DYT1 KI and transgenic mice were observed regardless of genotype, indicating that they are a consequence of the genetic background of the mice. To eliminate the potential confounding factor introduced by the DYT1 mutation and transgene expression, we looked specifically at control mice (i.e., nontransgenic, non-KI), detecting significant differences in mortality between 129/SvEv and C57BL/6 mice that received the same U6shHtorA(ΔE) and U6shMis-encoding viral preps (see control mice in figure 1C–D).
Abnormal motor function caused by striatal expression of U6shRNA
Baseline behavior analysis of motor function was performed in both DYT1 KI and D9.hTorA(WT or ΔE) mice before injection. A comparison of results from both open field behavior and rotarod analysis revealed no significant changes in performance between wild type and KI mice (not shown). Due to the high levels of mortality observed in DYT1 KI mice, post-injection behavior analysis could not be completed.
Owing to the lower and delayed mortality observed in D9.hTorA mice, post-injection behavior could be completed in surviving animals. We recorded their performance on rotarod and open field before and at two time points (6–8 and 14–16 weeks) post-injection. Motor dysfunction was observed in mice injected with either U6shHtorA(ΔE) or U6shMis, independent of transgene expression, with increased locomotion in open field (figure 2A) and worse performance on the rotarod (figure 2B) when compared to mice receiving AAV2/1.GFP encoding no shRNA.
Figure 2. Striatal dysfunction in mice injected with U6shRNAs.

Comparison of motor activity and function before, 6–8 weeks, and 14–16 weeks post injection. (A) Total distance traveled over one hour in an open field apparatus (mean ± SEM) (*P<0.05, **P<0.01, *** P<0.001). In all three genotypes, mice injected with either U6shMis or U6shHtorA(ΔE) showed increased motor activity at both 6–8 weeks and 14–16 weeks post injection that was not observed in mice injected with no shRNA. (B) Motor coordination as measured by a rotarod apparatus over 3 days showing latency to fall (mean ± SEM for trials 1–3 for each group per day) (*P<0.05, **P<0.01, *** P<0.001). In all three genotypes, mice injected with either shMis or shHtorA(ΔE) showed reduced motor coordination at both 6–8 weeks and 14–16 weeks post injection when compared to no shRNA controls. rpm = revolutions per minute.
U6shRNA induces striatal toxicity
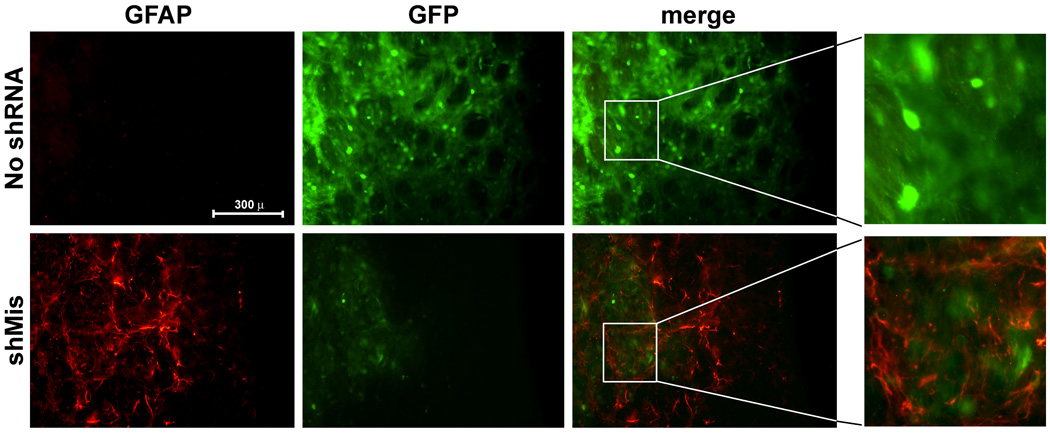
To determine what effect these toxic U6shRNAs at the tissue level we harvested brains from D9.hTorA and control mice 6–9 months post injection. Survey of striatal sections revealed that multiple U6shRNA-injected mouse brains exhibited enlarged ventricles (figure 3A), indicating striatal atrophy. Immunohistochemical analysis was completed to detect DARPP-32 expression, a marker of striatal medium spiny neurons. We observed loss of DARPP-32 signal and reduced GFP signal in the transduced striatum of mice injected with either of the U6shRNAs, but not in brains that received control virus (figure 3B), suggesting loss of transduced neurons. Investigation of DARPP-32 levels in DYT1 KI mice revealed a similar pattern suggesting neuronal loss (not shown). The regions transduced by the shRNA-endoding vectors, but not with control virus, also demonstrated decreased cresyl