

Abstract
p73 is a tumor suppressor belonging to the p53 family of transcription factors. Distinct isoforms are transcribed from the p73 locus. The use of 2 promoters at the N-terminus allows the expression of an isoform containing (TAp73) or not containing (ΔNp73) a complete N-terminal transactivation domain, with the latter isoform capable of a dominant negative effect over the former. In addition, both N-terminal variants are alternatively spliced at the C-terminus. TAp73 is a bona fide tumor suppressor, being able to induce cell death and cell cycle arrest; conversely, ΔNp73 shows oncogenic properties, inhibiting TAp73 and p53 functions. Here, we discuss the latest findings linking p73 to cancer. The generation of isoform specific null mice has helped in dissecting the contribution of TA versus ΔNp73 isoforms to tumorigenesis. The activity of both isoforms is regulated transcriptionally and by posttranslational modification. p73 dysfunction, particularly of TAp73, has been associated with mitotic abnormalities, which may lead to polyploidy and aneuploidy and thus contribute to tumorigenesis. Although p73 is only rarely mutated in cancer, the tumor suppressor actions of TAp73 are inhibited by mutant p53, a finding that has important implications for cancer therapy. Finally, we discuss the expression and role of p73 isoforms in human cancer, with a particular emphasis on the neuroblastoma cancer model. Broadly, the data support the hypothesis that the ratio between TAp73 and ΔNp73 is crucial for tumor progression and therapeutic response.
Keywords: p73, mutant p53, mitosis, knockout mice, neuroblastoma, rhabdomyosarcoma
p73: Gene Structure and Isoforms
p73 is a transcription factor and a member of the p53-family, which includes p53 itself and p63. The human Trp73 gene was cloned more than 10 years ago by McKeon’s group, who identified its chromosomal location at 1p36, a region frequently deleted in tumors such as neuroblastoma and other late stage human cancers,1 thus immediately linking p73 to cancer.
Because of alternative promoter usage and C-terminal alternative splicing, all p53 family members are expressed in a number of isomeric forms. All p53 family genes contain the same modular domain structure, including an amino-terminal transactivation domain (TA), a DNA-binding domain, and a carboxy-terminal oligomerization domain.2 The particular complexity of the p73 locus results both from the use of 2 major transcriptional start sites and from C-terminal splicing. Biologically, the most distinctive characteristic arises from alternative promoter usage: transcription from the most upstream P1 promoter generates the transactivating (TAp73) isoforms, whereas the second downstream promoter P2, situated within an alternative intron 3′, yields amino-terminal truncated proteins that lack the TA domain (δNp73).3-5 Another group of N-terminal truncated p73 isoforms, collectively called ΔTAp73, stems from alternative splicing targeting the N-terminus of the transcript generated from the P1 promoter.5 These products include ΔEx2p73 (lacking exon 2), ΔEx2/3p73 (lacking exons 2 and 3), and ΔN′p73. This latter isoform aberrantly contains the 3′ promoter of ΔNp73 within the transcript of TAp73 and produces a protein indistinguishable from ΔNp73, because its translation starts from the ATG on intron 3′.6 ΔTAp73 isoforms are mainly but not exclusively present in primary tumors and cancer cell lines.
Within the C-terminus of p73 are 2 further domains; an additional TA domain and a transcriptional inhibitory (TI) domain, although because of the C-terminal splicing, these are absent from some C-terminal variants. At least 7 different transcripts (α, β, γ, ζ, δ, ϵ, η) are known to be generated at the C-terminus, expressed on either a TA or ΔN N-terminal sequence, although it is still debatable how many of these are truly expressed as proteins. The longest C-terminal isoform (α), which has been detected as a protein, contains a sterile-α-motif (SAM) domain that mediates protein–protein interactions that are essential for tetramerization and thus for the formation of the active molecule.7 This domain, absent in p53, is important for intermolecular aggregation in both p63 and p73, as best shown for p63.8 Here, the p63-TI domain can bind the N-terminal TA domain and inhibit its activity in cis.8,9 As a consequence, the ability of TAp63α to trigger transcription of target genes is reduced when compared with shorter C-terminal isoforms that lack a TI domain. It is clearly important to verify that this internal inhibitory mechanism is also conserved in p73.
