

Abstract
Evasion of apoptosis is a significant problem affecting an array of cancers. In order to identify novel regulators of apoptosis, we performed an RNA interference (RNAi) screen against all kinases and phosphatases in the human genome. We identified MK-STYX (STYXL1), a catalytically inactive phosphatase with homology to the mitogen-activated protein kinase (MAPK) phosphatases. Despite this homology, MK-STYX knockdown does not significantly regulate MAPK signaling in response to growth factors or apoptotic stimuli. Rather, RNAi-mediated knockdown of MK-STYX inhibits cells from undergoing apoptosis induced by cellular stressors activating mitochondrion-dependent apoptosis. This MK-STYX phenotype mimics the loss of Bax and Bak, two potent guardians of mitochondrial apoptotic potential. Similar to loss of both Bax and Bak, cells without MK-STYX expression are unable to release cytochrome c. Proapoptotic members of the BCL-2 family (Bax, Bid, and Bim) are unable to trigger cytochrome c release in MK-STYX-depleted cells, placing the apoptotic deficiency at the level of mitochondrial outer membrane permeabilization (MOMP). MK-STYX was found to localize to the mitochondria but is neither released from the mitochondria upon apoptotic stress nor proximal to the machinery currently known to control MOMP, indicating that MK-STYX regulates MOMP using a distinct mechanism.
INTRODUCTION
Evasion of apoptosis has long been known to be one of the hallmarks of cancer (9). While the downregulation of proapoptotic pathways is critical in the establishment of most primary lesions, it is also crucial for tumor persistence through current therapeutic strategies, such as chemotherapy and radiation. Intrinsic apoptosis is a tightly regulated process in which a cell undergoes a suicide program after a critical level of damage or cellular insult, deemed irreparable, has occurred. The mitochondrion is intimately associated with intrinsic apoptosis, and many of the proteins housed in the organelle are critical for the initiation of and commitment to this process (8, 12). The Bcl-2 family of proteins, containing both proapoptotic and anti- apoptotic members, are master regulators of the cellular decision to undergo apoptosis (3). Bax and Bak, two proapoptotic Bcl-2 family members, are considered the “gatekeepers” of mitochondrion-dependent apoptosis (2, 28). Upon sufficient cellular insult, Bax and Bak are activated and proceed to destabilize the outer mitochondrial membrane (OMM), allowing the release of apoptotic proteins to the cytosol and triggering a cascade of death-inducing events. The best-characterized of these proapoptotic proteins, cytochrome c, interacts with the adaptor protein Apaf-1 to trigger the formation of the apoptosome, a heptameric complex that recruits and facilitates the activation of caspase 9, the initiator caspase of the intrinsic apoptotic pathway (30). Activated caspase 9 is then able to cleave effector caspases, such as caspase 3, which cleave cellular substrates, inducing the morphological hallmarks of apoptosis, such as membrane blebbing and DNA fragmentation.
During the sequence of events in the initiation and execution of intrinsic apoptosis, there are numerous critical junctures that could be regulated at the molecular level. Inactivation of caspases is sufficient to block the apoptotic pathway, though most cell types undergo what is known as “caspase-independent cell death” after a sufficient cellular insult, even in the presence of continuous caspase inhibition (24). Thus, the commitment to cell death is a cellular decision upstream of caspase activation. Bax and Bak, two proapoptotic proteins within the Bcl-2 family, control a specific process termed mitochondrial outer membrane permeabilization (MOMP), which is critical in the initiation of apoptosis. During MOMP, homo-oligomerization of Bax (or Bak) destabilizes the mitochondrial outer membrane, allowing the release of proteins contained within the mitochondria. Due to its disruptive cellular mechanism, MOMP is considered to be the “point of no return” in the apoptotic process (8). The simultaneous knockout of both Bax and Bak prevents MOMP and results in a phenotype in which cells are highly desensitized to a variety of death-inducing agents (13, 28).
In an attempt to characterize novel signaling pathways that contribute to the regulation of cellular survival and apoptosis, we performed a high-throughput RNA interference (RNAi) screen against all of the known and putative kinases and phosphatases in the human genome (16). In this study, we identified a small group of genes whose RNAi-mediated knockdown caused cells to become largely desensitized to chemotherapeutic agents. Of the enzymes assayed, the most striking resistance to drug-induced cell death came from the knockdown of the dual-specificity phosphatase (DUSP) MK-STYX, with almost complete protection from apoptosis compared to control cells when treated with chemotherapeutics, each having distinct mechanisms of action. As the knockdown of MK-STYX leads to dramatic protection from apoptosis, we hypothesized that the expression of MK-STYX is required for cells to appropriately respond to intrinsic apoptosis. Here, we have investigated the function of MK-STYX as a protein required for intrinsic apoptosis. Knockdown of MK-STYX is sufficient to block apoptosis in the presence of specific chemotherapeutics, as well as cellular insults known to induce the intrinsic apoptotic pathway. While the knockdown of MK-STYX phenocopies the RNAi phenotype of both caspase 9 and Bax/Bak, we have shown that the loss of MK-STYX inhibits cytochrome c release in response to intrinsic cellular stressors and proapoptotic Bcl-2 family members. In this study, we have identified a novel protein whose downregulation phenocopies the dual loss of Bax/Bak and have characterized MK-STYX as a gatekeeper of mitochondrial cell death.
MATERIALS AND METHODS
Cells and reagents.
HeLa cells (ATCC) and 293FT cells (Invitrogen) were maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS). Control and MK-STYX small interfering RNAs (siRNAs) (Qiagen) and Bax, Bak1, caspase 8, and caspase 9 pooled siRNAs (Dharmacon) were transfected into cells with oligofectamine (Invitrogen). Knockdown levels were determined using quantitative reverse transcription-PCR (qRT-PCR) as previously described (16). MK-STYX cDNA was cloned into pRK7 with an N-terminal V5 tag. Proapoptotic cDNAs were obtained from different sources: mouse full-length Bax was purchased from Open Biosource, tBid was a kind gift of Stanley Korsemeyer (Addgene 8780), and BimS was supplied by Doug Green. Each plasmid was transfected into cells using FuGene 6 (Roche). Paclitaxel (Taxol), actinomycin D, tunicamycin (Sigma-Aldrich), cisplatin (EMD/Calbiochem), and tumor necrosis factor alpha (TNF-α) (Peprotech) were diluted and used according to the manufacturer's instructions. UV irradiation was induced using a Stratalinker (Stratagene) according to the manufacturer's instructions. Endogenous MK-STYX was probed with a mouse monoclonal antibody generated at Van Andel Research Institute (VARI) (for immunofluorescence), and a rabbit polyclonal antibody toward STYXL1 (Sigma-Aldrich) was used for Western blotting. Other antibodies used were total caspase 9, phosphorylated ERK (p-ERK), total ERK, p-p38, total p38, p-JNK, total JNK, human Bid, total Bim, apoptosis-inducing factor (AIF), calnexin, total S6, Bcl-2, Bcl-xL, Smac, GRP75, and Hsp60 (Cell Signaling Technologies), as well as NDUFS3 (MitoSciences), V5 (Invitrogen), AF555-conjugated cytochrome c (BD biosciences), tubulin (Sigma-Aldrich), and total Bax (Millipore).
Cell viability and apoptosis assays.