violet staining, further suggesting neurodegeneration (figure 3C). Taken together, these results suggest that expression of U6shRNAs caused the loss of striatal medial spiny neurons. Furthermore, immunohistochemical staining for a glial marker, GFAP, demonstrated astroglial activation in the areas transduced by the toxic U6shRNA, but not with control AAV2/1 (supplemental figure 1). As described for the DARPP-32 staining, reduced GFP signal was observed together with enhanced GFAP staining, suggesting loss of transduced neurons and replacement by astroglial cells. However, we did not observe differences in Iba-1 expression (not shown), a marker of microglial activation4.
Figure 3. Striatal neurotoxity in mice injected with U6shRNAs.

(A) Brightfield pictures of C57 BL/6 control (nontransgenic) mice revealed enlarged lateral ventricles (indicated by arrowheads) in mice injected with shRNAs, suggesting striatal atrophy. (B) Immunohistological analysis revealed significant loss of DARPP-32 signal in mice injected with U6shRNAs in a region that overlapped with viral transduction (indicated by GFP), but not in control (i.e., no U6shRNA) virus. (C) Representative micrographs showing cresyl violet staining of transduced striatal tissue, demonstrating pallor (decreased staining) in areas transduced with shRNA-expressing but not with control vectors.
Analysis of the endogenous microRNA pathway
To determine whether significant variation in miRNA expression or processing underlie the different susceptibility of both strains to U6shRNA-induced toxicity, we analyzed the expression of two candidate miRNAs and key components of the miRNA pathway in striatal tissue obtained from control animals from both mouse strains. Expression and processing of a ubiquitous (miR30a) and a neuronal specific microRNA (miR9) (figure 4A) did not differ between both strains. We then measured expression levels of key component of the miRNA pathway by western blot analysis, demonstrating similar levels of Ago2 in both strains and a non-significant trend towards lower levels of exportin52 in 129/SvEv mice (figure 4B). Similar results were obtained in cortical tissue obtained from the same animals (not shown). We next asked whether the expression of toxic shRNAs influences the levels of miRNA expression and processing. For these experiments we used C57BL/6 mice due to their enhanced survival after the injections. We found that levels of unprocessed pre-miRNA were not significantly changed by the overexpression of shRNAs, but the levels of the mature strand were highly variable with a tendency to be increased (figure 5A). Interestingly, while exportin5 expression was mostly unchanged, the levels of Ago2 were inversely correlated with the increment in mature miRNA observed by Northern blotting for each animal (figure 5B). Quantitative RT-PCR for mature miR9 in an independent set of animals confirmed the northern blot findings (figure 5C). Levels of exportin5, Ago2, unprocessed pre-miRNA and mature miRNA in non-transduced cortical areas of the same animals were not changed (not shown). These observations could derive from an effect of the shRNAs on the endogenous miRNA pathway, or simply reflect variable cell death and astroglial reaction caused by the toxic shRNAs.
Figure 4. Analysis of the microRNA pathway in 29/SvEv and C57BL/6 mice.

