

Abstract
Carbonic anhydrases (CAs, EC 4.2.1.1) are metalloenzymes which catalyze the hydration of carbon dioxide to bicarbonate and protons. Many pathogenic bacteria encode such enzymes belonging to the α-, β-, and/or γ-CA families. In the last decade, the α-CAs from Neisseria spp. and Helicobacter pylori as well as the β-class enzymes from Escherichia coli, H. pylori, Mycobacterium tuberculosis, Brucella spp., Streptococcus pneumoniae, Salmonella enterica, and Haemophilus influenzae have been cloned and characterized in detail. For some of these enzymes the X-ray crystal structures were determined, and in vitro and in vivo inhibition studies with various classes of inhibitors, such as anions, sulfonamides and sulfamates reported. Although efficient inhibitors have been reported for many such enzymes, only for Neisseria spp., H. pylori, B. suis, and S. pneumoniae enzymes it has been possible to evidence inhibition of bacterial growth in vivo. Thus, bacterial CAs represent promising targets for obtaining antibacterials devoid of the resistance problems of the clinically used such agents but further studies are needed to validate these and other less investigated enzymes as novel drug targets.
Keywords: carbonic anhydrase, alpha-class, beta-class, bacterial enzyme, sulfonamide, antibacterials, overcome resistance
Introduction
Resistance to antibiotics belonging to several different classes is escalating and represents a worldwide problem (Ginsberg, 2008; Dye, 2009; Furtado and Nicolau, 2010), as both Gram-negative and Gram-positive bacteria (such as among others Staphylococcus aureus, Mycobacterium tuberculosis, Helicobacter pylori, Brucella suis, Streptococcus pneumoniae, etc.,) no longer respond to many such drugs (Cloeckaert and Schwarz, 2001; Nickerson and Schurr, 2006; Bush and Macielag, 2010). Cloning of the genomes of many bacterial pathogens offers however the possibility to explore alternative pathways for inhibiting virulence factors or proteins essential for their life cycle (Suerbaum and Michetti, 2002; Payne et al., 2007; Showalter and Denny, 2008; Tsolis et al., 2008). Among the many such new possible drug targets explored recently, are a class of enzymes catalyzing a simple but physiologically relevant process, carbon dioxide hydration to bicarbonate and protons (Smith et al., 1999; Supuran, 2008, 2010a,b). These enzymes are denominated carbonic anhydrases (CAs, EC 4.2.1.1), and they are all metalloenzymes. Five different genetically distinct CA families are known to date, the α-, β-, γ-, δ-, and ζ-CAs (Pastorekova et al., 2004; Supuran, 2008, 2010a). Whereas α-, β-, and δ-CAs use Zn(II) ions at the active site, the γ-CAs are probably Fe(II) enzymes (but they are active also with bound Zn(II) or Co(II) ions), whereas the ζ-class uses Cd(II) or Zn(II) to perform the physiologic reaction catalysis (Supuran, 2008, 2010a,b). The 3D fold of the five enzyme classes are very different from each other (a nice example of convergent evolution – Supuran, 2010b), as it is their oligomerization state: α-CAs are normally monomers and rarely dimmers; β-CAs are dimers, tetramers, or octamers; γ-CAs are trimers, whereas the δ- and ζ-CAs are probably monomers but in the case of the last family, three slightly different active sites are present on the same protein backbone which is in fact a pseudotrimer (Supuran, 2008, 2010a). Many representatives of all these enzyme classes have been crystallized and characterized in detail, except the δ-CAs (Supuran, 2008, 2010a,b). The mammalian CAs and their inhibition/activation have been recently reviewed (Supuran, 2008, 2010a,b) and no detailed discussion of these enzymes are presented in this review.
The α-CAs are present in vertebrates, protozoa, algae, and cytoplasm of green plants and in some Bacteria; the β-CAs are predominantly found in Bacteria, algae, and chloroplasts of both mono- as well as dicotyledons, but also in many fungi and some Archaea (Smith et al., 1999). In bacteria and fungi they are homodimers, as shown in Figure 1 for one of the enzymes from Salmonella enterica, stCA 1 (Brunzelle et al., submitted; Vullo et al., 2011). The γ-CAs were found in Archaea and some Bacteria, whereas the δ- and ζ-CAs seem to be present only in marine diatoms (Smith et al., 1999; Supuran, 2008, 2010a,b). In most organisms these enzymes are involved in crucial physiological processes connected with respiration and transport of CO2/bicarbonate, pH and CO2 homeostasis, electrolyte secretion in a variety of tissues/organs, biosynthetic reactions (such as gluconeogenesis, lipogenesis, and ureagenesis), bone resorption, calcification, tumorigenicity, and many other physiologic or pathologic processes (thoroughly studied in vertebrates), whereas in algae, plants and some bacteria they play an important role in photosynthesis and biosynthetic reactions. In diatoms δ- and ζ-CAs play a crucial role in carbon dioxide fixation (Smith et al., 1999; Zimmerman et al., 2007; Supuran, 2008, 2010a,b).
Figure 1.

