

Abstract
FAT10, also known as diubiquitin, has been implicated in the regulation of diverse cellular processes, including mitosis, immune response, and apoptosis. We seek to identify FAT10-targeted proteins, an essential step in elucidating the physiological function of FAT10. To this end, human FAT10 or its non-conjugatable derivative, FAT10ΔGG, was overexpressed in HEK293 cells. We observed a number of high molecular weight FAT10 conjugates in cells expressing wild-type FAT10, but not in FAT10ΔGG. The FAT10 conjugates are inducible by TNF-α and accumulated significantly when cells were treated with proteasome inhibitor, MG132. Among them, tumor suppressor p53 was found to be FATylated. The p53 transcriptional activity was found to be substantially enhanced in FAT10-overexpressing cells. In addition, overexpressing FAT10 in HEK293 cells also reduced the population of p53 which cross reacted with monoclonal anti-p53 antibody, PAB240, known to recognize only the transcriptionally inactive p53. FAT10 in the nucleus was found co-localized with p53 and altered its subcellular compartmentalization. Furthermore, overexpressing FAT10 led to a reduction in the size of promyelocytic leukemia nuclear bodies (PML-NBs) and altered their distribution in the nucleus. Based on these observations, a potential mechanism which correlates FATylation of p53 to its translocation and transcriptional activation is discussed.
Introduction
The ubiquitin-like modifiers (UBLs) is a family of proteins homologous to ubiquitin, which can covalently modify target substrates [1]. Besides the relatively well-studied SUMO and NEDD8, there are at least seven other UBLs: FAT10, ISG15, FUB1, UBL5, URM1, ATG8, and ATG12. The major substrates and enzymatic pathways for FAT10, FUB1 and UBL5 remain to be elucidated [2].
FAT10, also known as diubiquitin, is an 18 kDa protein sharing 29% and 36% sequence identity with ubiquitin at the N- and C-termini, respectively [3]. FAT10 is originally identified as a gene encoded in the major histocompatibility complex class I locus and is inducible with TNF-α and interferon-γ [4]. It is regulated in a cell-cycle dependent fashion with the highest expression at the S phase [5], and is negatively regulated by p53 [6]. Although the functions of FAT10 are unclear, it has been implicated to play important roles in many cellular processes. Upregulation of FAT10 gene expression was observed in hepatocellular carcinoma and several epithelial cancers, including gastrointestinal and gynecological cancers [7]. Overexpression of FAT10 induces apoptosis in a caspase-dependent manner [8]. FAT10 also plays a role in the regulation of chromosomal stability [9]. It has also been shown to interact non-covalently with MAD2, a spindle-assembly checkpoint protein [9, 10]. In addition, FAT10 can interact non-covalently with NEDD8 Ultimate Buster-1L (NUB-1L) based on results from yeast two-hybrid screening [11]. Several studies demonstrated that wild-type, but not non-conjugatable FAT10 forms a 35kDa conjugate resistant to boiling in the present of SDS and β-mercaptoethanol, indicating that FAT10 can form covalent linkage to target proteins [8, 12]. However, to date, no covalently-conjugated target protein has been identified. Intriguingly, knockout of the FAT10 gene in mice caused minimal phenotypic changes, though lymphocytes of FAT10-deficient mice were more susceptible to spontaneous apoptotic death [13].
Using our proteomic approach [14, 15, 16] we demonstrated that p53 can form a covalent conjugate with FAT10. This is the first identified FAT10 target protein. Overexpressing FAT10 leads to p53 conformational change and transcription activation. Notably, we observed that the up-regulation of p53 activity by overexpressing FAT10 may be mediated via its effect on PML nuclear body functions.
Materials and Methods
Antibodies and Plasmids
The monoclonal anti-Myc (9E10), monoclonal anti-p53 (DO-1), polyclonal anti-p53 (FL-393), anti-Ubiquitin, agarose conjugated anti-p53, TRITC conjugated anti-p53, FITC-conjugated anti-PML and FITC conjugated anti-Myc antibodies were purchased from Santa Cruz; monoclonal anti-β-actin antibody from Sigma; monoclonal anti-p53 (PAB 240) antibody from Abcam. The preparation of purified anti-FAT10 antibody was previously described [7]. The cDNAs encoding FAT10GG ( aa 1–165), and FAT10ΔGG( aa 1–163) were amplified by PCR. A 6×His-tag sequence immediately upstream of the start codon of the FAT10 cDNA sequence was designed in the amplifying primers. The PCR amplified cDNAs were inserted into the pTRE2hyg2-Myc vector (Clontech) as NheI/ClaI fragments to generate pTRE2hyg2-Myc-His-FAT10GG and pTRE2hyg2-Myc-His-FAT10ΔGG plasmids, respectively. The engineering of pTRE2hyg2-Myc-His-SUMO-1/2/3GG plasmids had been described previously [14, 15]. The pTER2hyg2-Myc-Luciferase (LUC) control plasmid was purchased from Clontech. The p53 response reporter plasmids, pRGC-Luc and pG13-Luc were generous gifts from Dr. Seung J. Baek (The University of Tennessee).