The Cell Titer Blue reagent (Promega) was used to detect cellular viability according to the manufacturer's directions. Real-time cell viability was determined using the xCelligence system, which utilizes an electric current to determine cellular attachment to an electrode-containing plate. The electrical impedance, caused by cell adhesion to the plate, is directly proportional to cellular viability and is what is numerically reported. Data from the xCelligence was collected using software provided with the system, and values were normalized at the point of treatment with compounds for all cell lines. DNA fragmentation was determined by an enzyme-linked immunosorbent assay (ELISA) as previously described (16). Propidium iodide (PI) positivity was determined after collection of total cell populations following apoptotic stimulus. Cells were collected, washed, and incubated in PI for 10 min in the absence of light. PI positivity was determined with a FACSCalibur flow cytometer using the FL2 channel to determine PI fluorescence, with at least 10,000 events captured. Data were analyzed with CellQuest software. Caspase 3/7 activity was assayed using the Caspase Glo assay (Promega) according to the manufacturer's directions. Crystal violet staining was performed as previously described (16). Terminal deoxynucleotidyltransferase-mediated dUTP-biotin nick end labeling (TUNEL) analysis was performed using the Apo-BrdU TUNEL assay kit (Invitrogen) according to the manufacturer's instructions. TUNEL positivity was gauged on a FACSCalibur flow cytometer as indicated above.
Microscopy and image analysis.
Mitochondria were stained using Mitotracker Red CMXRos (Invitrogen), which was added to cells at 20 nM for 30 min prior to fixation. The cells were fixed with 4% paraformaldehyde (methanol free), washed, and permeabilized with 0.5% Triton X-100. The cells were then blocked in 3% bovine serum albumin (BSA) and stained with primary antibodies according to the manufacturer's recommended dilutions. When necessary, Alexa Fluor-conjugated secondary antibodies (Invitrogen) were used at 1:1,000. Hoechst dye was used to visualize nuclei. For cytochrome c release assays, cells were preincubated with 20 μM zVAD (EMD-Calbiochem) for 2 h before treatment with death-inducing agents. The cells were treated with compound for 20 to 24 h before being fixed as described above. AF-555-conjugated cytochrome c antibody (BD Biosciences) was used at 1:100 for 1 h. Imaging was performed on an epifluorescence microscope (Nikon Eclipse Ti), and images were captured and analyzed using the NIS Elements AR 3.10 software. To quantify cytochrome c release, plates were scored as punctate (0) or diffuse (1) for at least 100 different cells in random fields, and the mean percentage of diffusely stained cells is reported. Quantitation is representative of at least three independent experiments.
Fractionation and mitochondrial purification.
Cells were fractionated as previously described (6). Briefly, HeLa or HeLa S3 cells were pelleted and resuspended in homogenization buffer (10 mM HEPES, pH 7.9, 1.5 mM MgCl2, 10 mM KCl, 1 mM dithiothreitol [DTT], 1 mM phenylmethylsulfonyl fluoride [PMSF]). The cells were allowed to swell for 10 min on ice and were then homogenized using 30 to 50 strokes in a standard glass Dounce homogenizer. A nuclear fraction was collected through centrifugation at 800 × g for 10 min at 4°C. The supernatant was collected and spun at 16,000 × g for 30 min at 4°C. The resulting pellet, the heavy membrane fraction (HMF), contained endoplasmic reticulum (ER), as well as mitochondria. To further isolate pure mitochondria, the crude pellet was soaked in 0.25% trypsin-EDTA for 10 min at 30°C and placed on a density gradient of 31% and 17% Histodenz overlaid with 6% Percoll. The fraction was spun at 100,000 × g for 3 h at 4°C, and pure mitochondria were isolated from the 31%-17% interface.
Digitonin permeabilization and proteinase K (PK) digestion.
Pure mitochondria were isolated as described above. For digitonin permeabilization experiments, mitochondria were resuspended and divided into equivalent fractions. Digitonin was added to the mitochondria to the indicated concentrations, and this mixture was incubated on ice for 10 min before being spun down at 16,000 × g at 4°C. The supernatants were collected and run on an SDS-PAGE gel, transferred to nitrocellulose, and probed with appropriate antibodies. For proteinase K digestion, mitochondria were isolated, resuspended, and divided as described above. Proteinase K (Sigma-Aldrich) was added to the mitochondria to the indicated concentrations, and fractions were placed at 16°C for 30 min. Proteinase K was inactivated through heat inactivation (95°C; 5 min) before samples were prepared and run on SDS-PAGE gels as described above.
Gene set enrichment analysis (GSEA).
Gene expression data generated by the Expression Project for Oncology from a variety of tumors and data from nondiseased tissue samples were obtained from the Gene Expression Omnibus (GSE2109 and GSE3526). All subsequent data processing and analysis were performed using software available from the BioConductor Project (version 2.5) and the R statistical environment v. 2.10.1. Expression values were generated using the robust multichip average (RMA), as implemented in the Affy package (1.24.2) using updated probe set mapping. Gene sets (n = 5,120) were obtained from the Molecular Signatures Database (MsigDB; Broad Institute), and the parametric gene set enrichment method as implemented in the PGSEA package was used to generate enrichment scores for each gene set using corresponding nondiseased tissue as a reference. Gene set enrichment scores that associated with STYXL1 were identified using Spearman's rank correlation. To visualize the fraction of genes that overlapped between the identified gene sets, we calculated a pairwise dissimilarity (D) score between each pair of identified gene sets. The dissimilarity score was used to compute a hierarchical-clustering dendrogram using Euclidean distance with average linkage.
RESULTS
MK-STYX modulates the cellular response to chemotherapeutics.
In an effort to identify enzymes that play a critical role in cell survival, we performed an RNAi screen that targeted all known and putative kinases and phosphatases in the human genome (16). In this screen, we identified a small group of phosphatases whose siRNA-mediated knockdown caused cancer cells to become desensitized to multiple chemotherapeutic agents, each with three different modes of action (paclitaxel, cisplatin, and etoposide). The top hit from this screen was a dual-specificity phosphatase, mitogen-activated protein kinase (MAPK) phosphatase-like serine threonine tyrosine interaction domain (MK-STYX). MK-STYX knockdown was achieved using three unique, nonoverlapping siRNAs, with two of three siRNAs providing effective knockdown of >90% at the mRNA level (Fig. 1 B). Each of these three siRNAs was tested for the ability to protect cells from death after treatment with the microtubule inhibitor paclitaxel, and all three siRNAs provided significant protection against cell death compared to cells transfected with a control siRNA (Fig. 1D). Importantly, the amount of protection from cell death correlates with MK-STYX knockdown; siRNA1 yields only a 60% knockdown and results in approximately 40% cell death in response to paclitaxel (10 nM), while siRNA2 and -3 are more effective at knocking down MK-STYX (96% and 90%, respectively) and promote maintenance of over 90% viability in the presence of paclitaxel (Fig. 1D and E).
Fig. 1.

Knockdown of MK-STYX promotes chemoresistance. (A) HeLa cells were transfected with siRNAs against all human kinases and phosphatases. The cells were treated with chemotherapeutics, and apoptosis was measured by a DNA fragmentation ELISA. Blue dots represent genes whose RNAi knockdown sensitized cells to apoptosis, red dots represent those that decreased the apoptotic response, and purple dots represent no change. MK-STYX is highlighted in teal. (B) Quantitative RT-PCR analysis of MK-STYX mRNA levels after knockdown with three siRNAs targeting unique regions of MK-STYX. The error bars indicate standard deviations. (C) MK-STYX siRNA2 provides potent knockdown of endogenous MK-STYX in the mitochondrial fraction of HeLa cells at the protein level. A nonspecific band serves as a loading control. (D and E) HeLa cells were transfected with control siRNA or three unique MK-STYX siRNA duplexes and assayed for cell viability using the cell titer blue assay (D) or the xCelligence real-time cell viability assay (E) after 10 nM paclitaxel treatment.
