

Abstract
Cells of vertebrates remove DNA double-strand breaks (DSBs) from their genome predominantly utilizing a fast, DNA-PKcs-dependent form of non-homologous end joining (D-NHEJ). Mutants with inactive DNA-PKcs remove the majority of DNA DSBs utilizing a slow, DNA-PKcs-independent pathway that does not utilize genes of the RAD52 epistasis group, is error-prone and can therefore be classified as a form of NHEJ (termed basic or B-NHEJ). We studied the role of DNA ligase IV in these pathways of NHEJ. Although biochemical studies show physical and functional interactions between the DNA-PKcs/Ku and the DNA ligase IV/Xrcc4 complexes suggesting operation within the same pathway, genetic evidence to support this notion is lacking in mammalian cells. Primary human fibroblasts (180BR) with an inactivating mutation in DNA ligase IV, rejoined DNA DSBs predominantly with slow kinetics similar to those observed in cells deficient in DNA-PKcs, or in wild-type cells treated with wortmannin to inactivate DNA-PK. Treatment of 180BR cells with wortmannin had only a small effect on DNA DSB rejoining and no effect on cell radiosensitivity to killing although it sensitized control cells to 180BR levels. This is consistent with DNA ligase IV functioning as a component of the D-NHEJ, and demonstrates the unperturbed operation of the DNA-PKcs-independent pathway (B-NHEJ) at significantly reduced levels of DNA ligase IV. In vitro, extracts of 180BR cells supported end joining of restriction endonuclease-digested plasmid to the same degree as extracts of control cells when tested at 10 mM Mg2+. At 0.5 mM Mg2+, where only DNA ligase IV is expected to retain activity, low levels of end joining (∼10% of 10 mM) were seen in the control but there was no detectable activity in 180BR cells. Antibodies raised against DNA ligase IV did not measurably inhibit end joining at 10 mM Mg2+ in either cell line. Thus, in contrast to the situation in vivo, end joining in vitro is dominated by pathways with properties similar to B-NHEJ that do not display a strong dependence on DNA ligase IV, with D-NHEJ retaining only a limited contribution. The implications of these observations to studies of NHEJ in vivo and in vitro are discussed.
INTRODUCTION
Double-strand breaks (DSBs) are induced in the genome of higher eukaryotes by exogenous agents such as ionizing radiation (IR) but also endogenously during DNA replication or from the action of by-products of the cellular metabolism. To counter the deleterious effects of these highly toxic lesions, cells have developed efficient mechanisms to remove DSBs and restore integrity in their genome. The kinetics of DNA DSB-rejoining in cells exposed to IR indicate two major components operating with half-times of minutes and several hours, respectively, that are thought to reflect different biochemical pathways (1).
Two fundamentally different biochemical processes may be utilized to remove DSBs from the genome of eukaryotic cells; homologous recombination repair (HRR) and non-homologous end joining (NHEJ) (2–4). Because mutants deficient in several genes implicated in HRR rejoin IR-induced DNA DSBs with kinetics indistinguishable from those of wild-type cells, we proposed that the two components of rejoining discerned by in vivo studies reflect different pathways of NHEJ (5).
Genetic studies in vertebrate cells provide information regarding the molecular nature of these pathways of NHEJ. Several laboratories have identified the DNA-dependent protein kinase (DNA-PK), a complex of the catalytic subunit DNA-PKcs and Ku autoantigen, as a crucial factor in NHEJ (reviewed in 4,6,7). A mutation in any of the DNA-PK subunits sensitizes cells to IR-induced killing and reduces the efficiency of DNA DSB rejoining. While early studies indicated a general defect in DNA DSB rejoining in cells deficient in components of DNA-PK (8–13), more recent studies in DNA-PKcs-deficient cells suggest a rather specific defect that reduces the fraction of DNA DSBs rejoined with fast kinetics (1,14). Because the fast component of rejoining shows a strong DNA-PKcs dependence it is termed here D-NHEJ, whereas the slow, DNA-PKcs-independent component is termed here basic or B-NHEJ. It is particularly relevant that when D-NHEJ becomes inactivated, B-NHEJ takes over and removes a similar load of DNA DSBs from the genome. Thus, DNA-PK deficiency does not have a profound effect on the proportion of DNA DSBs removed, but decreases almost 30-fold the kinetics with which this occurs (1).
Other studies have implicated DNA ligase IV, and its tightly associated, stabilizing co-factor Xrcc4, in NHEJ (15–24). Because this complex has ligase activity that can be assessed in vitro and because DNA-PK has been shown to phosphorylate Xrcc4 in vitro, it is thought that the ligase IV–Xrcc4 complex operates, together with the DNA-PK complex, in the same NHEJ pathway (21,24–30). This is further supported by recent biochemical studies demonstrating functional and physical interactions between DNA-PKcs and Ku on the one hand and DNA ligase IV on the other hand (31,32). However, genetic studies formally proving this point in mammalian cells are lacking. Furthermore, it is not clear whether DNA ligase IV can also function independently of DNA-PK in the slow, DNA-PK-independent pathway of rejoining, or whether it has a dedicated function in the DNA-PK-dependent pathway.
To begin addressing these questions, we studied the contributions of the two components of NHEJ in 180BR primary human fibroblasts, derived from a radiation-sensitive leukemia patient (33). Earlier studies associated the extreme radiosensitivity of 180BR fibroblasts with a severe defect in the rejoining of DNA DSBs and interphase chromosome breaks (17,18,34). No defects were detected in any of the factors constituting DNA-PK, or in other DNA damage-sensing proteins (20,35). Recently, a mutation has been reported within a highly conserved motif encompassing the active site in DNA ligase IV that severely compromises, without eliminating, the ability of the protein to form a stable enzyme–adenylate complex (22). With the molecular characterization of their genetic defect, 180BR cells define a useful system for genetic and biochemical studies on the role of DNA ligase IV in NHEJ and its functional interactions with other proteins, such as DNA-PK.
In these experiments, we also included an evaluation of the effect of wortmannin, an inhibitor of the PI3 family of kinases shown to radiosensitize cells to killing and to inhibit DNA DSB rejoining mainly by inhibiting DNA-PK (1,36). We reasoned that if DNA ligase IV acts exclusively within the D-NHEJ, inactivating mutations will have a specific effect on the fast component of rejoining. Furthermore, we reasoned that if DNA ligase IV and DNA-PK operate in the same pathway of NHEJ, inhibition of DNA-PK by wortmannin will be ineffective in 180BR cells. Our results are in line with these expectations and suggest that DNA ligase IV functions in the DNA-PK-dependent pathway of NHEJ.