Cell Culture, Transfection, and Cell Lines
The stable HEK 293 Tet-On cell lines overexpressing Myc-His-FAT10GG, Myc-His-FAT10ΔGG, Myc-His-SUMO-1/2/3GG, Myc-His-SUMO-1ΔGG, Myc-LUC and pTRE2hyg2-Myc vector were established using the protocol previously described [14]. FuGENE6 (Roche) was used for transient transfection.
Purification of 6×His-tagged SUMO substrates
The 6×His-tagged proteins were purified under denaturing conditions using Ni-NTA agarose according to the manufacturer’s instructions (Qiagen) with some modifications. The detailed method has been previously described [15].
Immunoprecipitation, immunoblotting, and immunofluorescence
Cells were lysed in lysis buffer [50 mM Tris-HCl (pH 7.5), 150 mM NaCl, 1% Nonidet P-40, 5 mM EDTA, 20 mM N-ethylmaleimide, 1 mM PMSF, 10 μg/ml pepstatin, 20 μg/ml leupeptin, 10 μg/ml aprotinin] to obtain whole cell extracts (WCE). The nuclear and cytosolic protein separation was carried out using NE-PER kit from PIERCE. Immunoprecipitation and immunoblotting were carried out as described [14, 15]. For immunofluorescence, HEK 293 Tet-On cells harboring pTRE2hyg2-Myc-His-FAT10GG, pTRE2hyg2-Myc-His-FAT10ΔGG or pTRE2hyg2-Myc-LUC were seeded onto 2-well Lab-Tek chamber slide (Nalge Nuc International) and induced with 2 μg/ml Dox for 48 h. Cells were fixed in 3.7% formaldehyde and permeabilized with 0.1% Triton X-100. The fixed cells were first stained with TRITC conjugated anti-p53 antibody and washed for 5 times with 1×PBS, then stained with either FITC conjugated anti-Myc antibody or FITC conjugated anti-PML antibody and washed with 1×PBS for 5 times. The fluorescent images were captured on a Zeiss LSM-5 Pascal laser scanning confocal microscope (Carl Zeiss, Jena, Germany). Images were analyzed with the 3D for LSM software package from Zeiss.
p53 transcriptional transactivation activity assay
The transfection with p53 response reporter plasmid (pRGC-Luc) was used to assess the transcriptional activity of p53 in vivo as previously described (16). The luciferase activity was determined with the Luciferase Reporter Assay Kit (BioVision) using a Turner Design Luminometer (Promega). The values obtained were normalized with protein concentration in each sample.
Results
Establishing Cell lines stably expressing FAT10 and its non-conjugatable form
Stable cell lines overexpressing Myc and 6xHis tagged wild-type FAT10 (Myc-His-FAT10GG, abbreviated as FAT10GG) and its non-conjugatable mutant (Myc-His-FAT10ΔGG, abbreviated as FAT10ΔGG) were established in HEK293 cells by transfection with pTRE2hyg2-Myc-His-FAT10GG and pTRE2hyg2-Myc-His-FAT10ΔGG, respectively. Fig. 1A shows that FAT10GG, FAT10ΔGG and luciferase (LUC) control were successfully expressed in stably transfected HEK 293 cell lines. The expression of FAT10GG and FAT10ΔGG was inducible with 2 μg/ml of doxycycline (Dox). The successful expression of FAT10GG in HEK cells allowed examination of associated cellular response. Interestingly, the FAT10GG and some of its conjugates, but not FAT10ΔGG and Luciferase (LUC), are also inducible with TNF-α. When cells were treated with proteasome inhibitor, MG132, both FAT10GG and FAT10ΔGG as well as FAT10 conjugates were accumulated.. The ubiquitinylated proteins were also accumulated as predicted when cells were treated with MG132 (Fig. S1). More importantly, many high molecular weight conjugates were found in cells expressing FAT10GG, but not in cells expressing FAT10ΔGG or LUC. Even without the treatment of MG132, the FAT10GG can form high molecular weight conjugates (See Fig. 1B, obtained with longer exposure time than that shown in Fig. 1A). Together, these indicate that FAT10 formed covalent conjugates with its target proteins, because these conjugates are resistant to boiling in the present of SDS and β-mercaptoethanol. It should be pointed out that Fig. 1A shows that, the FAT10GG and its conjugates expression level is apparently higher than that of FAT10ΔGG for unknown reason. Nevertheless this observation is consistent with a previous report [8]. To demonstrate that the observed conjugates were not caused by elevated FAT10GG expression relative to that of FAT10ΔGG, we utilized the advantage of the adjustable Tet-On cell line by reducing the Dox concentration to 0.5 μg/ml for the FAT10GG overexpressing cells, to yield a similar expression level for FAT10GG and FAT10ΔGG. The results as shown in Fig. 1B, reveal that the observed FAT10 conjugated proteins are not an artifact due to differential expression of FAT10GG vs FAT10ΔGG, but due to the ability of FAT10GG and not FAT10ΔGG to form protein conjugates.
Fig. 1. Cell lines stably expressing FAT10 are established.