View of the dimeric stCA 1 as obtained by X-ray crystallography (PDB file 3QY1). The polypeptide chains are represented as ribbons. The Zn(II) ions (gray spheres) and their ligands (Cys42, Asp44, His98, and Cys101) are shown as stick representation. The two active sites are identical and consist of a long channel at the bottom of which is found the Zn(II) ion in a tetrahedral geometry.
The classical CA inhibitors (CAIs) are the primary sulfonamides, RSO2NH2, which are in clinical use for more than 50 years as diuretics and systemically acting antiglaucoma drugs (Supuran et al., 2003; Supuran, 2008, 2010a,b). In fact there are around 30 clinically used drugs (or agents in clinical development) belonging to the sulfonamide or sulfamate class, which show significant CAI inhibitory activity (Supuran, 2008). However, it has emerged in the last years that sulfonamide/sulfamate CAIs have potential as anticonvulsant, antiobesity, anticancer, antipain, and antiinfective drugs (Supuran et al., 2003; Supuran, 2008, 2010a,b). All these drugs target in fact mammalian CAs, of which 16 different isoforms are known so far (Supuran et al., 2003; Supuran, 2008, 2010a,b).
Except vertebrates in which they have been extensively studied for decades as shown above, CAs are present in many human pathogens such as the malaria provoking protozoa Plasmodium falciparum (Krungkrai and Supuran, 2008; Krungkrai et al., 2008), bacteria such as Escherichia coli (Cronk et al., 2001), H. pylori (Nishimori et al., 2006, 2007, 2008), M. tuberculosis (Suarez Covarrubias et al., 2005, 2006; Carta et al., 2009; Güzel et al., 2009; Minakuchi et al., 2009; Nishimori et al., 2009, 2010; Davis et al., 2011), Brucella spp. (Joseph et al., 2010, 2011; Vullo et al., 2010; Winum et al., 2010), S. pneumoniae (Burghout et al., 2011), S. enterica (Vullo et al., 2011), and Haemophilus influenzae (Cronk et al., 2006; Hoffmann et al., 2011) as well as pathogenic fungi (Schlicker et al., 2009). Inhibition of these enzymes started to be investigated with sulfonamide/sulfamate inhibitors, but several other chemotypes were also explored, such as phenols, boronic acids, metal complexing anions, and other similar small molecules. As bacteria predominantly encode for β-class CAs, which are not present in vertebrates (Smith et al., 1999; Supuran, 2008), these enzymes started to be considered as possible drug targets for obtaining antibacterials devoid of the resistance problems mentioned above, which affect most classes of antibiotics in clinical use (Nishimori et al., 2010; Supuran, 2010a,b; Winum et al., 2010).
Here we review the current state-of-the art regarding the bacterial CAs cloned and characterized so far, as well as the in vitro and in vivo inhibition studies of these enzymes, which may reply to this stringent question: are the bacterial CAs future drug targets for obtaining conceptually novel antibiotics?
Bacterial α-Carbonic Anhydrases and Their Inhibition
Table 1 shows the α-CAs cloned and characterized so far from pathogenic bacteria. The first one is an enzyme from Neisseria gonorrhoeae (Chirică et al., 1997; Elleby et al., 2001), although older report mention a similar CA in N. sicca and related species (which have not been cloned so far; Sanders, 1967; Adler et al., 1972). The N. gonorrhoeae CA contains 252 amino acid residues and has a molecular mass of 28 kDa, being quite homologous to mammalian CAs (Chirică et al., 1997). A comparison with the amino acid sequences of human isoforms hCA I and II suggested that the secondary structures are essentially identical in the bacterial enzyme but several loops are much shorter than in the human isoforms (Chirică et al., 1997). This has been confirmed thereafter by resolving the X-ray crystal structure of this enzyme (Elleby et al., 2001). Most of the active-site residues are indeed identical to those found in hCA II, the crucial Zn(II) ion being coordinated by three His residues and a water molecule/hydroxide ion, being placed at a bottom of a rather deep and large active site. The bacterial enzyme showed a high CO2 hydrase activity, with a kcat of 1.1 × 106 s−1 and Km of 20 mM (at pH 9 and 25°C; Chirică et al., 1997). The enzyme also showed esterase activity for the hydrolysis of 4-nitrophenyl acetate, similarly to the mammalian isoforms hCA I and II.
Table 1.
CAs from pathogenic bacteria cloned and characterized so far, and their inhibition studies.