Three 4-month-old 129/SvEv and C57BL/6 male mice were sacrificed, and their brains extracted, the striatum dissected, and assayed for expression of candidate miRNAs and component of the miRNA machinery as described in the methods section. (A) Northern blot analysis displaying expression of miR9 and miR30a in 129/SvEv and C57BL/6 striatum. U6 served as a loading control. (B) Western blot analysis showing expression levels of exportin5 (exp5), Argonaut 2 (Ago2). α-tubulin (α-tub) is a loading control. Each lane represents miRNA or protein obtained from a different animal. Quantification of the northern and western blot analysis (mean ± SEM) is shown. *p= 0.07.
Figure 5. Analysis of the microRNA pathway in transduced striatum.

The striata of animals injected with vectors encoding shMis, shHtorA(ΔE) or control virus were extracted. (A) The right striatum was used for northern blot analysis of miR9 expression and processing by detecting pre-miR9 and mature miR9. Note that both bands are from the same blot, but with different exposure. U6 is shown as a loading control. (B) The left striatum of the same animal was used for western blot analysis to detect expression of exportin5, Ago2 and α-tubulin as a loading control. The samples for the northern and western blot analyses shown in the top and bottom panels were run in the same order. (C) Q-RT-PCR of miR9, normalized to U6 expression, in striata expressing of AAV2/1.eGFP (reference), AAV2/1.eGFP.U6shMis or AAV2/1.eGFP.U6shHtorA(ΔE). Differences were non-significant.
Discussion
In this work, we demonstrate lethal toxicity caused by the application of therapeutic RNAi-mediating vectors to mouse models of neurological disease. Our studies show that pol-III transcribed shRNAs applied to the murine striatum can cause neuronal loss, behavioral dysfunction and animal demise. Furthermore, the genetic background of the animals employed modifies susceptibility to this adverse effect, indicating that this factor needs to be considered and controlled for when designing RNAi trials. This work adds to our current knowledge of therapeutic gene silencing.
RNAi therapy has shown great promise toward the treatment of neurological disorders. Therapeutic trials have been completed or are underway for treatment of several non-neurological diseases, and preclinical studies in different animal models of various neurological diseases have been reported1. The clinical, genetic and biological characteristics of DYT1 dystonia make it an optimal disease model to explore the potential of therapeutic RNAi. Several studies in cellular models of DYT1 paved the road for trials in vivo8,10,11,13. The shRNAs used for this work were previously tested in cultured mammalian neurons demonstrating efficacy and, most importantly, no evidence of toxicity8. Thus, the results of this study indicate that lack of cell death or inflammatory responses in cultured cells does not translate into safety of the targeting sequence in vivo, stressing the need to evaluate the presence of adverse effects in the appropriate model.
A landmark study by Kay and colleagues2, in which they encountered animal demise when targeting genes in mouse hepatocytes, brought general attention to the potential toxicity of shRNAs in non-nervous tissues. Similarly to our study, this occurred in wild type mice. Most trials in mouse models of neurological disease employed viral delivery of shRNAs and focused mainly on the demonstration of efficacy1. However, hints of potential toxicity by shRNAs were already observed. For instance, when targeting mutant huntingtin in HD transgenic mice, Rodriguez-Lebron et al14 detected toxicity using a shRNA previously shown to be effective in cultured cells. Furthermore, Davidson and colleagues found that wild type mice injected with one of their therapeutic shRNA for HD showed mild and transient phenotypical worsening15. The same group demonstrated toxicity with microglial activation upon intrastriatal delivery of shRNAs in a mouse model of HD, thought to derive from off-targeting effects4. More recently, AAV-mediated expression of shRNAs has been show to cause neuronal death16. Our work further extends these reports by demonstrating striatal atrophy with associated behavioral dysfunction and death. It is important to emphasize that we encountered the same levels of toxicity in wild type that in DYT1 mice. This has important implications for preclinical tolerability studies for optimizing shRNAs to minimize induced toxicity, regardless of the model. Even more, because these toxic constructs caused cell loss in non-disease mice, it is possible that some diseases characterized by neurodegeneration could be accelerated by the application of these vectors. Collectively, these studies strongly suggest that the first generation of vectors, based on highly expressed pol-III driven shRNAs, are not optimal for therapeutic development and should be optimized and carefully evaluated for potential toxicity. Different strategies have been developed to abolish the toxic side effects of highly expressed shRNAs, such as decreasing levels of expression by placing the shRNA under a different pol-III promoter like H1 or a pol-II promoter, or enhancing processing efficiency of the targeting sequence by embedding it in a miRNA backbone4,5,17. Similar modifications should be adapted to advance in the development of therapeutic RNAi-based constructs for DYT1 dystonia and other neurological diseases.