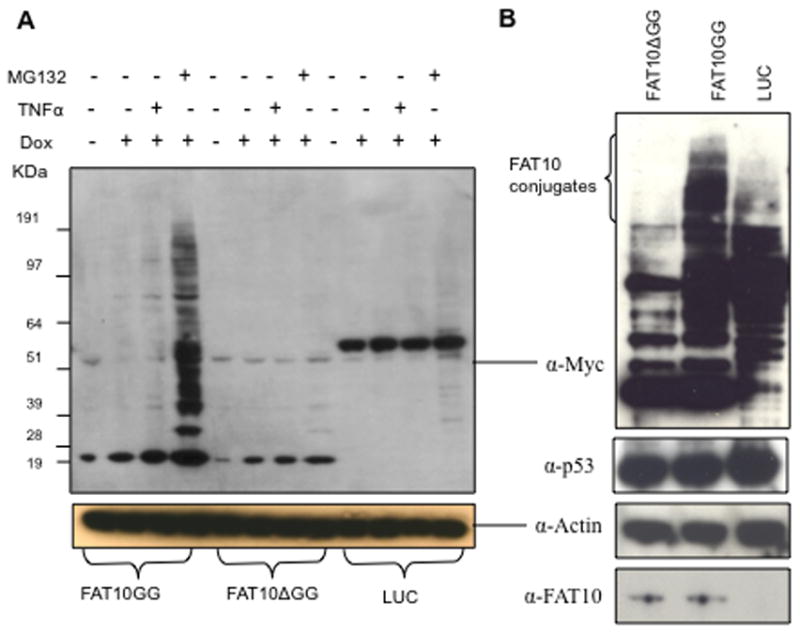
HEK 293 Tet-On cells were transfected with pTRE2hyg2-Myc-His-FAT10GG, pTRE2hyg2-Myc-His-FAT10ΔGG or pTRE2hyg2-Myc-Luciferase (LUC) plasmid as indicated. Cells stably expressing Myc-His-FAT10GG, Myc-His-FAT10ΔGG or Myc-LUC were selected with 300 μg/ml hygromycin. A, after a 48 h incubation with or without 2 μg/ml Dox and/or 5 μM MG132 and/or 100 unit per ml of TNF-α as indicated, whole cell extracts were resolved using NuPage gels and probed with anti-Myc antibody. B, after a 48 h incubation with 2 μg/ml Dox (0.5 μg/ml for Myc-His-FAT10GG), whole cell extracts were resolved using NuPAGE gels and probed with anti-Myc, anti-β-actin, anti-p53 or anti-FAT10 antibody, respectively. β-actin was used to show equal loading of whole cell extracts. To better visualize the FAT10 conjugated bands, the upper gel in B was exposed about 5 times longer than that in A.
Overexpressing FAT10GG enhances p53 transcriptional activity
Rassi et al reported that overexpressing FAT10 induces apoptosis [8]. We investigate whether the transcriptional activity of p53, a pivotal protein of apoptosis, is affected in cells overexpressing FAT10GG. The luciferase reporter plasmids, containing binding sites for p53 at the promoter regions, were used to analyze the transcriptional activity of p53. When stable cell lines were transfected with the pRGC-Luc p53 reporter plasmid, the luciferase activity in cells expressing FAT10GG was found to be elevated about 10-fold over those expressing FAT10ΔGG or mock vector (Fig. 2), demonstrating that overexpressing FAT10GG upregulates p53 transcriptional activity in HEK 293 cells. Similar results were also obtained with another p53 reporter construct pG13-Luc (Fig. S2).
Fig. 2. Overexpression of FAT10 increases p53 transcriptional activity.
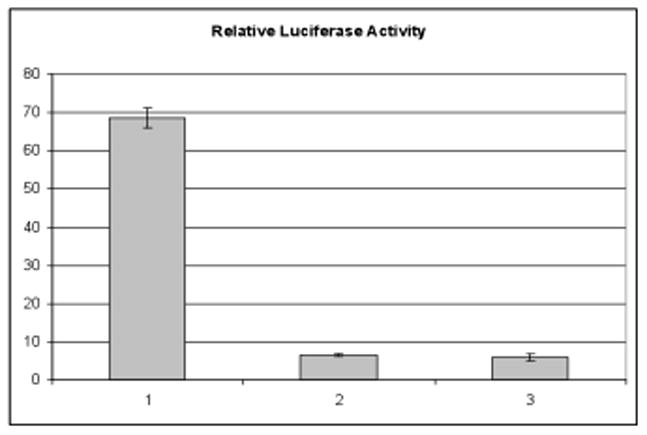
HEK 293 Tet-On cells stably expressing Myc-His-FAT10GG (1), Myc-His-FAT10ΔGG (2) or pTRE2hyg2-Myc vector (3) were transfected with pRGC-luc, a p53 response reporter plasmid, 1 μg per well in 12-well plate. Twenty-four hours after transfection, luciferase reporter activity was assayed, and the values obtained were normalized by protein concentration in each sample. The data shown represent the mean (arbitrary units) from three independent transfections with error bar indicating standard deviation.
