

Abstract
In E. coli, secA expression is regulated at the translational level by an upstream gene (secM) that encodes a presecretory protein. SecM contains a C-terminal sequence motif that induces a transient translation arrest. Inhibition of SecM membrane targeting prolongs the translation arrest and increases SecA synthesis by concomitantly altering the structure of the secM-secA mRNA. Here we show that the SecM signal peptide plays an essential role in this regulatory process by acting as a molecular timer that coordinates membrane targeting with the synthesis of the arrest motif. We found that signal peptide mutations that alter targeting kinetics and insertions or deletions that change the distance between the SecM signal peptide and the arrest motif perturb the balance between the onset and release of arrest that is required to regulate SecA synthesis. Furthermore, we found that the strength of the interaction between the ribosome and the SecM arrest motif is calibrated to ensure the release of arrest upon membrane targeting. Our results strongly suggest that several distinctive features of the SecM protein evolved as a consequence of constraints imposed by the ribosome and the Sec machinery.
Keywords: membrane targeting, ribosome, SecM, signal peptide, translational regulation
INTRODUCTION
In E. coli, the vast majority of presecretory and membrane proteins are transported across or integrated into the inner membrane (IM) by the Sec pathway. The Sec machinery consists of a universally conserved transport channel (the SecYEG complex) and a bacterial-specific peripheral membrane protein (SecA) that uses the energy of ATP hydrolysis to promote translocation through the SecYEG complex (Driessen and Nouwen, 2008). Presecretory proteins are targeted to the Sec machinery by cleavable N-terminal signal peptides that are typically ~20–25 residues in length. Although signal peptides are highly degenerate, they consist of a basic N region, a ~7–13 residue hydrophobic H region, and a slightly polar C region (von Heijne, 1985). In contrast, most IM proteins lack cleavable signal peptides and use the first transmembrane segment as an internal targeting signal. Although the signal recognition particle (SRP) interacts primarily with transmembrane segments, it also recognizes a small fraction of signal peptides that are especially hydrophobic. SRP binds to its substrates as they emerge from ribosomes and then rapidly targets ribosome-nascent chain (RNC) complexes to the SecYEG complex cotranslationally (Keenan et al., 2001). Because most signal peptides are bypassed by SRP, however, presecretory proteins are generally fully synthesized and targeted to the IM post-translationally. These proteins interact with trigger factor at an early stage of translation and subsequently interact with other chaperones such as SecB which preserve them in a loosely folded conformation (Cross et al., 2009). Both co- and post-translational targeting pathways converge at the IM, and SecA facilitates the translocation of proteins targeted by both mechanisms as well as the integration of IM proteins that have large periplasmic segments (Driessen and Nouwen, 2008; Neumann-Haefelin et al., 2000; Schierle et al., 2003).
While the components of the E. coli SecYEG complex appear to be produced constitutively, the synthesis of SecA is tightly regulated. SecA expression is controlled at the level of translation by a cotranscribed gene called secM which encodes a 170 amino acid presecretory protein (Oliver et al., 1998). SecM contains a C-terminal sequence motif (150FxxxxWIxxxxGIRAGP166) whose recognition inside the ribosome tunnel inhibits further translation (Nakatogowa and Ito, 2002). Under normal conditions, ribosomes stall transiently at P166. This stalling briefly denatures a hairpin in the secM-secA mRNA that masks the secA Shine-Dalgarno sequence and thereby facilitates the synthesis of a basal level of SecA which is required for cell viability (Murakami et al., 2004). The stalling appears to be released by a physical force exerted by the Sec machinery during translocation (Butkus et al., 2003). After SecM is transported into the periplasm it is rapidly degraded by Prc and other unidentified proteases. Conditions that impair the targeting of SecM to the Sec machinery, such as mutations in the Sec apparatus or the SecM signal peptide cause a prolonged translation arrest that increases the exposure of the Shine-Dalgarno sequence and leads to an overproduction of SecA (Nakatogawa and Ito, 2001; Oliver et al., 1998; Sarker and Oliver, 2002). Presumably this regulatory system evolved to monitor and adapt to changes in the secretion status of the cell.
Recent studies have indicated that the conformation of the SecM C terminus inside the ribosome tunnel is critical for recognition of the arrest motif (Woolhead et al., 2006; Yap and Bernstein, 2009). Although the mutation of any of the nine residues that constitute the E. coli SecM arrest motif leads to translation arrest defects, an analysis of genetic suppressors and naturally occurring SecM homologs showed that the introduction of multiple C-terminal mutations can completely restore arrest activity (Yap and Bernstein, 2009). The data suggest that tunnel components recognize only a single residue, R163, but that a group of secondary residues that vary in position, number and side chain chemistry are also required to provide conformational information that positions R163 in a highly specific location inside the tunnel. Presumably the force exerted on a polypeptide chain by the Sec machinery releases arrest by disrupting the secondary structure of the SecM nascent chain and/or dislodging R163 from the tunnel walls. Given that the translocation of SecM is SecA-dependent and that SecA has been suggested both by biochemical and structural studies to push segments of a polypeptide chain across the IM (Schiebel et al., 1991; Zimmer et al., 2008), it seems likely that SecA is the component of the Sec machinery that acts on SecM.
Although the ability of SecM to fine-tune secA expression is strongly influenced by the efficiency of membrane targeting, it is unclear whether other factors such as targeting pathway selection or the rate of translocation might also affect its regulatory function. In this regard it is noteworthy that the depletion of SRP prolongs translation arrest (Nakatogawa and Ito, 2001). This observation suggests that SecM is targeted cotranslationally. Consistent with this possibility, the hydrophobicity of the H region of the SecM signal peptide is near the threshold for SRP recognition (Peterson et al., 2003; Huber et al., 2005). If SecM were targeted by SRP at an early stage of translation, however, it would likely monitor the status of a few presecretory proteins and IM proteins rather than the bulk of presecretory proteins. Furthermore, the SecM signal peptide contains an unusually long N region that is far more highly conserved than the mature region of the protein (Fig. 1A). Mutations in this region increase the basal level of secA expression and reduce its inducibility under secretion-deficient conditions (Sarker and Oliver, 2002). Conserved sequences in signal peptides have been shown to influence a variety of post-targeting steps and can even direct proteins to specific subsets of Sec channels (Hegde and Bernstein, 2006; Carlsson et al., 2007). In the one case where a conserved N-terminal extension has been shown to affect targeting pathway selection, it exerts a strong inhibitory effect on SRP recognition (Peterson et al., 2006). Indeed it is unclear why SecM would contain an unusual signal peptide if its regulation of secA expression only requires rapid targeting by the SRP pathway.
Fig. 1.

The conserved N region of the SecM signal peptide plays a key role in the release of translation arrest. A. The signal peptides of representative SecM homologs were predicted using SignalP 3.0 (Bendtsen et al., 2004) and aligned using ClustalW. The N, H, and C regions of each signal peptide are shown. Signal peptide cleavage sites were predicted using Invariant residues are highlighted in red and highly conserved residues are highlighted in blue. The length of each signal peptide (SP) and each full-length protein is indicated. B. The sequences of the SecM,Δ5 and Δ10 signal peptides are shown. C. MNY3 (MC4100 Δprc::cat) transformed with a plasmid encoding wild-type SecM (pMY1), Δ5SP–SecM (pMY3) or Δ10SP–SecM (pMY4) or the corresponding arrest-deficient derivative (P166A) were subjected to pulse-chase labeling after the addition of IPTG to induce expression of the plasmid-borne gene. Full-length (f), translation-arrested (a) and mature (m) forms of SecM were immunopreciptated and resolved by SDS-PAGE. D. Cells were grown as in part C and pulse-labeled. Samples were then fractionated by CTABr precipitation and SecM-containing polypeptides were immunoprecipitated. T, unfractionated sample; P, CTABr pellet; S, CTABr supernatant.