MATERIALS AND METHODS
Cell culture
180BR primary human fibroblasts were established from a patient with acute lymphoblastic leukemia who died from radiation morbidity (33). Cells were kindly provided by Drs Malaise and Arlett (Institut Gustave-Roussy, Vilejuif, France and University of Sussex, Sussex, UK, respectively). 180BRM is a cell line derived in our laboratory from 180BR cells by transfection with plasmid pRNS-1 containing a replication-deficient form of SV40 virus and the neo gene that confers resistance to G418 (70 µg/ml). 180BRM cells have an extended life span that allows maintenance in culture under active growth for several months (30–40 passages), but are not immortalized. MRC5 are primary human fibroblasts. MRC5sv is an immortalized cell line derived from MRC5 by transfecting with the SV40 virus. MRC5 and MRC5sv cells were obtained from Dr Arlett. 180BR and MRC5 cells were grown in MEM supplemented with 10% fetal bovine serum, penicillin and streptomycin; 180BRM and MRC5sv cells were grown in similar media supplemented with 10% bovine calf serum. All cells were maintained at 37°C in 95% air plus 5% CO2.
Repair of DNA DSBs
Cells for DNA DSB repair experiments were plated in 60 mm dishes at 2 × 105 cells/dish and were labeled with 0.01 µCi/ml [14C]thymidine plus 2.5 µM cold thymidine for the entire period of growth. Cells were irradiated after growing to a cell density of ∼106 cells per dish. When required by the experimental protocol, cells were treated with wortmannin by replacing the growth medium with fresh medium containing 20 µM wortmannin (Sigma) from a 1 mM stock solution prepared in DMSO. Cells were irradiated after a 1 h pre-incubation in the presence of wortmannin. The procedures utilized to evaluate DNA DSB repair have been described previously (1)
Pulsed-field gel electrophoresis
Asymmetric field inversion gel electrophoresis (AFIGE) (1,37) was carried out in 0.5% Seakem agarose (FMC), cast in the presence of 0.5 µg/ml ethidium bromide, in 0.5× TBE (45 mM Tris pH 8.2, 45 mM boric acid, 1 mM EDTA) at 10°C for 40 h. During this time, cycles of 1.25 V/cm for 900 s in the direction of DNA migration alternated with cycles of 5.0 V/cm for 75 s in the reverse direction. The agarose gels were quantified to estimate DNA damage by means of a PhosphorImager (Molecular Dynamics). Gels were dried and exposed to radiation-sensitive screens for 48–96 h. DNA DSBs were quantitated by calculating the fraction of activity released (FAR) from the well into the lane in irradiated and non-irradiated samples. The FAR measured in non-irradiated cells (background) was subtracted from the results shown with irradiated cells. Gel images were obtained either by photographing ethidium bromide-stained gels under UV light, or from the PhosphorImager.
Repair kinetics were fitted assuming two exponential components of rejoining according to the equation FAR = Ae–bt + Ce–dt (1). The first term in the equation was fitted to the fast, and the second to the slow component of rejoining. Fitting was achieved using the non-linear regression analysis routines of a commercially available software package (SigmaPlot). Parameters A and C describe the amplitudes and parameters b and d the rate constants of the fast and the slow component of rejoining, respectively. From these parameters the half-times for the rejoining of the fast and the slow component can be calculated as t50,fast = ln2/b, and t50,slow = ln2/d, respectively. The fraction of DSBs rejoined by the fast and slow component of rejoining can be calculated as Ffast = A/(A+C) and Fslow = C/(A+C), respectively. Although formalisms assuming more components may also fit the data, the one used here is the simplest that can satisfactorily describe our observations. In the results shown, the parameters for the half-times have been constrained to the average of the values obtained by freely fitting each individual set of data, as it was observed that they did not differ very much from each other. This approach, when possible (1), reduces the number of variables and facilitates interpretation.
Irradiation
Cells were irradiated using a Pantak X-ray machine operated at 310 kV, 10 mA with a 2 mm Al filter (effective photon energy ∼90 kV), at a distance of 50 cm and a dose rate of 2.7 Gy/min. Dosimetry was performed with a Victoreen dosimeter that was used to calibrate an in-field ionization monitor.
Cell extract preparation
180BRM and MRC5sv cells were maintained as a monolayer in the exponential phase of growth in 100 mm tissue culture dishes. Extracts were prepared using previously described procedures (38). To prepare whole cell extracts, the preparation was adjusted to 400 mM KCl after homogenization, and centrifuged at 15 000 r.p.m. for 30 min (Sorval RC-5B, SS-34 Rotor). Nucleic acids were removed by binding to DEAE cellulose, and the extract was dialyzed overnight against 25 mM HEPES pH 7.5, 100 mM KCl, 1 mM EDTA, 0.2 mM PMSF, 0.5 mM DTT and 10% glycerol. Protein concentration was measured using the Bradford assay (Bio-Rad).
Protein and DNA substrates
DNA ligase IV antibodies were kindly provided by Dr Stephen Jackson (University of Cambridge, Cambridge, UK). Plasmid pSP65 (3.0 kbp) (kindly provided by Dr Petra Pfeiffer, Universitaet zu Koeln, Koeln, Germany) was prepared using a caesium chloride–ethidium bromide gradient, and was digested with SalI before use in rejoining reactions. Wortmannin (Sigma) (10 mM stock) was prepared in DMSO and diluted (in 10% DMSO) immediately before use. It was added to the reactions prior to ATP/DNA and was allowed to react for 10 min at 25°C before DNA and ATP were added to start the reactions.
Plasmid end-joining
Reactions were performed in 20 mM HEPES–KOH pH 7.5, 10 mM MgCl2, 80 mM KCl, 1 mM ATP, 1 mM DTT, 0.25 µg of DNA and different amounts of cell extracts as indicated in a final volume of 20 µl at 25°C for 1 h. In some experiments the concentration of Mg2+ was reduced to 0.5 mM. Reactions were stopped by the addition of 2 µl 5% SDS, 2 µl 0.5 M EDTA and 1 µl proteinase K (5 mg/ml) and were incubated for 1 h at 37°C. Half of the reaction was loaded onto a 0.7% agarose gel and electrophoresed at 45 V (2 V/cm) for 5 h. Gels were stained in SYBR Gold, scanned in a FluorImager (Molecular Dynamics) and quantitated using the ImageQuant software.
RESULTS
DNA ligase IV deficiency compromises the fast component of NHEJ
We investigated whether DNA ligase IV is involved in both forms of NHEJ identified by the in vivo kinetics of DNA DSB rejoining, or whether it mainly contributes to the fast, DNA-PK-dependent component (D-NHEJ). For this purpose, the kinetics of DNA DSB rejoining were examined in 180BR cells and the results compared to those obtained with MRC5 cells. Figures 1 and 2 show the results obtained. The upper panel in each figure shows a typical pulsed-field gel, whereas the lower panel shows results obtained from the quantification of similar gels from three independent experiments. The lines drawn through the data points reflect fitting to the sum of two exponential functions as outlined in Material and Methods. Two components characterized by widely different half-times can be identified and adequate fitting is possible using the same half-times for each of these components in 180BR and MRC5 cells (Table 1). According to this analysis, the initial fast component removes DNA DSBs with a half-time of 53 min, whereas the final, slow component removes DNA DSBs with nearly 20 times slower kinetics characterized by a half-time of 17 h. The adequate fitting achieved in both types of cells with the same half-time values indicates that defects in DNA ligase IV have only relatively small effects on the half-times of each of the NHEJ components.