To further characterize the chemoresistance induced by knocking down MK-STYX, we determined the 50% inhibitory concentrations (IC50s) of two chemotherapeutics in HeLa cells transfected with a control or MK-STYX-specific siRNA. In control cells, the IC50 for paclitaxel is 25 nM, and that for cisplatin is 20 μM (Fig. 2 A and B). Loss of MK-STYX in HeLa cells confers striking chemoresistance at high doses of chemotherapeutics, with substantial cell viability maintained in doses of chemotherapeutics exceeding 10 times the IC50 for both paclitaxel and cisplatin (Fig. 2A and B). These data demonstrate that loss of MK-STYX imparts a highly chemoresistant phenotype.
Fig. 2.

MK-STYX expression regulates the cellular response to chemotherapeutics. (A and B) HeLa cells transfected with control or MK-STYX-specific siRNA were treated with a dose response of paclitaxel (A) or cisplatin (B), and cell viability was measured 24 h posttreatment using the cell titer blue assay. The error bars indicate standard deviations. (C) Control or MK-STYX siRNA-transfected HeLa cells were treated with 100 nM paclitaxel or 50 μM cisplatin for 24 h, and apoptosis was assayed by a DNA fragmentation ELISA. (D) Control or MK-STYX siRNA-transfected HeLa cells were treated with 100 nM paclitaxel or 50 μM cisplatin for 24 h, and caspase 9 activation was assayed through the appearance of cleaved isoforms via immunoblotting, indicating the initiation of the intrinsic apoptotic cascade. The asterisk indicates a constitutive form of cleaved caspase 9 found even in healthy cells. (E and F) HeLa cells were transfected with MK-STYX cDNA for 24 h. Cell death was measured by propidium iodide uptake (E) and quantified (F). (G) Introduction of exogenous MK-STYX drives caspase 3/7 activation in HeLa cells. (H and I) Introduction of MK-STYX in 293FT cells increases cell death in the presence of both paclitaxel (H) and cisplatin (I), as measured using the cell titer blue assay.
While having distinct mechanisms of action, both paclitaxel and cisplatin are effective anticancer agents due to their abilities to induce intrinsic apoptosis (1, 26). As MK-STYX knockdown cells fail to respond to each of these chemotherapeutics, we predicted that the RNAi-mediated knockdown diminishes the apoptotic response. To confirm that these drugs were inducing apoptosis, we looked at distinct hallmarks of the process, such as DNA fragmentation and caspase activation. In control cells, paclitaxel and/or cisplatin induce both DNA fragmentation and caspase 9 activation, characteristic of apoptotic induction (Fig. 2C and D). In comparison, cells transfected with MK-STYX siRNAs fail to induce caspase 9 activation and do not undergo DNA fragmentation in response to high doses of paclitaxel and cisplatin. These data demonstrate that knockdown of MK-STYX inhibits the apoptotic response in cells treated with chemotherapeutics.
Due to the striking phenotype seen with MK-STYX knockdown, we predicted that the overexpression of MK-STYX would sensitize cells to chemotherapeutic agents. We found that overexpression of MK-STYX is sufficient to induce cell death, as determined by propidium iodide positivity (Fig. 2E and F). Additionally, MK-STYX overexpression is sufficient to induce the activation of caspase 3, an effector caspase critical for the progression of apoptosis (Fig. 2G). Thus, as MK-STYX knockdown inhibits apoptosis in response to paclitaxel and cisplatin, the overexpression of MK-STYX is sufficient to induce an apoptotic program. To address our original hypothesis that the overexpression of MK-STYX could sensitize cells to chemotherapeutics, we moved to the more chemoresistant 293 cell line, which has an IC50 of approximately 125 nM paclitaxel and 125 μM cisplatin. Overexpression of MK-STYX sensitized 293 cells to both paclitaxel and cisplatin (Fig. 2H and I), dropping the IC50 4-fold for each of these drugs. These data further support a role for the expression levels of MK-STYX in the modulation of the cellular response to chemotherapeutics.
MK-STYX is a catalytically inactive phosphatase with a unique function.
We have shown that the knockdown of MK-STYX provides strong desensitization to a variety of chemotherapeutics. As a preliminary attempt to understand its cellular function, we looked to proteins with homology to its coding sequence as a starting point for characterization. MK-STYX was first identified bioinformatically due to its sequence similarity to the MAP kinase phosphatases (MKPs) (29). Like the MKPs, MK-STYX contains both an N-terminal CH2 domain, which is thought to act as a protein-protein interaction motif, and a C-terminal DUSP domain, which carries out the catalysis of dephosphorylation. All DUSPs are characterized by a signature motif, DXnCX5R, in which the cysteine is the critical residue for catalytic activity (25). Interestingly, MK-STYX lacks this critical cysteine and instead contains a conservative substitution of a serine residue within its active site, rendering MK-STYX catalytically inactive. The field of phosphatase biology has exploited this C-to-S substitution as a method for the identification of phosphatase substrates (5). This point mutation converts a phosphatase into a “substrate-trapping mutant” that binds to its appropriate substrates due to conservation of the binding pocket but is unable to dephosphorylate its substrate because of the absence of the catalytic cysteine residue. Due to the natural occurrence of the C-to-S substitution in MK-STYX, we hypothesized that MK-STYX could function as an endogenous substrate-trapping mutant, binding to and altering the activities of phosphoproteins that modulate cell survival pathways.
Early characterization of the MKPs utilized the C-to-S mutation to infer the biological function of these enzymes in the regulation of MAPK signaling. The C-to-S mutation in MKP-1 is sufficient to reverse its role in MAPK regulation; while wild-type MKP-1 counteracts MAPK signaling through the dephosphorylation of ERK1/2 (22), C-to-S-mutated MKP-1 promotes sustained MAPK activation and subsequent downstream MAPK signaling, even in the absence of serum (22). Constitutive ERK1/2 activation is a potent survival signal that is often dysregulated in cancers (21) and has been previously implicated in chemoresistance (15, 18). Given the homology to the MKPs, we sought to determine whether MK-STYX could act in a fashion similar to that of the C-to-S-mutated MKP-1, asking whether MK-STYX could sustain the phosphorylation and activation of ERK1/2 and lead to increased survival and chemoresistance. To test this, HeLa cells were transfected with control or MK-STYX siRNAs and assayed for ERK activation in the presence or absence of epidermal growth factor (EGF). Surprisingly, we found that siRNAs to MK-STYX had little effect on the activation of ERK1/2 (Fig. 3 A). As EGF stimulation of ERK1/2 is also known to modulate cellular proliferation, we sought to determine whether MK-STYX knockdown affected basal proliferation or acute proliferation in response to serum stimulation. Real-time cell proliferation measurements demonstrated no difference in cellular proliferation between control and MK-STYX knockdown cells, either under basal growth conditions (Fig. 3B) or following acute-phase serum stimulation (Fig. 3C). To confirm similar proliferation rates between control and MK-STYX knockdown cells, we measured bromodeoxyuridine (BrdU) incorporation, which remained unchanged (Fig. 3B and C) in cells with loss of MK-STYX.
Fig. 3.