In this study we sought to elucidate the significance of the unusual SecM signal peptide. We found that altering the native signal peptide or replacing it with other signal peptides strongly impaired the regulatory activity of SecM. Signal peptides that promote cotranslational targeting prevented the transient stalling that is necessary for the production of a basal level of SecA, whereas those that ordinarily promote post-translational targeting failed to target SecM to the IM effectively and thereby caused a prolonged arrest. These results, together with the observation that the effects of the signal peptide mutations could be mimicked by altering the distance between the signal peptide and the arrest motif, strongly suggest that the SecM signal peptide acts as a molecular timer that targets the protein to the IM immediately after synthesis of the arrest motif. Our results show that previously unrecognized constraints imposed on the SecM regulatory switch by the translation and translocation machineries led to the evolution of a unique mode of protein targeting that enables SecM to specifically monitor the fate of post-translationally targeted presecretory proteins.
RESULTS
The SecM signal peptide facilitates the establishment and release of translation arrest by regulating the timing of membrane targeting
In initial experiments we transformed E. coli strain MNY3 (MC4100 Δprc::cat) with a plasmid encoding wild-type SecM or a SecM mutant (P166A) that lacks translation arrest activity under the control of the trc promoter. After the addition of IPTG to induce secM expression, cells were subjected to pulse-chase labeling. To maintain consistency with previous studies (Nakatogawa and Ito, 2001; Yap and Bernstein, 2009), we pulse labeled cells for 1 min. Subsequently, full-length (170 residue), arrested (166 residue) and mature (133 residue) forms of SecM were immunoprecipitated. Because full-length SecM is not observed in secretion-deficient cells (Nakatogawa and Ito, 2001; Yap and Bernstein, 2009), we expected that it would be produced only when the Sec machinery exerts a force on the nascent chain and releases (or prevents) translation arrest. Based on previous work (Nakatogawa and Ito, 2001), we also expected that the absence of the Prc protease would not only partially stabilize the mature form of the protein, but would also increase the relative amount of full-length SecM.
The results of these experiments strongly suggested that translation arrest occurs before SecM RNCs are targeted to the IM. We observed roughly equal amounts of full-length, arrested and mature forms of SecM in pulse-labeled cells (Fig. 1C, lane 1). The arrested form, however, was found exclusively in the cytoplasm (Fig. 2, lanes 1–3). The isolation of a large amount of full-length protein was striking and strongly suggested that the signal peptide is cleaved relatively slowly. Consistent with previous results (Nakatogawa and Ito, 2001), the translation-arrested form of wild-type SecM gradually disappeared and had a half-life of <1 min (Fig. 1C, lanes 1–4). Full-length and mature SecM also disappeared in parallel. The P166A mutant exhibited a similar decay profile, but due to the lack of translation arrest only full-length SecM and the mature form of the protein were observed (Fig. 1C, lanes 5–8). To clarify the relationship between the different forms of SecM, we repeated these experiments using a shorter (30 sec) pulse. The data clearly show that the arrested and full-length forms of the protein appeared first and were slowly converted into the mature form (Fig. S1). Moreover, SecM and SecM (P166A) and a control presecretory protein (an HA-tagged version of OmpA) were translocated into inverted membrane vesicles (INVs) and converted to the mature form in cell-free translation assays (Fig. 3A and Fig. 3B, lanes 1, 2 and 5–8). Finally, we found that the pattern of SecM polypeptides, the level of SecA, and the secretion of a control presecretory were unaffected by the overexpression of secM (Fig. S2). Taken together, these results suggest a scenario in which translation-arrested RNCs are targeted to the IM, where arrest is released. Subsequently the protein is translocated across the IM and degraded by periplasmic proteases that substitute for Prc.
Fig. 2.
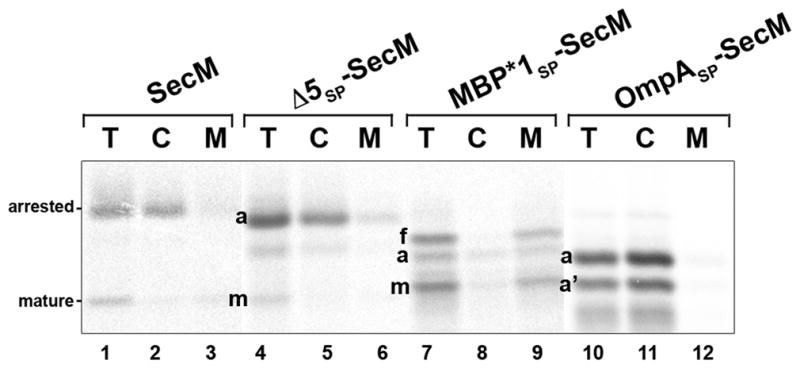
Localization of translation-arrested SecM RNCs. MNY3 transformed with a plasmid that encodes the indicated SecM derivative were pulse labeled. Cells were converted to spheroplasts, which were then separated into cytoplasmic and membrane fractions, and full-length (f), arrested (a) and mature (m) forms of SecM were immunoprecipitated from each fraction. A SecM fragment that was presumably a nascent chain attached to the ribosome situated behind the stalled ribosome (a′) was also immunoprecipitated from some of the samples. T, total spheroplast extract; C, cytoplasm; M, membranes.
Fig. 3.

Translocation of SecM but not Δ10SP-SecM, OmpA SP-SecM or SecM(Δ38–127) into INVs. Coupled in vitro transcription-translation reactions programmed with PCR products that encode OmpA-HA (A) or the indicated SecM derivative (B and C) were performed and INVs were added to a portion of each reaction. PK was subsequently added to digest protein that was not translocated into the INVs. Polypeptides that were synthesized in the in vitro reactions were immunoprecipitated with the indicated antiserum and resolved by SDS-PAGE. In part B, 1.5 times as much protein was loaded in lanes 5–12 as in lanes 1–4, and in part C, 1.5 times as much protein was loaded in lanes 3–6 as in lanes 1–2. A non-specific background band is denoted with an asterisk.
To examine the role of the SecM signal peptide in translation arrest, we first deleted 5 or 10 amino acids from the conserved N region to create the Δ5SP and Δ10SP mutations (Fig. 1B). MNY3 were transformed with a plasmid encoding Δ5SP–SecM, Δ10SP–SecM, Δ5SP–SecM (P166A) or Δ10SP–SecM (P166A) and pulse-chase labeling was conducted as described above. Surprisingly, the mutations significantly prolonged translation arrest (Fig. 1C, lanes 9–12 and 17–20). The single predominant band that was observed in pulse-labeled cells could be identified as the arrested form of SecM based both on its size and its precipitation by cetyl trimethylammonium bromide (CTABr), a reagent that selectively precipitates tRNA-linked polypeptides (Fig. 1D). Unlike the arrested form of wild-type SecM, the arrested form of Δ5SP–SecM and Δ10SP–SecM persisted through the entire time course. Although the mature form of the protein was also observed, its appearance was delayed. These results suggest that the signal peptide deletions prevent the release of arrest by impairing the membrane targeting of the arrested SecM polypeptide. Consistent with this interpretation, the arrested form of Δ5SP–SecM was localized to the cytoplasm (Fig. 2, lanes 4–6). Furthermore, Δ10SP–SecM synthesized in vitro was not translocated into INVs (Fig. 3B, lanes 3 and 9–10). The mutations did not impair the protein translocation activity of the signal peptide, however, because Δ5SP–SecM (P166A) and Δ10SP–SecM (P166A) were exported and degraded in the periplasm at essentially the same rate as SecM (P166A) (Fig. 1C, compare lanes 5–8 to lanes 13–16 and 21–24).