p53 is modified by FAT10
The Myc-6xHis-tagged FAT10 and its modified proteins were purified from FAT10GG and FAT10ΔGG overexpressing cells using Ni-NTA agarose as described [15], and the purified proteins were probed with anti-p53 antibody by western blot. As shown in the left panel of Fig. 3A, a protein band corresponding to the size of the FATylated p53 was observed in cells expressing FAT10GG, but not in those expressing FAT10ΔGG. To further confirm that p53 is indeed modified by FAT10, we conducted the reciprocal experiment where p53 was immunoprecipitated from the cell extracts and the immunoprecipitates were probed with anti-Myc antibody (right panel of Fig. 3A). These results strongly support the notion that p53 can be covalently modified by FAT10. To compare the relative quantity of p53 modified by FAT10 to that by SUMO family members, the stable cell lines expressing SUMO-1/2/3GG or a non-conjugatable SUMO-1 GG were used. As shown in Fig. 3B, the molecular weight of FATylated p53 was slightly higher than that of SUMOylated p53, but the amount of FATylated p53 was significantly lower than SUMOylated p53. It should be pointed out that the data shown represent the results from one of three independent studies.
Fig. 3. p53 is covalently modified by FAT10.
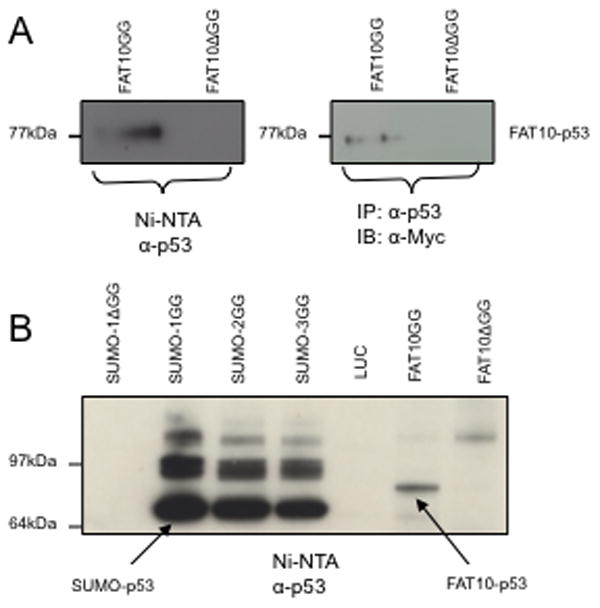
A, HEK 293 cells stably expressing Myc-His-FAT10GG and Myc-His-FAT10ΔGG were lysed in denaturing buffer containing guanidinium-HCl. His-tagged FAT10 and its conjugated proteins were purified by Ni-NTA agarose beads and probed with anti-p53 antibody (left panel). Or the whole cell lysates were immuoprecipitated with anti-p53 antibody and precipitants were probed with anti-Myc antibody (right panel). B, HEK 293 cells stably expressing Myc-His-SUMO1ΔGG, Myc-His-SUMO-1/2/3GG, Myc-LUC, Myc-His-FAT10GG or Myc-His-FAT10ΔGG were lysed in denaturing buffer containing guanidinium-HCl, His-tagged SUMO or FAT10 conjugated proteins were purified by Ni-NTA agarose beads and probed with anti-p53 antibody. The arrows indicate the band corresponding to the size of mono-SUMOylated p53 or FATylated p53.
p53 co-localizes with FAT10 and PML
Subcellular localization of FAT10 and p53 was probed by immunofluorescence using anti-Myc and anti-p53 antibodies followed by confocal microscopy. As shown in Fig. 4A, a significant quantity of FAT10GG is co-localized with p53 in the nucleus; however, FAT10ΔGG and LUC do not co-localize with p53. This suggests that co-localization of FAT10 and p53 depends on a covalent linkage between them. Interestingly, there is a significant change in p53 immunofluorescence when cells overexpressing FAT10GG were compared to those overexpressing FAT10ΔGG or LUC. The majority of p53 in cells overexpressing FAT10ΔGG or LUC localized in a few large nuclear compartments. In contrast, the p53 spread evenly in the nuclei in cells overexpressing FAT10GG (Fig. 4A). It has been reported that p53 co-localizes with PML nuclear bodies [17]. We therefore carried out co-immunostaining with both p53 and PML to investigate whether the large nuclear compartments in nuclei are PML nuclear bodies (NBs). As shown in Fig. 4B, p53 indeed co-localizes with PML in the large nuclear compartments in cells overexpressing FAT10ΔGG or LUC; however, the PML-NBs in cells overexpressing FAT10GG are smaller by a factor of 2–3 and they are distributed almost evenly. In addition, we observed on average 5 to 10 PML-NB in cells overexpressing FAT10GG, but only 1 or 2 PML-NB in cells overexpressing FAT10ΔGG or LUC.
Fig. 4. p53 co-localizes with FAT10 and PML.
