

Preface
Osteoporosis, a syndrome characterized by thin bones and fractures, has become more prevalent in both women and men. Established therapies for this disorder consist primarily of drugs that prevent bone loss, such as the bisphosphonates and selective estrogen receptor modulators. Although these drugs have been shown to reduce fractures in randomized trials, there is an urgent need for treatments that could lower fracture risk further without additional adverse effects. The introduction of parathyroid hormone (teriparatide), which significantly increases bone mineral density, albeit for a relatively short duration, raised expectations that drugs which stimulate bone formation might cure osteoporosis. After outlining current approaches to treating osteoporosis, this review focuses on emerging therapeutic opportunities for osteoporosis that are based on recent insights into skeletal physiology. Such novel strategies offer promise for not only reducing age-related bone loss and the associated risk of fractures, but restoring bone mineral density to healthy levels.
Introduction
Osteoporosis affects almost 44 millions of men and women aged 50 and older in the US, and evidence indicates its prevalence is rising, particularly as life expectancy continues to increase (http://www.nof.org/). Similar statistics have been noted in Europe, Asia and Australia. Osteoporosis is manifested by fractures and enhanced skeletal fragility due to micro-architectural changes in the trabecular and cortical skeleton. Osteoporotic fractures cause significant morbidity and in many individuals leads to loss of productivity and reduced quality of life. The mortality rate in the first year after a hip fracture is 20–30% and more than half of those individuals sustaining a hip fracture will not return to their previous lifestyle1. Primarily as a result of these fractures, health care costs in the U.S. alone from osteoporosis exceed 15 billion dollars annually.2
There is a long latency period until the clinical onset of osteoporosis, and during this time there are profound alterations in bone remodeling, the basic homeostatic process that controls calcium balance and skeletal integrity 3. Imbalances in the ‘bone remodeling unit’, often due to accelerated bone turnover, are caused by increased bone resorption or reduced bone formation relative to resorption. The end result is a net loss of bone mass and qualitative changes in skeletal architecture, both of which greatly increase an individual’s risk of fracture.
Until recently, treatment options for osteoporosis were limited to agents that prevented bone loss and partially reduced fracture risk by suppressing bone resorption, thereby recalibrating the remodeling unit to a new steady state in which bone turnover slowed and bone mineral density was maintained. Although the number of approved therapies for osteoporosis has more than doubled in the last 15 years, the level of fracture-risk reduction has not improved dramatically. Indeed, the currently available treatments for osteoporosis, assuming optimal compliance, reduce the risk of non-vertebral fractures by at best only 30–40% 3,4,5,6.
Newer pharmacological agents offer possibilities for building on earlier treatment successes through actions on bone formation. This has led some to speculate that these drugs can restore old skeletons to more youthful states. Such guarded optimism is due in part to a greater understanding of the molecular and cellular events that occur during acquisition of peak bone mass in adolescence and bone remodeling in adults. Emerging from these findings is a new therapeutic paradigm in osteoporosis medicine; i.e. a targeted molecular approach to enhance the inherent capacity of the skeleton to augment bone mass regardless of age.
In this review, we will outline the physiology of the skeleton and discuss recent discoveries that provide new insight into bone remodeling and its relationship to energy and metabolic status. We will briefly overview the evidence supporting current therapeutic approaches to osteoporosis, but then focus on pharmacological targeting of specific pathways that offer new possibilities for intervention, especially those that could stimulate bone formation. Finally, we will discuss the potential commercial and regulatory challenges that must be overcome before final approval and marketing of these agents
Overview of skeletal physiology
Trauma is the principal cause of fractures at any stage of life, but factors that alter the quality or quantity of bone mass further predispose a susceptible individual to fractures. The bone remodeling sequence is a critical determinant of both the amount and type of bone that is laid down across the lifespan. Remodeling is essential for preserving serum calcium and maintaining bone strength during adulthood. This cyclic process occurs within microscopic elements called remodeling or basic multicellular units (BMU)5. Remodeling begins with bone dissolution or resorption and ends with bone formation, although the precise sequence of initiation is still not fully understood.
Bone resorption is carried out by osteoclasts, which originate from the hematopoietic monocyte-macrophage lineage. New bone is formed by osteoblasts, cells of the fibroblast-stromal lineage that produce bone matrix proteins and synthesize a lattice for subsequent mineralization. Stromal cells are found in the marrow and are destined to become either fat, bone or cartilage under the influence of differentiation factors within the marrow milieu. Osteocytes are osteoblasts that have completed their task of bone formation and are entombed within the bone matrix. Although these cells were originally considered inert, it is now clear that they communicate with resting surface osteoblasts (bone lining cells) and, under the proper circumstances (e.g. micro cracks, gravitary forces, fluid shifts), initiate bone remodeling. Micro canaliculi connect the osteoblast to the osteocyte and likely serve as the conduit for signals that originate from osteocytes in response to gravitational forces5. Remodeling begins when osteocytes are activated by local factors (as noted in Figure 1) or systemic modulators such as PTH, interleukins, estrogen withdrawal, or hormones. Marrow stromal cells synthesize and release two cytokines, mCSF and RANKL, which enhance the recruitment and differentiation of osteoclasts. These cells cause bone resorption and in the process release growth factors such as TGF-β from the matrix to recruit more osteoblasts5a The outcome of these events is resorption followed by formation in a coupled remodeling sequence.
Figure 1. Regulation of osteoclast development by RANKL and other cytokines in the bone marrow microenvironment.
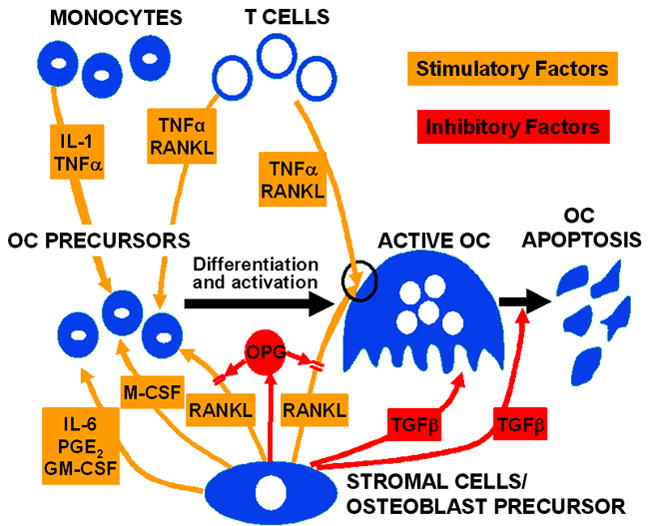
Modified from Riggs et al. with permission 22. Remodeling begins with the elaboration of cytokines from osteoblast precursors leading to the recruitment and differentiation of multinucleated osteoclasts that subsequently attach to the endosteal bone surface. Resorption of the skeletal matrix releases growth factors such as IGF-I and TFG-beta which recruit lining cells and early osteoblast precursors that eventually form new bone. Osteoblasts that do not undergo programmed cell death are entombed within the skeletal matrix and become osteocytes that can signal lining cells and respond to gravitary forces to initiate remodeling.] <Added on page 5>
In adulthood, each remodeling unit is balanced—resorption equals formation—and the process lasts between 90 and 130 days. Maintenance of bone density during remodeling ensures a ready source of calcium for the body and a persistent reservoir of stored calcium. Prior to adult remodeling, acquisition of peak bone mass occurs, usually between 12 and 15 years of age. During this time, there is a tremendous increase in bone mass as a result of growth and modeling of the skeleton.. Pubertal surges of gonadal steroids and growth hormone are considered critical for the increase in bone mass during adolescence. Longitudinal studies suggest that several factors regulate peak bone density, including dietary intake of specific nutrients, physical activity, and, most importantly, genetic determinants. This view was established in both human and mouse models. However, despite intense efforts over the past decade to identify heritable determinants of bone mass, progress has been slow in part because it has been recognized that polymorphic differences in many genes, including the vitamin D and estrogen receptors, parathyroid hormone (PTH), collagen A1a, receptor activator of NF-κB ligand (RANKL) and others, all contribute to skeletal acquisition.
Bone loss occurs after peak acquisition when resorption exceeds formation. During the early menopause estrogen deficiency contributes to rapid skeletal loss. In addition, with advanced age, bone formation is often compromised by multiple factors including further loss of gonadal steroids, reduced vitamin D production and action, increased PTH secretion, and local generation of cytokines and growth factors that are inhibitory to osteoblasts. Less work has focused on the genetics of bone loss, although this is likely to be the major pathogenic process in the development of osteoporosis.
Current therapies for osteoporosis
Current therapies for osteoporosis target adult bone loss and are centered on restoring the balance between formation and resorption. Estrogens, selective estrogen receptor modulators (SERMs), bisphosphonates, calcitonin and denosumab are all anti-resorptives, although their sites of action may differ. Only one anabolic agent for the treatment of osteoporosis has been approved by American regulatory agencies: teriparatide, a recombinant form of the human parathyroid hormone. European agencies have also approved the use of PTH (1–84) for postmenopausal osteoporosis. Below, we provide a brief overview of the these therapies, and a summary is also provided in Tables 1,2.
Table 1.
Mechanism of action of current therapies for osteoporosis
| Drug class | Examples | Mechanism of action | Advantages | Disadvantages | Refs |
|---|---|---|---|---|---|
| Anti-resorptive agents | |||||
| Oestrogen | Inhibit cytokine release | Hip fracture risk reduction | Non skeletal adverse events | 8,9 | |
| SERMs | Raloxifene | Inhibit cytokine release | Breast cancer risk reduction | Hot flashes | 12,16 |
| Bisphosphonates | Alendronate, risedronate, ibandronate, zolendronic acid | Inhibits farnesyl pyrophosphate synthetase | Hip and spine fracture reduction | Long term risk unknown but rare ONJ and sub-troch fractures possible | 18,21,22 |
| Antibodies | Denosumab | Inhibits RANKL | Significant fracture risk reduction | Long term safety not known | 29,30 |
| Anabolic agents | |||||
| PTH | Teriparatide | Recombinant PTH | Fracture risk reduction | Compliance, cost | 40,41 |
Table 2.
Clinical aspects of current therapies for osteoporosis. All these agents show vertebral fracture risk reduction.
| Drug | Examples | Dosage regimen | Increase in BMD (%) | Fracture risk reduction-non vertebral | Refs |
|---|---|---|---|---|---|
| Anti-resorptive agents | |||||
| Oestrogen | Conjugated equine estrogen | 0.625 mg | 3–4%/yr | 20–30% | 7,8,9 |
| SERMs | Raloxifene | 60 mg | 2%/yr | 10% not significant | 11,12,13,16 |
| Bisphosphonates | Alendronate, risedronate, ibandronate, zolendronic acid | 70 once wk 35 once wk 150 once mo 5mg IV/yr |
4%/yr 3%/yr 3%/yr 4%/yr |
20–30% non vert for all except ibandronate | 18,20 |
| Antibodies | Denosumab | 60 mg SC/6mos | 5% | 20 non vert | 32 |
| Anabolic agents | |||||
| PTH | Teriparatide | 20 ug SC/day | 6–8%/yr | 20–40% non vert | 40–42 |
Estrogen
Estrogen was extensively used for the prevention and treatment of osteoporosis prior to the development of bisphosphonates, because it was effective in improving bone mineral density (BMD). However, the most definitive data for the anti-fracture efficacy of estrogen only came in 2002, published from the Women’s Health Initiative (WHI) 7. In that study, combined therapy (0.625 mg/d conjugated equine estrogen, plus medroxyprogesterone acetate, 2.5 mg/d) was associated with a significant reduction in hip fractures as well as all osteoporotic fractures7. Ironically, although the WHI was the most definitive trial on the anti-fracture efficacy of estrogen therapy, this study also led to a marked reduction in the use of estrogen for fracture risk reduction due to the increased risk of cardiovascular events and breast cancer7,8.
Given the risks associated with conventional doses of estrogen (0.625 mg/d of equine conjugated estrogens [equivalent to approximately 1.0 mg/d of oral micronized 17β-estradiol or 0.05 mg/d of transdermal 17β-estradiol]), a number of studies have examined the use of very low dose estrogen in women at risk for fractures. Doses of 0.25 mg/d of oral micronized 17β-estradiol or unopposed transdermal estradiol, 0.014 mg/d increased BMD at multiple sites and reduced markers of bone resorption 9,10. Importantly, for doses of 0.25 mg/d of 17β-estradiol6, the adverse effect profile was similar in the estrogen and placebo groups, with no statistically significant differences in breast tenderness, abnormal mammograms, or changes in endometrial thickness. Thus low dose estrogen therapy, which keeps serum estradiol levels well below the premenopausal range, is effective at improving BMD. However, it remains to be seen whether large scale studies examining fracture and safety endpoints (breast cancer, cardiovascular events, others) will ever be initiated using this approach, given the growing list of alternative agents to prevent and treat osteoporosis.
Selective Estrogen Receptor Modulators (SERMs)
There has long been interest in identifying compounds with beneficial skeletal effects, but without the adverse breast, endometrial, and cardiovascular effects associated with estrogen treatment. SERMs are non-steroidal compounds with tissue-specific actions, which are believed to be due to the fact that these drugs induce a different conformation of the ER than estradiol 11. Raloxifene at a dose of 60 mg/d is the only SERM currently approved by the US FDA for the treatment of postmenopausal osteoporosis. It has been shown to reduce vertebral fracture risk by 30–50% relative to placebo in postmenopausal women with osteoporosis, but has not been shown to protect against non-vertebral or hip fractures12. Raloxifene also reduces the risk of developing breast cancer 13, is neutral with regards to cardiovascular events, but does lead to an increase in the risk of venous thromboembolic disease, fatal stroke, and hot flushes14.
Several additional SERMs (idoxifene, levormeloxifene, arzoxifene) have been studied but their clinical development abandoned due either to lack of efficacy for reducing non-vertebral fractures or to increases in endometrial thickness. More recently, a large, randomized placebo-controlled trial with lasofoxifene demonstrated that this drug significantly reduced the risk of vertebral as well as non-vertebral fractures, ER-positive breast cancer, coronary heart disease, and stroke15. However, lasofoxifene was also associated with an increased risk of venous thromboembolic events and hot flushes. In addition, one of the two doses tested (0.25 mg/d) resulted in a 37% increase in all cause mortality; thus, the future status of this drug remains uncertain.