To obtain further insight into the function of the SecM signal peptide, we next examined the effect of replacing it with the OmpA or maltose binding protein (MBP) signal peptide (Fig. 4A). The OmpA signal peptide directs post-translational targeting. While the MBP signal peptide also directs post-translational targeting, there is evidence that the translocation of at least a fraction of the 43 kD MBP begins at a very late stage of translation (Randall, 1983). MNY3 transformed with plasmids encoding the chimeras were subjected to pulse-chase labeling. Like Δ5SP–SecM and Δ10SP–SecM, the majority of the radiolabeled OmpASP-SecM was in a translation-arrested (150 residue) form (Fig. 4B, lanes 1–4; Fig. 4C, lanes 1–3). In addition to this polypeptide, we detected a ~135 residue SecM fragment (arrested’ or a’) that persisted through the time course. This fragment was precipitated by CTABr and likely corresponds to a nascent SecM chain tethered to the ribosome that is situated behind the leading stalled ribosome. Because essentially no full-length OmpASP-SecM or mature SecM was observed, the signal peptide substitution appeared to completely abolish membrane targeting. Consistent with this notion, the arrested form of OmpASP-SecM was localized in the cytoplasm and OmpASP-SecM synthesized in vitro was not translocated into INVs (Fig. 2, lanes 10–12; Fig. 3B, lanes 4 and 11–12). Furthermore, unlike overexpressed proteins that “jam” the Sec machinery, OmpASP-SecM did not compromise the export of endogenous presecretory proteins or trigger the proteolysis of SecY (Fig. S3; Szabady et al., 2005; van Stelten et al., 2009). MBPSP-SecM also underwent prolonged translation arrest (Fig. 4B, lanes 9–12; Fig. 4C, lanes 7–9). More of the protein was secreted than OmpASP-SecM, possibly because it was targeted to the IM slightly faster. As expected, the OmpA and MBP signal peptides remained competent for targeting and translocation because OmpASP–SecM (P166A) and MBPSP–SecM (P166A) were exported rapidly (Fig. 4B, lanes 5–8 and 13–16). Taken together with the results described above, the data imply that neither mutant SecM signal peptides nor typical post-translational signal peptides effectively target stalled SecM RNCs to the Sec machinery and permit the release of arrest that is required to modulate SecA levels.
Fig. 4.

Heterologous signal peptides differentially affect SecM-mediated translation arrest. A. The sequences of post-translational (OmpASP and MBPSP) and cotranslational (MBP*1SP) signal peptides that were used to replace the native SecM signal peptide (SecMSP) are shown. The mutations that were introduced into MBPSP to create MBP*1SP are underlined. B. MNY3 was transformed with a plasmid that encodes native SecM (pMY1), OmpASP-SecM (pMY6), MBPSP-SecM (pMY7), MBP*1SP-SecM (pMY8), or the corresponding arrest-deficient derivative (P166A). Cells were subjected to pulse-chase labeling after the addition of IPTG and SecM-containing polypeptides were immunoprecipitated. C. Cells were grown as in part B and pulse-labeled. Samples were then fractionated by CTABr precipitation and SecM-containing polypeptides were immunoprecipitated. T, unfractionated sample; P, CTABr pellet; S, CTABr supernatant.
A phenomenon that we term “ribosome crowding” may provide a partial explanation for the inability of post-translational signal peptides to target arrested SecM RNCs to the IM. Consistent with the observation that SecM-arrested ribosomes are not a substrate of SsrA-mediated trans-translation (Garza-Sanchez et al., 2006), we found that the retention of OmpASP-SecM RNCs in the cytoplasm was not due to the binding of a component of the SsrA pathway (data not shown). We also could not identify another factor that binds to OmpASP-SecM RNCs that might hinder their interaction with the Sec complex. We were intrigued, however, by the observation that stalling led to the synthesis of a fragment (the OmpASP-SecM arrested’ polypeptide) that is only slightly smaller than OmpASP-SecM. Based on the size of this fragment, the ribosome behind the stalled ribosome was ~45 nucleotides away. This distance is considerably smaller than the average distance between ribosomes in E. coli (97 nucleotides; see Underwood et al., 2005) and only slightly greater than the length of mRNA that is protected by the ribosome (~30 nucleotides; see Steitz, 1969). The close spacing may result from the ability of trailing ribosomes to continue protein synthesis briefly after the lead ribosome has undergone translation arrest. We surmised that ribosome crowding might hinder the targeting of the lead RNC. To test this idea, MNY3 that produced OmpASP-SecM were pulse-labeled, and puromycin was added at the time of the chase. This aminoacyl-tRNA mimetic causes the premature release of nascent chains, but does not bind to stalled SecM-RNCs (Muto et al., 2006; Woolhead et al., 2006). Consistent with our hypothesis, we found that puromycin promoted the membrane targeting and translocation of a fraction of the OmpASP-SecM stalled at P166 (Fig. S4). By releasing the penultimate ribosome, puromycin also promoted the post-translational export of the arrested’ fragment. The export of OmpASP-SecM nascent chains in this experiment suggests that they do not lose their translocation-incompetence, but instead are retained in the cytoplasm due to the spatial organization of the polysome created by translation arrest.
Interestingly, while signal peptides that direct post-translational targeting prolonged SecM translation arrest, a signal peptide that directs cotranslational targeting had the opposite effect. Previous work has shown that the highly hydrophobic MBP*1 signal peptide (Fig. 4A) is recognized by SRP (Lee and Bernstein, 2001). Replacement of the SecM signal peptide with the MBP*1 signal peptide strongly suppressed the production of translation-arrested SecM (Fig. 4B, lanes 17–20; Fig. 4C, lanes 13–15). Indeed a MBP*1SP-SecM derivative containing the P166A mutation produced a similar pattern of polypeptides (Fig. 4B, lanes 21–24; Fig. 4C, lanes 16–18). Because MBP*1SP and other cotranslational signal peptides are generally removed before the completion of protein synthesis and the precursor form is not observed (Lee and Beckwith, 1986; Peterson et al., 2003), it is curious that full-length MBP*1-SecM was converted to the mature form relatively slowly. Cell fractionation experiments showed that both full-length and arrested forms of MBP*1-SecM were associated with the IM, however, and confirmed that membrane targeting occurred relatively early, before the onset of translation arrest (Fig. 2, lanes 7–9). Thus, it is likely that the presence of the mature region of SecM perturbs signal peptide cleavage (and accounts for the apparently slow cleavage of the wild-type SecM signal peptide described above). Indeed the relatively long half-life of the full-length form of wild-type SecM is consistent with this interpretation (Fig. 1C, lanes 1–4). In any case, the data suggest that the Sec machinery exerts a force on the nascent chain too soon when SecM RNCs are targeted to the IM at an early stage of translation and thereby prevents the establishment of the arrested state and/or causes its premature release.