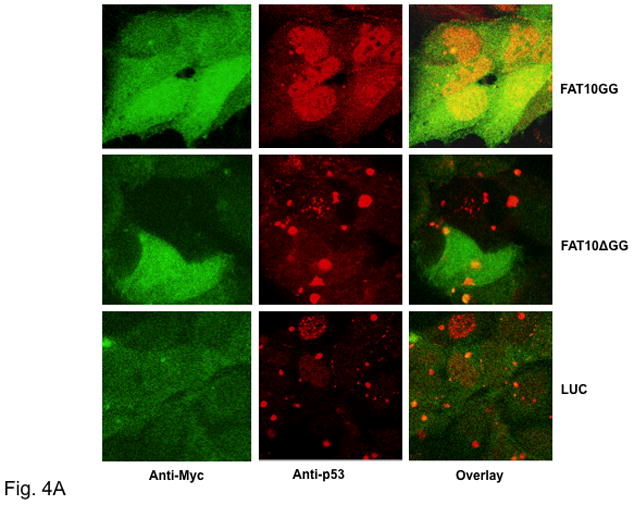
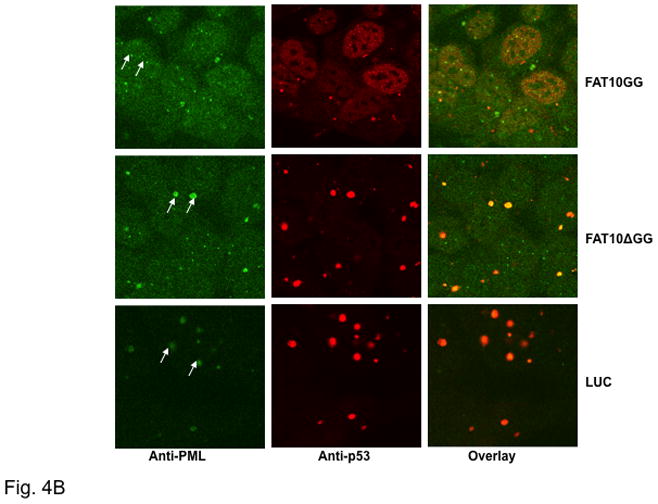
Following induction with 2 μg/ml Dox for 48 h, HEK 293 Tet-On Cells stably expressing Myc-tagged FAT10GG, FAT10ΔGG or LUC were fixed with 3.7% formaldehyde and then stained with TRITC conjugated anti-p53 antibody and FITC conjugated anti-Myc (A) or stained with TRITC conjugated anti-p53 antibody and FITC conjugated anti-PML (B). Images were captured using a Zeiss LSM-5 confocal microscope. Arrow to highlight PML-NB. Note that these results represent one of three independent studies.
Overexpressing FAT10 does not alter the p53 nuclear translocation but change its conformation
To investigate whether the elevation of p53 transcriptional activity in cells overexpressing FAT10GG is caused by increased translocation of p53 from cytoplasm to the nucleus, we separated nuclear and cytosolic proteins and probed with anti-p53 antibody or anti-Myc antibody. As shown in Fig. 5A, the majority of p53 was in the nucleus and this distribution was not changed when cells overexpress FAT10GG or FAT10ΔGG. The majorities of Myc-tagged FAT10GG, FAT10ΔGG and LUC were in the cytosol (Fig. 5A). The monoclonal PAB240 anti-p53 antibody, specifically recognizing mutant and transcriptionally inactive conformation of p53 in immunoprecipitation [18], was used to determine whether the conformation of p53 could be altered in cells overexpressing FAT10. As shown in Fig. 5B, the population of p53 which could be immunoprecipitated by PAB240 decreased significantly in cells overexpressing FAT10GG compared to that in cells overexpressing FAT10ΔGG or LUC. These data suggest that overexpressing FAT10 enhances p53 activity by reducing its PAB240 sensitive conformation instead of altering its nuclear proportion.
Fig. 5. FATylation does not alter the nuclear localization of p53, but change its conformation.
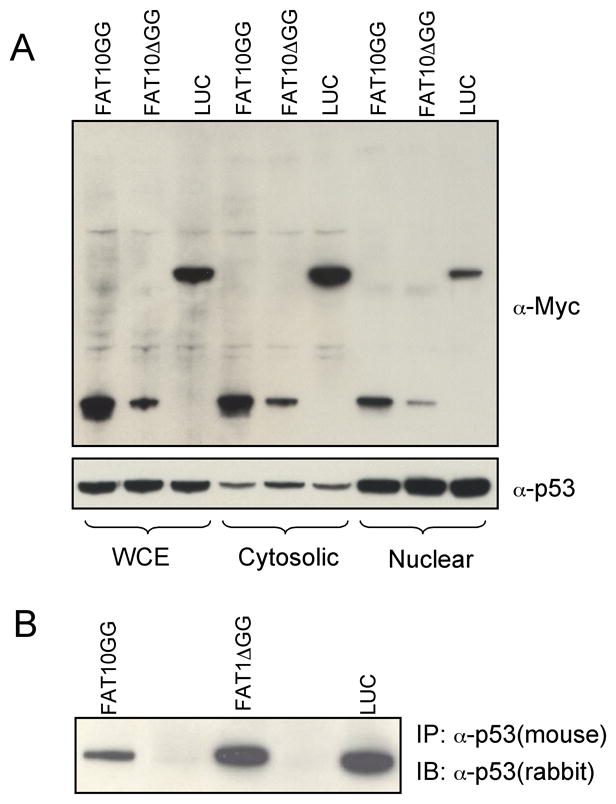
A, 20 μg of Whole cell extracts (WCE), Cytosolic or Nuclear proteins from cells stably expressing Myc-tagged FAT10GG, FAT10ΔGG or LUC were resolved in NuPAGE gel and probed with anti-Myc or anti-p53 respectively. B, Whole cell extracts from cells stably expressing Myc-tagged FAT10GG, FAT10ΔGG or LUC were immunoprecipitated with monoclonal anti-p53 antibody (PAB 240) and probed with polyclonal anti-p53 antibody. Note that these results represent one of three independent studies.