Another SERM, basedoxifene, has been tested in a somewhat novel approach in combination with conjugated equine estrogens. The rationale was that such a combination would improve BMD and reduce hot flushes, but without some of the other adverse effects on the endometrium and breast associated with estrogen therapy alone. In a study of 3997 postmenopausal women, Lindsay et al. 16 found that the combination of basedoxifene and estrogen increased BMD at multiple sites over two years, reduced the number of hot flushes, and improved measures of vaginal atrophy, lipid parameters, homocysteine levels, with a similar incidence of breast pain and other adverse events compared to placebo 17. However, in the three year fracture trial, new vertebral fractures were reduced with basedoxifene, but non-vertebral fractures were not relative.
Bisphosphonates
Bisphosphonates are currently the most widely used drugs for the prevention and treatment of osteoporosis due to their potent effects in inhibiting bone resorption, ease of frequency of administration, and lack of serious long term non-skeletal effects. Structurally, they are chemically stable derivatives of inorganic pyrophosphate (PPi) and like PPi, bisphosphonates have a very high affinity for bone mineral because they bind to hydroxyapatite crystals 18. These drugs are potent inhibitors of bone resorption, and they do so by binding to and inhibiting the activity of farnesyl pyrophosphate synthetase, a key regulatory enzyme in the mevalonic acid pathway critical to the production of cholesterol, other sterols, and isoprenoid lipids 19. As a result, the isoprenylation of proteins (including the small guanosine triphosphate-binding proteins Rab, Rac, and Rho, which regulate key osteoclast cellular activities such as stress fiber assembly, membrane ruffling, and survival) is inhibited, resulting in varying degrees of apoptosis of osteoclasts 20.
There are currently four bisphosphonates approved in the US for the prevention and/or treatment of osteoporosis: alendronate, risedronate, ibandronate, and zolendronic acid 18. Alendronate, risedronate, and ibandronate are administered orally (daily, weekly, or monthly), whereas ibandronate can also be administered intravenously every 3 months and zolendronic acid (5 mg) is administered intravenously either annually (for treatment) or every other year (for prevention). All of these drugs are potent inhibitors of bone resorption and reduce the risk of vertebral fractures by 40–70% and hip fractures by 40–50%. Overall non-vertebral fractures (i.e. fractures that do not include spine or hip) are suppressed by only 20–40%18 with the use of alendronate, risedronate or zoledronate. Ibandronate has not been shown to have non-vertebral fracture efficacy. In respect to cost, yearly zoledronic acid is much more expensive than generic alendronate whereas risedronate and ibandronate are intermediate in price.
While the bisphosphonates are extremely useful agents for reducing fracture risk, concerns have been raised regarding their long term safety. Osteonecrosis of the jaw, a painful condition with exposed bone in the oral cavity, has received perhaps the greatest attention 21. The risk of this complication appears to be greatest in patients with cancer receiving high monthly doses of zolendronic acid or pamidronate, but is in the range of 1 in 10,000 to 1 in 100,000 per patient treatment-years in patients receiving doses of bisphosphonates for the treatment of osteoporosis. More recently, a specific type of femoral fracture (subtrochanteric) has been associated with bisphosphonate use 22. The risk of this complication remains uncertain, 23, and further studies are needed to define its true incidence as well as the pathogenesis of both osteonecrosis of the jaw and sub-trochanteric femoral fractures. Notwithstanding the rare occurrence of these complications, significant concerns have been raised about long term treatment with the bisphosphonates. This in turn has led to a greater urgency for considering alternative approaches to osteoporosis therapy.
Denosumab
Osteoclast maturation is dependent on on receptor activator of NF-κB ligand (RANKL) 24,25, which is expressed on the surface of bone marrow stromal/osteoblast precursor cells, T-cells, as well as B-cells 26 (Figure 1). RANKL binds its cognate receptor, RANK, on osteoclast lineage cells, and is neutralized by the soluble, decoy receptor, osteoprotegerin (OPG), which is also produced by osteoblastic lineage cells 27,28. Denosumab is a fully human monoclonal antibody that binds to RANKL, thereby reducing osteoclast differentiation, activity and survival, and leading to a decrease in bone resorption. Multiple trials using denosumab have been done in humans, the largest being the phase III FREEDOM trial 29, in which treatment with denosumab resulted in a relative decrease in vertebral fractures of 68%, in hip fractures of 40%, and in non-vertebral fractures of 20% compared with placebo.
Since oral bisphosphonates are currently the first-line therapy for osteoporosis, Brown et al. compared the efficacy and safety of denosumab versus oral alendronate in the DECIDE study 30. Postmenopausal women receiving denosumab treatment had significantly greater increases in BMD at all skeletal sites assessed (total hip, lumbar spine, femoral neck, trochanter and the distal one-third radius). Decreases in levels of bone-turnover markers were also significantly greater in denosumab- versus alendronate-treated patients. Analysis by high-resolution peripheral quantitative CT (HR-pQCT) demonstrated that while alendronate reduced bone loss, denosumab partially restored cortical bone density 31. These findings suggest that denosumab may reduce cortical porosity by allowing filling or partial filling of cortical pores. A subsequent study, the STAND trial, included postmenopausal women that were previously treated with alendronate and switched to denosumab 32. In subjects transitioning to denosumab, total hip BMD increased by 1.90% compared to a 1.05% increase in subjects continuing on alendronate. Serum CTX levels decreased further in the subjects transitioning to denosumab versus continuous alendronate treatment. In sum denosumab suppresses bone resorption to a greater degree than the bisphosphonates, thereby leading to a bigger increase in bone mineral density.
Based on all the available data, the FDA recently approved denosumab (Prolia, Amgen) for the treatment of postmenopausal women who have a high risk of osteoporotic fractures, including those with a history of fracture or multiple risk factors for fracture, or those who have failed or are intolerant to other osteoporosis therapies. Like bisphosphonates 18, denosumab has also been used to reduce pain and other skeletal events in patients with metastatic cancer 33. Of interest, a recent study by Thomas et al. found that high, frequent doses of denosumab (120 mg monthly) resulted in near complete or complete elimination of giant-cell tumors as evaluated by histology, suggesting that at least for this tumor type, denosumab may have direct anti-tumor effects34. Recently Denosumab gained approval from the U.S. FDA for the prevention of skeletal related events in patients with bone metastases from solid tumors.
As noted above, the use of bisphosphonates, particularly in cancer patients administered high doses, has been associated with the development of osteonecrosis of the jaw 21. Similarly, several cases of osteonecrosis of the jaw have now also been reported with denosumab use 35. Perhaps the major concern about long-term denosumab use relates to its possible effects on the skin and immune system, since TRAIL, (TNF-related apoptosis-inducing ligand) is suppressed by denosumab and is produced by not just on osteoblasts but also immune cells 24. In the large denosumab clinical trial, compared to placebo, the active treatment group had statistically significant increases in the rates of eczema and hospitalizations for cellulitus 29. In addition, it has been shown that patients treated with denosumab have a slightly greater risk of recurrent neoplasms 36, which, although not statistically significant, supports ongoing surveillance of such patients, particularly when used in the community setting in patients with co-morbidities that might have excluded them from participating in clinical trials.
Adherence of patients to prescribed drugs and the cost of treatment are important considerations in the selection of treatment options and response to therapy. In a recent study, 77% of patients preferred denosumab injections every 6 months over weekly oral alendronate 37. The annual cost for the use of denosumab (two injections per year) is approximately $1650, which is very comparable to the cost for the annual infusion of the bisphosphonate, zolendronic acid but much less than generic alendronate.
Strontium ranelate
Strontium ranelate is a chemical compound originally developed as an anabolic agent for the treatment of postmenopausal osteoporosis. Strontium is taken up by bone, primarily through a physical mechanism, and associated with limited calcium exchange in hydroxyapatite. Although its mechanism of action is not fully elucidated it has both anti-resorptive and anabolic properties. Strontium ranelate (2 grams/daily) was found to reduce vertebral fractures by 40% in a phase III trial and non-vertebral fractures by 16% in a second large randomized study It is well tolerated with few adverse events 37a,b. It is approved in Europe and Asia but not in the United States for the treatment of postmenopausal osteoporosis.
Teriparatide and PTH (1–84)
A new class of anti-osteoporosis drugs was introduced in 2002 with the approval of Forteo (teriparatide, a recombinant version of human PTH1–34) for the treatment of postmenopausal osteoporosis. PTH is a naturally occurring hormone secreted by the parathyroid glands in response to low circulating calcium levels. It acts directly on bone to stimulate resorption and preserve serum calcium, but also indirectly influence calcium metabolism by acting on the kidney to enhance the activity of the 1 alpha hydroxylase enzyme that converts 25OH vitamin D to 1,25 dihydroxyvitamin D, thereby enhancing absorption of calcium and phosphate from the intestine. Thus, PTH has both direct and indirect activity on calcium metabolism (i.e. direct on bone to mobilize calcium; indirect through the kidney to promote calcium reabsorption and 1, alpha hydroxylase activity, and through the gut to promote calcium absorption via 1,25 dihydroxyvitamin D) 38.
However, the mechanism of the anabolic action of PTH has been debated for nearly as long as the first animal studies were completed with PTH in the 1930s. The ultimate paradox of PTH relates to the longstanding observation that primary and secondary hyperparathyroidism is associated with cortical bone loss and fractures, whereas intermittent PTH administration enhances trabecular bone mass and reduces fractures with very modest effects on cortical bone. PTH targets lining cells and osteoblasts generating a number of growth factors including IGF-I and RANKL 38. RANKL is a critical modulator of osteoclastogenesis and is almost certainly responsible for the coupling of resorption with formation during PTH therapy. Recent studies have shown that PTH also targets osteocytes and inhibits sclerostin thereby further promoting bone formation through the Wnt/β-catenin signaling pathway 39.
In clinical studies, intermittent subcutaneous injections of PTH not only increased bone mineral density but also reduce fractures of the spine and non-vertebral bones. In the largest randomized, placebo-controlled clinical trial using teriparatide in postmenopausal women with severe osteoporosis, 20 μg/day of PTH, administered subcutaneously, reduced spinal and nonvertebral fractures but not hip fractures by more than 50% while it substantially increased (i.e., 8%/year) lumbar BMD 40. Similar findings were noted in men with osteoporosis who were treated for 11 months. Unfortunately, the PTH trial in postmenopausal women was stopped after 20 months because of concerns related to the development of osteosarcoma in rats treated with high doses of PTH1–34. However, retrospective studies have found no association between osteosarcoma and primary or secondary hyperparathyroidism, and only 2 cases of osteosarcoma in PTH-treated patients have been reported in more than 9 years of market use40a.
More recently, recombinant human full-length PTH (PTH1–84) has shown similar benefits on the spine and hip and is approved in most European countries but not in the United States. The risk profile is identical to teriparatide except for a slightly greater risk of hypercalcemia 41. Currently, it is recommended that PTH therapy should be limited to those individuals with moderate to severe osteoporosis, and then only for 2 years. PTH is also approved for the treatment of glucocorticoid-induced osteoporosis. In general, PTH is well tolerated, although nausea, flushing, hypotension, and mild but asymptomatic hypercalcemia (i.e., serum calcium <11.0 mg/dL) can occur. Unlike the bisphosphonates, discontinuation of PTH can result in bone loss of 3 to 4% in the first year after PTH cessation 42. This post-treatment effect is prevented by adding an antiresorptive drug after PTH is stopped. Despite the appeal of using an anabolic with an antiresorptive, no evidence so far indicates that combinations of the current classes of drugs have additive or synergistic effects 41,42, although this certainly does not preclude the possibility that more efficacious combinations of antiresorptive and anabolic agents may be found in the future. Finally it should be noted that the marked increase in bone mineral density with intermittent PTH use plateaus after two years of treatment. Whether there is resistance at the osteoblast level to continued administration of PTH, or a reset remodeling balance based on the increase in bone resorption is not clear, but is important when considering new anabolic approaches to osteoporosis treatment. Cost and compliance with daily injections have been major limiting factors of for PTH therapies for osteoporosis.
Newer formulations of PTH, including transdermal patches, have been studied in short-term dose-ranging phase II trials. Also, oral agents that provoke intermittent PTH release by acting on the calcium sensing receptor in the parathyroids, i.e. calciolytics, are also under investigation as a means of capitalizing on the anabolic window produced by PTH. However, preliminary reports suggest that the administration of calciolytics do not fully recapitulate the pharmacokinetics of subcutaneously administered PTH. Another potential anabolic osteoporosis agent that is closely related to PTH, is PTH related protein, PTHrp, a peptide hormone that also activates the PTH receptor and regulates a number of homeostatic processes including chondrocyte proliferation Intermittent PTHrp administration has been shown to increase bone mass in postmenopausal women, and one pharmaceutical group has completed a Phase II study with daily subcutaneous PTHrp for 12 months. Bone mineral density increased significantly in the spine and hip at both 6 and 12 months in that study. These findings were comparable to results in women from the same study treated with teriparatide for 12 months.
Future Anabolic Therapies for Osteoporosis
There is still a need for therapies that reduce fracture risk beyond that achievable with bone-resorbing agents, particularly since virtually all the currently available drugs do not eliminate the possibility of future fractures. Moreover, concern has grown about rare but important adverse effects of the bisphosphonates on skeletal sites such as the jaw (i.e. ONJ) and the proximal femur (i.e. subtrochanteric fractures). Drug discovery and development for osteoporosis is currently focused on several potential anabolic agents, with one major exception: cathepsin K inhibitors, one of which, odanacatib, is in Phase III trials (Box 2). The remainder of this article therefore focuses on advances in the understanding of osteoblast regulation and function that have revealed new opportunities for developing drugs to stimulate bone formation. These opportunities stem directly from ongoing benchand translational studies that have identified new pathways for regulating osteoblast differentiation and bone formation.
Box 2. Cathepsin K inhibitors.