Whereas several mechanisms can potentially cause shRNA-derived adverse effects, emerging evidence suggests that dysfunction of the endogenous miRNA pathway due to saturation from exogenous constructs is the most likely mechanism2,18. Overall, these studies suggest that very high overexpression of shRNAs from the highly active U6 promoter might cause toxicity by saturating the miRNA pathway. Our different shRNA constructs were expressed from the U6 promoter and, at least in cultured cells, they silence their targets very potently, suggesting high levels of overexpression. Here, we show that AAV2/1 delivery of U6shRNAs to the striatum can induce lethal toxicity in mice, with loss of neurons transduced by the different U6shRNA vectors. While animal demise from fulminant liver failure is predictable, death caused by the loss of a limited region of the striatum is not as intuitive. However, McManus and colleagues19 recently demonstrated that dysfunction of the miRNA pathway caused by the conditional deletion of dicer in postnatal murine striatal medium spiny neurons results in progressive motor dysfunction, reduced brain size, wasting and death at 10–12 weeks of age. Thus, dysfunction of the miRNA pathway in striatal neurons can cause progressive motor dysfunction and death. Why toxic shRNAs led to increased locomotor activity is unclear, but it could perhaps due to differences in transduction efficiency or susceptibility to this toxicity between medium spiny neurons that participate in the direct or indirect pathway.
An interesting and unexpected result of our studies is the different susceptibility to shRNA-induced adverse effects between two commonly used mouse genetic backgrounds that exhibit significant differences at the level of gene expression in brain tissue20. We speculated that the basis of this finding could be different efficiency in miRNA processing between striatal neurons of both strains, and completed studies that show a trend toward lower levels of exportin5 expression in the more susceptible strain, although the significance of this finding is unclear. Expression of toxic shRNAs did not alter exportin5 levels, but lead to variability in endogenous Ago2 and miR9 expression. Interestingly, levels of Ago2 inversely correlated with the accumulation of mature miRNA. In their initial report of liver toxicity, Grimm et al.2 explored the underlying cause, reaching the conclusion that saturation of the putative rate limiting step of this pathway, the nuclear export of pre-miRNAs by exportin5, was the responsible event. However, the same group recently identified Ago2 as a rate-limiting determinant of RNAi efficacy, toxicity and persistence 18. In light of this report, it is tempting to speculate that the variations in Ago2 levels leads to impaired processing of endogenous miRNAs and is implicated in shRNA-toxicity in neurons. Why Ago2 levels are variable upon the expression of toxic shRNAs and why this correlates with higher miR9 levels is unclear, and might simply reflects the changes in tissue architecture caused by neuronal degeneration and glial reaction. Furthermore, how reduced Ago2 expression would affect miR9 levels is unclear. Whether differences in exportin5 and/or Ago2 expression or function between 129/SvEv and C57BL/6 mice are responsible for their different susceptibility to the toxic effects of shRNAs remains to be proven. Manipulation of exportin5 and Ago2 expression upon the neuronal application of toxic shRNAs would help test this hypothesis. Regardless what the final cause for these differences proves to be, though, researchers should carefully consider the background of the animal model employed for preclinical RNAi therapy trials. 129/SvEv mice might be an optimal strain when aiming to exclude shRNA-induced toxicity in preclinical studies.
In summary, this study demonstrates the presence of lethal toxicity caused by striatal expression of shRNA and illustrates differences derived from the genetic background of the model employed for therapeutic silencing studies. These findings have important implications for the design of preclinical therapeutic RNAi trials in models of human neurological disease.