To obtain further insight into the properties of the SecM signal peptide, we compared the targeting of HA-tagged versions of native OmpA, OmpA containing the SecM signal peptide, and OmpA containing the MBP*1 signal peptide. MC4100 transformed with a plasmid encoding an OmpA derivative were subjected to pulse-chase labeling and immunopreciptations were performed with an anti-HA antiserum. Because the targeting of OmpA is the rate-limiting step in the export process (Lee and Bernstein, 2002), the kinetics of targeting could be evaluated by comparing the relative amount of precursor and mature forms of the protein at each time point. Targeting was very rapid and the precursor form of native OmpA was detected primarily in pulse-labeled cells (Fig. 5, top panel, lanes 2–5). A small amount of the SecMSP-OmpA precursor could also be observed, but only the mature form of the cotranslationally targeted MBP*1SP-OmpA protein was seen (Fig. 5, top panel, lanes 6–13). In contrast, the signal peptide of a control protein (OmpC) was cleaved at the same rate in cells that produced each of the OmpA derivatives (Fig. 5, bottom panel). These results indicate that the SecM signal peptide mediates a relatively slow targeting reaction that is clearly distinguishable from conventional cotranslational targeting.
Fig. 5.

The SecM signal peptide specifies intermediate targeting kinetics. MC4100 were transformed with a plasmid (pHL36, pMY10-11) encoding the indicated HA-tagged OmpA derivative. Cells were subjected to pulse-chase labeling after the addition of IPTG and immunoprecipitations were performed using anti-HA and anti-OmpC antisera. p, precursor; m, mature. A separate aliquot of cells transformed with pHL36 was removed and treated with 2 mM sodium azide (NaN3) 2 min prior to labeling to show the position of the OmpC precursor.
The regulatory function of SecM depends on its length
Our analysis of signal peptide mutations and substitutions provided evidence that the timing of membrane targeting is inextricably linked to the regulatory function of SecM. Rapid cotranslational targeting impairs the transient translation arrest that is required for the synthesis of a basal level of SecA. In contrast, slow targeting is ineffective, and by causing the retention of arrested SecM RNCs in the cytoplasm would lead to constitutive SecA overproduction. The data strongly suggested that the native SecMsignal peptide specifies an intermediate rate of targeting and thereby acts as a timer that ensures that the SecYEG complex engages nascent SecM immediately after the synthesis of the arrest motif. This hypothesis predicts that any experimental condition that promotes SecM targeting prior to the synthesis of the arrest motif or that increases the time window between the synthesis of the arrest motif and membrane targeting would duplicate the effect of modifying the signal peptide. To test this prediction, we kept the SecM signal peptide intact, but systematically increased and decreased the length of the mature region to either delay or accelerate the synthesis of the arrest motif.
Consistent with our hypothesis, we found that changes in the length of SecM perturbed the establishment or release of arrest. Pulse-chase experiments in MNY3 showed that translation arrest mediated by SecM(Δ38–127) and SecM(Δ56–127), derivatives that contain 84 and 101 residues, respectively, was extremely stable. None of the full-length or mature forms of the protein was detected after a 5 min chase (Fig. 6, lanes 9–12 and 17–20). We also did not observe release of the arrested form of SecM(Δ38–127) or its translocation into INVs in vitro (Fig. 3C). The introduction of the P166A mutation, however, led to complete secretion and degradation of the protein in the periplasm (Fig. 6, lanes 13–16 and 21–24). Like the addition of a post-translational signal peptide, truncation of the protein appeared to allow ribosomes to reach the arrest motif too far in advance of the targeting reaction. Translation arrest was established and released normally when the length of SecM was increased to 130 residues (Fig. 6, compare lanes 1–4 and 25–28). This observation shows that the function of SecM is compatible with modest changes in its length and suggests that a loss of targeting-competence occurs relatively slowly after synthesis of the arrest motif (see Discussion). In contrast, when the length of SecM was increased to 235 residues by duplicating part of the mature region, we observed a significant defect in translation arrest that mimicked the effect of attaching a cotranslational signal peptide to the protein and targeting it to the IM before the arrest motif was synthesized (Fig. 6, lanes 33–36; compare to Fig. 4B, lanes 17–20). Presumably because the synthesis of the arrest motif was delayed, a 224-residue derivative of OmpASP-SecM that has an extended mature region was targeted to the IM more efficiently than OmpASP-SecM (Fig. S5). Taken together, these results not only suggest that the E. coli SecMsignal peptide calibrates the rate of membrane targeting to the rate of translation, but also help to explain why the signal peptide and the length of the SecM orthologs produced by the Enterobacteriales are coordinately conserved (Fig. 1A).
Fig. 6.

The length of the SecM polypeptide affects the onset and release of translation arrest. MNY3 transformed with a plasmid (pMY15-17; pMY19) that encodes the indicated SecM mutant or the corresponding arrest-deficient derivative (P166A) were subjected to pulse-chase labeling after the addition of IPTG, and full-length (f), arrested (a), and mature (m) forms of SecM were immunoprecipitated. Each derivative contained a three amino acid linker that was fortuitously introduced during plasmid construction.
The stability of SecM-RNCs is calibrated to ensure efficient release of translation arrest upon membrane targeting
In a previous study we used a genetic screen to isolate suppressor mutations that restore the translation arrest activity of the arrest-deficient SecM Q160P mutation (Yap and Bernstein, 2009). While the arrest motif of one suppressor (Sup 1) spans only seven residues (Fig. 7A), it induces translation arrest as effectively as the native E. coli SecM arrest motif. In the same study we showed that a chimeric protein containing the C-terminus of the distantly related Mannheimia succiniciproducens SecM homolog [SecM(C-Ms); Fig. 7A] also undergoes translation arrest in E. coli. Pulse-chase labeling in MNY3, however, indicated that the arrest mediated by Sup1 and SecM(C-Ms) was considerably more stable than that mediated by E. coli SecM and was released more slowly (Fig. 7B, top panel, lanes 2–9 and 14–17). The efficient export of a control protein (OmpA) in all of the cells confirmed that the increased stability of the arrested state was not simply due to a targeting defect that resulted from the synthesis of the SecM variants (Fig. 7B, bottom panel). The data suggested that the force exerted by the Sec machinery is insufficient to release Sup1- and SecM(C-Ms)-mediated translation arrest rapidly, possibly because the arrest motif adopts a relatively stable conformation or interacts more strongly with components of the ribosome tunnel. While the arrest mediated by these SecM isoforms appeared to be especially stable, we found that the arrest mediated by a stalling peptide (WPPPSI) that was isolated in a genetic screen from a random peptide library (Tanner et al., 2009) is relatively weak. Arrest mediated by SecM(WPPPSI) was released before SecM reached the IM in a secY+ strain (Fig. S6, lanes 1–4) and in the absence of a force exerted by the Sec machinery in a secY mutant strain (Fig. S6, lanes 9–12).
Fig. 7.

Cotranslational targeting of a SecM isoform suppresses a defect in the release of arrest. A. The C-terminal 21 residues of native E. coli SecM and two isoforms [Sup1 and SecM(C-Ms)] are shown. The residues that are required for effective translation arrest are shaded. B. MNY3 transformed with a plasmid (pMY1-2; pMY9) encoding the indicated SecM was subjected to pulse-chase labeling after the addition of IPTG and immunoprecipitations were performed using anti-SecM and anti-OmpA antisera. Full-length (f), arrested (a) and mature (m) forms of SecM are indicated. A separate aliquot of cells transformed with pMY1 was removed and treated with 2 mM sodium azide (NaN3) 2 min prior to labeling to show the position of the OmpA precursor.