A schematic representation of the p73 gene structure is shown in Figure 1.
Figure 1.
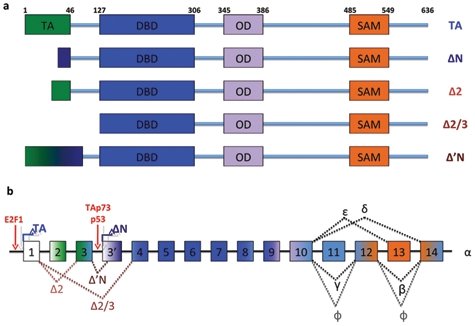
Scheme of the p73 gene structure. Matured spliced isoforms and protein domains positions are shown (a). The structures of the Trp73 locus are depicted, and the exons originating the domains (shown in Figure 3a) are represented (b). Note the binding of E2F on P1 promoter and activation of ΔNp73 by p53 or TAp73. TA = transactivation domain; DBD = DNA binding domain; OD = oligomerization domain.
p73 Isoforms: Tumor Suppressor or Oncogene?
In general, TAp73 and ΔNp73 show opposing roles. Thus, TAp73 is an inducer of cell cycle arrest and apoptosis and largely mimics p53 activities,10,11 and its tumor suppressor function has been confirmed by studies in selective TAp73 null mice (see below).12 This function is inhibited, and consequently survival is promoted, by the dominant negative property of ΔN isoforms (which may therefore be considered oncogenes), exerted either by heteroduplex formation with TAp73 variants or by competition for promoter binding with both p53 and TAp73.13-15 In this context, it is noteworthy that p73 and p53 have structurally dissimilar oligomerization domains.16 As a consequence, p53 and p73 do not form heterotetramers, explaining why ΔNp73 inhibits p53 by competition for promoter binding and not by direct protein–protein interaction. This negative regulation by ΔNp73 forms an autoregulatory feedback loop, since both TAp73 and p53 can induce expression of ΔN isoforms by direct binding to the P2 promoter.17,18
However, in apparent contradiction to this alternative suppressor/oncogene hypothesis, the first total p73 knockout mouse model showed developmental abnormalities but no spontaneous tumors (Fig. 2).3 This is in striking contrast with p53 knockout mice, which show high susceptibility to spontaneous and induced carcinogenesis,19 but this has to be interpreted with some caution due to the limited lifespan of the p73 null mice. It later emerged that the picture was not quite so clear-cut. Indeed, Tyler Jacks’s group found that heterozygosity of p73 substantially shortened the animal’s life span, and half of the p73+/− cohort developed malignant tumors such as lung adenocarcinomas, thymic lymphomas, and hemangiosarcomas by 15 months of age. In addition, p73 heterozygosity led to an increase in benign lesions, such as squamous cell hyperplasia and lung adenomas. Importantly, these tumors show loss of heterozygosity of the remaining WT p73 allele, which is a typical hallmark of tumor suppressor inactivation in cancer. p53+/− mice also develop spontaneous tumors and exhibit a median survival of 10 months. Moreover, survival of double heterozygote p53+/−;p73+/− mice is further reduced (from 10 to 6 months), due to an increased tumor burden and more aggressive metastatic disease. In many of the tumor specimens analyzed, the loss of heterozygosity of p73 was more frequent than the loss of the remaining p53 allele, revealing a potent tumor suppressor function for p73.20
Figure 2.