| Bacterium | Family | Name | Inhibition study | Reference | |
|---|---|---|---|---|---|
| In vitro | In vivo | ||||
| Neisseria gonorrhoeae | α | – | Sulfonamides, anions | Sulfonamides | Chirică et al. (1997), Elleby et al. (2001) |
| Neisseria sicca | α | – | Sulfonamides | Sulfonamides | Adler et al. (1972), Sanders (1967) |
| Helicobacter pylori | α | hpαCA | Sulfonamides, anions | Sulfonamides | Chirică et al. (2002), Nishimori et al. (2006, 2007), Marcus et al. (2005), Shahidzadeh et al. (2005) |
| H. Pylori | β | hpβCA | Sulfonamides, anions | Sulfonamides | Chirică et al. (2002), Nishimori et al. (2006, 2007) |
| Escherichia coli | β | – | NI | NI | Cronk et al. (2006) |
| Haemophilus influenzae | β | HICA | Bicarbonate | NI | Cronk et al. (2006), Hoffmann et al. (2011) |
| Mycobacterium tuberculosis | β | mtCA 1 | Sulfonamides | NA | Suarez Covarrubias et al. (2005, 2006) |
| β | mtCA 2 | Sulfonamides | NA | Minakuchi et al. (2009), Nishimori et al., 2009, 2010 | |
| β | mtCA 3 | Sulfonamides | NA | Güzel et al. (2009), Carta et al. (2009), Davis et al. (2011), Winum et al. (2010) | |
| Brucella suis | β | bsCA 1 | Sulfonamides | Sulfonamides | Joseph et al. (2010), Vullo et al. (2010) |
| β | bsCA 2 | Sulfonamides | Sulfonamides | Joseph et al. (2011), Winum et al. (2010) | |
| Streptococcus pneumoniae | β | PCA | Sulfonamides, anions | NI | Burghout et al. (2010, 2011) |
| Salmonella enterica | β | stCA 1 | Sulfonamides, anions | NI | Vullo et al. (2011) |
| β | stCA 2 | Sulfonamides, anions | NI | Vullo et al. (2011) | |
| Vibrio cholerae | Unknown | – | Sulfonamide | Sulfonamide | Kovacikova et al. (2010), Abuaita and Withey (2009) |
– Means not named; NA, no activity in vivo (presumably due to penetration problems); NI, not investigated.
Several studies showed in fact much earlier that the activity and the growth of N. sicca and related species (N. meningitides, N. gonorrhoeae, and N. lactamica among others) were inhibited by the sulfonamide CAIs used clinically acetazolamide and ethoxzolamide (MacLeod and DeVoe, 1981; Vaneechoutte et al., 1988; Nafi et al., 1990). Such inhibition was completely overcome by the addition of exogenous bicarbonate, proving that the process was indeed mediated by the bacterial CA. Nafi et al. (1990) also observed that a number of bacterial strains including members of the genera Pseudomonas, Staphylococcus, Streptococcus, Serratia, and Proteus also strongly expressed gene products immunologically related to the N. sicca CA, but these enzymes were not characterized at that time (and except the S. pneumoniae one, see later in the text, even today.
But the best studied bacterial α-CA is the one from the gastric pathogen provoking ulcer and gastric cancer, H. pylori, hpαCA (Chirică et al., 2002; Marcus et al., 2005; Shahidzadeh et al., 2005; Nishimori et al., 2006, 2007) – see Table 1. The genome project of H. pylori identified in fact two different classes of CAs, with different subcellular localization: a periplasmic α-class CA (hpαCA) and a cytoplasmic β-class CA (hpβCA; Nishimori et al., 2008). These two CAs were shown to be catalytically efficient with almost identical activity to that of the human isoform hCA I, for the CO2 hydration reaction, and highly inhibited by many sulfonamides/sulfamates, including acetazolamide, ethoxzolamide, topiramate, and sulpiride, all clinically used drugs (Nishimori et al., 2008). Furthermore, certain CAIs, such as acetazolamide and methazolamide, were shown to inhibit the bacterial growth in cell cultures (Nishimori et al., 2008). Since the efficacy of H. pylori eradication therapies currently employed has been decreasing due to drug resistance and side effects of the commonly used drugs, the dual inhibition of α- and/or β-CAs of H. pylori could be applied as an alternative therapy in patients with H. pylori infection or for the prevention of gastroduodenal diseases provoked by this widespread pathogen (Nishimori et al., 2008). In fact, in a pilot study Shahidzadeh et al. (2005) showed the efficacy of acetazolamide in the treatment of gastric ulcer. This compound (as well as ethoxzolamide) were in fact widely used as antiulcer agents in the 70- and 80-s, although their mechanism of action was not properly understood at that time (Puscas, 1984).
Bacterial β-Carbonic Anhydrases and Their Inhibition
As mentioned above, the β-CA class is the most widespread in bacteria (Smith et al., 1999; Supuran, 2008, 2010a,b). The proof-of-concept study that such an enzyme may be a drug target has been published recently by Nishimori et al. (2007) who cloned and purified the H. pylori enzyme (hpβCA), showing that it is highly susceptible to be inhibited by sulfonamides and sulfamates (see Discussion above for the in vivo data). Afterward, a rather large number of other β-CAs were cloned, purified, and characterized from other pathogens (Table 1). The X-ray crystal structures are also available for the E. coli (Cronk et al., 2001), H. influenzae (Cronk et al., 2006), two of the three M. tuberculosis enzymes (Suarez Covarrubias et al., 2005, 2006), and one S. enterica (stCA 1) β-CA (Brunzelle et al., submitted). The 3D folds of these enzymes are rather conserved and similar to the stCA 1 shown in Figure 1. The two active sites are identical, being rather long channels at the bottom of which is found the catalytic zinc ion, tetrahedrally coordinated by Cys42, Asp44, His98, and Cys101 (in stCA 1). This is the so called “closed active site,” since these enzymes are not catalytically active (at pH values < 8.3; Suarez Covarrubias et al., 2005, 2006). However, at pH values > 8.3, the “closed active site” is converted to the “open active site” (with gain of catalytic activity), this being associated with a movement of the Asp residue from the catalytic Zn(II) ion, with the concomitant coordination of an incoming water molecule approaching the metal ion. This water molecule (as hydroxide ion) is in fact responsible for the catalytic activity, as for the α-CAs investigated in much greater detail (Suarez Covarrubias et al., 2005, 2006). It should be also mentioned that some β-CAs possess and open active site at all pH values (Schlicker et al., 2009).