A new class of drugs to treat osteoporosis is cathepsin inhibitors. Cathepsin K is the most abundantly expressed cysteine protease in osteoclasts and exhibits collagenolytic activity in bone (mainly type 1 collagen) under acidic conditions; however, it is also expressed in skin and skin-derived cells. The most specific cathepsin K inhibitor developed so far is odanacatib. In a Phase II trial 166, the 50 mg dose of odanacatib was associated with an increase in spine (5.5%) and total hip (3.2%) bone mineral density (BMD) at 24 months of treatment. This study has now been extended for an additional 12 months [Odanacatib] was not associated with any increase in skin or upper respiratory tract infections during the treatment period, a difference from earlier cathepsin K inhibitors. A Phase III fracture trial of odanacatib including 16,200 postmenopausal osteoporotic women is currently underway (http://clinicaltrials.gov/ct2/results?term=odanacatib+) - NCT00529373. Of interest, the decreases in bone formation markers following odanacatib therapy were modest and transient as compared to those seen with other antiresorptive therapies (Unpublished data, McClung et al. the 37th European Symposium on Calcified Tissues, 2010). These results were consistent with findings in bone biopsies that were available from a subset of the study participants 171 in which there was a non-significant decrease in bone formation rate and mineralizing surface. In addition, TRAP5b, a serum marker thought to reflect osteoclast numbers, did not change even with the highest dose of odanacatib. These data reflect a potentially important difference between odanacatib and other anti-resorptive agents. Odanacatib selectively inhibits the removal of matrix proteins, but it permits persistent osteoclast viability and cellular activity, including acid secretion. In general, cathepsin K inhibitors do not result in apoptosis of osteoclasts; therefore, osteoclasts are available to produce chemokines and growth factors such as the Wnts that are responsible for coupling with osteoblasts to maintain bone formation 115. Also, the dramatic decrease in bone turnover with other anti-resorptive treatments may carry long-term risks, including the accumulation of microcracks that cannot be repaired and over time leading to an accumulation of microfractures that weaken bone and could result in fractures. Whether odanacatib would have a similar effect can only be demonstrated in long term clinical trials.
Targeting the Wnt/β-catenin pathway
The identification of patients with high bone mass due to mutations in the Wnt signaling pathway in 2002 led to an explosion of interest in modulating this pathway as a novel approach for treating for osteoporosis. Wnts are secreted glycoproteins crucial for the development and homeostatic renewal of many tissues, including bone. Figure 2 depicts the central role played by the canonical Wnt pathway in regulating osteoblast development 43,44. Wnts stimulate several signaling pathways by binding a receptor complex consisting of LRP5/6 and one of ten Frizzled (Fz) molecules 45. The canonical Wnt signaling pathway, which involves stabilization of β-catenin and regulation of Lef/Tcf transcription factors, has been the most extensively studied. The Wnt pathway in osteoblasts is active in all osteoblast lineage cells, including pre-osteoblasts, lining cells and osteocytes46. Notably, Wnt/β-catenin signaling may be a normal physiological response to mechanical loading 47 and is certain to be participate in the fracture healing process 48. Wnt signaling has three major functions in osteoblast lineage cells: dictating osteoblast specification from osteo/chondro-progenitors, stimulating osteoblast proliferation, and enhancing osteoblast and osteocyte survival. Within the BMU, Wnts also suppress bone resorption through suppression of osteoclast formation at least in part by regulating RANKL levels in pre-osteoblasts 49. Wnt ligands are ubiquitous and can have significant pro-proliferative effects on both normal and neoplastic cells. Presumably owing to concerns about potential carcinogenicity there has been no effort to develop experimental Wnt-like ligands for stimulating bone formation. However, other aspects of the Wnt/β-catenin pathway have been targeted, and selected examples are discussed below.
Figure 2. Osteoblast lineage specification, expansion, and terminal differentiation as well as the central role of canonical (ca) Wnts in regulating these processes.
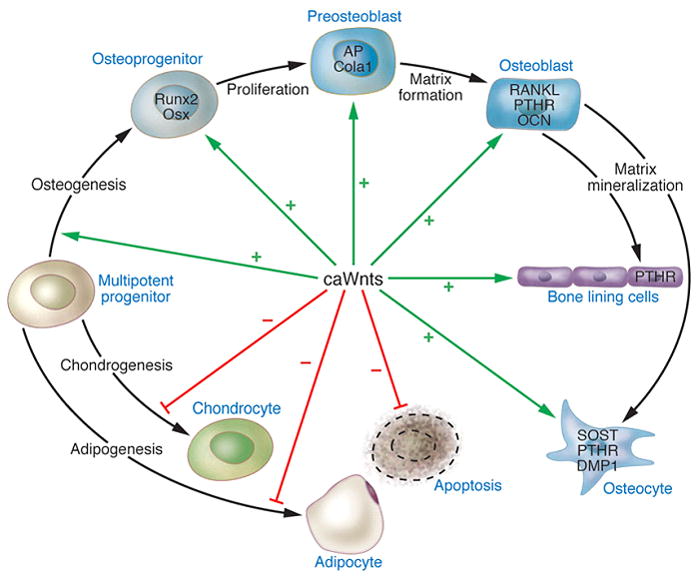
Osteoblasts are derived from multi-potent mesodermal or neural crest progenitors. Activation of the caWnt pathway through β-catenin stabilization prevents chondrogenesis. Wnt10b prevents adipogenesis. The caWnt pathway promotes survival of all cells of the osteoblast lineage and induces proliferation of preosteoblasts. Reproduced from Khosla et al. with permission 39.
Sclerostin
A target in the Wnt pathway for which drug development efforts are the most advanced is sclerostin, a secreted Wnt antagonist produced almost exclusively by osteocytes. It regulates bone mass by binding to LRP5 and LRP6 to inhibit the canonical Wnt/β-catenin signaling pathway 50–54. The biological importance of sclerostin in humans is highlighted by sclerosteosis and van Buchem’s disease—two genetic diseases caused by loss of sclerostin function— which are associated with markedly increased bone mass 55–58. Since sclerostin is expressed almost exclusively in osteocytes, inhibitors are anticipated to stimulate bone formation through Wnt pathway activation while having reduced carcinogenic potential compared with broader Wnt pathway activators. Moreover, in sclerostin related genetic disorders bone formation is high but bone resorption is also reduced suggesting that agents which suppress sclerostin might have dual properties in the remodeling unit.
Sclerostin-neutralizing monoclonal antibodies have been developed to inhibit the binding of sclerostin to the LRP5 receptor, resulting in the activation of the canonical Wnt signaling pathway 59. Initial studies in ovariectomized rats as a model of postmenopausal osteoporosis showed strong anabolic effects of the sclerostin antibody, with marked increases in bone formation on trabecular, periosteal, endocortical, and intracortical surfaces following short-term treatment (5 weeks) 60 (Figure 3). A subsequent study in adolescent gonad-intact, non-human primates for 2 months using three different doses of the sclerostin-neutralizing monoclonal antibody demonstrated a significant increase in bone mineral content (BMC) and BMD at several skeletal sites (femoral neck, radial metaphysis, and tibial metaphysis). Overall, there was an 11 to 29% increase in BMD from baseline compared to the vehicle-treated animals. There were also significant increases in trabecular thickness and bone strength at the lumbar vertebrae in the highest-dose group 61.
Figure 3. Sclerostin monoclonal antibody treatment in osteopenic rats restores trabecular BMD and bone volume back to sham levels at the distal femur.
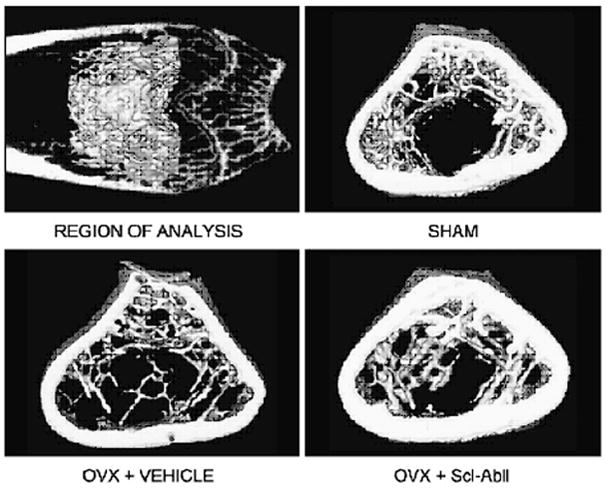
Shown is the distal femur region of analysis (top left panel) and representative 3D μCT images of a 1-mm central section (with attached cortices) from sham operated, ovariectomized rates treated with vehicle, and ovariectomized rats treated with the sclerostin antibody. Reproduced from Li et al. with permission 55.
A Phase I trial was conducted with 74 healthy men and postmenopausal women in a randomized, double-blind, placebo controlled ascending single-dose study to evaluate the safety, tolerability, pharmacokinetics and pharmacodynamics of the sclerostin antibody 62. The study demonstrated a dose-related increase in BMD of up to 5.3% at the lumbar spine and 2.8% at the total hip with a single subcutaneous dose of 10 mg/kg compared to placebo. Furthermore, the sclerostin-neutralizing antibody not only increased bone formation (P1NP, bone specific alkaline phosphatase, and osteocalcin) but also decreased bone resorption (sCTx) markers, resulting in a large anabolic window. Osteoblast and osteoclast activity was uncoupled following the treatment with the sclerostin-neutralizing antibody, a finding similar to sclerostosis or von Buchem’s disease. A Phase II dose escalation trial has recently been initiated with a total of 419 postmenopausal women with a T-score between −2 and −3.5 by DXA. In this study, different doses will be given once a month or once every 3 months. The primary endpoint is the percentage change from baseline in spine BMD. This study will be particularly informative since investigators will compare the sclerostin antibody with alendronate and teriparatide treatment. However, the long term safety of a sclerostin monoclonal antibody, particularly in respect to the development of neoplasms will await larger phase III trials.
Wnt binding proteins
Other secreted Wnt antagonists whose action might be blocked to stimulate the Wnt signaling pathway and increase bone mass include secreted frizzled-related proteins (sFRPs) and Wnt inhibitory factor 1 (Wif1), since both bind directly to Wnt proteins and inhibit Wnt signaling. Mesenchymal cells express endogenous Wif1 and its expression level decreases during osteoblast differentiation, but increases during adipogenesis. Furthermore, over-expression of Wif1 in mesenchymal cells resulted in a significant decrease in alkaline phosphatase activity and an increase in adipogenesis. By contrast, complete knockdown of Wif1 leads to an increase in alkaline phosphatase activity in vitro 63. Thus, either monoclonal antibodies or antisense RNA molecules against Wif1 or sFRPs would be expected to lead to an increase in Wnt activity and result in an anabolic bone response.
However, much like the Wnts, attention needs to be paid to the possible oncogenic effects of pharmacological stimulation of the Wnt pathway by inhibiting these inhibitors. For example, Kansara et al. showed that Wif1 suppressed β-catenin levels, indirectly though its binding to the Wnts, and inhibited growth of mouse and human osteosarcoma cells 64. Deletion of Wif1 accelerated the development of radiation-induced osteosarcomas in vivo. In primary osteosarcomas, silencing of WIF1 by promoter hypermethylation was associated with loss of differentiation, increased β-catenin levels, and increased cell proliferation. Compared with normal human osteobasts, Wif1 mRNA and protein levels were significantly down-regulated in several osteosarcoma cell lines. The down-regulation of WIF1 mRNA expression was also associated with its promoter hypermethylation in several cell lines 65. Therefore, the development and use of Wnt antagonists, as well as all agents that target the Wnt/β-catenin pathway must proceed with extreme caution due to the potential for carcinogenicity. Further studies will be required to fully uncover the regulatory mechanisms involving Wnts and tumor development.
DKK1 antagonists
DKK1 is another negative regulator of the Wnt signaling pathway, which acts by direct binding to Lrp5/6, similar to sclerostin. Blocking the receptor leads to inhibition of osteoblastogenesis in various osteogenic cell lines and in vivo, mice deficient in the DKK1 gene develop extra digits 66. The loss of a single allele of DKK1 resulted in an overall increase in bone mass 67. Conversely, mice over-expressing DKK1 develop osteopenia 68. To date, most studies inhibiting DKK1 have been done in association with different types of cancer cell lines or cancer mouse models. DKK1 is highly expressed in patients with multiple myeloma (MM) as well as in patients with breast cancer, and this contributes to the development and progression of myeloma and breast osteolytic bone metastases. In a study by Bu et al. 69, while conditioned media from breast cancer cells blocked Wnt-3a-induced C2C12 osteoblast differentiation and OPG expression, conditioned media from cells in which DKK1 expression had been silenced was unable to do so. Serum from patients with MM inhibited osteoblast differentiation in vitro, and this effect was blocked using a neutralizing antibody to DKK1. In vivo experiments treating SCID-rab mice transplanted with myeloma cells with an anti-DKK1 antibody stimulated bone formation and reduced bone loss and tumor growth 70. In addition, an increase in bone mineral density was observed in the control (i.e., not transplanted with the myeloma cells) mice treated with the DKK1 antibody. The neutralizing DKK1 antibody (BHQ880, which can neutralize both human and murine DKK1) prevented suppression of osteoblast numbers but did not prevent increases in osteoclast numbers. In addition, the anti-DKK1 antibody prevented osteolytic bone disease, but did not reduce overall tumor burden 71.
Thus, a possible alternative strategy to prevent estrogen deficiency-induced osteoporosis would be to inhibit the function of DKK1, as was demonstrated by Wang et al. 72. In that study, injection of sham or ovariectomized rats with end-capped phosphorothiate DKK1 antisense oligonucleotides resulted in an increase in bone volume and osteoblast numbers as well as a decrease in osteoclast numbers. In addition, DKK1 antisense oligonucleotide treatment decreased the expression of RANKL in the bone microenviroment. No studies in nonhuman primates or human studies using the DKK1 inhibitor have been initiated so far. Moreover, unlike sclerostin, which is expressed primarily in bone, DKK1 is expressed in multiple tissues 73–75. Thus, there are greater concerns regarding the use of DKK1 antagonists and potential side effects, such as an increased incidence of tumors in, for example, the colon, where increased Wnt signaling is associated with tumorigenesis 76–79.