Figure 1.
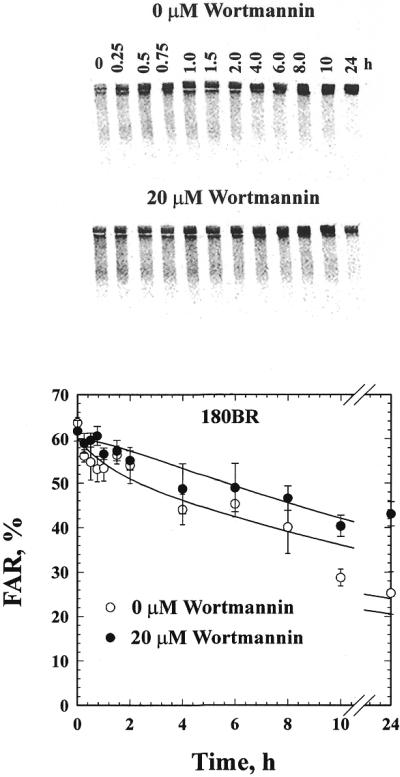
Rejoining of IR-induced DNA DSBs in 180BR primary fibroblasts in the presence or absence of 20 µM wortmannin. The upper panel shows a typical gel, while the lower panel shows the quantitative analysis from three independent experiments. Plotted are the mean and the standard error. The lines through the data points represent fitting to the sum of two exponential functions as described in Materials and Methods. The value of FAR measured in non-irradiated cells has been subtracted from all data points.
Figure 2.
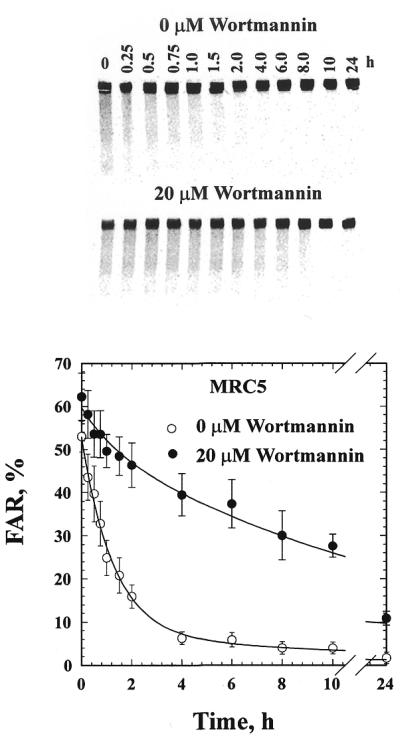
Rejoining of IR-induced DNA DSBs in MRC5 primary fibroblasts in the presence or absence of 20 µM wortmannin. Other details as in Figure 1.
Table 1. Half-times for the fast (t50, fast) and the slow (t50, slow) components of DNA DSB rejoining, as well as percent of breaks repaired by fast (Ffast) or slow (Fslow) kinetics in wild-type 180BR and MRC5 cells with and without wortmannin.
| Cells |
t50,fast
(min) |
t50,slow
(h) |
Ffast
(%) |
Fslow
(%) |
| 180BR | 53 | 17 | 10 | 90 |
| 180BR + wortmannin | 53 | 17 | 0 | 100 |
| MRC5 | 53 | 17 | 89 | 11 |
| MRC5 + wortmannin | 53 | 17 | 29 | 71 |
Despite the similarity in the half-times of DNA DSB rejoining, the relative contribution of each component differs significantly between 180BR and MRC5 cells. While the fast component removes 89% of the induced DNA DSBs in MRC5, it only removes 10% in 180BR cells and, similarly, the slow component only removes 11% of the DNA DSBs in MRC5 cells but it removes 90% in 180BR (Table 1). Thus, DNA ligase IV mutation drastically reduces the contribution of the fast component of DNA DSB rejoining. Yet it leaves the slow component unaffected since it proceeds with wild-type half-times, despite the fact that it now removes a significantly larger fraction of IR-induced DNA DSBs from the genome. The results in Figure 1 indicate that the slow component is capable of removing >50% of the DNA DSBs in 24 h. Thus, cells deficient in DNA ligase IV remove a significant proportion of DNA DSBs from their genome albeit with slow kinetics, suggesting the operation of a DNA ligase IV-independent pathway of DNA DSB rejoining.
Reduced inhibition by wortmannin of DNA DSB rejoining in 180BR cells
The above results are reminiscent of those obtained with cells deficient in DNA-PKcs (1,14) and indicate that the phenotypic manifestations of a deficiency in DNA ligase IV at the level of DNA DSB rejoining are similar to those associated with a defect in DNA-PKcs. This raises the possibility of epistatic relationships between the two repair factors. It also raises the question as to whether DNA ligase IV contributes to other NHEJ pathways (B-NHEJ), or whether it is dedicated to D-NHEJ.
To address this possibility we treated 180BR and MRC5 cells with wortmannin and measured DNA DSB rejoining. Wortmannin was initially identified as an inhibitor of PI-3 kinase but was later shown to also inhibit, at higher concentrations, other members of the PI-3 family of kinases including DNA-PK (1,36,39–42). While the rather broad spectrum of wortmannin inhibition raises questions regarding specificity, there is strong evidence that the effects of wortmannin on cell radiosensitivity to killing and rejoining of DNA DSBs derive almost exclusively from inhibition of DNA-PK (1,36). Thus, treatment with wortmannin can be used to inactivate DNA-PK in order to study, specifically, the role of this kinase in DNA DSB rejoining and cell radiosensitivity to killing on different genetic backgrounds. We reasoned that if DNA ligase IV functions in the DNA-PK-dependent pathway, wortmannin should have no effect on the kinetics of DNA DSB rejoining in 180BR cells. If, on the other hand, DNA-PK contributed to DNA DSB rejoining independently of DNA ligase IV, its inhibition by wortmannin should further compromise the ability of 180BR cells to rejoin DNA DSBs, by inhibiting the slow component of rejoining.
The results in Figure 1 demonstrate that treatment of 180BR cells with 20 µM wortmannin produces only a small effect on DNA DSB rejoining manifest mainly as an elimination of the fast component of rejoining. According to the analysis shown in Table 1, wortmannin leaves the half-time of the slow component unchanged, but reduces the contribution of the fast component from 10% to undetectable levels. This rather small effect could be attributed to some residual DNA ligase IV activity (22) that allows a marginal operation of the DNA-PK-dependent pathway.