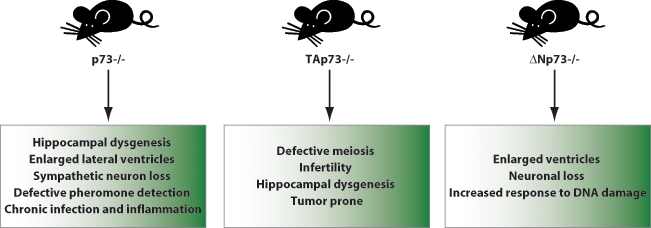
p73 mouse models. Three mouse models for p73 are available. The p73 knockout mouse was developed by targeting the DNA binding domain and thus lacks expression of all p73 isoforms and shows severe developmental defects. Recently, N-terminus selective knockouts have been developed as well: TAp73 and ΔNp73 knockout mice show less severe developmental phenotype, but, importantly, depletion of TAp73 predisposes the animal to spontaneous tumorigenesis.
Nonetheless, the p73 null and heterozygous mouse models used for these studies have a fundamental bias: they target all p73 isoforms, as they were developed by depletion of the common DNA binding domain. This bias has recently been overcome by the generation of N-terminal isoform specific knockouts that have allowed the relative contributions of TAp73 and ΔNp73 to tumorigenesis to be assessed (Figure 2).12,21
TAp73−/− mice indeed show an increased susceptibility to spontaneous and induced carcinogenesis, with more than 70% of the knockout animals developing tumors, mainly lung adenocarcinomas. In contrast, there is no evidence of tumor development in ΔNp73−/− mice, and E1A/Ras-transformed ΔNp73−/− mouse fibroblasts fail to form tumors when transplanted into immunocompromised recipient mice. These findings are in agreement with previously published data showing that overexpression of ΔNp73 in primary mouse fibroblasts facilitates Ras-induced transformation and favors tumor formation in transplantation assays.22 Moreover, overexpression of oncogenic Ras influences the TAp73/δNp73 ratio in favor of δNp73, and the resulting downregulation of TAp73 and upregulation of δNp73 are important for Ras transforming activity.23 Taken together, these findings support a tumor suppressive function for TAp73 and an oncogenic role for N-terminal truncated isoforms. Finally, a study by Nemajerova et al24 addresses the issue of p73 function in B-cell lymphomagenesis. The authors investigated whether p73 loss influences lymphoma development in Em-myc transgenic animals, which overexpress the oncogene c-Myc in B-cell progenitors under the control of the immunoglobulin heavy chain enhancer and develop pre–B-cell and/or B-cell lymphomas. The investigators found that p73 does not act as a prototype tumor suppressor in this setting, as tumor onset and overall mortality were not dependent on the p73 genotype. Nonetheless, depletion of p73 affected the phenotype of the disease by increasing extranodal dissemination. These data, together with the increased metastatic potential of p53+/−;p73+/− derived tumors,20 suggest that p73 may also regulate motility and invasion.
Transcriptional Regulators of p73 and Their Implication in Cancer
E2F1, member of the E2F family of transcription factor, is an important positive regulator of cell cycle progression and an inducer of apoptosis.25 In fact, E2F1 triggers the expression of p14ARF, a potent inhibitor of MDM2, and this results in accumulation of p53 and thereby induction of p53-dependent cell death. E2F1 is also able to induce cell death independently of p53, due to its ability to induce expression of TAp73.26-28 Indeed, the p73 P1 promoter contains 3 E2F1 binding sites, and in vivo binding of E2F-1 to the 2 proximal sites mediates the increased expression of TAp73 transcripts. Physiologically, this has important consequences. First, it has been shown that E2F1-dependent apoptosis was substantially impaired in p73−/− cells.27 Second, the E2F1/p73 axis mediates the response to DNA damage (DDR) and has been implicated in E2F1 therapeutic efficacy in pancreatic cancer. Indeed, treatment of cells with DNA damaging agents (i.e., camptothecin, Adriamycin, daunorubicin) leads to a 2- to 3-fold increase in TAp73 mRNA.29,30 In a pivotal study, Urist and colleagues30 showed that the kinases Chk1/Chk2, which are activated by DNA damage, phosphorylate and stabilize E2F1 that, in turn, induces TAp73, thus leading to DNA-damage–dependent apoptosis. Remarkably, this mechanism is independent of p53.30 Marabese and colleagues31 have added a further detail to this picture, showing that C/EBPα hinders E2F1 induction of p73. According to their data, C-EBPα binds the p73 P1 promoter and directly inhibits E2F1 transactivation. Following DNA damage, C/EBP1α is displaced from this repressor complex, allowing upregulation of TAp73 by E2F.31 Clinical data showing that there is a strong synergism between chemotherapy and E2F expression in the induction of apoptosis and that this also correlates with induction of p73 provide proof of concept that the E2F1/p73 axis may be of relevant therapeutic value.32