Lithium
Lithium has been used for decades to treat psychiatric disorders and clearly is not an emerging therapy for osteoporosis. But due to recent clinical and basic studies of the Wnt/β-catenin pathway, lithium has been re-examined as an anabolic skeletal factor. Osteoporosis is prevalent in patients with bipolar disorders and other psychiatric conditions. The first choice of long-term treatment for these disorders has been lithium salts. However, lithium has a complex effect on calcium homeostasis and it is unclear whether lithium is protective or detrimental to the skeleton. For example, lithium can stimulate PTH release and clinically it has been shown that lithium therapy may be associated with a mild, reversible hyperparathyroid state. 80–85 Lithium was recently shown to activate the canonical Wnt signaling pathway both in vitro and in vivo. Lithium inhibits glycogen synthase kinase-3β (GSK-3β), an enzyme that phosphorylates β-catenin in the cytoplasm, targeting it for ubiquitination and degradation. Using Lrp5 knock-out and normal C57Bl/6 mice, Clement-Lacroix et al. demonstrated that lithium activated Wnt signaling independent of the Wnt receptor and resulted in significant increased bone formation and bone mass in different lines of mice 86. However, administration of lithium carbonate caused bone loss in healthy sexually mature Wistar rats 87. Even though lithium treatment may lead to hyperparathyroidism, a recent study by Zamani et al. found a significant decrease in bone formation and resorption markers and a significant increase in bone density in treated patients compared to normal women 88. Consistent with these findings, evaluation of data from large cohort studies demonstrated a decreasing relative risk of any fracture with an increasing accumulated dose of lithium 89,90. These findings were confirmed by Wilting et al. who also found a significant increased risk of fractures after discontinuation of lithium treatment 91. Nevertheless, lithium has non-skeletal side effects that undoubtedly would limit its promise as an anabolic agent for osteoporosis.
Hormones, growth factors and cytokines
One approach to treat osteoporosis is by targeting extracellular molecules such as growth factors and cytokines. A number of extracellular factors have identified as potential anabolic factors for skeletal metabolism and some of them have been tested in clinical settings. In this section we will summarize the recent findings of extracellular factors which are involved in skeletal metabolism and discuss the potential usefulness for the treatment of osteoporosis.
IGF-I/IGF-II, IGFBPs/insulin
Regulatory systems governed by insulin-like growth factors (IGFs) and their binding proteins (IGFBPs) are operative in most tissues and exert profound effects on a wide range of metabolic processes 92–94. Accumulating evidence both in human and animal studies demonstrate the anabolic role of IGF-I and IGF-II on the skeleton in part through stimulating bone formation. For example, IGF-I serum levels are robustly increased at the periods of puberty when bone acquisition is maximized, then decline with age and are associated with bone loss in human. In genetically engineered animal models, the anabolic effect of circulating and skeletal IGF-I have been implicated in the physiological process of skeletal accrual, particularly for the cortical skeleton 95–103. The effects of IGF-I on stromal cell differentiation appear to be bimodal, with IGF-I exhibiting mitogenic properties during early osteoblast development. On the other hand, IGF-I also promotes terminal osteoblast differentiation (e.g. mineralization),
IGFBPs principally function as carriers for IGFs and regulate bone turnover by maintaining the proper IGFs concentration in the skeletal micro-environment104–106. Molar excess of the IGFBPs compared to the IGFs reduces bone mass in part through inhibiting the access of IGFs to their receptor, whereas the lack of IGFBPs relative to IGFs leads to the osteoporotic phenotype by increasing the turnover of IGFs in the pericellular space. Indeed, administration of IGF-II together with IGFBP2 has anabolic properties on the skeleton 107,108. However, IGFBPs may have intrinsic biological activity independent of bound IGFs and this may occur through their unique motifs including their heparin-binding domain and RGD sequence. For example, the heparin-binding domain of IGFBP2 has been implicated in the down-regulation of PTEN expression, which may be beneficial for IGF-I signaling.
Given the anabolic properties of the IGFs/IGFBPs system, these peptides have been considered as potential therapeutic agents for the treatment for osteoporosis. In line with this, several small studies suggest that administration of recombinant human IGF-I (rhIGF-I) could positively affect bone mass 109, 110. Ghiron et al. evaluated the bone formation and resorption markers in 16 healthy elderly women (71.9 +/− 1.3 years of age) administered a high dose of rhIGF-I (60ug/kg/day) or a low dose of rhIGF-I (30ug/kg/day) for 28 days 110. Bone formation markers, including osteocalcin, skeletal alkaline phosphatase (sALP), and type I procollagen carboxy-terminal extension peptide (CICP), were increased by rhIGF-I treatment, whereas bone resorption markers such as urine hydroxyproline (OHP), total pyridinolines (PYD), and N-telopeptide showed dose-dependent differences. High-dose rhIGF-I increased these resorption markers, but low-dose rhIGF-I did not appreciably affect bone resorption. These data are suggestive that low-dose rhIGF-I might be considered as an anabolic treatment since it increases bone formation with only minimal increases in bone resorption.
rhIGF-I has also been studied in patients with anorexia nervosa which is characterized by amenorrhea, low BMD, reduced body weight, low serum IGF-I levels, and GH resistance. Grinspoon et al. reported that rhIGF-I caused an increase in BMD in these patients and when combined with oral contraceptive pills rhIGF-I showed a greater increase in BMD than oral contraceptives alone 109. rhIGF-I has a very short half life, and hence combination therapy with its carrier such as rhIGFBP3 has been proposed111. Boonen et al. evaluated the effect of rhIGF-I/rhIGFBP3 combination therapy on the recovery of bone mass in patients with recent hip fracture after surgery (aged 65–90 yr). Interestingly, bone loss in the contralateral hip was recovered in patients with combinational therapy, but placebo-treated patients failed to regain lost bone. However, IGF-I also stimulates bone resorption, and hence it seems unlikely that therapy with this peptide could be continued indefinitely. In addition, metabolic adverse effects such as hypoglycemia and hypophosphatemia caused by the activation of the IGF receptor in other tissues may limit the widespread application of this therapy.
Insulin has been used as a treatment for Type I and II diabetes mellitus for nearly 90 years. However, two recent studies suggest that insulin has novel activity on the skeletal remodeling unit that cannot be duplicated by IGF-I. The Clemens laboratory demonstrated that absence of the insulin receptor in osteoblasts leads to markedly decreased bone formation and an obesity phenotype 112 The Karsenty laboratory showed that insulin mediates bone remodeling by suppressing Twist 2 and OPG, thereby leading to higher bone turnover 113. Moreover as noted below, osteocalcin, a bone specific protein, enhances insulin sensitivity and secretion, which in turn could lead to increased bone formation. Whether insulinotropic agents might be useful in the treatment of osteoporosis requires further studies.
TGF- β
TGF-β has complex effects on bone, but there has been considerable interest in this growth factor recently based on the demonstration by Tang et al. that active TGF-β1 released during bone resorption coordinates bone formation by inducing the migration of osteoblast precursors 114 This has led to the proposal that TGF-β1 is a factor that couples bone resorption to bone formation signaling Additional factors produced by osteoclasts that appear to serve a similar coupling function include sphingosine kinase I, Wnt10b, and BMP6 115. Collectively, these findings raise the possibility that regulating the production of TGF-β or these other coupling factors in the bone microenvironment may represent a novel approach to increasing osteoblast numbers and/or activity. However, like other growth factors, TGF-β is ubiquitously expressed and therefore using this peptide as a treatment modality might have adverse non-skeletal effects.
Osteocalcin
Osteocalcin (OCN) is an osteoblast-specific secretory protein which is the major non-collagenous protein in the extracellular matrix of bone 116. OCN undergoes γ-carboxylation which occurs on glutamic-acid residues post-translationally and converts under-carboxylated OCN to γ-carboxyglutamic acid (Gla). This process increases the affinity of OCN for the extracellular matrices, especially hydroxyapatite 117,118. OCN has been widely used as a marker for bone turnover and bone formation 119; however, the exact role of OCN still remains unknown 120. For example, OCN null mice are obese and insulin resistant suggesting that bone plays an integral role in energy metabolism in part through OCN 121–122 Un-carboxylated and under-carboxylated forms of OCN can circulate and function as endocrine factors increasing the production of insulin in the β-cells of pancreas and adiponectin in adipocytes 1116, 121, 122, thus exerting a systemic glucose-lowering effect. Interestingly, OCN bioactivity is regulated negatively by adipocytes through increased sympathetic nervous tone driven by leptin 116, 122,123, resulting in decreased insulin secretion from β-cells.
Recently, two independent laboratories have expanded our understanding of the metabolic and skeletal functions of OCN. Conditional deletion of the insulin receptor in osteoblasts leads to impaired bone formation and insulin resistance 112. This occurs through modulation of the ratio of under-carboxylated OCN relative to total OCN. Under-carboxylated OCN is presumed to be the active metabolic form and its release from the skeletal matrix is triggered by bone resorption through insulin signaling and suppression of OPG 113. Thus, modulating OCN activity could improve insulin sensitivity, which might also prove beneficial for skeletal health. However, whether this pathway is operative in humans still needs to be determined. Clinical studies have demonstrated that serum OCN and under-carboxylated OCN levels are inversely associated with glucose levels124–129. On the other hand, glucose metabolism in patients treated with warfarin, which blocks γ-carboxylation of several molecules including osteocalcin, has not been studied systematically. In addition, a receptor for osteocalcin has not been identified. Clearly, further clinical and translational studies are needed to clarify the effectiveness of OCN in the regulation of bone and glucose metabolism in humans. However, the possibility that one could administer an agent that enhances glucose sensitivity and is either neutral or anabolic for bone is certainly tantalizing.
Antagonists of PPARγ
Peroxisome proliferator-activated receptor-gamma (PPARγ ) is a member of the PPAR family of transcriptional factors and nuclear receptors and plays a critical role in many aspects of cellular activities, including cell differentiation, lipid and glucose metabolism, and neoplasm development 130–132. Alternative splicing generates PPARγ isoforms including PPARγ1 and PPARγ2, two major isoforms of PPARγ. PPARγ1 is expressed in most of the tissues, but expression of PPARγ2 is limited to adipogenic cells. PPARγ2 has been identified as a critical transcription factor regulating adipogenesis and adipocyte metabolic functions.
The synthetic class of compounds, the thiazolidinediones, TZDs, are ligands for PPARγ and have been used frequently for the treatment of type 2 diabetes mellitus because of their lipid and glucose lowering effects133–137. Recent data from the clinical and animal studies points to an adverse effects of these agents on skeletal metabolism such that bone loss and fractures may result138–140. In contrast, PPARγ heterozygous mice have high bone mass and reduced marrow adiposity141. Similarly, inhibition of a downstream target of PPARγ, nocturnin, results in enhanced osteogenesis and suppressed adipogenesis 142. This occurs because PPARγ is a master regulator of mesenchymal cell fate, favoring the adipogenic lineage when activated and suppressing osteoblastogenesis. In addition to changes in marrow stromal cell allocation, activation of PPARγ also increases osteoblast and osteocyte apoptosis, possibly through reduction of IGF-I and enhances osteoclastogenesis by stimulating c-fos expression in osteoclast precursors. Thus, inhibition of PPARγ activity could be a pharmacologic means of treating osteoporosis. This concept has been studied in experimental models. Krause et al. reported that the PPARγ inhibitor, GW9662, enhanced osteogenic markers in human mesenchymal stem cells143 However, another PPARγ antagonist, bisphenol-A-diglycidyl ether (BADGE) treatment did not prevent bone loss in streptozoticin-induced type 1 diabetic mice, although BADGE inhibited the development of marrow adiposity in these mice 144. Since PPARγ could be a positive regulator for osteoclastogenesis, it is possible that PPARγ antagonists increase skeletal mass in part through inhibiting bone resorption, although this concept has not been tested 145, 146. These lines of evidence demonstrate that targeting PPARγ activity could be a promising strategy for the treatment of osteoporosis. Indeed, several companies are exploring this target, although further animal, clinical and translational studies will be required (Figure 4).
Figure 4. Proposed effect of PPARγ antagonist in the bone marrow milieu.
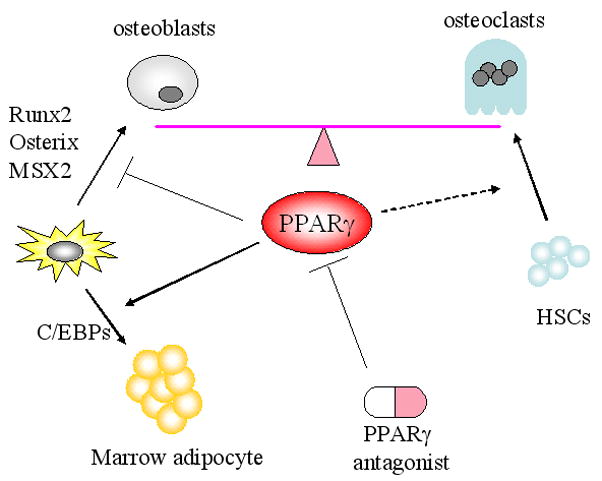
Bone remodeling is fine tuned by the balance between bone formation by osteoblasts and bone resorption by osteoclasts. PPARγ is involved in the cell fate determination of mesenchymal stem cells (MSCs) toward the adipogenic lineage and away from osteogenic lineage. In addition, PPARγ may be the positive regulator for osteoclastogenesis although this tenet needs to be clarified. Therefore, PPARγ antagonist may increase bone mass by switching the cell fate of MSCs toward osteogenic lineage and suppressing osteoclastogenesis. HSCs: hematopoietic stem cells, PPARγ: Peroxisome proliferator-activated receptor-gamma. C/EBP: CCAAT enhancer binding protein. Runx2: Runt-related transcription factor 2. Msx2: Muscle segment homeobox homolog of 2.