MK-STYX does not modulate MAPK signaling in response to growth factors or apoptotic stress. (A) HeLa cells were transfected with the indicated siRNAs for 48 h, serum deprived for 6 h, and treated with EGF (100 ng/ml) for the indicated times. Cell extracts were then subjected to immunoblotting with phospho-ERK1/2 and total ERK antibodies. (B) Proliferation rates of control and MK-STYX knockdown cells are similar under basal conditions (10% serum) via a real-time viability assay (xCelligence) and by BrdU incorporation (immunofluorescence). The error bars indicate standard deviations. (C) Similar experiments were performed, but after 6 h, the cells were serum deprived, followed by addition of 10% FBS to the growth medium. (D and E) HeLa cells transfected with the indicated siRNAs were exposed to 100 nM paclitaxel, 50 μM cisplatin, or 80 J/m2 of UV for 4 h. The cells were then lysed and immunoblotted with phospho-p38, total p38, phospho-JNK, and total JNK antibodies.
In addition to signaling in response to growth factor stimulation, MAPKs are known to be important regulators of apoptotic induction. Due to the strong association of MK-STYX knockdown and cellular apoptotic potential, we sought to determine whether MK-STYX knockdown affects the phosphorylation and activation of the stress response kinases p38 and JNK. We first looked at p38 and JNK activation in the context of paclitaxel and cisplatin treatment, using conditions similar to those in our apoptotic assays. In response to either paclitaxel or cisplatin, p38 is activated in similar fashions in control and MK-STYX knockdown cells (Fig. 3D). Although both paclitaxel and cisplatin treatments have been shown in the literature to induce JNK activation (19, 27), we found no significant activation of JNK in our experimental system concentrations similar to those used to activate p38. To determine whether JNK activation changes in the presence of MK-STYX knockdown, we UV irradiated cells, a well-characterized and potent activator of JNK signaling. As MK-STYX knockdown protects against UV irradiation-induced apoptosis (Fig. 4 C), we propose that this apoptotic stimulus is a relevant context in which to study JNK activation. Importantly, knockdown of MK-STYX does not change the induction of p38 or JNK phosphorylation in response to UV, demonstrating that these proapoptotic signaling pathways are still intact. Thus, despite significant homology to the MAPK phosphatases, knockdown of MK-STYX does not significantly alter MAPK signaling in growth factor or stress signaling.
Fig. 4.

Knockdown of MK-STYX protects against intrinsic, but not extrinsic, apoptosis. (A, B, D, E, and F) Control or MK-STYX-specific siRNAs were transfected into HeLa cells for 24 h, and the cells were then plated onto an electrode-containing plate (e-plate), where they adhered for 24 h and were then treated with 100 nM paclitaxel (A), 10 ng/ml actinomycin D (B), 0.5% serum (growth factor deprivation) (D), 10 μg/ml tunicamycin (E), and 10 ng/ml TNF-α cotreated with cycloheximide (F). (C) For UV irradiation, cells were transfected with siRNAs for 24 h before exposure to 80 J/m2 of UV irradiation. The cells were immediately collected and plated onto an e-plate, where they were allowed to adhere and proliferate. Cell viability was tracked using the xCelligence real-time viability system. The error bars indicate standard deviations.
MK-STYX protects against intrinsic, not extrinsic, apoptotic signals.
Our data suggest that MK-STYX modulates the chemoresistance potential of cancer cells, but through a mechanism that is distinct from the modulation of prosurvival and proapoptotic MAPK pathways. While we have shown that MK-STYX knockdown protects against three distinct chemotherapeutic agents, we realized that this panel of death-inducing agents had a relatively limited breadth in its ability to uncover mechanism. We predicted that if we could find a specific cytotoxic agent against which MK-STYX knockdown does not protect, this would give us valuable insight into how the loss of this single gene could have such a striking phenotype. To examine this, we sought to determine the viability profile of cells treated with death-inducing agents with distinct and well-characterized mechanisms. We utilized a real-time cell viability assay to determine the kinetics of death in cells treated with a variety of death-inducing agents, including a high dose of paclitaxel, a microtubule-stabilizing agent that induces intrinsic apoptosis; actinomycin D, a transcriptional inhibitor that activates the intrinsic pathway; UV irradiation, which induces single-strand DNA breaks and causes intrinsic apoptosis; growth factor deprivation through extended serum starvation; tunicamycin, an agent that promotes ER stress through the inhibition of N-linked glycosylation; and TNF-α, which activates the extrinsic or death-receptor-linked apoptotic pathway. While cells transfected with a control siRNA were sensitive to all of these treatments (Fig. 4A to F, red data points), knockdown of MK-STYX was sufficient to protect against each of the death-inducing agents that induced intrinsic, or mitochondrion-dependent, apoptosis (Fig. 4A to E, blue data points). Interestingly, however, the knockdown of MK-STYX does not protect against treatment with TNF-α, which is unique in this group, as it is the only experimental condition that triggers the extrinsic apoptotic pathway (Fig. 4F). As inducers of extrinsic apoptosis are the sole experimental condition that is able to trigger cell death in MK-STYX knockdown cells, we sought to further characterize this phenotype.
MK-STYX phenocopies the loss of Bax/Bak and caspase 9 by preventing intrinsic apoptosis.
The results of the intrinsic and extrinsic apoptosis processes are comparable due to the activation of effector caspases, caspases 3 and 7. However, critical upstream processes differ in the activation of these caspases, distinguishing them as two distinct forms of apoptosis. Caspase 8, the mediator of extrinsic apoptosis, is activated by the death-inducing signaling complex (DISC), which is assembled in response to the binding of extracellular proapoptotic ligands to membrane-associated death receptors (10). Caspase 9, the mediator of intrinsic apoptosis, is activated in response to the formation of the apoptosome (cytochrome c and Apaf-1). Importantly, for intrinsic apoptosis to occur, the integrity of the mitochondrial outer membrane must be compromised to allow the release of cytochrome c from within the mitochondrion to the cytosol (8). The proapoptotic proteins Bax and Bak are responsible for the destabilization of the outer mitochondrial membrane and thus act as a regulatory point upstream of initiator caspases during intrinsic apoptosis.
As Bax/Bak, caspase 9, and caspase 8 are critical proteins that control key steps in carrying out either intrinsic or extrinsic apoptosis, we predicted that the RNAi-mediated knockdown of these proteins would be able to differentially inhibit cell death induced by paclitaxel, an intrinsic apoptotic initiator, or TNF-α, an extrinsic apoptotic initiator. Due to the striking protection against intrinsic apoptotic stimuli but sensitivity to extrinsic stimuli, we predicted that loss of MK-STYX would phenocopy the increased cellular viability seen in the Bax/Bak and caspase 9 knockdowns but not that of the caspase 8 knockdown. As shown in Fig. 5 A, paclitaxel (10 nM) treatment causes significant death in cells transfected with a control siRNA. Strikingly, the knockdown of MK-STYX or caspase 9 or the dual knockdown of Bax and Bak was sufficient to completely inhibit this process (Fig. 5A), reaffirming their critical importance in the intrinsic apoptotic pathway. Importantly, the siRNA-mediated knockdown of caspase 8 was not sufficient to inhibit cell death in the presence of paclitaxel, demonstrating its dispensable nature in intrinsic apoptosis. However, RNAi-mediated knockdown of caspase 8 completely protects against cell death caused by TNF-α treatment (Fig. 5B). As expected, the knockdown of caspase 9 did not protect against this inducer of extrinsic apoptosis. Additionally, the knockdown of Bax/Bak or MK-STYX provided only slight, if any, protection against this death-inducing stimulus. These data support a specific role for MK-STYX as a regulator of intrinsic apoptosis and demonstrate that loss of MK-STYX phenocopies the inhibition of cell death provided by both caspase 9 and Bax/Bak knockdown.