Discussion
FAT10, a ubiquitin-like modifier, has been implicated in the regulation of diverse cellular processes, which includes mitosis, chromosome stability, apoptosis, immune control and 26S proteasome mediated protein turnover [4–9, 12]. Like other members of UBLs, the C-terminal GG residues are required for conjugating FAT10 to its substrate proteins [2]. Using an established protocol [14, 15], we established HEK293 cell lines stably overexpressing double tagged FAT10, and its non-conjugatable counterpart, FAT10ΔGG, with the intent to identify FAT10 protein substrates and their regulatory pathway. Our study reveals that (i), p53 is FATylated; (ii), the conformation of p53 is altered and its transcriptional activity is greatly elevated in FAT10 overexpressing HEK293 cells; and (iii), overexpressing FAT10 leads to a change in PML-NB population to a smaller in size and more homogenously distributed in nuclei.
Previous reports [8, 12] showed that FAT10 forms only a 35 kDa conjugate with an unknown target protein. In contrast, we found that in addition to the 35 kDa complex, FAT10GG, but not FATΔGG, covalently conjugated to a large number of proteins (Fig. 1A and 1B). This inconsistency could be attributed to the fact that a different cell line was used in this study and the FAT10 expression level was higher here. Recently, another group reported the observation of high-molecular-weight conjugates [19], suggesting that FAT10 indeed forms covalent conjugates with other proteins. Interestingly, nearly all the FATylated proteins and FAT10 are sensitive to proteasome inhibitor, MG132 (Fig. 1A), indicating that protein FATylation, like polyubiquitinylation, may also mark proteins for proteasome mediated degradation.
Overexpression of FAT10 in mouse fibroblasts has been shown to induce apoptosis [8]. In this context, we found p53, an extensively investigated pro-apoptotic protein, was activated by about ten-fold in cells overexpressing FAT10GG relative to those overexpressing FAT10ΔGG or mock vector (Fig. 2). This indicates that FAT10 may exert its biological effect, in parts, by activating p53 transcriptional activity. Intricately, FAT10 expression is up-regulated in many tumor tissues [7]. It remains unclear why the upregulation of FAT10 in those tumor cells does not induce apoptosis. Our finding provides some insight into this apparent conflict: overexpressing FAT10 induces apoptosis by activating p53; however, p53 is the most commonly mutated gene known in human cancer [20], so it is possible that upregulation of FAT10 fails to induce apoptosis in those tumor cells with mutated p53 genes. Because p53 negatively regulates FAT10 expression [6], FAT10 is likely upregulated in those tumor cells with mutant p53 (loss of function). On the contrary, in normal cells the activation of p53 by upregulating FAT10 will, in turn, reduces FAT10 expression as a feedback control to keep its expression at low level, as FAT10 is almost undetectable in most tissues except in spleen and thymus [10]. Interestingly, FAT10 has also been shown to activate NF-kappaB, an ubiquitous transcription factor, mediated via the expression of the proteasomal subunit, LMP2 (Low Molecular mass Polypeptide 2) (21). However, the mechanism by which FAT10 regulates LMP2 expression remains to be elucidated.
To investigate the mechanism by which overexpressing FAT10 upregulating p53 activity, we studied whether p53 is a protein substrate of FAT10. Our results show that p53 is covalently linked to FAT10 and the C-terminal GG of FAT10 is required for this interaction. The FATylated p53 is detected by either His-tagged protein pull-down assay or co-immunoprecipitation assay (Fig. 3A). However, the quantity of the FATylated p53 detected is lower than the SUMO-1/2/3 modified p53 (Fig. 3B). SUMO-1 and SUMO-2/3 had been shown to modify less than 5% of p53 protein in HEK293 cells [16, 22]. The data in Fig. 3B indicate that p53 was modified by FAT10 to an even lower extent. Note that the band above SUMO-p53 has been identified as disumoylated p53 [16]. Furthermore, overexpressing SUMO family members causes a 2 ∼ 4 folds increase in p53 transcriptional activity [16], while a ten-fold activation is observed in FAT10GG overexpressing cells. This observation suggests that FAT10 indirectly stimulates p53 activity and the FATylation of p53 is a dynamic process which may lead to its translocation and/or subsequent p53 structural change.