Serotonin
Serotonin is a neurotransmitter that affects appetite, energy balance and behavioral and emotional activity, and also functions to modulate gastrointestinal peristalsis, platelet contraction and hemostasis, depending on its site of synthesis 147–150. Serotonin synthesis is catalyzed by the rate-limiting enzyme, tryptophan hydroxylase (Tph)151. Circulating serotonin, which accounts for the 95 % of the total serotonin in the body, is generated in the enterochromaffin cells of the duodenum by Tph1152. Circulating serotonin is taken up principally by platelets through the 5-hydroxytrophan (serotonin) transporter (5-HTT) and does not cross the blood brain barrier. In contrast, serotonin in the brain is mainly produced in the brainstem by Tph2153.
Circulating and CNS serotonin may have opposing effects on skeletal metabolism. Yadav et al. reported that suppression of circulating serotonin by inhibiting Tph1 expression in the gut resulted in a high bone mass phenotype154, suggesting that circulating serotonin functions as a hormone that suppresses bone acquisition. In contrast, suppression of brain-derived serotonin by targeting the Tph2 gene decreased bone mass through the activation of the hypothalamus and stimulation of the sympathetic nervous system 155. This would indicate that brain serotonin is a neurotransmitter regulating bone mass in a positive direction.
Given the effects of serotonin as a modulator of behavioral and emotional activity, a number of serotonin-targeted drugs have been developed to treat psychiatric disorders. Two classes of drugs, second-generation antipsychotic agents (SGAs) and the selective serotonin reuptake inhibitors (SSRIs), have been widely prescribed to treat psychosis. Unexpectedly, however, these agents have been shown to have a deleterious effect on skeletal mass 90, 156–163. SGAs such as risperidone, which have antagonist activity at the 5-HT2A receptor, and to a lesser extent the dopamine D2 receptor164, have been implicated in bone loss and higher fracture risk156, 157, 163. Hyperprolactinemia caused by the inhibition of dopamine D2 receptor signaling could in part account for the bone loss caused by SGAs because hyperprolactinemia can stimulate bone resorption by suppressing gonadotropin secretion 165. However, it is still possible that the SGAs cause bone loss by inhibiting serotonin receptor signaling. Indeed, male mice treated with risperidone exhibit profound bone loss without affecting serum prolactin levels (personal communication, Rosen CJ).
SSRIs, which inhibit serotonin uptake by 5-HTT and are supposed to increase the local concentration of serotonin, have also been implicated in reduced bone mass and fractures 158–162. Because SSRIs could increase pericellular serotonin concentrations both in the central nervous system and skeletal microenvironment, it is still unclear as to the exact mechanisms whereby SSRIs reduce bone mass. Nevertheless, these lines of evidence clearly suggest that serotonin signaling is involved in skeletal metabolism and manipulating this pathway is a potential target for the treatment for osteoporosis. In fact, a small molecule which inhibits Tph1 (LP533401) has been shown to possess an anabolic effect on the skeleton of mice166 at least in one preliminary study. Further translational and pre-clinical studies will be required to shed light on this novel approach for osteoporosis treatment.
Outlook and challenges for anabolic agents
Drug discovery and development for osteoporosis is now largely focused on anabolic agents (with the notable exception of the cathepsin K inhibitors discussed in Box 2), and several of the approaches discussed above could lead to marketed therapies in the next decade. PTHrP (Box 1) and a monoclonal antibody to sclerostin show particular potential for moving into Phase III trials. However, the road to approval of new osteoporosis therapies has several important obstacles. First, the only endpoint recognized by European and American regulatory agencies for establishing the efficacy of new osteoporosis drugs is a reduction in fractures over a minimum of three years. Low bone mineral density is a risk factor for fracture, but an increase in density is not sufficient to gain regulatory approval. Indeed, several agents have been shown to increase bone mineral density but not reduce the number of fractures. After completing the requisite animal studies (two models are required) and the toxicology work, sponsors must have dose ranging phase II studies ready before embarking on very large and expensive phase III fracture trials. With the exception of PTHrP, which could potentially be studied in a non-inferiority fracture trial due its resemblance in structure and function to PTH1–34, any of the other agents mentioned above would currently have to be tested through randomized placebo-controlled trials. Even assuming that sufficient efficacy is achieved, demonstrating acceptable safety will be a major challenge particularly for newer agents where non-skeletal tissues may be involved (e.g. Wnt/β-catenin and neoplastic growth). Not only does the design of the trial have to include assurances that there is adequate power to assess fracture risk reduction, but adjudication of events from these other possible non-skeletal effects needs to be a major component of the research design.
Box 1. Parathyroid hormone–related protein.
Parathyroid hormone–related protein (PTHrP) is a naturally occurring polypeptide closely related to PTH that activates the same receptor as PTH (PTHR1)168. It is expressed in many tissues, but is found in high levels at the growth plate, in osteoblasts, and within mammary tissue. It was cloned from cancer cells and identified as the circulating cause of hypercalcemia of malignancy (HOM). Originally thought to be a potent stimulus of bone resorption and hence hypercalcemia, experimental studies using intermittent PTHrP have shown significant anabolic properties similar to PTH 1–34. PTHrP was first shown to be effective in treating low bone mass in laboratory animals, but later was studied for safety and efficacy in humans 169.
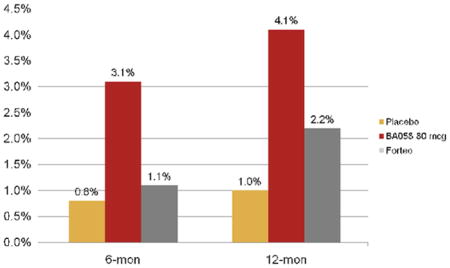
One study of PTHrP (1–34) was performed to determine if it could increase bone mass in postmenopausal women with osteoporosis when administered daily by subcutaneous injection for 3 months 165. This double-blind, placebo-controlled, randomized clinical study enrolled 16 postmenopausal women with osteoporosis between 50 and 75 years of age 170. All had been on hormone replacement therapy for an average of 8 years and still had osteoporosis. Women who had been taking any other type of osteoporosis medication were excluded from the study. Half self-administered PTHrP, 400 μg/d and the other half injected a placebo. The patients were followed up for 3 months and the participants tolerated the treatment without developing hypercalcemia, hypotension, nausea, flushing, or other adverse effects. The lumbar spine BMD increased by 4.7% during the 3-month treatment period, a larger increase in bone mass than seen with either anti-resorptives or PTH 1–34. Subsequently, a 3 week dose ranging study to determine the maximum tolerable dose was performed. Hypercalcemia developed only at the highest dose (750 μg/d) and in 1 of 6 individuals 169. Bone formation markers increased to 40% with the higher doses, and resorption indices at day 21 were only modestly increased for the 625 μg/d and 750 μg/d doses. A similar but longer phase II dose ranging study was performed by Radius Pharmaceuticals with PTHrP 1–34 (BA058) vs PTH 1–34 in more than 200 postmenopausal women. The results remain unpublished but at a meeting on anabolic treatments for osteoporosis, they reported increases in spine and hip BMD that equaled or exceeded teriparatide (i.e. PTH 1–34; Personal communication from Dr. O’Dea; see Figure 2). Thus, PTHrP remains a potentially important future anabolic treatment for osteoporosis, particularly if this agent increases bone formation more than resorption. However, the long-term safety and fracture efficacy will only be determined by a large phase III clinical trial.
Second, there are now several drugs to treat osteoporosis. This has led some individuals to propose a halt to randomized placebo controlled trials for new osteoporosis agents. A discussion of those issues is beyond the scope of this paper, but is highlighted in a recent perspective in New England Journal of Medicine167. Suffice it to say, recruitment into a phase III randomized placebo controlled trial for a new osteoporosis drug will be difficult and require larger numbers of subjects, particularly if women at highest risk are excluded from participating. Improving recruitment and retention of subjects will require more careful consideration of individual risk profiles and may include a more personalized approach such as a FRAX analysis for 10 year fracture risk, at the time of informed consent.
Third, even if approval is gained, there is a major push at the regulatory level for more in depth post marketing trials that are more than just observational analyses. These would include multi-year extensions, either open label or blinded, in a phase III trial design, and further scrutiny of adverse event reporting. Nonetheless, there are major opportunities for new drug development in osteoporosis. The number of targets has increased substantially and as our knowledge of bone biology expands, this list is sure to grow. Hence, cautious optimism pervades the world of osteoporosis medicine in respect to newer therapeutic opportunities for skeletal restoration.
Acknowledgments
Grant Support; This work is supported by US National Institutes of Health grants: NIH AR45433-13, AG004875, AG028936, and AR027965.
Abbreviations
- Basic multicellular units
the single physiologic unit of skeletal remodeling including osteoblasts, osteoclasts and osteocytes
- PTHrp
parathyroid hormone related peptide
- WHI
women’s health initiative- a large observational and randomized controlled trial performed in the United States
- SERMs
selective estrogen receptor modulators
- RANKL
receptor activator of NF-kB ligand
- PPARγ
peroxisome proliferator activated receptor gamma
- Stromal cells
connective tissue cells in the bone marrow that are multi-potent
- Microcanaliculi
small channels that connect the osteocyte with resting cells on the endosteal surface
- BMD, bone mineral density
areal bone mass as measured by dual energy x-ray absorptiometry
- β-catenin
an intracellular transcription factor activated by extracellular ligands such as the Wnts as well as other intracellular signaling peptides
- PTEN
phosphatase and tensin homolog deleted on chromosome 10 (PTEN) is a lipid phosphatase that opposes Insulin/IGF-I signaling by dephosphorylating phosphatidylinositol–3,4,5-trisphosaphate (PIP3) to phosphatidylinositol–4,5-diphosphate (PIP2)
- Wnts
a family of growth factors that bind to LRP5,6 and their own frizzled receptor
- Tph1
tryptophan hydoxylase enzyme 1, the rate limiting enzyme in the generation of serotonin
- OCN
osteocalcin
Footnotes
Disclosure; None
References
- 1.Becker DJ, Kilgore ML, Morrisey MA. The Societal Burden of osteoporosis. Curr Rehumatol Rep. 2010;12:186–191. doi: 10.1007/s11926-010-0097-y. [DOI] [PubMed] [Google Scholar]
- 2.Harralson RH, Zuckerman JD. Prevalence, Health Care Expenditures, and Orthopedic Surgery Workforce for Musculoskeletal Conditions. JAMA. 2009;302(14):1586–1587. doi: 10.1001/jama.2009.1489. [DOI] [PubMed] [Google Scholar]
- 3.Rosen CJ. Bone remodeling, energy metabolism, and the molecular clock. Cell Metab. 2008;7:7–10. doi: 10.1016/j.cmet.2007.12.004. [DOI] [PubMed] [Google Scholar]
- 4.Rosen CJ, Bouxsein ML. Mechansism of disease: is osteoporosis the obesity of bone? Nat Clin Pract Rheumaol. 2006;2:35–43. doi: 10.1038/ncprheum0070. [DOI] [PubMed] [Google Scholar]
- 5.Manolagas SC. Birth and death of bone cells: basic regulatory mechanisms and implications for the pathogenesis and treatment of osteoporosis. Endocr Rev. 2000;21:115–37. doi: 10.1210/edrv.21.2.0395. [DOI] [PubMed] [Google Scholar]
- 5a.Tang Y, Wu X, Lei W, Pang L, Wan C, Shi Z, Zhao L, Nagy TR, Peng X, Hu J, Feng X, Van Hul W, Wan M, Cao X. TGF-beta1-induced migration of bone mesenchymal stem cells couples bone resorption with formation. Nat Med. 2009 Jul;15(7):757–65. doi: 10.1038/nm.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jansen JP, Bergman GJ, Huels J, Olson M. The Efficacy of Bisphosphonates in the Prevention of Vertebral, Hip, and Nonvertebral-Nonhip Fractures in Osteoporosis: A Network Meta-Analysis. Semin Arthritis Rheum. 2010 Sep; doi: 10.1016/j.semarthrit.2010.06.001. [DOI] [PubMed] [Google Scholar]