Many of these enzymes displayed excellent activity for the physiologic CO2 hydration reaction and were inhibited (sometimes in the low nanomolar range) by sulfonamides and sulfamates (Nishimori et al., 2010; Supuran, 2010a,b; Winum et al., 2010). However, in vivo, it has been possible to observe inhibition of the bacterial growth only for H. pylori, S. penumoniae, and B. suis (Nishimori et al., 2008; Burghout et al., 2010; Winum et al., 2010). Especially in the case of M. tuberculosis, although nanomolar and sub-nanomolar in vitro inhibitors were detected (Güzel et al., 2009), no in vivo inhibition of growth has been observed, probably because the highly polar sulfonamides have difficulties to penetrate through the bacterial wall of these pathogens (Nishimori et al., 2010). Thus, much work is warranted in order to detect potent in vitro CAIs that also work in vivo, in order to validate these β-CAs as drug targets.
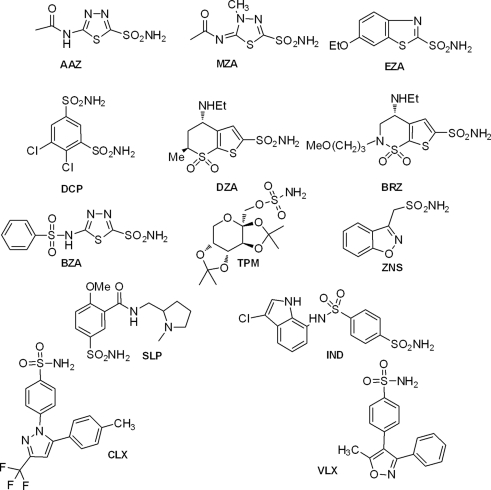
Table 2 shows the in vitro inhibition data of several of these enzymes with sulfonamide/sulfamates, which represent one of the main classes of CAIs (Supuran, 2008, 2010b). Such compounds are clinically used drugs, e.g., AAZ, acetazolamide; MZA, methazolamide; EZA, ethoxzolamide; DCP, dichorophenamide; DZA, dorzolamide; BRZ, brinzolamide; BZA, benzolamide; TPM, topiramate; ZNS, zonisamide; SLP, sulpiride; IND, indisulam; CLX, celecoxib; VLX, valdecoxib; as diuretics, antiepileptics, antiglaucoma, and antiinflammatory agents (Supuran, 2008, 2010b). It may be observed that most CAs from bacterial pathogenic organisms are inhibited in the micro – nanomolar range by many such sulfonamide/sulfamate drugs. It should be mentioned that no rational drug design campaigns have been done to detect better CAIs targeting bacterial CAs so far, but the preliminary screening results summarized in Table 2 are indeed promising, since a lot of effective led compounds have been detected. It is envisageable that more research in this area may lead to highly effective and bacterial CA selective compounds which may validate these enzymes as antibacterial drug targets.
Table 2.
In vitro inhibition data of bacterial CAs with sulfonamides and sulfamates, some of which are clinically used drugs (only the enzymes for which these data were reported in the literature are included).
| Compound | Ki (μM) | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| hpαCA | hpβCA | mtCA 1 | mtCA 2 | mtCA 3 | bsCA 1 | bsCA 2 | stCA1 | stCA2 | |
| AAZ | 0.021 | 0.040 | 0.481 | 0.009 | 0.104 | 0.063 | 0.303 | 0.059 | 0.084 |
| MZA | 0.225 | 0.176 | 0.781 | 0.660 | 0.562 | 0.054 | 0.642 | 0.134 | 0.068 |
| EZA | 0.193 | 0.033 | 1.03 | 0.027 | 0.594 | 0.017 | 0.420 | 0.528 | 0.721 |
| DCP | 0.378 | 0.105 | 0.872 | 2.01 | 0.611 | 0.058 | 0.112 | 0.090 | 0.095 |
| DZA | 4.36 | 0.073 | 0.744 | 0.099 | 0.137 | 0.021 | 0.923 | 0.445 | 0.607 |
| BRZ | 0.210 | 0.128 | 0.839 | 0.127 | 0.201 | 0.026 | 0.625 | 0.687 | 0.412 |
| BZA | 0.315 | 0.054 | 0.810 | 0.467 | 0.338 | 0.075 | 0.117 | 0.085 | 0.098 |
| TPM | 0.172 | 0.032 | 0.612 | 0.474 | 3.02 | 0.057 | 0.099 | 0.624 | 0.697 |
| ZNS | 0.231 | 0.254 | 28.68 | 0.876 | 0.208 | 1.85 | 0.406 | 5.43 | 5.70 |
| SLP | 0.204 | 0.035 | 2.30 | 0.266 | 7.92 | 0.019 | 0.084 | 5.64 | 8.73 |
| IND | 0.413 | 0.143 | 0.097 | 0.717 | 7.84 | 0.050 | 0.130 | 8.86 | 6.90 |
| CLX | nt | nt | 10.35 | 0.713 | 7.76 | 0.018 | 0.128 | 5.83 | 6.11 |
| VLX | nt | nt | 12.97 | 0.682 | 7.81 | 0.019 | 0.612 | 6.85 | 6.58 |
Nt, not tested.