In contrast to results with 180BR cells, and in full agreement with results previously reported for other cell lines (1), treatment with wortmannin of MRC5 cells strongly reduces (from 89 to 29%) the contribution of the fast and enhances (from 11 to 71%) the contribution of the slow component of rejoining without measurably altering the half-times of either component. DNA-PK activity was inhibited by wortmannin to a similar degree in 180BR and MRC5 cells (results not shown) suggesting that the difference in inhibition of DNA DSB rejoining cannot be attributed to differences in the level of DNA-PK inhibition.
Wortmannin treatment does not radiosensitize 180BR cells
The experiments described above suggest that DNA ligase IV functions within the DNA-PK-dependent pathway of NHEJ. We inquired whether this conclusion also holds for cell radiosensitivity to killing. Because the colony-forming ability of the primary cells was suboptimal, in these experiments we introduced two cell lines, MRC5sv and 180BRM, derived from MRC5 and 180BR, respectively, by transfection with SV40 DNA. 180BRM represents a pool of several G418-resistant colonies. Although these cells are not truly immortalized, they grow faster than 180BR, have a much better plating efficiency, and an extended life span that allows the production of relatively large numbers of cells that can be used for survival studies, but also for extract preparation (see below). DNA DSB rejoining experiments (data not shown) indicated that these cells have impaired ability to rejoin IR-induced DNA DSBs, and that wortmannin has no effect on this rejoining suggesting that they behave similarly to 180BR. Figure 3 shows survival curves for X-ray-induced cell killing in 180BRM cells and MRC5sv cells. As expected, 180BRM are significantly more radiosensitive than MRC5sv cells. However, after treatment with 20 µM wortmannin, MRC5sv cells are radiosensitized to the levels of 180BRM cells, whereas the radiosensitivity of 180BRM cells remains practically unchanged. This result further suggests that DNA ligase IV operates in the DNA-PK-dependent pathway of NHEJ and, as a result, inhibition of DNA-PK on a DNA ligase IV-deficient background is of no consequence on cell radiosensitivity to killing.
Figure 3.

Cell survival in 180BRM and MRC5sv cells after exposure to various doses of X-rays. Cells were irradiated in the presence or absence of 20 µM wortmannin, added 1 h before exposure, and plated for colony formation 4 h later. Results represent the mean and standard error from three independent experiments.
Extracts of 180BRM cells have a considerable end joining activity
The results presented above and those previously published (1) suggest the operation of a B-NHEJ pathway capable of removing the majority of IR-induced DNA DSBs when D-NHEJ is compromised by defects in DNA ligase IV, or DNA-PK. They also suggest that DNA ligase IV operates within the D-NHEJ pathway, and that its activity is a prerequisite for the function of this pathway. We inquired whether aspects of these genetic observations could be reproduced in vitro in a plasmid end joining assay (reviewed in 43).
We evaluated end joining of a SalI-digested plasmid by activities present in whole cell extracts of MRC5sv and 180BRM cells and the results are shown in Figure 4A. As expected, extracts of MRC5sv cells (lane 2) support end joining that leads to the generation of multimeric forms of the plasmid, suggesting intermolecular ligation. Circles (OC) are produced at very low amounts under these experimental conditions. Despite the low DNA ligase IV activity, reactions assembled with extracts of 180BRM cells (lane 7) show end joining activity that is very similar to that observed with MRC5sv cells. Thus, end joining in this plasmid-based assay, under the conditions employed, does not depend measurably on DNA ligase IV activity.
Figure 4.

End joining after incubation at 25°C for 1 h of SalI-digested plasmid DNA by 10 µg whole cell extract prepared from actively growing 180BRM or MRC5sv cells. (A) Reactions assembled at different Mg2+ concentrations as indicated. (B) Reactions assembled either at 10 or 0.5 mM Mg2+ with extracts pre-treated for 1 h at 4°C with an anti-ligase IV monoclonal antibody as indicated. Reactions were stopped by adding 2 µl 5% SDS, 2 µl 0.5 M EDTA and 1 µl 1 mg/ml proteinase K, and incubated at 37°C for 1 h before assaying by gel electrophoresis. Gels were stained with SYBR Gold and scanned in a FluorImager. Control reactions were assembled in the absence of cell extract.
Since DNA ligase IV, unlike DNA ligases III and I, is present as a pre-adenylated enzyme in the cells (44,45), it remains active at low concentrations of magnesium (Mg2+). An attempt was therefore made to evaluate its relative contribution to end joining by lowering the Mg2+ concentration. Figure 4A includes the results obtained. For MRC5sv cells, reduction in Mg2+ leads to a gradual reduction in end joining activity. Low but detectable end joining is observed at 0.5 mM Mg2+. Analysis using a FluorImager indicates that at this concentration end joining is ∼10% of the 10 mM control. A similar experiment using extracts of 180BRM cells shows a stronger dependence on Mg2+ and barely detectable end joining at 0.5 mM. This is compatible with end joining being dependent on DNA ligase IV only at low Mg2+ concentrations, which is significantly reduced in 180BRM cells. Thus, although end joining under the assay conditions employed here includes a measurable contribution from DNA ligase IV, this contribution is small. This is in contrast to in vivo data (see above), where its involvement in D-NHEJ allowed DNA ligase IV to dominate end joining in wild-type cells. Thus, under the in vitro conditions employed here, the natural balance between end joining pathways is disrupted.
As an alternative way of examining the dependence of end joining on DNA ligase IV, we used antibodies raised against this enzyme. Figure 4B shows the results obtained for reactions assembled either at 10 mM or 0.5 mM Mg2+. It is evident that treatment of extracts with the antibody has no effect on DNA end joining in MRC5sv cells at 10 mM Mg2+. The lack of effect under the same conditions in 180BRM cells, which lack the antigen, shows that the antibody exerts no secondary effects on the reaction. The lack of inhibition observed in this set of reactions is in line with the small contribution of DNA ligase IV to the overall in vitro end joining activity. It is relevant that when the same antibody was tested at 0.5 mM Mg2+ to specifically assay DNA ligase IV-dependent activity, inhibition was observed demonstrating that the antibody is indeed capable of inhibiting end joining by targeting DNA ligase IV (Fig. 4B). At low Mg2+ concentrations, the antibody inhibited a barely detectable end joining in 180BRM cells confirming the presence of residual activity in these mutants (22). Inhibition of end joining by the same antibody was also reported in an assay specifically developed to measure DNA ligase IV-dependent activity (46).