- 7.Rossouw JE, et al. Risks and benefits of estrogen plus progestin in healthy postmenopausal women: principal results From the Women’s Health Initiative randomized controlled trial. Jama. 2002;288:321–33. doi: 10.1001/jama.288.3.321. [DOI] [PubMed] [Google Scholar]
- 8.Cauley JA, et al. Effects of estrogen plus progestin on risk of fracture and bone mineral density: the Women’s Health Initiative randomized trial. Jama. 2003;290:1729–38. doi: 10.1001/jama.290.13.1729. [DOI] [PubMed] [Google Scholar]
- 9.Prestwood KM, Kenny AM, Kleppinger A, Kulldorff M. Ultralow-dose micronized 17beta-estradiol and bone density and bone metabolism in older women: a randomized controlled trial. Jama. 2003;290:1042–8. doi: 10.1001/jama.290.8.1042. [DOI] [PubMed] [Google Scholar]
- 10.Ettinger B, et al. Effects of ultralow-dose transdermal estradiol on bone mineral density: a randomized clinical trial. Obstet Gynecol. 2004;104:443–51. doi: 10.1097/01.AOG.0000137833.43248.79. [DOI] [PubMed] [Google Scholar]
- 11.McDonnell DP. Mining the complexities of the estrogen signaling pathways for novel therapeutics. Endocrinology. 2003;144:4237–40. doi: 10.1210/en.2003-0900. [DOI] [PubMed] [Google Scholar]
- 12.Seeman E, Crans GG, Diez-Perez A, Pinette KV, Delmas PD. Anti-vertebral fracture efficacy of raloxifene: a meta-analysis. Osteoporos Int. 2006;17:313–6. doi: 10.1007/s00198-005-2030-1. [DOI] [PubMed] [Google Scholar]
- 13.Vogel VG, et al. Effects of tamoxifen vs raloxifene on the risk of developing invasive breast cancer and other disease outcomes: the NSABP Study of Tamoxifen and Raloxifene (STAR) P-2 trial. Jama. 2006;295:2727–41. doi: 10.1001/jama.295.23.joc60074. [DOI] [PubMed] [Google Scholar]
- 14.Barrett-Connor E, et al. Effects of raloxifene on cardiovascular events and breast cancer in postmenopausal women. N Engl J Med. 2006;355:125–37. doi: 10.1056/NEJMoa062462. [DOI] [PubMed] [Google Scholar]
- 15.Cummings SR, et al. Lasofoxifene in postmenopausal women with osteoporosis. N Engl J Med. 362:686–96. doi: 10.1056/NEJMoa0808692. [DOI] [PubMed] [Google Scholar]
- 16.Lindsay R, Gallagher JC, Kagan R, Pickar JH, Constantine G. Efficacy of tissue-selective estrogen complex of bazedoxifene/conjugated estrogens for osteoporosis prevention in at-risk postmenopausal women. Fertil Steril. 2009;92:1045–52. doi: 10.1016/j.fertnstert.2009.02.093. [DOI] [PubMed] [Google Scholar]
- 17.Lobo RA, et al. Evaluation of bazedoxifene/conjugated estrogens for the treatment of menopausal symptoms and effects on metabolic parameters and overall safety profile. Fertil Steril. 2009;92:1025–38. doi: 10.1016/j.fertnstert.2009.03.113. [DOI] [PubMed] [Google Scholar]
- 17a.Silverman SL, Christiansen C, Genant HK, Vukicevic S, Zanchetta JR, de Villiers TJ, Constantine GD, Chines AA. Efficacy of bazedoxifene in reducing new vertebral fracture risk in post menopausal women with osteoporosis: results from a 3 year randomized placebo and active controlled trial. J Bone Miner Res. 2008 Dec;23(12):1923–34. doi: 10.1359/jbmr.080710. [DOI] [PubMed] [Google Scholar]
- 18.Drake MT, Clarke BL, Khosla S. Bisphosphonates: mechanism of action and role in clinical practice. Mayo Clin Proc. 2008;83:1032–45. doi: 10.4065/83.9.1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kavanagh KL, et al. The molecular mechanism of nitrogen-containing bisphosphonates as antiosteoporosis drugs. Proc Natl Acad Sci U S A. 2006;103:7829–34. doi: 10.1073/pnas.0601643103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Luckman SP, et al. Nitrogen-containing bisphosphonates inhibit the mevalonate pathway and prevent post-translational prenylation of GTP-binding proteins, including Ras. J Bone Miner Res. 1998;13:581–9. doi: 10.1359/jbmr.1998.13.4.581. [DOI] [PubMed] [Google Scholar]
- 21.Khosla S, et al. Bisphosphonate-associated osteonecrosis of the jaw: report of a task force of the American Society for Bone and Mineral Research. J Bone Miner Res. 2007;22:1479–91. doi: 10.1359/jbmr.0707onj. [DOI] [PubMed] [Google Scholar]
- 22.Lenart BA, et al. Association of low-energy femoral fractures with prolonged bisphosphonate use: a case control study. Osteoporos Int. 2009;20:1353–62. doi: 10.1007/s00198-008-0805-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Abrahamsen B, Eiken P, Eastell R. Subtrochanteric and diaphyseal femur fractures in patients treated with alendronate: a register-based national cohort study. J Bone Miner Res. 2009;24:1095–102. doi: 10.1359/jbmr.081247. [DOI] [PubMed] [Google Scholar]
- 24.Lacey DL, et al. Osteoprotegerin ligand is a cytokine that regulates osteoclast differentiation and activation. Cell. 1998;93:165–76. doi: 10.1016/s0092-8674(00)81569-x. [DOI] [PubMed] [Google Scholar]
- 25.Riggs BL. The mechanisms of estrogen regulation of bone resorption. J Clin Invest. 2000;106:1203–4. doi: 10.1172/JCI11468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Eghbali-Fatourechi G, et al. Role of RANK ligand in mediating increased bone resorption in early postmenopausal women. J Clin Invest. 2003;111:1221–30. doi: 10.1172/JCI17215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hsu H, et al. Tumor necrosis factor receptor family member RANK mediates osteoclast differentiation and activation induced by osteoprotegerin ligand. Proc Natl Acad Sci U S A. 1999;96:3540–5. doi: 10.1073/pnas.96.7.3540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Simonet WS, et al. Osteoprotegerin: a novel secreted protein involved in the regulation of bone density. Cell. 1997;89:309–19. doi: 10.1016/s0092-8674(00)80209-3. [DOI] [PubMed] [Google Scholar]
- 29.Cummings SR, et al. Denosumab for prevention of fractures in postmenopausal women with osteoporosis. N Engl J Med. 2009;361:756–65. doi: 10.1056/NEJMoa0809493. [DOI] [PubMed] [Google Scholar]
- 30.Brown JP, et al. Comparison of the effect of denosumab and alendronate on BMD and biochemical markers of bone turnover in postmenopausal women with low bone mass: a randomized, blinded, phase 3 trial. J Bone Miner Res. 2009;24:153–61. doi: 10.1359/jbmr.0809010. [DOI] [PubMed] [Google Scholar]
- 31.Seeman E, et al. Microarchitectural deterioration of cortical and trabecular bone: Differing effects of denosumab and alendronate. J Bone Miner Res. doi: 10.1002/jbmr.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kendler DL, et al. Effects of denosumab on bone mineral density and bone turnover in postmenopausal women transitioning from alendronate therapy. J Bone Miner Res. 25:72–81. doi: 10.1359/jbmr.090716. [DOI] [PubMed] [Google Scholar]
- 33.Neville-Webbe HL, Coleman RE. Bisphosphonates and RANK ligand inhibitors for the treatment and prevention of metastatic bone disease. Eur J Cancer. 46:1211–22. doi: 10.1016/j.ejca.2010.02.041. [DOI] [PubMed] [Google Scholar]
- 34.Thomas D, et al. Denosumab in patients with giant-cell tumour of bone: an open-label, phase 2 study. Lancet Oncol. 11:275–80. doi: 10.1016/S1470-2045(10)70010-3. [DOI] [PubMed] [Google Scholar]
- 35.Aghaloo TL, Felsenfeld AL, Tetradis S. Osteonecrosis of the jaw in a patient on Denosumab. J Oral Maxillofac Surg. 68:959–63. doi: 10.1016/j.joms.2009.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.McClung MR, et al. Denosumab in postmenopausal women with low bone mineral density. N Engl J Med. 2006;354:821–31. doi: 10.1056/NEJMoa044459. [DOI] [PubMed] [Google Scholar]
- 37.Kendler DL, et al. Preference and satisfaction with a 6-month subcutaneous injection versus a weekly tablet for treatment of low bone mass. Osteoporos Int. 21:837–46. doi: 10.1007/s00198-009-1023-x. [DOI] [PubMed] [Google Scholar]
- 37a.Marie PJ, Ammann P, Boivin G, Rey C. Mechanisms of action and therapeutic potential of strontium in bone. Calcif Tissue Int. 2001;69:121–129. doi: 10.1007/s002230010055. [DOI] [PubMed] [Google Scholar]
- 37b.Meunier PJ, Boux C, Seeman E, et al. The effects of strontium ranelate on the risk of vertebral fracture in women with postmenopausal osteoporosis. N ENgl J Med. 2004;350:459–468. doi: 10.1056/NEJMoa022436. [DOI] [PubMed] [Google Scholar]
- 38.Rosen CJ. The role of parathyroid hormone in the management of osteoporosis. Horm Res. 2005;64 (Suppl 2):81–5. doi: 10.1159/000087762. [DOI] [PubMed] [Google Scholar]
- 39.Paszty C, Turner CH, Robinson MK. Sclerostin: a gem from the genome leads to bone-building antibodies. J Bone Miner Res. 2010;25:1897–1904. doi: 10.1002/jbmr.161. [DOI] [PubMed] [Google Scholar]
- 40.Neer RM, et al. Effect of parathyroid hormone (1–34) on fractures and bone mineral density in postmenopausal women with osteoporosis. N Engl J Med. 2001;344:1434–41. doi: 10.1056/NEJM200105103441904. [DOI] [PubMed] [Google Scholar]
- 40a.Subbiah V, Madsen VS, Raymond AK, Benjamin RS, Ludwig JA. Of mice and men: divergent risks of teriparatide-induced osteosarcoma. Osteoporos Int. 2010 Jun;21(6):1041–5. doi: 10.1007/s00198-009-1004-0. Epub 2009 Jul 14. [DOI] [PubMed] [Google Scholar]
- 41.Black DM, et al. The effects of parathyroid hormone and alendronate alone or in combination in postmenopausal osteoporosis. N Engl J Med. 2003;349:1207–15. doi: 10.1056/NEJMoa031975. [DOI] [PubMed] [Google Scholar]
- 42.Black DM, et al. One year of alendronate after one year of parathyroid hormone (1–84) for osteoporosis. N Engl J Med. 2005;353:555–65. doi: 10.1056/NEJMoa050336. [DOI] [PubMed] [Google Scholar]
- 43.Boyden LM, Mao J, Belsky J, Mitzner L, Farhi A, Mitnick MA, Wu D, Insogna K, Lifton RP. High bone density due to a mutation in LDL-receptor-related protein 5. N Engl J Med. 2002;346:1513–1521. doi: 10.1056/NEJMoa013444. [DOI] [PubMed] [Google Scholar]
- 44.Khosla S, Westendorf JJ, Oursler MJ. Building bone to reverse osteoporosis and repair fractures. J Clin Invest. 2008;118:421–8. doi: 10.1172/JCI33612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Westendorf JJ, Kahler RA, Schroeder TM. Wnt signaling in osteoblasts and bone diseases. Gene. 2004;341:19–39. doi: 10.1016/j.gene.2004.06.044. [DOI] [PubMed] [Google Scholar]
- 46.Hens JR, et al. TOPGAL mice show that the canonical Wnt signaling pathway is active during bone development and growth and is activated by mechanical loading in vitro. J Bone Miner Res. 2005;20:1103–13. doi: 10.1359/JBMR.050210. [DOI] [PubMed] [Google Scholar]
- 47.Robinson JA, et al. Wnt/beta-catenin signaling is a normal physiological response to mechanical loading in bone. J Biol Chem. 2006;281:31720–8. doi: 10.1074/jbc.M602308200. [DOI] [PubMed] [Google Scholar]
- 48.Chen Y, et al. Beta-catenin signaling plays a disparate role in different phases of fracture repair: implications for therapy to improve bone healing. PLoS Med. 2007;4:e249. doi: 10.1371/journal.pmed.0040249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Spencer GJ, Utting JC, Etheridge SL, Arnett TR, Genever PG. Wnt signalling in osteoblasts regulates expression of the receptor activator of NFkappaB ligand and inhibits osteoclastogenesis in vitro. J Cell Sci. 2006;119:1283–96. doi: 10.1242/jcs.02883. [DOI] [PubMed] [Google Scholar]
- 50.Baron R, Rawadi G. Targeting the Wnt/beta-catenin pathway to regulate bone formation in the adult skeleton. Endocrinology. 2007;148:2635–43. doi: 10.1210/en.2007-0270. [DOI] [PubMed] [Google Scholar]
- 51.van Bezooijen RL, et al. Sclerostin is an osteocyte-expressed negative regulator of bone formation, but not a classical BMP antagonist. J Exp Med. 2004;199:805–14. doi: 10.1084/jem.20031454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Semenov M, Tamai K, He X. SOST is a ligand for LRP5/LRP6 and a Wnt signaling inhibitor. J Biol Chem. 2005;280:26770–5. doi: 10.1074/jbc.M504308200. [DOI] [PubMed] [Google Scholar]
- 53.Poole KE, et al. Sclerostin is a delayed secreted product of osteocytes that inhibits bone formation. Faseb J. 2005;19:1842–4. doi: 10.1096/fj.05-4221fje. [DOI] [PubMed] [Google Scholar]
- 54.van Bezooijen RL, ten Dijke P, Papapoulos SE, Lowik CW. SOST/sclerostin, an osteocyte-derived negative regulator of bone formation. Cytokine Growth Factor Rev. 2005;16:319–27. doi: 10.1016/j.cytogfr.2005.02.005. [DOI] [PubMed] [Google Scholar]
- 55.Brunkow ME, et al. Bone dysplasia sclerosteosis results from loss of the SOST gene product, a novel cystine knot-containing protein. Am J Hum Genet. 2001;68:577–89. doi: 10.1086/318811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Balemans W, et al. Identification of a 52 kb deletion downstream of the SOST gene in patients with van Buchem disease. J Med Genet. 2002;39:91–7. doi: 10.1136/jmg.39.2.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Balemans W, et al. Increased bone density in sclerosteosis is due to the deficiency of a novel secreted protein (SOST) Hum Mol Genet. 2001;10:537–43. doi: 10.1093/hmg/10.5.537. [DOI] [PubMed] [Google Scholar]