We surmised that rapid targeting of Sup1 to the IM by SRP might compensate for the increased stability of translation arrest by promoting an earlier interaction between the nascent polypeptide and the Sec complex. Indeed the Sup1 arrest motif resembles that of the Pasteurellale SecM orthologs, which are short and contain distinctive signal peptides that might accelerate targeting (Fig. 1A). Consistent with our hypothesis, we found that normal release of arrest was restored by replacing the native SecMsignal peptide with the MBP*1 signal peptide (Fig. 7B, top panel, lanes 10–13). Taken together, the results suggest that the regulatory function of SecM is compatible with conventional cotranslational targeting, but that there has been an evolutionary selection for both an unusual signal peptide that mediates an intermediate rate of targeting and a moderately strong arrest motif. As discussed below, this combination of features likely enables SecM to specifically monitor the status of post-translational export.
DISCUSSION
In this study we obtained evidence that the SecM signal peptide has a novel function. An initial analysis of wild-type SecM strongly suggested that translation arrest occurs in the cytoplasm, prior to the targeting of the protein to the IM. Subsequently, we found that the deletion of 5–10 residues from the long, conserved N region of the native signal peptide or the replacement of the signal peptide with a typical post-translational signal peptide impaired SecM targeting and thereby prevented the effective release of translation arrest. Because modification of the signal peptide did not impair the targeting or translocation of an arrest-deficient version of SecM, the data implied that the native signal peptide has a specialized function that is specifically required for the targeting of arrested SecM RNCs. Interestingly, we also found that replacement of the SecM signal peptide with a targeting signal that is recognized by SRP at an early stage of translation caused a translation arrest defect (or premature release of arrest) and therefore appeared to have the opposite effect. Taken together, the results suggested that the signal peptide targets SecM RNCs to the IM at an intermediate rate that is calibrated to ensure that translocation is initiated immediately after the synthesis of the arrest motif (Fig. 8). Consistent with the idea that the signal peptide acts as a molecular timer, we found that simply altering the distance between the signal peptide and the arrest motif (which alters the time between synthesis of the arrest motif and membrane targeting) perturbed the normal onset and release of arrest.
Fig. 8.

The SecM signal peptide functions as a molecular timer. In conventional cotranslational targeting (top), SRP binds to the signal peptide as soon as it emerges from a translating ribosome and rapidly guides RNCs to the SecYEG complex. In post-translational targeting (bottom), a presecretory protein is first completely synthesized and released from ribosomes. The protein is then targeted to the SecYEG complex by molecular chaperones such as SecB, which keep it in a translocation-competent conformation. In SecM targeting (middle), the SecM signal peptide specifies an intermediate rate of targeting that ensures that RNCs reach the IM immediately after synthesis of the arrest motif (residues 150–166).
In addition to obtaining insight into the targeting of SecM, we fortuitously obtained insight into the general phenomenon of translation arrest and the targeting of RNCs to the SecYEG complex. We found that the stability of the translation-arrested state conferred by different arrest motifs varies considerably. Consistent with previous results (Yap and Bernstein, 2009), this observation suggests that the conformation of even related arrest motifs inside the ribosome tunnel and/or the number or strength of interactions between the arrest motif and tunnel components can differ. Indeed the ability to regulate the duration of translation arrest may be crucial to the function of other regulatory peptides that have been identified in both bacteria and eukaryotes (Tenson and Ehrenberg, 2002). Perhaps more surprisingly, we also found that translation-arrested SecM RNCs that are located in the cytoplasm gradually lose their ability to interact with the Sec machinery. While this loss of targeting competence may be due in part to ribosome crowding, it may also be due to slow conformational changes in the ribosome that result from translation arrest or from the slow binding of unidentified trans-acting factors. Polysomes formed in the cytoplasm appear to have a distinct organization (Brandt et al., 2009) that might not be compatible with membrane targeting. Because most cotranslational targeting signals have an N terminal location, it is possible that SRP generally targets RNCs to the IM before a polysome can form. In that case the orientation of ribosomes that subsequently form membrane-bound polysomes might be influenced by interactions with the Sec machinery.
Our results indicate that the SecM regulatory switch is subject to far more constraints than previously anticipated. We found that wild-type SecM and a SecM isoform that contains both a typical cotranslational signal peptide and an unusually stable arrest motif are functionally equivalent. Presumably there was a selection against the use of a canonical cotranslational targeting signal because SecM was devised to monitor the status of presecretory proteins that are bypassed by SRP rather than those that are SRP substrates. Indeed it has long been assumed that SecM monitors post-translational translocation, but this idea has never been tested. In principle, the targeting of SecM by a conventional post-translational pathway would allow SecM to sense conditions that reduce the efficiency of post-translational export and to respond by upregulating secA expression. Because conventional post-translational targeting subjects SecM RNCs to a prolonged translation arrest that impairs their interaction with the Sec machinery, however, a unique mechanism that accelerates targeting and reduces the duration of arrest is required. Like canonical cotranslational targeting, this mechanism fortuitously targets secM-secA mRNA to the IM and thereby facilitates the localized synthesis of SecA that appears to maximize its functionality (Nakatogawa et al., 2005). By pegging the rate of targeting to the time required to synthesize the polypeptide segment between the signal peptide and the arrest motif, this targeting mechanism also restricted the length of SecM. Concomitantly, the arrest motif had to be designed to maintain a stable arrested state briefly in the cytoplasm, but to be immediately responsive to the force generated by the Sec machinery. In this regard it seems likely that the stalling of SecM synthesis while P166-tRNA remains bound to the A site (Muto et al., 2006) evolved to prevent the cytoplasmic degradation of the protein by the SsrA pathway. Taken together, all of these considerations strongly suggest that the creation of the SecM regulatory switch required that the signal peptide, arrest motif and length of the protein co-evolve and integrate information about the function of both the translation and translocation machineries.
There are several possible mechanisms by which the long N region of the SecM signal peptide might coordinate membrane targeting with the synthesis of the arrest motif. Because our results suggest that deletions in this region effectively convert the signal peptide into a post-translational targeting signal and thereby slow targeting, this segment might enhance or accelerate targeting or the initiation of translocation. For example, the N region might enable the SecM signal peptide to interact with or gate open the SecYEG complex more rapidly than a typical post-translational signal peptide, possibly by first binding to an unidentified trans-acting factor. Alternatively, the long N region might facilitate SRP binding at a relatively late stage of translation. The observation that N region deletions promote post-translational rather than cotranslational targeting implies that this segment does not inhibit SRP binding, but does not exclude the possibility that the N region facilitates the delayed recognition of a signal peptide that would otherwise be only a marginal SRP substrate. Indeed a delayed interaction between SRP and the SecM signal peptide would provide an explanation for the finding that the translocation of SecM is inhibited by SRP depletion (Nakatogawa and Ito, 2001). Given that SRP forms a high affinity interaction with signal peptides only when it is bound to ribosomal protein L23 (Dalley et al., 2008), however, it is unclear if SRP would bind effectively to a signal peptide that is distant from the tunnel exit site. Furthermore, the effects of SRP depletions should be interpreted with caution because the absence of SRP can indirectly impair the export of specific proteins that are targeted by SRP-independent mechanisms (Beha et al., 2003).