The dominant role of DNA ligase IV-independent end joining under these experimental conditions makes an examination of the operation of this enzyme within the D-NHEJ difficult without artificially compromising the DNA ligase IV-independent end joining pathway. Indeed, biochemical evidence for a function of DNA ligase IV within D-NHEJ is provided in studies carried out at low Mg2+ concentrations (24,46). These observations support the in vivo data presented here suggesting the operation of DNA ligase IV-independent pathways in DNA DSB end joining, and the function of DNA ligase IV within the DNA-PK-dependent pathway of NHEJ.
DISCUSSION
DNA ligase IV deficiency inhibits the fast component of NHEJ
The results presented in the previous section indicate that a defect in DNA ligase IV (22) is not associated with a general defect in DNA DSB repair, but rather with a specific inhibition of D-NHEJ. Despite this defect, 180BR cells remove a substantial proportion of IR-induced DNA DSBs utilizing B-NHEJ that remains unaffected by this particular mutation of DNA ligase IV (Fig. 1). This is similar to observations with cells deficient in DNA-PKcs (1,14) and suggests that both proteins function in D-NHEJ.
The time-frame of the experiments described here does not allow to determine conclusively whether DNA ligase IV-deficient cells are able to remove all DNA DSBs from their genome utilizing B-NHEJ, as was shown for DNA-PKcs-deficient cells (1,14). However, since the kinetics of DNA DSB rejoining do not show signs of plateau by 24 h, we speculate that B-NHEJ also has the potential to remove the majority of DNA DSBs from the 180BR genome. Similar trends were also observed in earlier work with these cells (18). Despite the ability of DNA-PK or DNA ligase IV-deficient cells to remove the majority of IR-induced DNA DSB from their genome, there is evidence that this pathway is error prone (47), and frequently leads to non-reciprocal translocations or genomic instability (48,49).
DNA ligase IV-deficient cells rejoin a small proportion (∼10%) of IR-induced DNA DSBs with fast kinetics. Similar observations were also made with cells deficient in DNA-PKcs (1). The rather small component of D-NHEJ may be due to residual activities (DNA ligase IV, or DNA-PK) in the cells (22,50), or to alternative pathways of rejoining. While the results presented do not allow a conclusive discrimination between these two possibilities, the sensitivity to wortmannin of the residual, fast component suggests that it may actually derive from residual activity of the DNA-PK-dependent pathway, or depend on a wortmannin-sensitive kinase other than DNA-PK. However, ataxia telangiectasia mutated (ATM) can be ruled out as this kinase, as AT cells have nearly normal kinetics of DNA DSB rejoining under most conditions (51,52).
DNA ligase IV functions within the DNA-PK-dependent pathway of NHEJ
The possibility that DNA ligase IV functions within the DNA-PK-dependent pathway of NHEJ is further corroborated by the observation that treatment with wortmannin is ineffective in 180BR cells. If inactivation of DNA-PK in a DNA ligase IV-deficient background has such a minor effect on the rejoining kinetics of DNA DSBs, one can conclude that the function of DNA-PK in DNA DSB rejoining requires DNA ligase IV. This observation places the two factors in the same pathway, in agreement with biochemical evidence for physical and functional interactions (see below). Since the slow component of DNA DSB rejoining remains unaltered in 180BR cells despite the defect in DNA ligase IV, it is possible to also suggest that DNA ligase IV is not involved in this pathway of NHEJ. Yet this conclusion is hampered by the fact that residual DNA ligase IV activity remains in 180BR capable of supporting V(D)J recombination (22). Also, if DNA ligase IV were rate limiting, as biochemical studies suggest, (24), a low level of activity could provide a significant contribution to B-NHEJ. Therefore, the question whether DNA ligase IV functions exclusively within the D-NHEJ pathway cannot be answered with confidence from the results presented and must await experiments with cells where DNA ligase IV activity is completely absent.
Similar conclusions can also be drawn from the results on cell radiosensitivity to killing. Treatment of 180BR cells with wortmannin has no radiosensitizing effect, whereas the same treatment in control cells radiosensitizes to the 180BR level. Thus, genetic inactivation of DNA ligase IV or drug-induced inactivation of DNA-PK have, individually, the same consequences on cell radiosensitivity to killing, whereas combined inactivation does not exceed the consequences of single inactivation.
In vivo and in vitro results show interesting parallels but also point to important differences
The results at the biochemical level using a plasmid-based assay for NHEJ are in partial agreement with genetic in vivo data and suggest that the assay reproduces some important aspects of the end joining process. Thus, active end joining is observed in the absence of DNA ligase IV, as would be expected if B-NHEJ remained active. However, contrary to the in vivo observations, B-NHEJ dominated the in vitro reaction even in extracts of wild-type cells. The reason for this difference is not presently understood, but it may derive from differences in the abundance and the spatial distribution of DNA ends in the irradiated cell in vivo and in the in vitro assay (1).
We have previously suggested that D-NHEJ may be an accelerated form of B-NHEJ (1). According to this model a constitutively active and evolutionarily conserved slowly operating basic NHEJ apparatus (B-NHEJ) is accelerated several fold in chromatin domains, termed DNA-PK surveillance domains, where the actions of Ku and DNA-PKcs are combined to facilitate end joining (D-NHEJ) (1). The results presented here suggest that DNA ligase IV, probably as a complex with Xrcc4, operates within the same system to enable fast rejoining of IR-induced DNA DSBs. The evolution of D-NHEJ from B-NHEJ, as proposed by this model, is in line with biochemical experiments suggesting that DNA-PK and DNA ligase IV are by themselves unable to support end joining and that additional activities are required (24,46 and data not shown). These additional and presently unknown activities could be components of the B-NHEJ apparatus.
DNA ligase IV in DNA DSB repair and genomic stability
The cooperation of the DNA ligase IV–Xrcc4 with the DNA-PK complex is also supported by recent biochemical studies. From the known DNA ligases only DNA ligase IV interacts with DNA-bound Ku, and it is thought that this interaction allows Ku to recruit the DNA ligase IV–Xrcc4 complex to the DNA ends, thus facilitating end joining (31). Furthermore, Xrcc4 has been shown to be a substrate for DNA-PK (26,53), and DNA ligase IV has been identified as a putative of DNA-PK substrate in a peptide screen (54).
Despite these interactions, genetic data also suggest independent functions of the different components of these repair complexes that may be unrelated to their involvement in NHEJ. Thus, while mice deficient in DNA-PKcs are immunodeficient and exhibit increased radiosensitivity, they develop normally and show no evidence of growth retardation (55–57). On the other hand, mice deficient in Ku subunits are immunodeficient and radiosensitive, but also display growth retardation and propensity to tumor development (58–61). Finally, DNA ligase IV or Xrcc4 deficiency leads to embryonic lethality as a result of massive neuronal apoptosis, arrested lymphogenesis and various cellular defects (62–64). Elucidation of the molecular basis of these differences will advance our knowledge on the function of these proteins and their role in DNA repair. It is important, however, that defects in any of the components of the DNA-PK pathway leads to pronounced genomic instability (48,49,64–66).