- 58.Staehling-Hampton K, et al. A 52-kb deletion in the SOST-MEOX1 intergenic region on 17q12-q21 is associated with van Buchem disease in the Dutch population. Am J Med Genet. 2002;110:144–52. doi: 10.1002/ajmg.10401. [DOI] [PubMed] [Google Scholar]
- 59.Ellies DL, et al. Bone density ligand, Sclerostin, directly interacts with LRP5 but not LRP5G171V to modulate Wnt activity. J Bone Miner Res. 2006;21:1738–49. doi: 10.1359/jbmr.060810. [DOI] [PubMed] [Google Scholar]
- 60.Li X, et al. Sclerostin antibody treatment increases bone formation, bone mass, and bone strength in a rat model of postmenopausal osteoporosis. J Bone Miner Res. 2009;24:578–88. doi: 10.1359/jbmr.081206. [DOI] [PubMed] [Google Scholar]
- 61.Ominsky MS, et al. Two doses of sclerostin antibody in cynomolgus monkeys increases bone formation, bone mineral density, and bone strength. J Bone Miner Res. 25:948–59. doi: 10.1002/jbmr.14. [DOI] [PubMed] [Google Scholar]
- 62.Padhi D, Jang G, Stouch B, Fang L, Posvar E. Single-dose, placebo-controlled, randomized study of AMG 785, a sclerostin monoclonal antibody. J Bone Miner Res. doi: 10.1002/jbmr.173. [DOI] [PubMed] [Google Scholar]
- 63.Cho SW, et al. Wnt inhibitory factor (WIF)-1 inhibits osteoblastic differentiation in mouse embryonic mesenchymal cells. Bone. 2009;44:1069–77. doi: 10.1016/j.bone.2009.02.012. [DOI] [PubMed] [Google Scholar]
- 64.Kansara M, et al. Wnt inhibitory factor 1 is epigenetically silenced in human osteosarcoma, and targeted disruption accelerates osteosarcomagenesis in mice. J Clin Invest. 2009;119:837–51. doi: 10.1172/JCI37175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Rubin EM, et al. Wnt inhibitory factor 1 decreases tumorigenesis and metastasis in osteosarcoma. Mol Cancer Ther. 9:731–41. doi: 10.1158/1535-7163.MCT-09-0147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Adamska M, MacDonald BT, Sarmast ZH, Oliver ER, Meisler MH. En1 and Wnt7a interact with Dkk1 during limb development in the mouse. Dev Biol. 2004;272:134–44. doi: 10.1016/j.ydbio.2004.04.026. [DOI] [PubMed] [Google Scholar]
- 67.Morvan F, et al. Deletion of a single allele of the Dkk1 gene leads to an increase in bone formation and bone mass. J Bone Miner Res. 2006;21:934–45. doi: 10.1359/jbmr.060311. [DOI] [PubMed] [Google Scholar]
- 68.Li J, et al. Dkk1-mediated inhibition of Wnt signaling in bone results in osteopenia. Bone. 2006;39:754–66. doi: 10.1016/j.bone.2006.03.017. [DOI] [PubMed] [Google Scholar]
- 69.Bu G, et al. Breast cancer-derived Dickkopf1 inhibits osteoblast differentiation and osteoprotegerin expression: implication for breast cancer osteolytic bone metastases. Int J Cancer. 2008;123:1034–42. doi: 10.1002/ijc.23625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Yaccoby S, et al. Antibody-based inhibition of DKK1 suppresses tumor-induced bone resorption and multiple myeloma growth in vivo. Blood. 2007;109:2106–11. doi: 10.1182/blood-2006-09-047712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Heath DJ, et al. Inhibiting Dickkopf-1 (Dkk1) removes suppression of bone formation and prevents the development of osteolytic bone disease in multiple myeloma. J Bone Miner Res. 2009;24:425–36. doi: 10.1359/jbmr.081104. [DOI] [PubMed] [Google Scholar]
- 72.Wang FS, et al. Knocking down dickkopf-1 alleviates estrogen deficiency induction of bone loss. A histomorphological study in ovariectomized rats. Bone. 2007;40:485–92. doi: 10.1016/j.bone.2006.09.004. [DOI] [PubMed] [Google Scholar]
- 73.Qian J, et al. Dickkopf-1 (DKK1) is a widely expressed and potent tumor-associated antigen in multiple myeloma. Blood. 2007;110:1587–94. doi: 10.1182/blood-2007-03-082529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Gregory CA, et al. Dkk-1-derived synthetic peptides and lithium chloride for the control and recovery of adult stem cells from bone marrow. J Biol Chem. 2005;280:2309–23. doi: 10.1074/jbc.M406275200. [DOI] [PubMed] [Google Scholar]
- 75.Glinka A, et al. Dickkopf-1 is a member of a new family of secreted proteins and functions in head induction. Nature. 1998;391:357–62. doi: 10.1038/34848. [DOI] [PubMed] [Google Scholar]
- 76.Gonzalez-Sancho JM, et al. The Wnt antagonist DICKKOPF-1 gene is a downstream target of beta-catenin/TCF and is downregulated in human colon cancer. Oncogene. 2005;24:1098–103. doi: 10.1038/sj.onc.1208303. [DOI] [PubMed] [Google Scholar]
- 77.Aguilera O, et al. The Wnt antagonist DICKKOPF-1 gene is induced by 1alpha,25-dihydroxyvitamin D3 associated to the differentiation of human colon cancer cells. Carcinogenesis. 2007;28:1877–84. doi: 10.1093/carcin/bgm094. [DOI] [PubMed] [Google Scholar]
- 78.Aguilera O, et al. Epigenetic inactivation of the Wnt antagonist DICKKOPF-1 (DKK-1) gene in human colorectal cancer. Oncogene. 2006;25:4116–21. doi: 10.1038/sj.onc.1209439. [DOI] [PubMed] [Google Scholar]
- 79.Sato H, et al. Frequent epigenetic inactivation of DICKKOPF family genes in human gastrointestinal tumors. Carcinogenesis. 2007;28:2459–66. doi: 10.1093/carcin/bgm178. [DOI] [PubMed] [Google Scholar]
- 80.Christiansen C, Baastrup PC, Transbol I. Development of ‘primary’ hyperparathyroidism during lithium therapy: longitudinal study. Neuropsychobiology. 1980;6:280–3. doi: 10.1159/000117770. [DOI] [PubMed] [Google Scholar]
- 81.Christiansen C, Baastrup PC, Lindgreen P, Transbol I. Endocrine effects of lithium: II. ‘Primary’ hyperparathyroidism. Acta Endocrinol (Copenh) 1978;88:528–34. doi: 10.1530/acta.0.0880528. [DOI] [PubMed] [Google Scholar]
- 82.Mak TW, Shek CC, Chow CC, Wing YK, Lee S. Effects of lithium therapy on bone mineral metabolism: a two-year prospective longitudinal study. J Clin Endocrinol Metab. 1998;83:3857–9. doi: 10.1210/jcem.83.11.5269. [DOI] [PubMed] [Google Scholar]
- 83.Mallette LE, Eichhorn E. Effects of lithium carbonate on human calcium metabolism. Arch Intern Med. 1986;146:770–6. [PubMed] [Google Scholar]
- 84.Mallette LE, Khouri K, Zengotita H, Hollis BW, Malini S. Lithium treatment increases intact and midregion parathyroid hormone and parathyroid volume. J Clin Endocrinol Metab. 1989;68:654–60. doi: 10.1210/jcem-68-3-654. [DOI] [PubMed] [Google Scholar]
- 85.Nordenstrom J, Elvius M, Bagedahl-Strindlund M, Zhao B, Torring O. Biochemical hyperparathyroidism and bone mineral status in patients treated long-term with lithium. Metabolism. 1994;43:1563–7. doi: 10.1016/0026-0495(94)90017-5. [DOI] [PubMed] [Google Scholar]
- 86.Clement-Lacroix P, et al. Lrp5-independent activation of Wnt signaling by lithium chloride increases bone formation and bone mass in mice. Proc Natl Acad Sci U S A. 2005;102:17406–11. doi: 10.1073/pnas.0505259102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Lewicki M, Paez H, Mandalunis PM. Effect of lithium carbonate on subchondral bone in sexually mature Wistar rats. Exp Toxicol Pathol. 2006;58:197–201. doi: 10.1016/j.etp.2006.06.003. [DOI] [PubMed] [Google Scholar]
- 88.Zamani A, Omrani GR, Nasab MM. Lithium’s effect on bone mineral density. Bone. 2009;44:331–4. doi: 10.1016/j.bone.2008.10.001. [DOI] [PubMed] [Google Scholar]
- 89.Vestergaard P, Rejnmark L, Mosekilde L. Reduced relative risk of fractures among users of lithium. Calcif Tissue Int. 2005;77:1–8. doi: 10.1007/s00223-004-0258-y. [DOI] [PubMed] [Google Scholar]
- 90.Bolton JM, et al. Fracture risk from psychotropic medications: a population-based analysis. J Clin Psychopharmacol. 2008;28:384–91. doi: 10.1097/JCP.0b013e31817d5943. [DOI] [PubMed] [Google Scholar]
- 91.Wilting I, et al. Lithium use and the risk of fractures. Bone. 2007;40:1252–8. doi: 10.1016/j.bone.2006.12.055. [DOI] [PubMed] [Google Scholar]
- 92.Kawai M, Rosen CJ. The IGF-I regulatory system and its impact on skeletal and energy homeostasis. J Cell Biochem. doi: 10.1002/jcb.22678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Kawai M, Rosen CJ. Insulin-like growth factor-I and bone: lessons from mice and men. Pediatr Nephrol. 2009;24:1277–85. doi: 10.1007/s00467-008-1040-6. [DOI] [PubMed] [Google Scholar]
- 94.Niu T, Rosen CJ. The insulin-like growth factor-I gene and osteoporosis: a critical appraisal. Gene. 2005;361:38–56. doi: 10.1016/j.gene.2005.07.016. [DOI] [PubMed] [Google Scholar]
- 95.Bikle D, et al. The skeletal structure of insulin-like growth factor I-deficient mice. J Bone Miner Res. 2001;16:2320–9. doi: 10.1359/jbmr.2001.16.12.2320. [DOI] [PubMed] [Google Scholar]
- 96.Liu JP, Baker J, Perkins AS, Robertson EJ, Efstratiadis A. Mice carrying null mutations of the genes encoding insulin-like growth factor I (Igf-1) and type 1 IGF receptor (Igf1r) Cell. 1993;75:59–72. [PubMed] [Google Scholar]
- 97.Yakar S, et al. Normal growth and development in the absence of hepatic insulin-like growth factor I. Proc Natl Acad Sci U S A. 1999;96:7324–9. doi: 10.1073/pnas.96.13.7324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Yakar S, et al. Circulating levels of IGF-1 directly regulate bone growth and density. J Clin Invest. 2002;110:771–81. doi: 10.1172/JCI15463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Zhang M, et al. Osteoblast-specific knockout of the insulin-like growth factor (IGF) receptor gene reveals an essential role of IGF signaling in bone matrix mineralization. J Biol Chem. 2002;277:44005–12. doi: 10.1074/jbc.M208265200. [DOI] [PubMed] [Google Scholar]
- 100.Zhao G, et al. Targeted overexpression of insulin-like growth factor I to osteoblasts of transgenic mice: increased trabecular bone volume without increased osteoblast proliferation. Endocrinology. 2000;141:2674–82. doi: 10.1210/endo.141.7.7585. [DOI] [PubMed] [Google Scholar]
- 101.Wang Y, et al. Role of IGF-I signaling in regulating osteoclastogenesis. J Bone Miner Res. 2006;21:1350–8. doi: 10.1359/jbmr.060610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Mathews LS, et al. Growth enhancement of transgenic mice expressing human insulin-like growth factor I. Endocrinology. 1988;123:2827–33. doi: 10.1210/endo-123-6-2827. [DOI] [PubMed] [Google Scholar]
- 103.He J, Rosen CJ, Adams DJ, Kream BE. Postnatal growth and bone mass in mice with IGF-I haploinsufficiency. Bone. 2006;38:826–35. doi: 10.1016/j.bone.2005.11.021. [DOI] [PubMed] [Google Scholar]
- 104.Hwa V, Oh Y, Rosenfeld RG. Insulin-like growth factor binding proteins: a proposed superfamily. Acta Paediatr Suppl. 1999;88:37–45. doi: 10.1111/j.1651-2227.1999.tb14349.x. [DOI] [PubMed] [Google Scholar]
- 105.Jones JI, Clemmons DR. Insulin-like growth factors and their binding proteins: biological actions. Endocr Rev. 1995;16:3–34. doi: 10.1210/edrv-16-1-3. [DOI] [PubMed] [Google Scholar]
- 106.Firth SM, Baxter RC. Cellular actions of the insulin-like growth factor binding proteins. Endocr Rev. 2002;23:824–54. doi: 10.1210/er.2001-0033. [DOI] [PubMed] [Google Scholar]
- 107.Khosla S, et al. Insulin-like growth factor system abnormalities in hepatitis C-associated osteosclerosis. Potential insights into increasing bone mass in adults. J Clin Invest. 1998;101:2165–73. doi: 10.1172/JCI1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Conover CA, et al. Subcutaneous administration of insulin-like growth factor (IGF)-II/IGF binding protein-2 complex stimulates bone formation and prevents loss of bone mineral density in a rat model of disuse osteoporosis. Growth Horm IGF Res. 2002;12:178–83. doi: 10.1016/s1096-6374(02)00044-8. [DOI] [PubMed] [Google Scholar]
- 109.Grinspoon S, Thomas L, Miller K, Herzog D, Klibanski A. Effects of recombinant human IGF-I and oral contraceptive administration on bone density in anorexia nervosa. J Clin Endocrinol Metab. 2002;87:2883–91. doi: 10.1210/jcem.87.6.8574. [DOI] [PubMed] [Google Scholar]
- 110.Ghiron LJ, et al. Effects of recombinant insulin-like growth factor-I and growth hormone on bone turnover in elderly women. J Bone Miner Res. 1995;10:1844–52. doi: 10.1002/jbmr.5650101203. [DOI] [PubMed] [Google Scholar]
- 111.Boonen S, et al. Musculoskeletal effects of the recombinant human IGF-I/IGF binding protein-3 complex in osteoporotic patients with proximal femoral fracture: a double-blind, placebo-controlled pilot study. J Clin Endocrinol Metab. 2002;87:1593–9. doi: 10.1210/jcem.87.4.8426. [DOI] [PubMed] [Google Scholar]
- 112.Fulzele K, et al. Insulin Receptor Signaling in Osteoblasts Regulates Postnatal Bone Acquisition and Body Composition. Cell. 142:309–319. doi: 10.1016/j.cell.2010.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Ferron M, et al. Insulin Signaling in Osteoblasts Integrates Bone Remodeling and Energy Metabolism. Cell. 142:296–308. doi: 10.1016/j.cell.2010.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Tang Y, et al. TGF-beta1-induced migration of bone mesenchymal stem cells couples bone resorption with formation. Nat Med. 2009;15:757–65. doi: 10.1038/nm.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Pederson L, Ruan M, Westendorf JJ, Khosla S, Oursler MJ. Regulation of bone formation by osteoclasts involves Wnt/BMP signaling and the chemokine sphingosine-1-phosphate. Proc Natl Acad Sci U S A. 2008;105:20764–9. doi: 10.1073/pnas.0805133106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Kawai M, Devlin MJ, Rosen CJ. Fat targets for skeletal health. Nat Rev Rheumatol. 2009;5:365–72. doi: 10.1038/nrrheum.2009.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Hauschka PV, Lian JB, Cole DE, Gundberg CM. Osteocalcin and matrix Gla protein: vitamin K-dependent proteins in bone. Physiol Rev. 1989;69:990–1047. doi: 10.1152/physrev.1989.69.3.990. [DOI] [PubMed] [Google Scholar]