Our results add to the growing evidence that signal peptides have a myriad of specific functions and are not simply generic targeting signals (Hegde and Bernstein, 2006). Although only a few prokaryotic and eukaryotic signal peptides that have long N regions or N terminal extensions that create a bipartite domain organization have been studied to date, in each case the extended region has been shown to harbor a unique co- or post-targeting function. Because the existence of numerous uncharacterized long signal peptides has been predicted by in silico methods (Hiss et al., 2008; Hiss and Schneider, 2009), it seems likely that additional new functions will emerge.
EXPERIMENTAL PROCEDURES
Bacterial strains and plasmids
E. coli strains MC4100 (F− araD139 Δ(argF-lac)205 flb-5301 pstF25 rpsL150 deoC1 relA1), CU164 (MC4100 secY39cs zhd::tet) (Baba et al., 1990), MNY3 (MC4100 Δprc::cat), MNY28 (MC4100 Δprc ΔssrA::cat) and C41 (Miroux and Walker, 1996) were used in all experiments. To construct MNY3, a 1.1 kb DNA fragment encoding the cat gene was amplified by PCR using oligonucleotides P257/P258 (all oligonucleotide sequences are listed in Table S1) and pKD3 (Datsenko and Wanner, 2000) as a template. The DNA fragment was introduced into the MC4100 chromosome by homologous recombination using the Lambda Red method (Datsenko and Wanner, 2000). To generate MNY28, the cat gene was removed from MNY3 by FRT-mediated excision (Datsenko and Wanner, 2000), and the ΔssrA::cat allele from strain SG12084 (Munavar et al, 2005) was introduced by P1 transduction. In plasmids pMY1, which encodes full-length secM, and pHL36, which encodes HA-tagged ompA (Peterson et al, 2006), the presecretory protein is under the control of the trc promoter.
Plasmid construction
To generate plasmid pMY1, the secM gene was first amplified by PCR using the oligonucleotides P390 and P008 and plasmid pSecM (Woolhead et al., 2006) as a template. The PCR product was then digested with Nco I and Xba I and cloned into the cognate sites of pTrc99A (Pharmacia). Finally, a SecM mutation (S2G) that was created in the cloning process was removed using the oligonucleotide pair P515/P516. The QuikChange Mutagenesis Kit (Stratagene) was used to make this change and all other specific mutations.
To construct pMY3, which encodes Δ5SP-SecM, secM was amplified using oligonucleotides P400/P008 and pMY1 as a template and cloned into the Nco I and Xba I sites of pTrc99A. A SecM mutation (R2G) that was created during the cloning process was then removed using oligonucleotides P401/P402. Plasmid pMY4, which encodes Δ10SP-SecM, was made using oligonucleotides P425/P426 and pMY3 as a template. To make plasmid pMY5, a BamH I site was introduced into the secM coding region in pMY1 using oligonucleotides P357/P358. DNA fragments encoding OmpASP and MBPSP were amplified by PCR using the primer pairs P381/P382 and P359/P360, respectively, and MC4100 genomic DNA as a template. The PCR fragment encoding OmpASP was digested with Nco I and BamH I and cloned into the cognate sites of pMY5. An extra valine residue after the initiation codon was eliminated using the oligonucleotides P511/P512 to generate pMY6, which encodes OmpASP-SecM. A V18C mutation was introduced into pMY6 using the oligonucleotides P509/P510 to make pMY28. The PCR fragment encoding MBPSP was digested with Kpn I and BamH I and ligated to the cognate sites of pMY5 to generate a plasmid that encodes MBPSP-SecM (pMY7). The triple mutation S13F/T16M/T17L was introduced into pMY7 using the oligonucleotides P394/P395 to make a plasmid that encodes MBP*1SP-SecM (pMY8). To attach SecMSP to an HA-tagged version of OmpA (pMY10), a DNA fragment encoding SecMSP was first amplified by PCR using the oligonucleotides P361/P362 and pSecM (Woolhead et al., 2006) as a template and then cloned into the Nde I and Eag I sites of pHL36 (Peterson et al., 2006). To make pMY11 (MBP*1SP-OmpA), a fragment encoding MBP*1SP was amplified by PCR using the oligonucleotides P484/P485 and pMY8 as a template and cloned into the Nde I and Eag I sites of pHL36.
Truncated and enlarged versions of SecM were constructed as follows: SecM DNA fragments A, B, C and D were first generated by PCR using primer pairs P037/P458, P459/P008, P037/P488 and P489/P008 and pMY1 as a template. pMY15 [which encodes SecM(Δ38–127)] was constructed by ligating fragments A and B via an EcoR I site. The ligated product was reamplified by PCR using primers P037/P008 and cloned into the Nco I and Xba I sites of pTrc99A. pMY16 and pMY17 [which encode SecM(Δ56–127) and SecM(Δ56–98), respectively] were constructed in a similar way except that fragment pairs B/C and C/D were used. pMY18 was constructed by introducing a BamH I site into the secM coding region in pMY1 using oligonucleotides P460/P461. A DNA fragment encoding SecM residues 82–144 was then excised from pSecM(BamH) (Yap and Bernstein, 2009) using EcoR I and BamH I and cloned into the cognate sites of pMY18 to yield a plasmid that encodes SecM(rep. 82–144) (pMY19). To construct pMY20, which encodes OmpASP-SecM(rep. 82–146), an EcoR I-Xba I fragment derived from pMY19 was cloned into the cognate sites of pMY6. All of the SecM derivatives described above contain three extra amino acids that were generated during the cloning process.
To construct plasmids that encode Sup1 (pMY2) and MBP*1SP-Sup1 (pMY9), the Q160P/G161P mutation was introduced into pMY1 and pMY8, respectively, using the oligonucleotides P043/P044. A plasmid that encodes SecM(C-Ms) (pMY27) was generated by first amplifying a fragment of the M. succiniciproducens SecM homolog by PCR using the oligonucleotides P134/P135 and the ultramer P136 as a template. The PCR product was then cloned into the BamH I and Xba I sites of pSecM(BamH). To make a plasmid encoding a SecM derivative that contains the WPPPSI motif (pMY26), a small fragment encoding the motif was first amplified by PCR using the oligonucleotides P554/P555 and the ultramer P553 as a template. The PCR product was then cloned into the BamH I and Xba I sites of pSecM(BamH).
Pulse-chase and cell fractionation experiments
E. coli harboring the appropriate plasmid were grown at 37°C in M9 medium containing 0.2% glycerol, 100μg/mlampicillin, and all the L-amino acids (40μg/ml) except methionine and cysteine. Overnight cultures were washed and diluted into fresh medium at OD550=0.02. When the cultures reached OD550=0.2, synthesis of plasmid-borne genes was induced by the addition of 200 μM IPTG (SecM) or 50 μM IPTG (OmpA) unless otherwise noted. Following a 30 min incubation pulse-chase labeling (using a 1 min pulse, unless otherwise noted) and CTABr fractionation were conducted as described (Ulbrandt et al., 1997; Nakatogawa and Ito, 2001). Cell fractionation experiments were conducted as described (Peterson et al., 2006). In these experiments a spheroplast lysate (designated as “total”) was centrifuged at 100,000 × g for 30 min to obtain cytoplasmic and membrane fractions. Proteins were collected by TCA precipitation, and immunoprecipitations were conducted using polyclonal rabbit antisera raised against SecM peptides (Nakatogawa and Ito, 2001), a SecY peptide (Arkowitz and Wickner, 1994), an OmpC peptide (CQSKGKNLGRGYDDEDILKYVD), OmpA (a gift from P.C. Tai), SecA (a gift from Don Oliver), MBP (New England BioLabs), or the HA epitope (Santa Cruz Biotechnology). Immunoprecipitated proteins were then resolved by SDS-PAGE.