Acknowledgments
ACKNOWLEDGEMENTS
The authors are indebted to Drs Colin Arlett and Edmond Malaise for cells, Drs Steven Jackson and Susan Critchlow for DNA ligase IV–Xrcc4 and to Dr Petra Pfeiffer for the pSP65 plasmid. Special thanks go to Ms Nancy Mott for help in the preparation of the manuscript. This work was supported by NCI Grants 42026, 56706 and P30-CA56036 awarded from NIH, DHHS.
References
- 1.DiBiase S.J., Zeng,Z.-C., Chen,R., Hyslop,T., Curran,W.J.,Jr and Iliakis,G. (2000) DNA-dependent protein kinase stimulates an independently active, nonhomologous, end-joining apparatus. Cancer Res., 60, 1245–1253. [PubMed] [Google Scholar]
- 2.Thacker J. (1999) A surfeit of RAD51-like genes? Trends Genet., 15, 166–168. [DOI] [PubMed] [Google Scholar]
- 3.Thompson, L.H. and Schild,D. (1999) The contribution of homologous recombination in preserving genome integrity in mammalian cells. Biochimie, 81, 87–105. [DOI] [PubMed] [Google Scholar]
- 4.Jeggo P.A. (1998) DNA breakage and repair. Adv. Genet., 38, 186–218. [DOI] [PubMed] [Google Scholar]
- 5.Wang H., Zeng,Z.-C., Bui,T.-A., Sonoda,E., Takata,M., Takeda,S. and Iliakis,G. (2001) Efficient rejoining of radiation-induced DNA double-strand breaks in vertebrate cells deficient in genes of the RAD52 epistasis group. Oncogene, in press. [DOI] [PubMed] [Google Scholar]
- 6.Dynan W.S. and Yoo,S. (1998) Interaction of ku protein and DNA-dependent protein kinase catalytic subunit with nucleic acids. Nucleic Acids Res., 26, 1551–1559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Smith ,G.C.M. and Jackson,S.P. (1999) The DNA-dependent protein kinase. Genes Dev., 13, 916–934. [DOI] [PubMed] [Google Scholar]
- 8.Lees-Miller S.P., Godbout,R., Chan,D.W., Weinfeld,M., Day,R.S.,III, Barron,G.M. and Allalunis-Turner,J. (1995) Absence of p350 subunit of DNA-activated protein kinase from a radiosensitive human cell line. Science, 267, 1183–1185. [DOI] [PubMed] [Google Scholar]
- 9.Biedermann K.A., Sung,J., Giaccia,A.J., Tosto,L.M. and Brown,J.M. (1991) scid mutation in mice confers hypersensitivity to ionizing radiation and a deficiency in DNA double-strand break repair. Proc. Natl Acad. Sci. USA, 88, 1394–1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Allalunis-Turner M.J., Zia,P.K.Y., Barron,G.M., Mirzayans,R. and Day,R.S.,III (1995) Radiation-induced DNA damage and repair in cells of a radiosensitive human malignant glioma cell line. Radiat. Res., 144, 288–293. [PubMed] [Google Scholar]
- 11.Hendrickson E.A., Qin,X.-Q., Bump,E.A., Schatz,D.G., Oettinger,M. and Weaver,D.T. (1991) A link between double-strand break-related repair and V(D)J recombination: the scid mutation. Proc. Natl Acad. Sci. USA, 88, 4061–4065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chang C., Biedermann,K.A., Mezzina,M. and Brown,J.M. (1993) Characterization of the DNA double strand break repair defect in scid mice. Cancer Res., 53, 1244–1248. [PubMed] [Google Scholar]
- 13.Stackhouse M.A. and Bedford,J.S. (1994) An ionizing radiation-sensitive mutant of CHO cells: irs-20. III. Chromosome aberrations, DNA breaks and mitotic delay. Int. J. Radiat. Biol., 65, 571–582. [DOI] [PubMed] [Google Scholar]
- 14.Nevaldine B., Longo,J.A. and Hahn,P.J. (1997) The scid defect results in much slower repair of DNA double-strand breaks but not high levels of residual breaks. Radiat. Res., 147, 535–540. [PubMed] [Google Scholar]
- 15.Giaccia A., Weinstein,R., Hu,J. and Stamato,T.D. (1985) Cell cycle-dependent repair of double-strand DNA breaks in a γ-ray-sensitive chinese hamster cell. Somat. Cell Mol. Genet., 11, 485–491. [DOI] [PubMed] [Google Scholar]
- 16.Taccioli G.E., Rathbun,G., Oltz,E., Stamato,T., Jeggo,P.A. and Alt,F.W. (1993) Impairment of V(D)J recombination in double-strand break repair mutants. Science, 260, 207–210. [DOI] [PubMed] [Google Scholar]
- 17.Badie C., Iliakis,G., Foray,N., Alsbeih,G., Pantelias,G.E., Okayasu,R., Cheong,N., Russell,N.S., Begg,A.C., Arlett,C.F. and Malaise,E.P. (1995) Defective repair of DNA double-strand breaks and chromosome damage in fibroblasts from a radiosensitive leukemia patient. Cancer Res., 55, 1232–1234. [PubMed] [Google Scholar]
- 18.Badie C., Iliakis,G., Foray,N., Alsbeih,G., Cedervall,B., Chavaudra,N., Pantelias,G., Arlett,C. and Malaise,E.P. (1995) Induction and rejoining of DNA double-strand breaks and interphase chromosome breaks after exposure to X rays in one normal and two hypersensitive human fibroblast cell lines. Radiat. Res., 144, 26–35. [PubMed] [Google Scholar]
- 19.Wei Y.-F., Robins,P., Carter,K., Caldecott,K., Pappin,D.J.C., Yu,G.-L., Wang,R.-P., Shell,B.K., Nash,R.A., Schar,P., Barnes,D.E. et al. (1995) Molecular cloning and expression of human cDNAs encoding a novel DNA ligase IV and DNA ligase III, an enzyme active in DNA repair and recombination Mol. Cell. Biol., 15, 3206–3216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Badie C., Goodhardt,M., Waugh,A., Doyen,N., Foray,N., Calsou,P., Singleton,B., Gell,D., Salles,B., Jeggo,P. et al. (1997) A DNA double-strand break defective fibroblast cell line (180BR) derived from a radiosensitive patient represents a new mutant phenotype. Cancer Res., 57, 4600–4607. [PubMed] [Google Scholar]
- 21.Grawunder U., Zimmer,D., Fugmann,S., Schwarz,K. and Lieber,M.R. (1998) DNA ligase IV is essential for V(D)J recombination and DNA double-strand break repair in human precursor lymphocytes. Mol. Cell, 2, 477–484. [DOI] [PubMed] [Google Scholar]