As mentioned above, it is very probable that many β-CAs (or enzymes belonging to other CA families) are present in other bacterial pathogens. For example, in Vibrio cholerae it has been recently shown that sodium bicarbonate induces cholera toxin (CT) expression (Abuaita and Withey, 2009). Although the mechanism for bicarbonate-mediated CT induction has not been defined in detail, it has been demonstrated that bicarbonate stimulates virulence gene expression by enhancing ToxT (a regulatory protein that directly activates transcription of the genes encoding CT) activity (Abuaita and Withey, 2009). The sulfonamide CAI ethoxzolamide, inhibited bicarbonate-mediated virulence induction, suggesting that conversion of CO2 into bicarbonate by a CA plays a role in virulence induction in V. cholerae. Thus, bicarbonate was the first positive effector for ToxT activity to be identified. Given that bicarbonate is present at high concentration in the upper small intestine where V. cholerae colonizes, bicarbonate is likely an important chemical stimulus that V. cholerae senses and that induces virulence during the natural course of infection (Abuaita and Withey, 2009; Kovacikova et al., 2010). However, the CA involved in these processes was not yet cloned and characterized, and as a consequence, their inhibition not properly understood.
Conclusion
By catalyzing the simple but highly important hydration of carbon dioxide to bicarbonate and protons, bacterial CAs are probably involved in critical steps of the bacterial life cycle, some of which are important for survival, invasion, and pathogenicity. Bacteria encode such enzymes belonging to the α-, β-, and/or γ-CA families, but up to now only the first two classes have been investigated in some detail in different species. Indeed, the α-CAs from Neisseria spp. and H. pylori as well as the β-class enzymes from E. coli, H. pylori, M. tuberculosis, Brucella spp., S. pneumoniae, S. enterica, and H. influenzae have been cloned and characterized. For some of these enzymes the X-ray crystal structures were determined at rather high resolution, allowing for a good understanding of the catalytic/inhibition mechanisms. However no adducts with inhibitors of these enzymes have been characterized so far, although in vitro and in vivo inhibition studies with various classes of inhibitors, such as anions, sulfonamides, and sulfamates have been reported. Efficient in vitro inhibitors have been reported for many such enzymes, but only for Neisseria spp., H. pylori, B. suis, and S. pneumoniae CAs it has been possible to evidence inhibition of bacterial growth in vivo. Thus, bacterial CAs represent at this moment very promising targets for obtaining antibacterials devoid of the resistance problems of the clinically used such agents but further studies are needed to validate these and other less investigated enzymes as novel drug targets.
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Work from the author’s laboratory is financed by two FP7 EU project (Metoxia and Gums and Joints grants). Thanks are addressed to Dr. G. De Simone (CNR, Naples, Italy) for generating Figure 1.
References
- Abuaita B. H., Withey J. H. (2009). Bicarbonate induces Vibrio cholerae virulence gene expression by enhancing ToxT activity. Infect. Immun. 77, 4111–4120 10.1128/IAI.00983-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adler L., Brundell J., Falkbring S. O., Nyman P. O. (1972). Carbonic anhydrase from Neisseria sicca, strain 6021. I. Bacterial growth and purification of the enzyme. Biochim. Biophys. Acta 284, 298–310 [DOI] [PubMed] [Google Scholar]
- Burghout P., Cron L. E., Gradstedt H., Quintero B., Simonetti E., Bijlsma J. J., Bootsma H. J., Hermans P. W. (2010). Carbonic anhydrase is essential for Streptococcus pneumoniae growth in environmental ambient air. J. Bacteriol. 192, 4054–4062 10.1128/JB.00151-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burghout P., Vullo D., Scozzafava A., Hermans P. W. M., Supuran C. T. (2011). Inhibition of the β-carbonic anhydrase from Streptococcus pneumoniae by inorganic anions and small molecules: towards innovative drug design of anti infectives? Bioorg. Med. Chem. 19, 243–248 10.1016/j.bmc.2010.11.031 [DOI] [PubMed] [Google Scholar]