In vitro protein translocation assays
S-30 extracts were made from strain C41 and INVs were prepared from MC4100 as described (Lesley et al., 1991; Moser et al., 2007). To remove residual membranes and generate S-135 extracts, S-30 extracts were centrifuged in a Beckman TLA100 rotor (285,600 × g, 10 min, 4°C). In vitro coupled transcription-translation reactions (150 μl total volume) were performed at 37°C essentially as described using the S-135 extracts (Woolhead et al., 2006). Reactions were programmed with PCR products that were amplified using oligonucleotides P008 and P037 and an appropriate plasmid encoding OmpA-HA, SecM, or a SecM derivative as a template. After 15 min, one fourth of each reaction (which was designated “-INVs”) was placed into a separate tube and 4 μl of INVs (40 U/ml as measured by A280) was added to the remainder (which was designated “+INVs”). All samples were then incubated for an additional 30 min. Subsequently, the −INVs samples and half of each +INVs sample were subjected to TCA precipitation. The second half of each +INVs sample was incubated with 500 μg/ml proteinase K (PK) at 25°C for 25 min and then subjected to TCA precipitation. Immunoprecipitations were performed with anti-SecM and anti-HA antisera and proteins were resolved by SDS-PAGE.
SDS-PAGE
In experiments that involved an analysis of SecM or its derivatives, proteins were resolved by SDS-PAGE on 14% Tris-glycine gels (15 cm × 15 cm). In all other experiments proteins were resolved on 8–16% Tris-glycine minigels (Invitrogen). Radiolabeled proteins were visualized using a Fuji BAS-2500 phosphorimager.
Supplementary Material
Acknowledgments
We thank Yihong Ye for critical reading of the manuscript. We also thank Wanyoike Kang’ethe, Don Oliver and P.C. Tai for providing antisera. This work was supported by the Intramural Research Program of the National Institute of Diabetes and Digestive and Kidney Diseases and by an NIH K99/R00 award (GM094212) to M.-N.Y.
References
- Arkowitz RA, Wickner W. SecD and SecF are required for the proton electrochemical gradient stimulation of preprotein translocation. EMBO J. 1994;13:954–963. doi: 10.1002/j.1460-2075.1994.tb06340.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baba T, Jacq A, Brickman E, Beckwith J, Taura T, Ueguchi C, Akiyama Y, Ito K. Characterization of cold-sensitive secY mutants of Escherichia coli. J Bacteriol. 1990;172:7005–7010. doi: 10.1128/jb.172.12.7005-7010.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beha D, Deitermann S, Müller M, Koch HG. Export of β-lactamase is independent of the signal recognition particle. J Biol Chem. 2003;278:22161–22167. doi: 10.1074/jbc.M300929200. [DOI] [PubMed] [Google Scholar]
- Bendtsen JD, Nielsen H, von Heijne G, Brunak S. Improved prediction of signal peptides: SignalP 3.0. J Mol Biol. 2004;340:783–795. doi: 10.1016/j.jmb.2004.05.028. [DOI] [PubMed] [Google Scholar]
- Brandt F, Etchells SA, Ortiz JO, Elcock AH, Hartl FU, Baumeister W. The native 3D organization of bacterial polysomes. Cell. 2009;136:261–271. doi: 10.1016/j.cell.2008.11.016. [DOI] [PubMed] [Google Scholar]
- Butkus ME, Prundeanu LB, Oliver DB. Translocon “pulling” of nascent SecM controls the duration of its translational pause and secretion-responsive secA regulation. J Bacteriol. 2003;185:6719–6722. doi: 10.1128/JB.185.22.6719-6722.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlsson F, Stalhammar-Carlemalm M, Flardh K, Sandin C, Carlemalm E, Lindahl G. Signal sequence directs localized secretion of bacterial surface proteins. Nature. 2006;442:943–946. doi: 10.1038/nature05021. [DOI] [PubMed] [Google Scholar]
- Cross BC, Sinning I, Luirink J, High S. Delivering proteins for export from the cytosol. Nat Rev Mol Cell Biol. 2009;10:255–26. doi: 10.1038/nrm2657. [DOI] [PubMed] [Google Scholar]
- Dalley JA, Selkirk A, Pool MR. Access to ribosomal protein Rpl25p by the signal recognition particle is required for efficient cotranslational translocation. Mol Biol Cell. 2008;19:2876–2884. doi: 10.1091/mbc.E07-10-1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datsenko KA, Wanner BL. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci U S A. 2000;97:6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Driessen AJ, Nouwen N. Protein translocation across the bacterial cytoplasmic membrane. Annu Rev Biochem. 2008;77:643–667. doi: 10.1146/annurev.biochem.77.061606.160747. [DOI] [PubMed] [Google Scholar]
- Garza-Sanchez F, Janssen BD, Hayes CS. Prolyl-tRNA(Pro) in the A-site of SecM-arrested ribosomes inhibits the recruitment of transfer-messenger RNA. J Biol Chem. 2006;281:34258–34268. doi: 10.1074/jbc.M608052200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegde RS, Bernstein HD. The surprising complexity of signal sequences. Trends Biochem Sci. 2006;31:563–571. doi: 10.1016/j.tibs.2006.08.004. [DOI] [PubMed] [Google Scholar]
- Hiss JA, Resch E, Schreiner A, Meissner M, Starzinski-Powitz A, Schneider G. Domain organization of long signal peptides of single-pass integral membrane proteins reveals multiple functional capacity. PLoS One. 2008;3:e2767. doi: 10.1371/journal.pone.0002767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiss JA, Schneider G. Architecture, function and prediction of long signal peptides. Brief Bioinform. 2009;10:569–578. doi: 10.1093/bib/bbp030. [DOI] [PubMed] [Google Scholar]
- Huber D, Boyd D, Xia Y, Olma MH, Gerstein M, Beckwith J. Use of thioredoxin as a reporter to identify a subset of Escherichia coli signal sequences that promote signal recognition particle-dependent translocation. J Bacteriol. 2005;187:2983–2991. doi: 10.1128/JB.187.9.2983-2991.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keenan RJ, Freymann DM, Stroud RM, Walter P. The signal recognition particle. Annu Rev Biochem. 2001;70:755–775. doi: 10.1146/annurev.biochem.70.1.755. [DOI] [PubMed] [Google Scholar]
- Lee C, Beckwith J. Cotranslational and posttranslational protein translocation in prokaryotic systems. Annu Rev Cell Biol. 1986;2:315–336. doi: 10.1146/annurev.cb.02.110186.001531. [DOI] [PubMed] [Google Scholar]
- Lee HC, Bernstein HD. The targeting pathway of Escherichia coli presecretory and integral membrane proteins is specified by the hydrophobicity of the targeting signal. Proc Natl Acad Sci U S A. 2001;98:3471–3476. doi: 10.1073/pnas.051484198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HC, Bernstein HD. Trigger factor retards protein export in Escherichia coli. J Biol Chem. 2002;277:43527–43535. doi: 10.1074/jbc.M205950200. [DOI] [PubMed] [Google Scholar]