Materials and Methods
Animals
DYT1 knockin mice, generated by Dauer and colleagues21 were maintained on a 129/SvEv background. D9 transgenic mice that express one copy of either human TorA(ΔE) (D9.hTorA(ΔE) mice) or TorA(WT) (D9.hTorA(WT) mice) under the control of a DARPP-32 promoter were maintained on a C57 BL/6 background. The generation and characterization of this model will be described elsewhere (Ehrlich and colleagues, manuscript in preparation). Mice were housed in controlled temperature rooms with 12hr light and dark cycles. Food and water were provided ad libitum. All experimental protocols were approved by the University of Iowa Animal Care and Use Committee.
Viral Expression Vectors
Recombinant AAV serotype 2/1 vectors expressing shRNAs were generated as described previously4 by the Gene Transfer Core at the University of Iowa. Briefly, U6-shRNA expression cassettes were cloned into AAV2/1 shuttle plasmids and AAV2/1 recombinant virus was generated using a well established calcium phosphate transfection method.22 RT-PCR was use to determine viral titers with concentrations between 5×1012 − 2.5×1013vg/mL. The targeting sequences employed in this study were previously reported by Gonzalez-Alegre et al8, although their identification names have been modified for simplicity as follows: U6shTorA in the present study corresponds to shTAcom in8, U6shHtorA(ΔE) to shTAmut5 and U6shMis to shTAmis.
Striatal Injections
Three to six month old mice were anesthetized with ketamine/xylazine and placed into a David Kopf stereotaxic frame (Model 900, David Kopf Instruments, Tujunga, CA, USA). Following a sterile technique, bilateral striatal injections of 2 µl (100nl/min) of AAV2/1 were performed through a burr hole, using a Hamilton syringe and programmable UltraMicroPumpII (World Precision Instruments, Sarasota, FL, USA) at the following coordinates with respect to Bregma: 0.9mm anterior, 1.8mm lateral to the midline, 2.8mm ventral to the dura. Once completed, the needle was raised 0.3mm then left in for 5 additional minutes to allow diffusion and avoid spread of the solution along the pipette track.
Behavioral Analysis
Rotarod performance
In brief, mice were first acclimated to the rotarod (model 47600 Ugo Basile, Italy) for 3 minutes at a steady rate of 4rpm then at an accelerating rate from 4rpm to 16rpm over 2 min. Mice were tested for 3 days with 3 trials per day on a rotarod set to accelerate from 4 rpm to 40 rpm over a period of 5 minutes. Latency to fall was recorded on each day with a maximum cutoff at 500 sec. Mice who rode the rotating rod 3 complete revolutions without moving forward were removed due to inactivity. Mice that were unable to stay on the apparatus for at least 120 seconds were not placed into injection groups.
Open field
After acclimation, four mice were recorded simultaneously by placing them in an open field arena with 4 separate 25cm × 25cm fields. We employed an automated tracking system (Viewpoint, Videotrak, Lyon, France) to collect and analyze data (distance traveled) in 5 min bins for 60 minutes.
Immunohistochemical Analyses
Mice were sacrificed 6–9 months post injection then perfused transcardially with ice cold 0.9% NaCl then fixed with 4% paraformaldehyde. Brains were excised, postfixed overnight, the stored in a 30% sucrose solution at 4°C until ready for use. Immunohistochemical analysis was performed as previously described.5 Briefly, free floating coronal sections of brain (40µm) were incubated with antibodies to detect DARPP-32 (Cell Signaling Technology, Danvers, MA, USA) or Iba-1 (Wako Chemicals USA, Richmond, VA, USA) for 24 hours at room temperature. Brain sections were next incubated in biotinylated secondary antibody (1:200) for 1 hour, then placed in ABC reagent (both from Vector Laboratories, Burlingame, CA, USA) and developed in a 3-3’ diaminobenzidine solution. Finally, sections were mounted to SuperFrost Plus slides and coverslipped with Permount (Thermo-Fisher Scientific, Waltham, MA, USA). GFP autofluorescence of transduced regions were captured prior to staining and DARPP-32 images after using an Olympus SZX12 fluorescent stereoscope with an Olympus DP70 camera (Olympus America, Center Valley, PA, USA) using accompanying software. For indirect immunofluorescence, sections were incubated with Cy3-conjugated monoclonal GFAP antibodies (Sigma, St. Louis, MO, USA) and processed as described8 before visualization. Cresyl violet staining was completed following standard protocols.