- Bush K., Macielag M. J. (2010). New β-lactam antibiotics and β-lactamase inhibitors. Expert Opin. Ther. Pat. 20, 1277–1293 10.1517/13543776.2010.515588 [DOI] [PubMed] [Google Scholar]
- Carta F., Maresca A., Suarez Covarrubias A., Mowbray S. L., Jones T. A., Supuran C. T. (2009). Carbonic anhydrase inhibitors. Characterization and inhibition studies of the most active β-carbonic anhydrase from Mycobacterium tuberculosis, Rv3588c. Bioorg. Med. Chem. Lett. 19, 6649–6654 10.1016/j.bmcl.2009.10.009 [DOI] [PubMed] [Google Scholar]
- Chirica L. C., Elleby B., Jonsson B. H., Lindskog S. (1997). The complete sequence, expression in Escherichia coli, purification and some properties of carbonic anhydrase from Neisseria gonorrhoeae. Eur. J. Biochem. 244, 755–760 10.1111/j.1432-1033.1997.00755.x [DOI] [PubMed] [Google Scholar]
- Chirica L. C., Petersson C., Hurtig M., Jonsson B. H., Borén T., Lindskog S. (2002). Expression and localization of alpha- and beta-carbonic anhydrase in Helicobacter pylori. Biochem. Biophys. Acta 1601, 192–199 [DOI] [PubMed] [Google Scholar]
- Cloeckaert A., Schwarz S. (2001). Molecular characterization, spread and evolution of multidrug resistance in Salmonella enterica typhimurium DT104. Vet. Res. 32, 301–310 10.1051/vetres:2001126 [DOI] [PubMed] [Google Scholar]
- Cronk J. D., Endrizzi J. A., Cronk M. R., O’Neill J. W., Zhang K. Y. (2001). Crystal structure of E. coli β-carbonic anhydrase, an enzyme with an unusual pH-dependent activity. Protein Sci. 10, 911–922 10.1110/ps.46301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cronk J. D., Rowlett R. S., Zhang K. Y., Tu C., Endrizzi J. A., Lee J., Gareiss P. C., Preiss J. R. (2006). Identification of a novel noncatalytic bicarbonate binding site in eubacterial β-carbonic anhydrase. Biochemistry 45, 4351–4361 10.1021/bi052272q [DOI] [PubMed] [Google Scholar]
- Davis R. A., Hofmann A., Osman A., Hall R. A., Mühlschlegel F. A., Vullo D., Innocenti A., Supuran C. T., Poulsen S. A. (2011). Natural product-based phenols as novel probes for Mycobacterial and fungal carbonic anhydrases. J. Med. Chem. 54, 1682–1692 10.1021/jm1013242 [DOI] [PubMed] [Google Scholar]
- Dye C. (2009). Doomsday postponed? Preventing and reversing epidemics of drug-resistant tuberculosis. Nat. Rev. Microbiol. 7, 81–87 10.1038/nrmicro2048 [DOI] [PubMed] [Google Scholar]
- Elleby B., Chirica L. C., Tu C., Zeppezauer M., Lindskog S. (2001). Characterization of carbonic anhydrase from Neisseria gonorrhoeae. Eur. J. Biochem. 268, 1613–1619 10.1046/j.1432-1327.2001.02031.x [DOI] [PubMed] [Google Scholar]
- Furtado G. H., Nicolau D. P. (2010). Overview perspective of bacterial resistance. Expert Opin. Ther. Pat. 20, 1273–1276 10.1517/13543776.2010.507193 [DOI] [PubMed] [Google Scholar]
- Ginsberg A. M. (2008). Emerging drugs for active tuberculosis. Semin. Respir. Crit. Care Med. 29, 552–559 10.1055/s-0028-1085706 [DOI] [PubMed] [Google Scholar]
- Güzel Ö., Maresca A., Scozzafava A., Salman A., Balaban A. T., Supuran C. T. (2009). Discovery of low nanomolar and subnanomolar inhibitors of the mycobacterial β-carbonic anhydrases Rv1284 and Rv3273. J. Med. Chem. 52, 4063–4067 10.1021/jm9004016 [DOI] [PubMed] [Google Scholar]
- Hoffmann K. M., Samardzic D., Heever K. V., Rowlett R. S. (2011). Co(II)-substituted Haemophilus influenzae β-carbonic anhydrase: spectral evidence for allosteric regulation by pH and bicarbonate ion. Arch. Biochem. Biophys. 511, 80–87 10.1016/j.abb.2011.04.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joseph P., Ouahrani-Bettache S., Montero J. L., Nishimori I., Vullo D., Scozzafava A., Winum J. Y., Köhler S., Supuran C. T. (2011). A new Brucella suis β-carbonic anhydrase, bsCA II: inhibition of bsCA I and II with sulfonamides and sulfamates inhibits the pathogen growth. Bioorg. Med. Chem. 19, 1172–1178 10.1016/j.bmc.2010.12.048 [DOI] [PubMed] [Google Scholar]
- Joseph P., Turtaut F., Ouahrani-Bettache S., Montero J. L., Nishimori I., Minakuchi T., Vullo D., Scozzafava A., Köhler S., Winum J. Y., Supuran C. T. (2010). Cloning, characterization and inhibition studies of a β-carbonic anhydrase from Brucella suis. J. Med. Chem. 53, 2277–2285 10.1021/jm901855h [DOI] [PubMed] [Google Scholar]