- Lesley SA, Brow MD, Burgess RR. Use of in vitro protein synthesis from polymerase chain reaction-generated templates to study interaction of Escherichia coli transcription factors with core RNA polymerase and for epitope mapping of monoclonal antibodies. J Biol Chem. 1991;266:2632–2638. [PubMed] [Google Scholar]
- Miroux B, Walker JE. Over-production of proteins in Escherichia coli: mutant hosts that allow synthesis of some membrane proteins and globular proteins at high levels. J Mol Biol. 1996;260:289–298. doi: 10.1006/jmbi.1996.0399. [DOI] [PubMed] [Google Scholar]
- Moser M, Panahandeh S, Holzapfel E, Müller M. In vitro analysis of the bacterial twin-arginine-dependent protein export. Methods Mol Biol. 2007;390:63–79. doi: 10.1007/978-1-59745-466-7_5. [DOI] [PubMed] [Google Scholar]
- Munavar H, Zhou Y, Gottesman S. Analysis of the Escherichia coli Alp phenotype: heat shock induction in ssrA mutants. J Bacteriol. 2005;187:4739–4751. doi: 10.1128/JB.187.14.4739-4751.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami A, Nakatogawa H, Ito K. Translation arrest of SecM is essential for the basal and regulated expression of SecA. Proc Natl Acad Sci U S A. 2004;101:12330–12335. doi: 10.1073/pnas.0404907101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muto H, Nakatogawa H, Ito K. Genetically encoded but nonpolypeptide prolyl-tRNA functions in the A site for SecM-mediated ribosomal stall. Mol Cell. 2006;22:545–552. doi: 10.1016/j.molcel.2006.03.033. [DOI] [PubMed] [Google Scholar]
- Nakatogawa H, Ito K. Secretion monitor, SecM, undergoes self-translation arrest in the cytosol. Mol, Cell. 2001;7:185–192. doi: 10.1016/s1097-2765(01)00166-6. [DOI] [PubMed] [Google Scholar]
- Nakatogawa H, Ito K. The ribosomal exit tunnel functions as a discriminating gate. Cell. 2002;108:629–636. doi: 10.1016/s0092-8674(02)00649-9. [DOI] [PubMed] [Google Scholar]
- Nakatogawa H, Murakami A, Mori H, Ito K. SecM facilitates translocase function of SecA by localizing its biosynthesis. Genes Dev. 2005;19:436–444. doi: 10.1101/gad.1259505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann-Haefelin C, Schafer U, Müller M, Koch HG. SRP-dependent co-translational targeting and SecA-dependent translocation analyzed as individual steps in the export of a bacterial protein. EMBO J. 2000;19:6419–6426. doi: 10.1093/emboj/19.23.6419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliver D, Norman J, Sarker S. Regulation of Escherichia coli secA by cellular protein secretion proficiency requires an intact gene X signal sequence and an active translocon. J Bacteriol. 1998;180:5240–5242. doi: 10.1128/jb.180.19.5240-5242.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson JH, Woolhead CA, Bernstein HD. Basic amino acids in a distinct subset of signal peptides promote interaction with the signal recognition particle. J Biol Chem. 2003;278:46155–46162. doi: 10.1074/jbc.M309082200. [DOI] [PubMed] [Google Scholar]
- Peterson JH, Szabady RL, Bernstein HD. An unusual signal peptide extension inhibits the binding of bacterial presecretory proteins to the signal recognition particle, trigger factor, and the SecYEG complex. J Biol Chem. 2006;281:9038–9048. doi: 10.1074/jbc.M508681200. [DOI] [PubMed] [Google Scholar]
- Randall LL. Translocation of domains of nascent periplasmic proteins across the cytoplasmic membrane is independent of elongation. Cell. 1983;33:231–240. doi: 10.1016/0092-8674(83)90352-5. [DOI] [PubMed] [Google Scholar]
- Sarker S, Oliver D. Critical regions of secM that control its translation and secretion and promote secretion-specific secA regulation. J Bacteriol. 2002;184:2360–2369. doi: 10.1128/JB.184.9.2360-2369.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiebel E, Driessen AJM, Hartl FU, Wickner W. DmH+ and ATP function at different steps of the catalytic cycle of preprotein translocase. Cell. 1991;64:927–939. doi: 10.1016/0092-8674(91)90317-r. [DOI] [PubMed] [Google Scholar]
- Schierle CF, Berkmen M, Huber D, Kumamoto C, Boyd D, Beckwith J. The DsbA signal sequence directs efficient, cotranslational export of passenger proteins to the Escherichia coli periplasm via the signal recognition particle pathway. J Bacteriol. 2003;185:5706–5713. doi: 10.1128/JB.185.19.5706-5713.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steitz JA. Polypeptide chain initiation: nucleotide sequences of the three ribosomal binding sites in bacteriophage R17 RNA. Nature. 1969;224:957–964. doi: 10.1038/224957a0. [DOI] [PubMed] [Google Scholar]
- Szabady RL, Peterson JH, Skillman KM, Bernstein HD. An unusual signal peptide facilitates late steps in the biogenesis of a bacterial autotransporter. Proc Natl Acad Sci U S A. 2005;102:221–226. doi: 10.1073/pnas.0406055102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanner DR, Cariello DA, Woolstenhulme CJ, Broadbent MA, Buskirk AR. Genetic identification of nascent peptides that induce ribosome stalling. J Biol Chem. 2009;284:34809–34818. doi: 10.1074/jbc.M109.039040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tenson T, Ehrenberg M. Regulatory nascent peptides in the ribosomal tunnel. Cell. 2002;108:591–594. doi: 10.1016/s0092-8674(02)00669-4. [DOI] [PubMed] [Google Scholar]
- Ulbrandt ND, Newitt JA, Bernstein HD. The E. coli signal recognition particle is required for the insertion of a subset of inner membrane proteins. Cell. 1997;88:187–196. doi: 10.1016/s0092-8674(00)81839-5. [DOI] [PubMed] [Google Scholar]
- Underwood KA, Swartz JR, Puglisi JD. Quantitative polysome analysis identifies limitations in bacterial cell-free protein synthesis. Biotechnol Bioeng. 2005;91:425–435. doi: 10.1002/bit.20529. [DOI] [PubMed] [Google Scholar]
- van Stelten J, Silva F, Belin D, Silhavy TJ. Effects of antibiotics and a proto-oncogene homolog on destruction of protein translocator SecY. Science. 2009;325:753–756. doi: 10.1126/science.1172221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Heijne G. Signal sequences. The limits of variation. J Mol Biol. 1985;184:99–105. doi: 10.1016/0022-2836(85)90046-4. [DOI] [PubMed] [Google Scholar]
- Woolhead CA, Johnson AE, Bernstein HD. Translation arrest requires two-way communication between a nascent polypeptide and the ribosome. Mol Cell. 2006;22:587–598. doi: 10.1016/j.molcel.2006.05.021. [DOI] [PubMed] [Google Scholar]
- Yap MN, Bernstein HD. The plasticity of a translation arrest motif yields insights into nascent polypeptide recognition inside the ribosome tunnel. Mol Cell. 2009;34:201–211. doi: 10.1016/j.molcel.2009.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmer J, Nam Y, Rapoport TA. Structure of a complex of the ATPase SecA and the protein-translocation channel. Nature. 2008;455:936–943. doi: 10.1038/nature07335. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.