As mentioned, apoptosis is not the only outcome of E2F transcriptional activity,25 and the ability of E2F1 to discriminate between cell proliferation or cell death depends on posttranscriptional modification. Indeed, DNA damage induces stabilization and acetylation of E2F1 and this event promotes E2F1 binding to the promoters of proapoptotic genes, including the p73 P1 promoter.33
The first intron of the p73 locus contains a 1Kb sequence, including a consensus element bound by the transcriptional repressor ZEB1. ZEB1 binding to this element can dampen transcription from the p73 promoter, even in the presence of active E2F1.34 Recently, this region has been linked to chemotherapeutic sensitivity depending on BRCA1 status. Indeed, whereas TAp73 mediates cisplatin toxicity in BRCA1-negative cancer cell lines, it has a negligible effect on BRCA1-positive ovarian tumors. This is probably dependent on selective epigenetic methylation of the ZEB1 target sequence in BRCA1-negative tumors. This methylation, whose mechanism remains unknown, abrogates ZEB1 binding, thus allowing TAp73 upregulation in response to chemotherapy.35
Regulation of p73 by Phosphorylation
Three independent studies published in Nature in 1999 established c-Abl as a nonreceptor tyrosine kinase responsible for p73 phosphorylation and stabilization upon DNA damage.36-38 Indeed, DNA damage induces c-Abl phosphorylation of Tyrosine 99 of p73. This event leads to stabilization and activation of p73 and elicits TAp73-induced apoptosis in response to chemotherapy. Indeed, TAp73 fails to accumulate and to induce cell death in c-Abl−/− cells or upon reconstitution with a kinase defective c-Abl. The interaction between p73 and c-Abl is mediated by the src homology 2 (SH2) domain of the kinase and is increased upon Tyr99 phosphorylation. Interestingly, the direct interaction between the 2 proteins is an additional prerequisite for the efficient stabilization of p73.39 Despite the crucial importance of this posttranslational regulation in triggering cell death in response to chemotherapy, the mechanism underpinning p73 stabilization is yet to be clarified. In addition, c-Abl can phosphorylate and stabilize ΔNp73,39 hinting at a paradoxical prosurvival role for c-Abl. However, the significance of this has never been thoroughly investigated. The MAPK pathway is also required for c-Abl–dependent stabilization of p73. Thus, c-Abl activates p38 kinase, and inhibition of p38 impairs p73 stabilization, even upon tyr99 phosphorylation. Importantly, p38 targets a yet unidentified threonine residue on p73, and this is per se sufficient to promote p73 stabilization and to enhance its transcriptional activity.40
In contrast, p73 phosphorylation by cyclin/CDK complexes inhibits p73 activity. Cyclin/CDK complexes are well-known regulators of cell cycle progression and can phosphorylate p73 on a threonine residue (Thr 86), and Thr 86 phosphorylation impairs p73 transcriptional activity by a yet unknown mechanism. The cycD/CDK4 complex, which is involved in G1 progression, is unable to phosphorylate p73, whereas CycA/CDK2, CycE/CDK2, and CycB/cdc2 (involved in S-phase and G2/M progression) phosphorylate p73 both in vivo and in vitro. In consequence, Thr 86 phosphorylation of p73 is cell cycle dependent and is detectable at S phase with a peak during M phase.41 Strikingly, p73 has recently been implicated in regulation of genomic stability and cell survival during mitosis (see below), but a role for Cyc/CDK-dependent phosphorylation of p73 in this setting has not been reported.