- Kovacikova G., Lin W., Skorupski K. (2010). The LysR-type virulence activator AphB regulates the expression of genes in Vibrio cholerae in response to low pH and anaerobiosis. J. Bacteriol. 192, 4181–4191 10.1128/JB.00193-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krungkrai J., Krungkrai S. R., Supuran C. T. (2008). Carbonic anhydrase inhibitors: inhibition of Plasmodium falciparum carbonic anhydrase with aromatic/ heterocyclic sulfonamides-in vitro and in vivo studies. Bioorg. Med. Chem. Lett. 18, 5466–5471 10.1016/j.bmcl.2008.09.030 [DOI] [PubMed] [Google Scholar]
- Krungkrai J., Supuran C. T. (2008). The alpha-carbonic anhydrase from the malaria parasite and its inhibition. Curr. Pharm. Des. 14, 631–640 10.2174/138161208783877901 [DOI] [PubMed] [Google Scholar]
- MacLeod M. N., DeVoe I. W. (1981). Localization of carbonic anhydrase in the cytoplasmic membrane of Neisseria sicca (strain 19). Can. J. Microbiol. 27, 87–92 10.1139/m81-014 [DOI] [PubMed] [Google Scholar]
- Marcus E. A., Moshfegh A. P., Sachs G., Scott D. R. (2005). The periplasmic alpha-carbonic anhydrase activity of Helicobacter pylori is essential for acid acclimation. J. Bacteriol. 187, 729–738 10.1128/JB.187.12.4222-4228.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minakuchi T., Nishimori I., Vullo D., Scozzafava A., Supuran C. T. (2009). Molecular cloning, characterization and inhibition studies of the Rv1284 ß-carbonic anhydrase from Mycobacterium tuberculosis with sulfonamides and a sulfamate. J. Med. Chem. 52, 2226–2232 10.1021/jm9000488 [DOI] [PubMed] [Google Scholar]
- Nafi B. M., Miles R. J., Butler L. O., Carter N. D., Kelly C., Jeffery S. (1990). Expression of carbonic anhydrase in Neisseriae and other heterotrophic bacteria. J. Med. Microbiol. 32, 1–7 10.1099/00222615-32-1-1 [DOI] [PubMed] [Google Scholar]
- Nickerson C. A., Schurr M. J. (2006). Molecular Paradigms of Infectious Disease: A Bacterial Perspective. Munich: Springer – Verlag, 1–617 [Google Scholar]
- Nishimori I., Minakuchi T., Kohsaki T., Onishi S., Takeuchi H., Vullo D., Scozzafava A., Supuran C. T. (2007). Carbonic anhydrase inhibitors. The β-carbonic anhydrase from Helicobacter pylori is a new target for sulfonamide and sulfamate inhibitors. Bioorg. Med. Chem. Lett. 17, 3585–3594 10.1016/j.bmcl.2006.11.028 [DOI] [PubMed] [Google Scholar]
- Nishimori I., Minakuchi T., Morimoto K., Sano S., Onishi S., Takeuchi H., Vullo D., Scozzafava A., Supuran C. T. (2006). Carbonic anhydrase inhibitors: DNA cloning and inhibition studies of the alpha-carbonic anhydrase from Helicobacter pylori, a new target for developing sulfonamide and sulfamate gastric drugs. J. Med. Chem. 49, 2117–2126 10.1021/jm0512600 [DOI] [PubMed] [Google Scholar]
- Nishimori I., Minakuchi T., Vullo D., Scozzafava A., Innocenti A., Supuran C. T. (2009). Carbonic anhydrase inhibitors. cloning, characterization, and inhibition studies of a new β-carbonic anhydrase from Mycobacterium tuberculosis. J. Med. Chem. 52, 3116–3120 10.1021/jm9003126 [DOI] [PubMed] [Google Scholar]
- Nishimori I., Minakuchi T., Maresca A., Carta F., Scozzafava A., Supuran C. T. (2010). The β-carbonic anhydrases from Mycobacterium tuberculosis as drug targets. Curr. Pharm. Des. 16, 3300–3309 10.2174/138161210793429814 [DOI] [PubMed] [Google Scholar]
- Nishimori I., Onishi S., Takeuchi H., Supuran C. T. (2008). The and classes carbonic anhydrases from Helicobacter pylori as novel drug targets. Curr. Pharm. Des. 14, 622–630 10.2174/138161208783877875 [DOI] [PubMed] [Google Scholar]
- Pastorekova S., Parkkila S., Pastorek J., Supuran C. T. (2004). Carbonic anhydrases: current state of the art, therapeutic applications and future prospects. J. Enzyme Inhib. Med. Chem. 19, 199–229 10.1080/14756360410001689540 [DOI] [PubMed] [Google Scholar]
- Payne D. J., Gwynn M. N., Holmes D. J., Pompliano D. L. (2007). Drugs for bad bugs: confronting the challenges of antibacterial discovery. Nat. Rev. Drug Discov. 6, 29–40 10.1038/nrd2201 [DOI] [PubMed] [Google Scholar]
- Puscas I. (1984). Treatment of gastroduodenal ulcers with carbonic anhydrase inhibitors. Ann. N. Y. Acad. Sci. 429, 587–591 10.1111/j.1749-6632.1984.tb12392.x [DOI] [PubMed] [Google Scholar]