The immunostaining data reveal that a significant number of p53 molecules are co-localized with FAT10GG relative to those found in cells overexpressing the non-conjugatable FAT10ΔGG or LUC (Fig. 4A). Furthermore we found that p53 are mainly accumulated in some large nuclear bodies in cells overexpressing FAT10ΔGG and LUC, but not in those overexpressing FAT10GG where p53 accumulated in much smaller subcellular bodies and evenly distributed in nuclei (Fig. 4A and 4B). Considering that p53 is known to co-localize with PML nuclear bodies [17], we co-stained these cells with both p53 and PML antibodies. The results (Fig. 4B) showed that the subcellular bodies in which p53 accumulated are PML nuclear bodies (NBs). There are only 1 or 2 large PML-NBs in cells overexpressing FAT10ΔGG and LUC, and about 5 ∼ 10 small PML-NBs in cells overexpressing FAT10GG (Fig. 4B). It should be pointed out that DAPI staining experiments, using Hg lamp and a DAPI filter set, to identify the nucleus were also carried out and the results are similar to the nuclear images revealed by anti-p53 antibody shown in Fig. 4A and 4B. However, the DAPI confocal image were not recorded because the laser confocal microscope used was not equipped with a UV laser required. Furthermore, western blot analysis revealed that there is no significant difference in the amount of PML proteins found in these three overexpressing cell lines (Data not shown). In addition, as shown in Fig. 5A, overexpression of FAT10GG, FAT10ΔGG and mock vector did not significant change the quantity of p53 in the nuclei. Thus, the activation of p53 transcriptional activity observed in cells overexpressing FAT10GG may be attributed to the alteration of p53 conformation due to FAT10GG induced changes in PML-NBs. To address this issue, we probed the conformation of p53 using a conformation specific monoclonal anti-p53 antibody (PAB 240), which recognizes and precipitates mutant p53 conformation or wild type p53 with abnormal conformation, but not wild type normal conformation [18, 23]. As shown in Fig. 5B, overexpressing FAT10GG significantly reduces the level of p53 with abnormal conformation. Normally, there are 5 ∼ 10 PML-NBs in each HEK293 cell [24], but there are only 1 or 2 abnormally large PML-NBs in HEK 293 cells, indicating that the PML-NB may not function normally. In normal cells, recruiting p53 to PML-NBs activates its transcriptional activity [25]. Accumulation of p53 in these abnormally large PML-NBs may affect its conformation and hamper its transcriptional activity. FAT10 appears to mediate the normalization of the structure and distribution of PML-NBs and leads to the activation of p53.
The observation that FAT10 modifies p53 and stimulates its transcriptional activity is in agreement with the potential roles of FAT10 in the regulation of cell-cycle, apoptosis, proliferation arrest, and tumorigenesis [5, 7, 8]. The consequence of p53 stimulation is defined by ‘cellular context’ or particular cell genotype [26]. Raasi et al encountered with cell death and poor proliferation when they overexpressed FAT10 in HeLa cell [8]. This may be due to FAT10 activation of wild-type p53 in HeLa cells which induced apoptosis. However, we did not observe any obvious impairment in the proliferation of HEK293 cells overexpressing FAT10GG. Note that HEK293 cells are immortalized with viral E1A and E1B proteins that inhibit apoptosis via their attenuation of p53 activity [27], and FAT10 may affect the interaction between p53 and the viral proteins E1A, E1B 55K, and/or E1B 19K.
In summary, we demonstrated that p53 is FATylated and that overexpressing FAT10 in HEK293 cells led to a significant increase in p53 transcriptional activity, as well as changes in the conformation of p53, and in the size and distribution of PML-NBs. While there is no direct evidence showing FATylated p53 exhibits higher transcriptional activity, our results suggest a dynamic mechanism for FAT10-mediated p53 activation which involves FATylation of p53 and possibly other PML proteins, and leads to the translocation of p53 into functional PML-NBs where p53 undergoes conformational change and activation.
Supplementary Material
Research highlights.
Our study reveals that:
P53 is FATylated.
Overexpression of FAT10 in HEK293 cells leads to elevation of p53 transcriptional activity.
FAT10 overexpression leads to changes in p53 conformation and in the size and disbribution of PML-NB.
Acknowledgments
We thank Dr. Seung J. Baek (The University of Tennessee) for providing the pRGC-Luc p53 and pG13-Luc p53 response reporter plasmids.