Fig. 5.
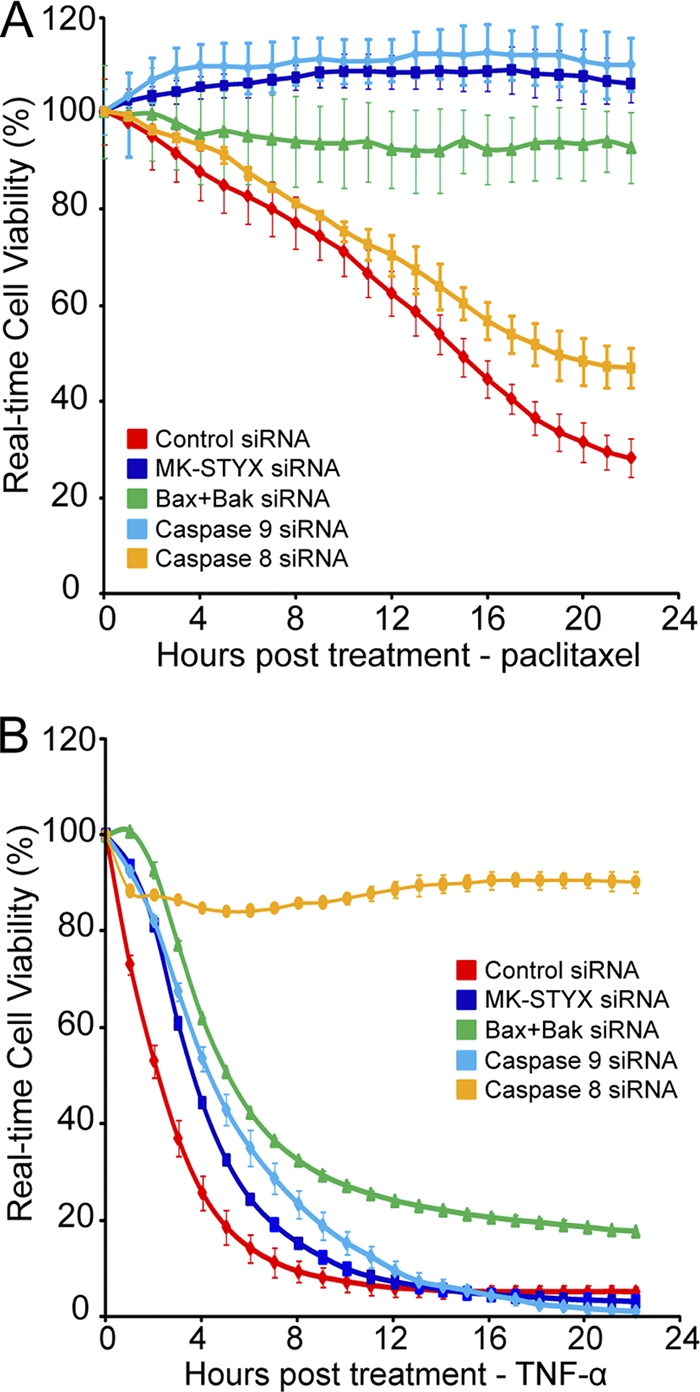
MK-STYX knockdown phenocopies Bax/Bak and caspase 9 knockdown. (A and B) HeLa cells were transfected with control or MK-STYX, Bax plus Bak1, caspase 8, or caspase 9 siRNA for 24 h. The cells were plated onto a 96-well e-plate and allowed to adhere and proliferate for 24 h. The cells were treated with either 10 nM paclitaxel (A) or 1 μg/ml TNF-α and cycloheximide (B) to induce intrinsic or extrinsic apoptosis, respectively. Cell viability was tracked in real time using the xCelligence system. The error bars indicate standard deviations.
MK-STYX knockdown prevents cytochrome c release.
While knockdown of MK-STYX resulted in sustained viability similar to both Bax/Bak and caspase 9 knockdowns, these proteins have critically different roles in the onset of apoptosis. We sought to determine whether the loss of MK-STYX performed a role similar to that of Bax/Bak, by inhibiting apoptosis at the level of cytochrome c release, or similar to that of caspase 9, through the inhibition of apoptosome formation and/or the activation of downstream caspases. This is a critical distinction, as the release of cytochrome c is considered the “point of no return” in the apoptotic process.
We initially hypothesized that the loss of MK-STYX blocks cytochrome c release. To test this, we examined the localization of endogenous cytochrome c using immunofluorescence. We transfected cells with a control or MK-STYX-specific siRNA for 48 h and followed this with treatment of a cisplatin dose response. We followed the release of cytochrome c in both control cells kinetically at 0, 4, 8, 16, and 24 h posttreatment. No significant release of cytochrome c was seen in either control or MK-STYX knockdown cells at 4 or 8 h after cisplatin treatment at any dose (data not shown). However, 16 h after cisplatin (25 μM) treatment, a significant population of control cells had released cytochrome c (Fig. 6 A), whereas the majority of MK-STYX knockdown cells (Fig. 6B) had not undergone cytochrome c release (54% versus 8% of cells, respectively). This result was extended to 24 h posttreatment, with 74% of control cells releasing cytochrome c compared with only 17% of MK-STYX knockdown cells (Fig. 6C). Significant protection from cisplatin-induced cytochrome c release was also observed in MK-STYX knockdown cells in response to increasing doses of cisplatin (Fig. 6D). The population of control cells undergoing cytochrome c release grew in response to increasing cisplatin doses from a basal rate of 11% release to 24%, 71%, and 87% of cells releasing cytochrome c in response to 10, 25, and 50 μM cisplatin, respectively. MK-STYX knockdown cells had a lower basal rate of cytochrome c release, near 5%, with only modest increases in cytochrome c release in response to chemotherapeutics: to 8% after 10 μM treatment, 17% after 25 μM treatment, and 21% after 50 μM treatment. Similar inhibition of cytochrome c release was found in the presence of multiple inducers of intrinsic apoptosis (Fig. 6D), suggesting that the knockdown of MK-STYX phenocopies the loss of prosurvival proteins Bax and Bak by inhibiting mitochondrial outer membrane permeabilization.
Fig. 6.

Knockdown of MK-STYX blocks cytochrome c release under intrinsic apoptotic conditions. (A and B) HeLa cells were transfected with control (A) or MK-STYX (B) siRNA and preincubated with zVAD (20 μM) for 2 h before exposure to 25 μM cisplatin for the indicated times. Cytochrome c release was visualized by loss of punctate red fluorescence within the mitochondria, as indicated by the arrows. (C, D, and E) Quantification of cytochrome c-releasing cells in response to a kinetic time course of 25 μM cisplatin treatment (C; visualized in panels A and B); cytochrome c release in response to dose responses of 0, 10, 25, and 50 μM cisplatin for 24 h (D); and in response to 100 nM paclitaxel, 25 μM cisplatin, 10 ng/ml actinomycin D (act D), or 80 J/m2 UV (E) for 24 h. In all three panels, cytochrome c release was quantified through blind scoring of the number of cells per field (>100 cells). The error bars indicate standard deviations.
Loss of MK-STYX blocks BH3-only-induced apoptosis.
Though Bax and Bak are the main effector molecules regulating mitochondrial outer membrane permeabilization, regulation of Bax/Bak homo-oligomerization is tightly controlled through the induction of proapoptotic Bcl-2 family members known as BH3-only proteins. Although there is some controversy as to how, on a molecular level, BH3-only proteins are able to trigger Bax and Bak activation, there is little disagreement about their proapoptotic effects. It is well known that two distinct BH3-only proteins, Bim and Bid, can directly interact with and potently trigger MOMP through activation of Bax and Bak (7, 14). Additionally, overexpression of Bax itself is sufficient to trigger cytochrome c release through self-activation and homo-oligomerization (4).