- 118.Murshed M, Schinke T, McKee MD, Karsenty G. Extracellular matrix mineralization is regulated locally; different roles of two gla-containing proteins. J Cell Biol. 2004;165:625–30. doi: 10.1083/jcb.200402046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Delmas PD. Biochemical markers of bone turnover. J Bone Miner Res. 1993;8 (Suppl 2):S549–55. doi: 10.1002/jbmr.5650081323. [DOI] [PubMed] [Google Scholar]
- 120.Boskey AL, et al. Fourier transform infrared microspectroscopic analysis of bones of osteocalcin-deficient mice provides insight into the function of osteocalcin. Bone. 1998;23:187–96. doi: 10.1016/s8756-3282(98)00092-1. [DOI] [PubMed] [Google Scholar]
- 121.Lee NK, et al. Endocrine regulation of energy metabolism by the skeleton. Cell. 2007;130:456–69. doi: 10.1016/j.cell.2007.05.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Ferron M, Hinoi E, Karsenty G, Ducy P. Osteocalcin differentially regulates beta cell and adipocyte gene expression and affects the development of metabolic diseases in wild-type mice. Proc Natl Acad Sci U S A. 2008;105:5266–70. doi: 10.1073/pnas.0711119105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Hinoi E, et al. The sympathetic tone mediates leptin’s inhibition of insulin secretion by modulating osteocalcin bioactivity. J Cell Biol. 2008;183:1235–42. doi: 10.1083/jcb.200809113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Strapazzon G, De Toni L, Foresta C. Serum undercarboxylated osteocalcin was inversely associated with plasma glucose level and fat mass in type 2 diabetes mellitus. Osteoporos Int. doi: 10.1007/s00198-010-1184-7. [DOI] [PubMed] [Google Scholar]
- 125.Yeap B, et al. Reduced serum total osteocalcin is associated with metabolic syndrome in older men via waist circumference, hyperglycemia and triglyceride levels. Eur J Endocrinol. doi: 10.1530/EJE-10-0414. [DOI] [PubMed] [Google Scholar]
- 126.Saleem U, Mosley TH, Jr, Kullo IJ. Serum osteocalcin is associated with measures of insulin resistance, adipokine levels, and the presence of metabolic syndrome. Arterioscler Thromb Vasc Biol. 30:1474–8. doi: 10.1161/ATVBAHA.110.204859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Shea MK, et al. Gamma-carboxylation of osteocalcin and insulin resistance in older men and women. Am J Clin Nutr. 2009;90:1230–5. doi: 10.3945/ajcn.2009.28151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Pittas AG, Harris SS, Eliades M, Stark P, Dawson-Hughes B. Association between serum osteocalcin and markers of metabolic phenotype. J Clin Endocrinol Metab. 2009;94:827–32. doi: 10.1210/jc.2008-1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Kindblom JM, et al. Plasma osteocalcin is inversely related to fat mass and plasma glucose in elderly Swedish men. J Bone Miner Res. 2009;24:785–91. doi: 10.1359/jbmr.081234. [DOI] [PubMed] [Google Scholar]
- 130.Tontonoz P, Spiegelman BM. Fat and beyond: the diverse biology of PPARgamma. Annu Rev Biochem. 2008;77:289–312. doi: 10.1146/annurev.biochem.77.061307.091829. [DOI] [PubMed] [Google Scholar]
- 131.Rosen ED, MacDougald OA. Adipocyte differentiation from the inside out. Nat Rev Mol Cell Biol. 2006;7:885–96. doi: 10.1038/nrm2066. [DOI] [PubMed] [Google Scholar]
- 132.Kawai M, Sousa KM, MacDougald OA, Rosen CJ. The many facets of PPARgamma: novel insights for the skeleton. Am J Physiol Endocrinol Metab. 2010;299:E3–9. doi: 10.1152/ajpendo.00157.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Iwamoto Y, et al. Effect of new oral antidiabetic agent CS-045 on glucose tolerance and insulin secretion in patients with NIDDM. Diabetes Care. 1991;14:1083–6. doi: 10.2337/diacare.14.11.1083. [DOI] [PubMed] [Google Scholar]
- 134.Fujiwara T, Yoshioka S, Yoshioka T, Ushiyama I, Horikoshi H. Characterization of new oral antidiabetic agent CS-045. Studies in KK and ob/ob mice and Zucker fatty rats. Diabetes. 1988;37:1549–58. doi: 10.2337/diab.37.11.1549. [DOI] [PubMed] [Google Scholar]
- 135.Ciaraldi TP, Gilmore A, Olefsky JM, Goldberg M, Heidenreich KA. In vitro studies on the action of CS-045, a new antidiabetic agent. Metabolism. 1990;39:1056–62. doi: 10.1016/0026-0495(90)90166-a. [DOI] [PubMed] [Google Scholar]
- 136.Suter SL, Nolan JJ, Wallace P, Gumbiner B, Olefsky JM. Metabolic effects of new oral hypoglycemic agent CS-045 in NIDDM subjects. Diabetes Care. 1992;15:193–203. doi: 10.2337/diacare.15.2.193. [DOI] [PubMed] [Google Scholar]
- 137.Nolan JJ, Ludvik B, Beerdsen P, Joyce M, Olefsky J. Improvement in glucose tolerance and insulin resistance in obese subjects treated with troglitazone. N Engl J Med. 1994;331:1188–93. doi: 10.1056/NEJM199411033311803. [DOI] [PubMed] [Google Scholar]
- 138.Schwartz AV, et al. Thiazolidinedione use and bone loss in older diabetic adults. J Clin Endocrinol Metab. 2006;91:3349–54. doi: 10.1210/jc.2005-2226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Grey A, et al. The peroxisome proliferator-activated receptor-gamma agonist rosiglitazone decreases bone formation and bone mineral density in healthy postmenopausal women: a randomized, controlled trial. J Clin Endocrinol Metab. 2007;92:1305–10. doi: 10.1210/jc.2006-2646. [DOI] [PubMed] [Google Scholar]
- 140.Yaturu S, Bryant B, Jain SK. Thiazolidinedione treatment decreases bone mineral density in type 2 diabetic men. Diabetes Care. 2007;30:1574–6. doi: 10.2337/dc06-2606. [DOI] [PubMed] [Google Scholar]
- 141.Akune T, et al. PPARgamma insufficiency enhances osteogenesis through osteoblast formation from bone marrow progenitors. J Clin Invest. 2004;113:846–55. doi: 10.1172/JCI19900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Kawai M, et al. A circadian-regulated gene, Nocturnin, promotes adipogenesis by stimulating PPAR-gamma nuclear translocation. Proc Natl Acad Sci U S A. 2010;107:10508–13. doi: 10.1073/pnas.1000788107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Krause U, et al. Pharmaceutical modulation of canonical Wnt signaling in multipotent stromal cells for improved osteoinductive therapy. Proc Natl Acad Sci U S A. 107:4147–52. doi: 10.1073/pnas.0914360107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Botolin S, McCabe LR. Inhibition of PPARgamma prevents type I diabetic bone marrow adiposity but not bone loss. J Cell Physiol. 2006;209:967–76. doi: 10.1002/jcp.20804. [DOI] [PubMed] [Google Scholar]
- 145.Wan Y, Chong LW, Evans RM. PPAR-gamma regulates osteoclastogenesis in mice. Nat Med. 2007;13:1496–503. doi: 10.1038/nm1672. [DOI] [PubMed] [Google Scholar]
- 146.Wei W et al. PGC1beta mediates PPARgamma activation of osteoclastogenesis and rosiglitazone-induced bone loss. Cell Metab. 11:503–16. doi: 10.1016/j.cmet.2010.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Kawai M, Rosen CJ. Minireview: A skeleton in serotonin’s closet? Endocrinology. 2010;151:4103–8. doi: 10.1210/en.2010-0499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Garfield AS, Heisler LK. Pharmacological targeting of the serotonergic system for the treatment of obesity. J Physiol. 2009;587:49–60. doi: 10.1113/jphysiol.2008.164152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Berger M, Gray JA, Roth BL. The expanded biology of serotonin. Annu Rev Med. 2009;60:355–66. doi: 10.1146/annurev.med.60.042307.110802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Gaspar P, Cases O, Maroteaux L. The developmental role of serotonin: news from mouse molecular genetics. Nat Rev Neurosci. 2003;4:1002–12. doi: 10.1038/nrn1256. [DOI] [PubMed] [Google Scholar]
- 151.Jonnakuty C, Gragnoli C. What do we know about serotonin? J Cell Physiol. 2008;217:301–6. doi: 10.1002/jcp.21533. [DOI] [PubMed] [Google Scholar]
- 152.Furness JB, Costa M. Neurons with 5-hydroxytryptamine-like immunoreactivity in the enteric nervous system: their projections in the guinea-pig small intestine. Neuroscience. 1982;7:341–9. doi: 10.1016/0306-4522(82)90271-8. [DOI] [PubMed] [Google Scholar]
- 153.Walther DJ, et al. Synthesis of serotonin by a second tryptophan hydroxylase isoform. Science. 2003;299:76. doi: 10.1126/science.1078197. [DOI] [PubMed] [Google Scholar]
- 154.Yadav VK, et al. Lrp5 controls bone formation by inhibiting serotonin synthesis in the duodenum. Cell. 2008;135:825–37. doi: 10.1016/j.cell.2008.09.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Yadav VK, Karsenty G. Leptin-dependent co-regulation of bone and energy metabolism. Aging (Albany NY) 2009;1:954–6. doi: 10.18632/aging.100100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Howard L, Kirkwood G, Leese M. Risk of hip fracture in patients with a history of schizophrenia. Br J Psychiatry. 2007;190:129–34. doi: 10.1192/bjp.bp.106.023671. [DOI] [PubMed] [Google Scholar]
- 157.Halbreich U, Palter S. Accelerated osteoporosis in psychiatric patients: possible pathophysiological processes. Schizophr Bull. 1996;22:447–54. doi: 10.1093/schbul/22.3.447. [DOI] [PubMed] [Google Scholar]
- 158.Vestergaard P, Rejnmark L, Mosekilde L. Selective serotonin reuptake inhibitors and other antidepressants and risk of fracture. Calcif Tissue Int. 2008;82:92–101. doi: 10.1007/s00223-007-9099-9. [DOI] [PubMed] [Google Scholar]
- 159.Diem SJ, et al. Use of antidepressants and rates of hip bone loss in older women: the study of osteoporotic fractures. Arch Intern Med. 2007;167:1240–5. doi: 10.1001/archinte.167.12.1240. [DOI] [PubMed] [Google Scholar]
- 160.Liu B, et al. Use of selective serotonin-reuptake inhibitors or tricyclic antidepressants and risk of hip fractures in elderly people. Lancet. 1998;351:1303–7. doi: 10.1016/s0140-6736(97)09528-7. [DOI] [PubMed] [Google Scholar]
- 161.Richards JB, et al. Effect of selective serotonin reuptake inhibitors on the risk of fracture. Arch Intern Med. 2007;167:188–94. doi: 10.1001/archinte.167.2.188. [DOI] [PubMed] [Google Scholar]
- 162.Ziere G, et al. Selective serotonin reuptake inhibiting antidepressants are associated with an increased risk of nonvertebral fractures. J Clin Psychopharmacol. 2008;28:411–7. doi: 10.1097/JCP.0b013e31817e0ecb. [DOI] [PubMed] [Google Scholar]
- 163.Calarge CA, Zimmerman B, Xie D, Kuperman S, Schlechte JA. A cross-sectional evaluation of the effect of risperidone and selective serotonin reuptake inhibitors on bone mineral density in boys. J Clin Psychiatry. 71:338–47. doi: 10.4088/JCP.08m04595gre. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Meltzer HY, Matsubara S, Lee JC. Classification of typical and atypical antipsychotic drugs on the basis of dopamine D-1, D-2 and serotonin2 pKi values. J Pharmacol Exp Ther. 1989;251:238–46. [PubMed] [Google Scholar]
- 165.Bostwick JR, Guthrie SK, Ellingrod VL. Antipsychotic-induced hyperprolactinemia. Pharmacotherapy. 2009;29:64–73. doi: 10.1592/phco.29.1.64. [DOI] [PubMed] [Google Scholar]
- 166.Yadav VK, et al. Pharmacological inhibition of gut-derived serotonin synthesis is a potential bone anabolic treatment for osteoporosis. Nat Med. 16:308–12. doi: 10.1038/nm.2098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Rosen CJ, Khosla S. Placebo-controlled trials in osteoporosis--proceeding with caution. N Engl J Med. 363:1365–7. doi: 10.1056/NEJMsb1002227. discussion e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Strewler GJ. The physiology of parathyroid hormone-related protein. N Engl J Med. 2000;342:177–85. doi: 10.1056/NEJM200001203420306. [DOI] [PubMed] [Google Scholar]
- 169.Horwitz MJ, et al. Parathyroid hormone-related protein for the treatment of postmenopausal osteoporosis: defining the maximal tolerable dose. J Clin Endocrinol Metab. 95:1279–87. doi: 10.1210/jc.2009-0233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Horwitz MJ, Tedesco MB, Gundberg C, Garcia-Ocana A, Stewart AF. Short-term, high-dose parathyroid hormone-related protein as a skeletal anabolic agent for the treatment of postmenopausal osteoporosis. J Clin Endocrinol Metab. 2003;88:569–75. doi: 10.1210/jc.2002-021122. [DOI] [PubMed] [Google Scholar]
- 171.Bone HG, et al. Odanacatib, a cathepsin-K inhibitor for osteoporosis: a two-year study in postmenopausal women with low bone density. J Bone Miner Res. 25:937–47. doi: 10.1359/jbmr.091035. [DOI] [PubMed] [Google Scholar]