- 22.Riballo E., Critchlow,S.E., Teo,S.-H., Doherty,A.J., Priestley,A., Broughton,B., Kysela,B., Beamish,H., Plowman,N., Arlett,C.F. et al. (1999) Identification of a defect in DNA ligase IV in a radiosensitive leukaemia patient. Curr. Biol., 9, 699–702. [DOI] [PubMed] [Google Scholar]
- 23.Bryans M., Valenzano,M.C. and Stamato,T.D. (1999) Absence of DNA ligase IV protein in XR-1 cells: evidence for stabilization by XRCC4. Mutat. Res., 433, 53–58. [DOI] [PubMed] [Google Scholar]
- 24.Lee K-J., Huang,J., Takeda,Y. and Dynan,W.S. (2000) DNA ligase IV and XRCC4 form a stable mixed tetramer that functions synergistically with other repair factors in a cell-free end-joining system. J. Biol. Chem. ,275, 34787–34796. [DOI] [PubMed] [Google Scholar]
- 25.Teo S-H. and Jackson,S.P. (1997) Identification of Saccharomyces cerevisiae DNA ligase IV: involvement in DNA double-strand break repair. EMBO J., 16, 4788–4795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Critchlow S.E., Bowater,R.P. and Jackson,S.P. (1997) Mammalian DNA double-strand break repair protein XRCC4 interacts with DNA ligase IV. Curr. Biol., 7, 588–598. [DOI] [PubMed] [Google Scholar]
- 27.Wilson T.E., Grawunder,U. and Lieber,M.R. (1997) Yeast DNA ligase IV mediates non-homologous DNA end joining. Nature, 388, 495–498. [DOI] [PubMed] [Google Scholar]
- 28.Grawunder U., Wilm,M., Wu,X., Kulesza,P., Wilson,T.E., Mann,M. and Lieber,M.R. (1997) Activity of DNA ligase IV stimulated by complex formation with XRCC4 protein in mammalian cells. Nature, 388, 492–495. [DOI] [PubMed] [Google Scholar]
- 29.Grawunder U., Zimmer,D., Kulesza,P. and Lieber,M.R. (1998) Requirement for an interaction of XRCC4 with DNA ligase IV for wild-type V(D)J recombination and DNA double-strand break repair in vivo. J. Biol. Chem., 273, 24708–24714. [DOI] [PubMed] [Google Scholar]
- 30.Grawunder U., Zimmer,D. and Lieber,M.R. (1998) DNA ligase IV binds to XRCC4 via a motif located between rather than within its BRCT domains. Curr. Biol., 8, 873–876. [DOI] [PubMed] [Google Scholar]
- 31.Nick McElhinny S.A., Snowden,C.M., McCarville,J. and Ramsden,D. (2000) Ku recruits the XRCC4–ligase IV complex to DNA ends. Mol. Cell. Biol., 20, 2996–3003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen L., Trujillo,K., Sung,P. and Tomkinson,A.E. (2000) Interactions of the DNA ligase IV–XRCC4 complex with DNA ends and the DNA-dependent protein kinase. J. Biol. Chem., 275, 26196–26205. [DOI] [PubMed] [Google Scholar]
- 33.Plowman P.N., Bridges,B.A., Arlett,C.F., Hinney,A. and Kingston,J.E. (1990) An instance of clinical radiation morbidity and cellular radiosensitivity, not associated with ataxia-telagiectasia. Br. J. Radiol., 63, 624–628. [DOI] [PubMed] [Google Scholar]
- 34.Badie C., Arlett,C., Iliakis,G. and Malaise,E.P. (1995) In Hagen,U., Hardes,D., Jung,H. and Streffer,C. (eds), 10th International Congress of Radiation Research, Congress Lectures. Universitätsdruckerei H. Stürtz AG, Würzburg, Germany, Vol. 2, pp. 476–480.
- 35.Lu H., Song,Q., Arlett,C. and Lavin,M.F. (1998) The radiosensitive cell line 180BR is not defective in the major DNA damage-sensing proteins. Cancer Res., 58, 84–88. [PubMed] [Google Scholar]
- 36.Chernikova S.B., Wells,R.L. and Elkind,M.M. (1999) Wortmannin sensitizes mammalian cells to radiation by inhibiting the DNA-dependent protein kinase-mediated rejoining of double-strand breaks. Radiat. Res., 151, 159–166. [PubMed] [Google Scholar]
- 37.Iliakis G., Metzger,L., Denko,N. and Stamato,T.D. (1991) Detection of DNA double strand breaks in synchronous cultures of CHO cells by means of asymmetric field inversion gel electrophoresis. Int. J. Radiat. Biol., 59, 321–341. [DOI] [PubMed] [Google Scholar]
- 38.Wang Y. and Wang,H. (1999) A rapid preparation of extracts for DNA replication in vitro. Radiat. Res., 151, 59–64. [PubMed] [Google Scholar]
- 39.Powis G., Bonjouklian,R., Berggren,M.M., Gallegos,A., Abraham,R., Ashendel,C., Zalkow,L., Matter,W.F., Dodge,J., Grindley,G. and Vlahos,C.J. (1994) Wortmannin, a potent and selective inhibitor of phosphatidylinositol-3-kinase. Cancer Res., 54, 2419–2423. [PubMed] [Google Scholar]
- 40.Hartley K.O., Gell,D., Smith,G.C.M., Zhang,H., Divecha,N., Connelly,M.A., Admon,A., Lees-Miller,S.P., Anderson,C.W. and Jackson,S.P. (1995) DNA-dependent protein kinase catalytic subunit: a relative of phosphatidylinositol 3-kinase and the ataxia telangiectasia gene product. Cell, 82, 849–856. [DOI] [PubMed] [Google Scholar]
- 41.Wymann M.P., Bulgarelli-Leva,G., Zvelebil,M.J., Pirola,L., Vanhaesebroeck,B., Waterfield,M.D. and Panayotou,G. (1996) Wortmannin inactivates phosphoinositide 3-kinase by covalent modification of Lys-802, a residue involved in the phosphate transfer reaction. Mol. Cell. Biol., 16, 1722–1733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Okayasu R., Suetomi,K. and Ullrich,R.L. (1998) Wortmannin inhibits repair of DNA double-strand breaks in irradiated normal human cells. Radiat. Res., 149, 440–445. [PubMed] [Google Scholar]
- 43.Labhart P. (1999) Nonhomologous DNA end joining in cell-free systems. Eur. J. Biochem., 265, 849–861. [DOI] [PubMed] [Google Scholar]
- 44.Robins P. and Lindahl,T. (1996) DNA ligase IV from HeLa cell nuclei. J. Biol. Chem., 271, 24257–24261. [DOI] [PubMed] [Google Scholar]