As with all regulatory networks, there is some specificity as to which BH3-only proteins are activated in different apoptotic contexts. While Bim is activated in response to mostly intrinsic apoptotic stimuli (20, 23), Bid is activated only in the presence of an extrinsic apoptotic stimulus. Specifically, upon activation at the DISC, caspase 8 is able to cleave full-length Bid, allowing the release of its C-terminal fragment, cBid, which then translocates to the mitochondria to activate Bax and Bak and induce MOMP. Bid activation is required for extrinsic cell death only in certain cell types; in some cells, caspase 8 activation alone is capable of inducing a full apoptotic program through the direct activation of effector caspases, such as caspase 3.
MK-STYX knockdown cells are sensitive to extrinsic-apoptosis-inducing stimuli from the panel of death-inducing agents that were tested. Since MK-STYX knockdown cells are protected against cytochrome c release, we predicted that MK-STYX knockdown cells could be selectively sensitive to Bid-induced apoptosis, as this is a specific control point in the extrinsic apoptotic process at which MOMP is involved. To test this, we overexpressed three proteins known to induce apoptosis: BimS (a constitutively apoptotic isoform of Bim) and tBid (a truncated mutant equivalent to cBid), as well as Bax itself. Cells transfected with control siRNAs were highly sensitive to overexpression of each of these three proteins, as demonstrated by a striking loss of crystal violet staining relative to a vector-only control (Fig. 7 A, middle), as well as significant TUNEL positivity (Fig. 7D, black bars). Conversely, 48 h after knockdown of MK-STYX, HeLa cells had virtually no cell death after overexpression of any of the three prodeath molecules (Fig. 7A, right, and D, white bars). To ensure that these three proteins were still expressed, we blotted for Bax, Bid, and Bim after overexpressing these constructs in HeLa cells. MK-STYX knockdown cells showed equivalent, if not higher, expression levels of all three proteins. Surprisingly, a significantly higher level of Bax was found in MK-STYX knockdown cells, even at the endogenous level (Fig. 7B, top), suggesting that the apoptotic resistance provided to these cells by knockdown of MK-STYX allows higher expression of classically proapoptotic proteins.
Fig. 7.

Knockdown of MK-STYX protects against BH3-only-induced apoptosis. (A) HeLa cells were transfected with control or MK-STYX siRNA for 48 h before transfection with vector, Bax, tBid, or BimS cDNA. Twenty-four hours after transfection, crystal violet was used to stain only viable cells. (B) The cell lysates detailed in panel A were collected and immunoblotted for expression levels of each overexpressed protein; tubulin served as a loading control. (C) GFP-tagged tBid was transfected into control and MK-STYX knockdown HeLa cells for 24 h. Cytochrome c was visualized by immunofluorescence (red); loss of punctate cytochrome c staining is indicated by the arrows. (D) Cells transfected with siRNAs and the indicated cDNAs were assayed for apoptosis through TUNEL positivity. The error bars indicate standard deviations.
As BH3-only proteins ultimately function to activate Bax and Bak and thus trigger MOMP, we had two main hypotheses as to how MK-STYX knockdown could protect cells against the overexpression of proapoptotic Bcl-2 family members. As MK-STYX knockdown cells blocked cytochrome c release in response to chemotherapeutic agents, we first predicted that, despite high expression levels, BimS and tBid would be insufficient to induce MOMP in MK-STYX knockdown cells, and thus, cytochrome c would not be released. Additionally, it is known that BH3-only proteins, as well as Bax itself, are required to translocate to the mitochondrial outer membrane upon apoptotic stimulus. Thus, one potential outcome is that these BH3-only proteins would be unable to translocate to the mitochondrial outer membrane. To determine both the subcellular localization of the expressed BH3-only proteins and cytochrome c localization, we tagged tBid and BimS with green fluorescent protein (GFP) and expressed them in cells. As expected, GFP-positive cells expressing tagged tBid underwent cytochrome c release and had condensed chromatin indicative of cells undergoing apoptosis (Fig. 7C, top). Notably, the untransfected GFP-negative cells had intact mitochondrial staining (red), indicating mitochondrion-retained cytochrome c, and appeared nonapoptotic. Similarly, MK-STYX knockdown cells had a tubular network of cytochrome c staining that overlapped with GFP-tBid, resulting in yellow merged staining (Fig. 7C, bottom). This illustrates two important findings: first, MK-STYX knockdown cells do not release cytochrome c in response to overexpression of BH3-only proteins, and second, GFP-tagged tBid seems fully competent to localize to the mitochondria. Upon translocation to the outer mitochondrial membrane, it has been shown that tBid binds to Bax, leading to conformational changes in Bax that allow its insertion into the membrane, subsequent homo-oligomerization, and MOMP to proceed (14). The mitochondrial localization of GFP-tBid in MK-STYX knockdown cells suggests that a molecular event in this process is being disrupted, preventing the release of cytochrome c.
MK-STYX localizes to the mitochondria.
To gain insight into how, on a mechanistic level, MK-STYX could be modulating the apoptotic potential of a cancer cell, we looked at its subcellular localization. Based on our data showing that MK-STYX knockdown blocks mitochondrial outer membrane permeabilization, we predicted that MK-STYX may localize within or translocate to or from the mitochondrion. To test this, we expressed a V5 epitope-tagged MK-STYX cDNA in HeLa cells and isolated crude mitochondrial fractionations. As shown in Fig. 8 A, V5–MK-STYX specifically localizes to the mitochondrion-enriched fraction of cells. As the overexpression of MK-STYX promotes apoptosis, we predicted that MK-STYX may be able to translocate. To examine this, we again overexpressed V5–MK-STYX in HeLa cells and determined its subcellular localization 24 h after UV irradiation. After UV irradiation, most of the cells had undergone morphological changes associated with apoptosis (data not shown). Additionally, Bax, a protein located in the cytosol in healthy cells, had translocated to the mitochondria of the cells, where it presumably induced MOMP (Fig. 8A). V5–MK-STYX is retained in the mitochondrion-rich fraction of the UV-irradiated cells, suggesting that while MK-STYX localizes to the mitochondria, cytosolic translocation upon the induction of MOMP is not its proapoptotic mechanism of action.
Fig. 8.

MK-STYX localizes to the mitochondria. (A) V5–MK-STYX was transfected into HeLa cells, and cellular compartments were isolated using differential centrifugation: WCL, whole-cell lysate; HMF, heavy membrane fraction/mitochondrion enriched; MEF, microsome-enriched fraction; and S100, cytosol. The fractions were probed with specific markers of each cellular compartment: AIF (mitochondria), calnexin (microsomes), and S6 (cytosol). MK-STYX localization was examined basally (top four gels) and in the context of UV irradiation (bottom two gels). Apoptotic stress is indicated by the translocation of Bax from the cytosol (S100) to the mitochondria (HMF) (bottom gel). (B) The mitochondrial localization of MK-STYX was confirmed using an endogenous antibody raised against MK-STYX (green), which was overlaid with Mitotracker (red; overlay, yellow). (C) Mitochondria were purified from the heavy membrane fraction using a density gradient. DPM, density-purified mitochondria. Pure fractions were probed with calnexin (microsomes) and NDUFS3 (mitochondria) and an antibody against endogenous MK-STYX. (D) Plot of gene set enrichment statistics and STYXL1 expression across tumor gene expression data. MK-STYX has a significant correlation (P < 0.00005) with the “upregulated with PPARγ agonist” gene expression set (R = 0.68), which is highly enriched in mitochondrial genes. (E and F) MK-STYX sublocalization was assayed using increasing digitonin concentrations to differentially release mitochondrial proteins from different compartments (E) and using a dose response of proteinase K, which can access different mitochondrial compartments as concentrations increase (F).