Molecular Studies
RNA isolation
Mice were perfused transcardially with ice cold 0.9% NaCl, then brains were excised and placed in a matrix and cut into 1mm sections. Transduced striatal regions were surgically removed, weighed, then placed in RNA later (Qiagen, Valencia, CA, USA) and stored at −80°C until ready for use. RNA was isolated using TRIzol (Invitrogen, Carlsbad, CA, USA) following manufacturer’s instructions and resuspended in Ultrapure DNase/RNase free distilled water (Invitrogen). RNA quality and concentration was assessed using a NanoDrop ND-1000 spectrophotometer (Thermo-Fisher Scientific).
Northern blotting
Small transcript Northern blots were performed as described23. Briefly, 5µg of total RNA was run on a 15% PAGE-UREA gel, transferred to a Hybond NX nylon membrane (Amersham Pharmacia, Piscataway, NJ, USA), then crosslinked with 0.16M N-(3-Dimethylaminopropyl)-N-ethylcarbodiimide hydrochloride in 127 mM 1-methylimidazole (pH8) at 60°C for 1 hour. Membranes were then hybridized with γ[32P] ATP-labeled oligonucleotides at 35°C overnight, washed in 2x SSC, exposed to a phosphoimager screen and quantified on a Molecular Dynamics STORM PhosphorImager (Molecular Dynamics, Sunnyvale, CA). The probe sequences used were as follows: U6- 5’CGTTCCAATTTTAGTATATGTGCTGCC 3’, miR9- 5’TCATACAGCTAGATAACCAAAGA 3’, miR30a- 5’CTTCCAGTCGAGGATGTTTACA 3’
Q-RT-PCR
For relative miRNA expression, 350ng of RNA was reverse transcribed using TaqMan microRNA Reverse Transcription Kit with Megaplex RT Primer Rodent Pool A (Applied Biosystems, Foster City, CA), and then diluted RT product (1:10) was used to determine levels of mature miR9 by TaqMan miRNA Assays (Applied Biosystems) using manufacturer's protocols and normalized to U6 small nuclear RNA. Fold changes were determined using the ΔΔCT method, and calibrated to AAV2/1.eGFP-injected striata.
Western blotting
Mice were perfused transcardially with ice cold 0.9% NaCl, then transduced striatum was excised, weighed and snap frozen in liquid nitrogen then stored at −80°C until ready for use. Brains were homogenized and suspended in RIPA lysis buffer. Western blotting was performed as previously described.13 Briefly, protein samples were run on a 12% SDS-PAGE gel then transferred to a nitrocellulose membrane and probed with exportin 5, Argonaut 2 (Abcam, Cambridge, UK) and α-tubulin antibodies (Sigma, St. Louis, MO, USA). Blots were incubated with a Western Lighting Plus-ECL kit (Perkin-Elmer, Wellesley, MA, USA) and developed with Kodak BioMax MR Film (Kodak).
Blot quantification
We quantified Western and Northern blot signal as described13 using U6 and α-tubulin signals as loading controls for normalization, and expressing the results as a percentage of resulting signal compared with the control lanes.
Statistical Analysis
All statistical analyses were performed using SAS software (SAS Institute Inc, Cary, NC, USA). Survival curves were estimated by the Kaplan–Meier method and differences between groups were evaluated using the log-rank test. A linear mixed model for repeated measures was used to analyze the open field and rotarod data. A probability value of less than 0.05 was considered statistically significant.
Supplementary Material
Figure 6.