- Sanders E. (1967). Use of sulfonamide carbonic anhydrase inhibitors in treatment of meningococcal carriers: rationale and report of a clinical trial of ethoxzolamide. Am. J. Med. Sci. 254, 709–716 10.1097/00000441-196711000-00017 [DOI] [PubMed] [Google Scholar]
- Schlicker C., Hall R. A., Vullo D., Middelhaufe S., Gertz M., Supuran C. T., Mühlschlegel F. A., Steegborn C. (2009). Structure and inhibition of the CO2-sensing carbonic anhydrase Can2 from the pathogenic fungus Cryptococcus neoformans. J. Mol. Biol. 385, 1207–1220 10.1016/j.jmb.2008.11.037 [DOI] [PubMed] [Google Scholar]
- Shahidzadeh R., Opekun A., Shiotani A., Graham D. Y. (2005). Effect of the carbonic anhydrase inhibitor, acetazolamide, on Helicobacter pylori infection in vivo: a pilot study. Helicobacter 10, 136–138 10.1111/j.1523-5378.2005.00306.x [DOI] [PubMed] [Google Scholar]
- Showalter H. D., Denny W. A. (2008). A roadmap for drug discovery and its translation to small molecule agents in clinical development for tuberculosis treatment. Tuberculosis (Edinb.) 88(Suppl. 1), S3–S17 10.1016/S1472-9792(08)70032-5 [DOI] [PubMed] [Google Scholar]
- Smith K. S., Jakubzick C., Whittam T. S., Ferry J. G. (1999). Carbonic anhydrase is an ancient enzyme widespread in prokaryotes. Proc. Natl. Acad. Sci. U.S.A. 96, 15184–15189 10.1073/pnas.96.7.3348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suarez Covarrubias A., Bergfors T., Jones T. A., Hogbom M. (2006). Structural mechanics of the pH-dependent activity of the β-carbonic anhydrase from Mycobacterium tuberculosis. J. Biol. Chem. 281, 4993–4999 10.1074/jbc.M510756200 [DOI] [PubMed] [Google Scholar]
- Suarez Covarrubias A., Larsson A. M., Hogbom M., Lindberg J., Bergfors T., Bjorkelid C., Mowbray S. L., Unge T., Jones T. A. (2005). Structure and function of carbonic anhydrases from Mycobacterium tuberculosis. J. Biol. Chem. 280, 18782–18789 10.1074/jbc.M414348200 [DOI] [PubMed] [Google Scholar]
- Suerbaum S., Michetti P. (2002). Helicobacter pylori infection. N. Engl. J. Med. 347, 1175–1186 10.1056/NEJMra020542 [DOI] [PubMed] [Google Scholar]
- Supuran C. T. (2008). Carbonic anhydrases: novel therapeutic applications for inhibitors and activators. Nat. Rev. Drug Discov. 7, 168–181 10.1038/nrd2467 [DOI] [PubMed] [Google Scholar]
- Supuran C. T. (2010a). Carbonic anhydrase inhibitors. Bioorg. Med. Chem. Lett. 20, 3467–3474 10.1016/j.bmcl.2010.05.009 [DOI] [PubMed] [Google Scholar]
- Supuran C. T. (2010b). Carbonic anhydrase inhibition/ activation: trip of a scientist around the world in the search of novel chemotypes and drug targets. Curr. Pharm. Des. 16, 3233–3245 10.2174/138161210793429797 [DOI] [PubMed] [Google Scholar]
- Supuran C. T., Scozzafava A., Casini A. (2003). Carbonic anhydrase inhibitors. Med. Res. Rev. 23, 146–189 10.1002/med.10025 [DOI] [PubMed] [Google Scholar]
- Tsolis R. M., Young G. M., Solnick J. V., Bäumler A. J. (2008). From bench to bedside: stealth of enteroinvasive pathogens. Nat. Rev. Microbiol. 6, 883–892 10.1038/nrmicro2012 [DOI] [PubMed] [Google Scholar]
- Vaneechoutte M., Verschraegen G., Claeys G., van den Abeele A. M. (1988). Selective medium for Branhamella catarrhalis with acetazolamide as a specific inhibitor of Neisseria spp. J. Clin. Microbiol. 26, 2544–2548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vullo D., Nishimori I., Minakuchi T., Scozzafava A., Supuran C. T. (2011). Inhibition studies with anions and small molecules of two novel β-carbonic anhydrases from the bacterial pathogen Salmonella enterica serovar Typhimurium. Bioorg. Med. Chem. Lett. 21, 3591–3595 10.1016/j.bmcl.2011.04.105 [DOI] [PubMed] [Google Scholar]
- Vullo D., Nishimori I., Scozzafava A., Köhler S., Winum J. Y., Supuran C. T. (2010). Inhibition studies of a β-carbonic anhydrase from Brucella suis with a series of water soluble glycosyl sulfanilamides. Bioorg. Med. Chem. Lett. 20, 2178–2182 10.1016/j.bmcl.2010.02.042 [DOI] [PubMed] [Google Scholar]
- Winum J. Y., Kohler S., Supuran C. T. (2010). Brucella carbonic anhydrases: new targets for designing anti-infective agents. Curr. Pharm. Des. 16, 3310–3316 10.2174/138161210793429850 [DOI] [PubMed] [Google Scholar]
- Zimmerman S. A., Ferry J. G., Supuran C. T. (2007). Inhibition of the Archaeal β-Class (Cab) and γ-Class (Cam) carbonic anhydrases. Curr. Top. Med. Chem. 7, 901–908 10.2174/156802607780636753 [DOI] [PubMed] [Google Scholar]