To confirm that MK-STYX is localized to the mitochondria, we performed an immunofluorescence assay with an antibody specific to MK-STYX. Figure 8B confirms the mitochondrial localization of MK-STYX, with almost complete overlap of the endogenous antibody (green) with Mitotracker, a mitochondrion-specific dye (red; the overlap is yellow). Additionally, we performed density gradient purification to gather a pure mitochondrial fraction. Endogenous MK-STYX can be detected in the crude mitochondrially enriched fraction and is also enriched in a pure fraction of mitochondria devoid of microsomal contamination (Fig. 8C). Further, we utilized GSEA to determine whether a significant correlation exists between MK-STYX expression and over 1,850 previously curated gene expression signatures across over 30 different tissue types. We found that the gene sets associated with MK-STYX expression are positively correlated with mitochondrial function and biogenesis (R = 0.68; P < 0.00001) (Fig. 8D), suggesting that MK-STYX could be coregulated with mitochondrial gene sets.
The mitochondrial association of MK-STYX, along with its potent inhibition of chemotherapeutics- and BH3-only-induced cytochrome c release, suggested that MK-STYX may directly modulate the actions of Bax and/or Bak at the outer mitochondrial membrane. To further understand the submitochondrial compartment in which MK-STYX could localize, we performed biochemical experiments exploiting digitonin, a lipid that preferentially solubilizes the outer mitochondrial membrane at low concentrations. In the presence of 0.1 mg/ml digitonin, soluble intermitochondrial membrane space (IMS) proteins rapidly begin to release the mitochondria, with OMM-associated proteins solubilizing at 0.5 mg/ml (Fig. 8E). Notably, proteins contained within the mitoplast, defined as the inner mitochondrial membrane (IMM) and its internal matrix (M) compartment, are not released until very high levels of digitonin (2 mg/ml) are reached. MK-STYX is not released from mitochondria at low to mid-range digitonin levels, suggesting that it is neither localized in the OMM nor a soluble IMS protein.
To further confirm the internal mitochondrial localization of MK-STYX, we performed PK digestion, which degrades proteins in each mitochondrial compartment relative to its concentration. At low concentrations of PK, the enzyme is sufficient only to digest OMM proteins that are readily accessible. As concentrations increase, PK gains access to and digests proteins of the IMS, IMM, and matrix. As demonstrated by molecular markers of these compartments, the OMM protein Bcl-2 is digested at 50 ng/μl PK, while IMS and matrix components are preserved. At 100 ng/μl PK, the soluble IMS protein Smac is digested, but the matrix-associated protein Hsp60 is protected from digestion. Only at the highest concentration of PK used, 500 ng/μl, are both Hsp60 and MK-STYX completely digested (Fig. 8F).
The digitonin permeabilization data, as well as the proteinase K data, thus suggest that MK-STYX resides within the mitochondria, probably associated with the mitoplast (IMM plus matrix) fraction. While we cannot conclude that MK-STYX exclusively resides within either the IMM or the matrix, these data suggest that MK-STYX likely does not reside within the OMM or the IMS of the mitochondria.
DISCUSSION
Here, we report that MK-STYX is a mitochondrial dual-specificity phosphatase critical for controlling the apoptotic potential of the mitochondria. Our results show that the RNAi-mediated loss of MK-STYX provides a specific block during mitochondrion-dependent apoptosis, allowing cells to survive high doses of chemotherapeutics. Our data further demonstrate that the MK-STYX knockdown phenotype mirrors the knockdown of proapoptotic proteins critical to carrying out intrinsic apoptosis, such as Bax/Bak and caspase 9. Bax/Bak and caspase 9 are known to modulate different control points of apoptosis, specifically, the release of cytochrome c and the activation of effector caspases, respectively. This is an important distinction, as cells are able to recover from apoptotic stimuli only if they have not yet undergone MOMP; even in the continuous presence of caspase inhibitors, cells will undergo a nonclassical form of cell death, often referred to as caspase-independent cell death, if MOMP has occurred (24). This has important implications for chemoresistance: in order to maintain clonogenicity and retain the capacity to divide, the block in apoptosis would have to be at or above the level of MOMP. The knockdown of MK-STYX protects against UV irradiation for at least 5 days after initial insult (Fig. 4E), supporting the notion that these cells have, in fact, maintained their ability to grow and divide well after apoptotic insult, demonstrating that they retain their clonogenicity. This confirms that the knockdown of MK-STYX inhibits the release of cytochrome c and thus mechanistically phenocopies the dual knockdown of Bax/Bak. As the downregulation of apoptosis, specifically through the blocking of cytochrome c release, has been characterized as a mechanism of acquired drug resistance in tumors (11), this phenotype provides a mechanistic explanation of how loss of MK-STYX can render cells unresponsive to a variety of apoptosis-inducing stimuli and suggests that loss of MK-STYX in tumors could provide a potent mechanism to evade apoptosis.
The cellular decision to undergo MOMP is controlled at the level of Bcl-2 family members. Bax and Bak are the proapoptotic effectors of the Bcl-2 family, and their activation (or lack thereof) is the ultimate determination of whether a cell will undergo MOMP, thus committing to cell death. Whether these effector proteins are engaged depends mainly upon activation of the proapoptotic BH3-only proteins. The BH3-only proteins, Bim and Bid, have been demonstrated to be able to directly interact with Bax, initiating its translocation to the mitochondria and subsequent activation (7, 14). Elegant work utilizing fluorescence markers has demonstrated that tBid interacts with and activates Bax in an ordered manner (14). tBid first binds the outer mitochondrial membrane and, in doing so, recruits soluble Bax to the mitochondria. This is followed by Bax insertion into the membrane and homo-oligomerization, forming pores within the membrane to facilitate MOMP. We have shown that expression of tBid in MK-STYX knockdown cells is insufficient to cause cell death. Importantly, tBid is mitochondrion associated in these cells, but cytochrome c has failed to release. Given the mechanistic studies outlined above, this suggests that a deficiency in Bax recruitment, insertion into the membrane, and/or homo-oligomerization is responsible for this block in apoptosis.
Upon uncovering these data, we predicted that MK-STYX lay on or proximal to the outer mitochondrial membrane, and directly acted on the membrane and/or these proapoptotic Bcl-2 family members to facilitate MOMP. Biochemical assays, such as digitonin permeabilization and proteinase K digestion, suggest that MK-STYX is in neither the OMM nor the IMS, but rather, is contained in or tightly associated with the inner mitochondrial compartment.
It is unclear how a protein associated with the inner mitochondrial compartment could modulate such a specific block in the permeabilization of the outer mitochondrial membrane. To our knowledge, no proteins associated with the mitoplast have been definitively demonstrated to control the apoptotic process. Interestingly, a few studies have implicated proteins within the oxidative phosphorylation cascade, such as the F0F1-ATPase, as critical for the induction of apoptosis (4, 17). While the mechanisms by which these inner mitochondrial membrane proteins could be modulating the apoptotic process remain to be elucidated, it is interesting to speculate that MK-STYX could affect the apoptotic program of the cell in a similar fashion.