Acknowledgments
We thank Drs. Beverly Davidson, Kevin Glenn, Anton McCaffrey and Henry Paulson, and members of their laboratories for their help and suggestions, Nicole Bode for her assistance with cresyl violet staining, and Dr. Bridgett Zimmerman for aid with statistical analyses. This work was supported by NIH/NINDS (P01NS050210 and R21NS047432), Dystonia Medical Research Foundation and Tyler’s Hope For A Dystonia Cure.
Footnotes
Conflict of interest
Janine Martin, Nicolle Wolken and Timothy Brown declare no conflict of interest. Dr. Pedro Gonzalez-Alegre’s work has been funded by the NIH, Dystonia Medical Research Foundation and Tyler’s Hope for a Dystonia Cure. He co-owns a patent on the technology described in this report. Dr. William Dauer’s work has been funded by the NIH and Dystonia Medical Research Foundation. Dr. Michelle Ehrlich’s work has been funded by the NIH.
Supplementary information is available at www.nature.com/gt
References
- 1.Gonzalez-Alegre P, Paulson HL. Technology insight: therapeutic RNA interference--how far from the neurology clinic? Nat Clin Pract Neurol. 2007;3:394–404. doi: 10.1038/ncpneuro0551. [DOI] [PubMed] [Google Scholar]
- 2.Grimm D, Streetz KL, Jopling CL, Storm TA, Pandey K, Davis CR, et al. Fatality in mice due to oversaturation of cellular microRNA/short hairpin RNA pathways. Nature. 2006;441:537–541. doi: 10.1038/nature04791. [DOI] [PubMed] [Google Scholar]
- 3.Cao W, Hunter R, Strnatka D, McQueen CA, Erickson RP. DNA constructs designed to produce short hairpin, interfering RNAs in transgenic mice sometimes show early lethality and an interferon response. J Appl Genet. 2005;46:217–225. [PubMed] [Google Scholar]
- 4.McBride JL, Boudreau RL, Harper SQ, Staber PD, Monteys AM, Martins I, et al. Artificial miRNAs mitigate shRNA-mediated toxicity in the brain: implications for the therapeutic development of RNAi. Proc Natl Acad Sci U S A. 2008;105:5868–5873. doi: 10.1073/pnas.0801775105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Boudreau RL, Martins I, Davidson BL. Artificial microRNAs as siRNA shuttles: improved safety as compared to shRNAs in vitro and in vivo. Mol Ther. 2009;17:169–175. doi: 10.1038/mt.2008.231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gonzalez-Alegre P. The inherited dystonias. Semin Neurol. 2007;27:151–158. doi: 10.1055/s-2007-971170. [DOI] [PubMed] [Google Scholar]
- 7.Ozelius LJ, Hewett JW, Page CE, Bressman SB, Kramer PL, Shalish C, et al. The early-onset torsion dystonia gene (DYT1) encodes an ATP-binding protein. Nat Genet. 1997;17:40–48. doi: 10.1038/ng0997-40. [DOI] [PubMed] [Google Scholar]
- 8.Gonzalez-Alegre P, Bode N, Davidson BL, Paulson HL. Silencing primary dystonia: lentiviral-mediated RNA interference therapy for DYT1 dystonia. J Neurosci. 2005;25:10502–10509. doi: 10.1523/JNEUROSCI.3016-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Torres GE, Sweeney AL, Beaulieu JM, Shashidharan P, Caron MG. Effect of torsinA on membrane proteins reveals a loss of function and a dominant-negative phenotype of the dystonia-associated DeltaE-torsinA mutant. Proc Natl Acad Sci U S A. 2004;101:15650–15655. doi: 10.1073/pnas.0308088101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hewett JW, Nery FC, Niland B, Ge P, Tan P, Hadwiger P, et al. siRNA knock-down of mutant torsinA restores processing through secretory pathway in DYT1 dystonia cells. Hum Mol Genet. 2008;17:1436–1445. doi: 10.1093/hmg/ddn032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kock N, Allchorne AJ, Sena-Esteves M, Woolf CJ, Breakefield XO. RNAi blocks DYT1 mutant torsinA inclusions in neurons. Neurosci Lett. 2006;395:201–205. doi: 10.1016/j.neulet.2005.10.098. [DOI] [PubMed] [Google Scholar]