- 45.Tomkinson A. and Levin,D.S. (1997) Mammalian DNA ligases. Bioessays, 19, 893–901. [DOI] [PubMed] [Google Scholar]
- 46.Baumann P. and West,S.C. (1998) DNA end-joining catalyzed by human cell-free extracts. Proc. Natl Acad. Sci. USA, 95, 14066–14070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Loebrich M., Rydberg,B. and Cooper,P.K. (1995) Repair of x-ray-induced DNA double-strand breaks in specific NotI restriction fragments in human fibroblasts: joining of correct and incorrect ends. Proc. Natl Acad. Sci. USA, 92, 12050–12054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ferguson D.O., Sekiguchi,J.M., Chang,S., Frank,K.M., Gao,Y., DePinho,R.A. and Alt,F.W. (2000) The nonhomologous end-joining pathway of DNA repair is required for genomic stability and the suppression of translocations Proc. Natl Acad. Sci. USA, 97, 6630–6633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Karanjawala Z.E., Grawunder,U., Hsieh,C.-L. and Lieber,M.R. (1999) The nonhomologous DNA end joining pathway is important for chromosome stability in primary fibroblasts. Curr. Biol., 9, 1501–1504. [DOI] [PubMed] [Google Scholar]
- 50.Galloway A.M., Spencer,C.A., Anderson,C.W. and Allalunis-Turner,M.J. (1999) Differential stability of the DNA-activated protein kinase catalytic subunit mRNA in human glioma cells. Oncogene, 18, 1361–1368. [DOI] [PubMed] [Google Scholar]
- 51.Lavin M.F. (1998) Radiosensitivity and oxidative signalling in ataxia telangiectasia: an update. Radiother. Oncol., 47, 113–123. [DOI] [PubMed] [Google Scholar]
- 52.Rotman G. and Shiloh,Y. (1999) ATM: a mediator of multiple responses to genotoxic stress. Oncogene, 18, 6135–6144. [DOI] [PubMed] [Google Scholar]
- 53.Leber R., Wise,T.W., Mizuta,R. and Meek,K. (1998) The XRCC4 gene product is a target for and interacts with the DNA-dependent protein kinase. J. Biol. Chem., 273, 1794–1801. [DOI] [PubMed] [Google Scholar]
- 54.Kim S.-T., Lim,D.-S., Canman,C.E. and Kastan,M.B. (1999) Substrate specificities and identification of putative substrates of ATM kinase family members. J. Biol. Chem., 274, 37538–37543. [DOI] [PubMed] [Google Scholar]
- 55.Gao Y., Chaudhuri,Y., Zhu,C., Davidson,L., Weaver,D.T. and Alt,F.W. (1998) A targeted DNA-PKcs-null mutation reveals DNA-PK-independent functions for KU in V(D)J recombination. Immunity, 9, 367–376. [DOI] [PubMed] [Google Scholar]
- 56.Taccioli G.E., Amatucci,A.G., Beamish,H.J., Gell,D., Xiang,X.H., Arzayus,M.I.T., Priestley,A., Jackson,S.P., Rothstein,A.M., Jeggo,P.A. and Herrera,V.L.M. (1998) Targeted disruption of the catalytic subunit of the DNA-PK gene in mice confers severe combined immunodeficiency and radiosensitivity. Immunity, 9, 355–366. [DOI] [PubMed] [Google Scholar]
- 57.Wang S., Guo,M., Ouyang,H., Li,X., Cordon-Cardo,C., Kurimasa,A., Chen,D.J., Fuks,Z., Ling,C.C. and Li,G.C. (2000) The catalytic subunit of DNA-dependent protein kinase selectively regulates p53-dependent apoptosis but not cell-cycle arrest. Proc. Natl Acad. Sci. USA, 97, 1584–1588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Nussenzweig A., Chen,C., da Costa Soares,V., Sanchez,M., Kokol,K., Nussenzweig,M.C. and Li,G.C. (1996) Requirement for Ku80 in growth and immunoglobulin V(D)J recombination. Nature, 382, 551–555. [DOI] [PubMed] [Google Scholar]
- 59.Zhu C., Bogue,M.A., Lim,D.-S., Hasty,P. and Roth,D.B. (1996) Ku86-deficient mice exhibit severe combined immunodeficiency and defective processing of V(D)J recombination intermediates. Cell, 86, 379–389. [DOI] [PubMed] [Google Scholar]
- 60.Ouyang B.H., Nussenzweig,A., Kurimasa,A., da Costa Soares,V., Li,X., Cordon-Cardo,C., Li,W.-h., Cheong,N., Nussenzweig,M., Iliakis,G., Chen,D.J. and Li,G.C. (1997) Ku70 is required for DNA repair but not for T cell antigen receptor gene recombination in vivo. J. Exp. Med., 186, 921–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Li G.C., Ouyang,H., Li,X., Nagasawa,H., Little,J.B., Chen,D.J., Ling,C.C., Fuks,Z. and Cordon-Cardo,C. (1998) Ku70: A candidate tumor suppressor gene for murine T cell lymphoma. Mol. Cell, 2, 1–8. [DOI] [PubMed] [Google Scholar]
- 62.Barnes D.E., Stamp,G., Rosewell,I., Denzel,A. and Lindahl,T. (1998) Targeted disruption of the gene encoding DNA ligase IV leads to lethality in embryonic mice. Curr. Biol., 8, 1395–1398. [DOI] [PubMed] [Google Scholar]
- 63.Frank K.M., Sekiguchi,J.M., Seidl,K.J., Swat,W., Rathbun,G.A., Cheng,H.-L., Davidson,L., Kangaloo,L. and Alt,F.W. (1998) Late embryonic lethality and impaired V(D)J recombination in mice lacking DNA ligase IV. Nature, 396, 173–177. [DOI] [PubMed] [Google Scholar]
- 64.Gao Y., Ferguson,D.O., Xie,W., Manis,J.P., Sekiguchi,J.A., Frank,K.M., Chaudhuri,J., Horner,J., DePinho,R.A. and Alt,F.W. (2000) Interplay of p53 and DNA-repair protein XRCC4 in tumorigenesis, genomic stability and development. Nature, 404, 897–900. [DOI] [PubMed] [Google Scholar]
- 65.Difilippantonio M.J., Zhu,J., Chen,H.T., Meffre,E., Nussenzweig,N.C., Max,E.E., Ried,T. and Nussenzweig,A. (2000) DNA repair protein Ku80 suppresses chromosomal aberrations and malignant transformation. Nature, 404, 510–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Frank K.M., Sharpless,N.E., Gao,Y., Sekiguchi,J.M., Fergson,D.O., Zhu,C., Manis,J.P., Horner,J., DePinho,R.A. and Alt,F.W. (2000) DNA ligase IV deficiency in mice leads to defective neurogenesis and embryonic lethality via the p53 pathway. Mol. Cell, 5, 993–1002. [DOI] [PubMed] [Google Scholar]