Abbreviations
- FAT10
HLA-F Adjacent Transcript 10
- PML-NB
Promyelocytic leukemia nuclear body
- UBLs
Ubiquitin-like modifiers
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Welchman RL, Gordon C, Mayer RJ. Nat Rev Mol Cell Biol. 2005;6:599–609. doi: 10.1038/nrm1700. [DOI] [PubMed] [Google Scholar]
- 2.Kerscher O, Felberbaum R, Hochstrasser M. Annu Rev Cell Dev Biol. 2006;22:159–180. doi: 10.1146/annurev.cellbio.22.010605.093503. [DOI] [PubMed] [Google Scholar]
- 3.Fan W, Cai W, Parimoo S, Lennon GG, Weissman SM. Immunogenetics. 1996;44:97–103. doi: 10.1007/BF02660056. [DOI] [PubMed] [Google Scholar]
- 4.Raasi S, Schmidtke G, de Giuli R, Groettrup M. Eur J Immunol. 1999;12:4030–4036. doi: 10.1002/(SICI)1521-4141(199912)29:12<4030::AID-IMMU4030>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 5.Lim CB, Zhang D, Lee CGL. Cell Div. 2006;1:20. doi: 10.1186/1747-1028-1-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang DW, Jeang KT, Lee CGL. Oncogene. 2006;25:2318–2327. doi: 10.1038/sj.onc.1209220. [DOI] [PubMed] [Google Scholar]
- 7.Lee CGL, Ren J, Cheong IS, Ban KH, Ooi LL, Yong TS, Kan A, Nuchprayoon I, Jin R, Lee KH, Choti M, Lee LA. Oncogene. 2003;22:2592–2603. doi: 10.1038/sj.onc.1206337. [DOI] [PubMed] [Google Scholar]
- 8.Raasi S, Schmidtke G, Groettrup M. J Biol Chem. 2001;276:35334–35343. doi: 10.1074/jbc.M105139200. [DOI] [PubMed] [Google Scholar]
- 9.Ren J, Kan A, Leong SH, Ooi LL, Jeang KT, Chong SS, Kon OL, Lee CGL. J Biol Chem. 2006;281:11413–11421. doi: 10.1074/jbc.M507218200. [DOI] [PubMed] [Google Scholar]
- 10.Liu YC, Pan J, Zhang C, Fan W, Collinge M, Bender JR, Weissman SM. Proc Natl Acad Sci U S A. 1999;96:4313–4318. doi: 10.1073/pnas.96.8.4313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hipp MS, Raasi S, Groettrup M, Schmidtke G. J Biol Chem. 2004;279:16503–16510. doi: 10.1074/jbc.M310114200. [DOI] [PubMed] [Google Scholar]
- 12.Hipp MS, Kalveram B, Raasi S, Groettrup M, Schmidtke G. Mol Cell Biol. 2005;25:3483–3491. doi: 10.1128/MCB.25.9.3483-3491.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Canaan A, Yu X, Booth CJ, Lian J, Lazar I, Gamfi SL, Castille K, Kohya N, Nakayama Y, Liu YC, Eynon E, Flavell R, Weissman SM. Mol Cell Biol. 2006;26:5180–5189. doi: 10.1128/MCB.00966-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li T, Evdokimov E, Shen RF, Chao CC, Tekle E, Wang T, Stadtman ER, Yang DC, Chock PB. Proc Natl Acad Sci U S A. 2004;101:8551–8556. doi: 10.1073/pnas.0402889101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li T, Santockyte R, Shen RF, Tekle E, Wang G, Yang DC, Chock PB. Arch Biochem Biophys. 2006;453:70–74. doi: 10.1016/j.abb.2006.03.002. [DOI] [PubMed] [Google Scholar]
- 16.Li T, Santockyte R, Shen RF, Tekle E, Wang G, Yang DC, Chock PB. J Biol Chem. 2006;281:36221–36227. doi: 10.1074/jbc.M608236200. [DOI] [PubMed] [Google Scholar]
- 17.Pearson M, Carbone R, Sebastiani C, Cioce M, Fagioli M, Saito S, Higashimoto Y, Appella E, Minucci S, Pandolfi PP, Pelicci PG. Nature. 2000;406:207–210. doi: 10.1038/35018127. [DOI] [PubMed] [Google Scholar]
- 18.Gannon JV, Greaves R, Iggo R, Lane DP. EMBO J. 1990;9:1595–1602. doi: 10.1002/j.1460-2075.1990.tb08279.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chiu YH, Sun Q, Chen ZJ. Mol Cell. 2007;27:1014–1023. doi: 10.1016/j.molcel.2007.08.020. [DOI] [PubMed] [Google Scholar]
- 20.Stratton MR. Eur J Cancer. 1992;28:293–295. doi: 10.1016/0959-8049(92)90437-7. [DOI] [PubMed] [Google Scholar]
- 21.Gong P, Canaan A, Wang B, Leventhal J, Snyder A, Nair V, Cohen CD, Kretzler M, D’Agati V, Weissman S, Ross MJ. J Am Soc Nephrol. 2010;21:316–326. doi: 10.1681/ASN.2009050479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gostissa M, Hengstermann A, Fogal V, Sandy P, Schwarz SE, Scheffner M, Del Sal G. EMBO J. 1999;18:6462–6471. doi: 10.1093/emboj/18.22.6462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Grand RJ, Lecane PS, Owen D, Grant ML, Roberts S, Levine AJ, Gallimore PH. Virology. 1995;210:323–334. doi: 10.1006/viro.1995.1349. [DOI] [PubMed] [Google Scholar]
- 24.Zhong S, Salomoni P, Pandolfi PP. Nat Cell Biol. 2000;2:E85–90. doi: 10.1038/35010583. [DOI] [PubMed] [Google Scholar]
- 25.Fogal V, Gostissa M, Sandy P, Zacchi P, Sternsdorf T, Jensen K, Pandolfi PP, Will H, Schneider C, Del Sal G. EMBO J. 2000;19:6185–6195. doi: 10.1093/emboj/19.22.6185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Oren M. Cell Death Differ. 2003;10:431–442. doi: 10.1038/sj.cdd.4401183. [DOI] [PubMed] [Google Scholar]
- 27.Debbas M, White E. Genes and Dev. 1993;7:546–554. doi: 10.1101/gad.7.4.546. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.