- 12.Bogush AI, McCarthy LE, Tian C, Olm V, Gieringer T, Ivkovic S, et al. DARPP-32 genomic fragments drive Cre expression in postnatal striatum. Genesis. 2005;42:37–46. doi: 10.1002/gene.20118. [DOI] [PubMed] [Google Scholar]
- 13.Gonzalez-Alegre P, Miller VM, Davidson BL, Paulson HL. Toward therapy for DYT1 dystonia: allele-specific silencing of mutant TorsinA. Ann Neurol. 2003;53:781–787. doi: 10.1002/ana.10548. [DOI] [PubMed] [Google Scholar]
- 14.Rodriguez-Lebron E, Denovan-Wright EM, Nash K, Lewin AS, Mandel RJ. Intrastriatal rAAV-mediated delivery of anti-huntingtin shRNAs induces partial reversal of disease progression in R6/1 Huntington's disease transgenic mice. Mol Ther. 2005;12:618–633. doi: 10.1016/j.ymthe.2005.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Harper SQ, Staber PD, He X, Eliason SL, Martins IH, Mao Q, et al. RNA interference improves motor and neuropathological abnormalities in a Huntington's disease mouse model. Proc Natl Acad Sci U S A. 2005;102:5820–5825. doi: 10.1073/pnas.0501507102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ehlert EM, Eggers R, Niclou SP, Verhaagen J. Cellular toxicity following application of adeno-associated viral vector-mediated RNA interference in the nervous system. BMC Neurosci. 11:20. doi: 10.1186/1471-2202-11-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Giering JC, Grimm D, Storm TA, Kay MA. Expression of shRNA from a tissue-specific pol II promoter is an effective and safe RNAi therapeutic. Mol Ther. 2008;16:1630–1636. doi: 10.1038/mt.2008.144. [DOI] [PubMed] [Google Scholar]
- 18.Grimm D, Wang L, Lee JS, Schurmann N, Gu S, Borner K, et al. Argonaute proteins are key determinants of RNAi efficacy, toxicity, and persistence in the adult mouse liver. J Clin Invest. 120:3106–3119. doi: 10.1172/JCI43565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cuellar TL, Davis TH, Nelson PT, Loeb GB, Harfe BD, Ullian E, et al. Dicer loss in striatal neurons produces behavioral and neuroanatomical phenotypes in the absence of neurodegeneration. Proc Natl Acad Sci U S A. 2008;105:5614–5619. doi: 10.1073/pnas.0801689105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sandberg R, Yasuda R, Pankratz DG, Carter TA, Del Rio JA, Wodicka L, et al. Regional and strain-specific gene expression mapping in the adult mouse brain. Proc Natl Acad Sci U S A. 2000;97:11038–11043. doi: 10.1073/pnas.97.20.11038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Goodchild RE, Kim CE, Dauer WT. Loss of the dystonia-associated protein torsinA selectively disrupts the neuronal nuclear envelope. Neuron. 2005;48:923–932. doi: 10.1016/j.neuron.2005.11.010. [DOI] [PubMed] [Google Scholar]
- 22.Sandalon Z, Bruckheimer EM, Lustig KH, Rogers LC, Peluso RW, Burstein H. Secretion of a TNFR:Fc fusion protein following pulmonary administration of pseudotyped adeno-associated virus vectors. J Virol. 2004;78:12355–12365. doi: 10.1128/JVI.78.22.12355-12365.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Marquez RT, Wendlandt E, Galle CS, Keck K, McCaffrey AP. MicroRNA-21 is upregulated during the proliferative phase of liver regeneration, targets Pellino-1, and inhibits NF-kappaB signaling. Am J Physiol Gastrointest Liver Physiol. 298:G535–G541. doi: 10.1152/ajpgi.00338.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.