Overall, we have used a systems biology approach to identify a novel regulator of mitochondrion-dependent apoptosis. We initially performed a high-throughput RNAi screen against all kinases and phosphatases in the human genome. The gene with the most significant phenotype, MK-STYX, was characterized for its molecular function. Although the literature suggested that MK-STYX could function to regulate MAPK signaling, we have shown that this is unlikely. Rather, we have characterized MK-STYX as a mitochondrial gene that is essential for mitochondrial outer membrane permeabilization, and thus, a master regulator of cellular apoptotic potential. This collection of biological approaches highlights the potential for high-throughput experiments to identify and elucidate the functions of uncharacterized proteins in the genome. In the human genome postsequencing era, it is easy to appreciate the significant fractions of genes with no known cellular function. We propose that RNAi screens, combined with biochemical and cell biological methods, will allow elucidation of the functions of many of these uncharacterized genes.
ACKNOWLEDGMENTS
We thank the Van Andel Institute Systems Biology and Computational Biology laboratories for advice, analysis, and reagents.
This work was supported by award number R01CA138651 from the National Cancer Institute to J.P.M.
Footnotes
Published ahead of print on 24 January 2011.
REFERENCES
- 1. Bhalla K. N. 2003. Microtubule-targeted anticancer agents and apoptosis. Oncogene 22:9075–9086 [DOI] [PubMed] [Google Scholar]
- 2. Chipuk J. E., Green D. R. 2008. How do BCL-2 proteins induce mitochondrial outer membrane permeabilization? Trends Cell Biol. 18:157–164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Chipuk J. E., Moldoveanu T., Llambi F., Parsons M. J., Green D. R. 2010. The BCL-2 family reunion. Mol. Cell 37:299–310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Eskes R., et al. 1998. Bax-induced cytochrome C release from mitochondria is independent of the permeability transition pore but highly dependent on Mg2+ ions. J. Cell Biol. 143:217–224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Flint A. J., Tiganis T., Barford D., Tonks N. K. 1997. Development of “substrate-trapping” mutants to identify physiological substrates of protein tyrosine phosphatases. Proc. Natl. Acad. Sci. U. S. A. 94:1680–1685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Fritz D. T., Ford L. P., Wilusz J. 2000. An in vitro assay to study regulated mRNA stability. Sci. STKE 2000:pl1. [DOI] [PubMed] [Google Scholar]
- 7. Gavathiotis E., et al. 2008. BAX activation is initiated at a novel interaction site. Nature 455:1076–1081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Green D. R., Kroemer G. 2004. The pathophysiology of mitochondrial cell death. Science 305:626–629 [DOI] [PubMed] [Google Scholar]
- 9. Hanahan D., Weinberg R. A. 2000. The hallmarks of cancer. Cell 100:57–70 [DOI] [PubMed] [Google Scholar]
- 10. Kischkel F. C., et al. 1995. Cytotoxicity-dependent APO-1 (Fas/CD95)-associated proteins form a death-inducing signaling complex (DISC) with the receptor. EMBO J. 14:5579–5588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kojima H., et al. 1998. Abrogation of mitochondrial cytochrome c release and caspase-3 activation in acquired multidrug resistance. J. Biol. Chem. 273:16647–16650 [DOI] [PubMed] [Google Scholar]
- 12. Kroemer G., Galluzzi L., Brenner C. 2007. Mitochondrial membrane permeabilization in cell death. Physiol. Rev. 87:99–163 [DOI] [PubMed] [Google Scholar]
- 13. Lindsten T., et al. 2000. The combined functions of proapoptotic Bcl-2 family members bak and bax are essential for normal development of multiple tissues. Mol. Cell 6:1389–1399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lovell J. F., et al. 2008. Membrane binding by tBid initiates an ordered series of events culminating in membrane permeabilization by Bax. Cell 135:1074–1084 [DOI] [PubMed] [Google Scholar]
- 15. MacKeigan J. P., Collins T. S., Ting J. P. 2000. MEK inhibition enhances paclitaxel-induced tumor apoptosis. J. Biol. Chem. 275:38953–38956 [DOI] [PubMed] [Google Scholar]
- 16. MacKeigan J. P., Murphy L. O., Blenis J. 2005. Sensitized RNAi screen of human kinases and phosphatases identifies new regulators of apoptosis and chemoresistance. Nat. Cell Biol. 7:591–600 [DOI] [PubMed] [Google Scholar]
- 17. Matsuyama S., Xu Q., Velours J., Reed J. C. 1998. The Mitochondrial F0F1-ATPase proton pump is required for function of the proapoptotic protein Bax in yeast and mammalian cells. Mol. Cell 1:327–336 [DOI] [PubMed] [Google Scholar]
- 18. McCubrey J. A., et al. 2007. Roles of the Raf/MEK/ERK pathway in cell growth, malignant transformation and drug resistance. Biochim. Biophys. Acta 1773:1263–1284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Potapova O., et al. 1997. The Jun kinase/stress-activated protein kinase pathway functions to regulate DNA repair and inhibition of the pathway sensitizes tumor cells to cisplatin. J. Biol. Chem. 272:14041–14044 [DOI] [PubMed] [Google Scholar]
- 20. Puthalakath H., et al. 2007. ER stress triggers apoptosis by activating BH3-only protein Bim. Cell 129:1337–1349 [DOI] [PubMed] [Google Scholar]
- 21. Roberts P. J., Der C. J. 2007. Targeting the Raf-MEK-ERK mitogen-activated protein kinase cascade for the treatment of cancer. Oncogene 26:3291–3310 [DOI] [PubMed] [Google Scholar]
- 22. Sun H., Charles C. H., Lau L. F., Tonks N. K. 1993. MKP-1 (3CH134), an immediate early gene product, is a dual specificity phosphatase that dephosphorylates MAP kinase in vivo. Cell 75:487–493 [DOI] [PubMed] [Google Scholar]
- 23. Sunters A., et al. 2003. FoxO3a transcriptional regulation of Bim controls apoptosis in paclitaxel-treated breast cancer cell lines. J. Biol. Chem. 278:49795–49805 [DOI] [PubMed] [Google Scholar]
- 24. Tait S. W., Green D. R. 2008. Caspase-independent cell death: leaving the set without the final cut. Oncogene 27:6452–6461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Tonks N. K. 2006. Protein tyrosine phosphatases: from genes, to function, to disease. Nat. Rev. Mol. Cell Biol. 7:833–846 [DOI] [PubMed] [Google Scholar]
- 26. Wang D., Lippard S. J. 2005. Cellular processing of platinum anticancer drugs. Nat. Rev. Drug Discov. 4:307–320 [DOI] [PubMed] [Google Scholar]
- 27. Wang T. H., et al. 1999. Microtubule dysfunction induced by paclitaxel initiates apoptosis through both c-Jun N-terminal kinase (JNK)-dependent and -independent pathways in ovarian cancer cells. J. Biol. Chem. 274:8208–8216 [DOI] [PubMed] [Google Scholar]
- 28. Wei M. C., et al. 2001. Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science 292:727–730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wishart M. J., Dixon J. E. 1998. Gathering STYX: phosphatase-like form predicts functions for unique protein-interaction domains. Trends Biochem. Sci. 23:301–306 [DOI] [PubMed] [Google Scholar]
- 30. Zou H., Henzel W. J., Liu X., Lutschg A., Wang X. 1997. Apaf-1, a human protein homologous to C. elegans CED-4, participates in cytochrome c-dependent activation of caspase-3. Cell 90:405–413 [DOI] [PubMed] [Google Scholar]