p73 Inhibition by Mutant p53 and Regulation by ASPP
We have already discussed the potential interactions between wild-type p53 and p73. What can occur in tumors harboring mutant p53? There is now good evidence that some p53 mutants can bind and sequester p73, thus inhibiting its transactivation and proapoptotic activity.42-44 In this regard, p53 mutants fall into 2 categories. Some, such as 143A, 175H, and 173L, affect p53 conformation, whereas others, such as 179Q and 248W, have a negligible impact on protein conformation. It is interesting that the same 2 classes differentially affect p73: indeed, conformational mutants can bind to, and consequently inhibit, p73 more efficiently than nonconformational mutants. But structure is not the only variable influencing mutant-p53/p73 interaction. More specifically, a p53 polymorphism at codon 72 also dictates binding affinity with p73. Codon 72 of p53 encodes either an arginine or a proline residue (72R and 72P, respectively), and this polymorphic variance has been shown to influence the clinical outcome of cancer patients. Indeed, survival studies showed that patients with point mutations of p53 affecting the 72R variant have a reduced overall survival compared with patients bearing the same mutation but in a 72P polymorphic p53. DNA sequencing of heterozygous patients with both 72R and 72P alleles shows that mutations occur in the 72R allelic variant in the vast majority of subjects, thus indicating a selective advantage for cancer cell bearing p53 mutations within the 72R polymorphic allele. Intriguingly, this allele also shows the highest affinity for p73 and inhibits its transcriptional activity more efficiently than 72P mutants. Thus, Bergamaschi et al44 have shown that p53 mutants inhibit p73-dependent cell death induced by DNA damaging anticancer drugs (i.e., doxorubicin, cisplatinum, Taxol). This inhibition correlates with their binding affinity for p73 and also depends on the polymorphic variant, with codon 72R showing the highest degree of cell death inhibition. Remarkably, mutant p53 acts as a dominant negative toward WT p53 independently of the codon 72 polymorphism, strongly suggesting a central role for p73 in the adverse effects associated with the arginine residue.44 The potential impact of the mutant p53/p73 interaction has recently been reinforced by the findings of Bruno and colleagues45 on Che-1–dependent regulation of mutant p53 and cell survival. Che-1 is a RNA polymerase II binding protein involved in the regulation of gene expression, and previous findings showed that Che-1 also localizes to the p53 promoter and triggers its expression.46 Che-1 is necessary for the sustained expression of mutant p53 in a variety of cancer cell lines, as knock-down of Che-1 by siRNA potently reduces levels of mutant p53. In addition, the concomitant downregulation of DNA repair genes (such as Bloom Syndrome Helicase and Rad17) elicited by Che-1 depletion results in S-phase arrest, accumulation of DNA damage, and consequent stabilization of E2F1. These events unleash TAp73, which, by increasing expression of proapoptotic genes, such as Noxa and Puma, results in apoptosis of cancer cells.
One translational implication of these data is that the effect of any therapeutic approach targeting mutant p53 will depend on the status of p73 in the specific tumors.
In addition to interfamily interaction, other transcriptional regulators finely tune p73 activity. One crucial example is the apoptosis stimulating proteins of p53 (ASPP), which are able to regulate all p53 family members.47,48 The ASPP family consists of 3 members, ASPP1, ASPP2, and iASPP, and by interacting with all p53 family members can act as tumor suppressors.
The Role of p73 in Mitosis and Aneuploidy
Major genomic abnormalities, such as aneuploidy and polyploidy, are well-known features of established tumors, and it has often been implied that these are necessary for tumor development.
Several reports12,49-52 have consistently linked p73, especially TAp73, to mitosis and maintenance of genomic stability. In fact, in the absence of p73, primary cells rapidly become polyploid. The impact of this and its precise mechanism, however, remain debatable. Talos et al50 have reported that loss of p73 induces a rapid increase in cell ploidy that is counteracted by activation of p53, as the authors could detect persistent genomic instability only after depletion of both p73 and p53. When seeking a molecular mechanism to explain this phenotype, they failed to detect any obvious abnormality in mitotic function but found aberrant activation of cyclin/CDK complexes during S phase, which led to delayed S phase and endoreduplication and, thus, polyploidy. In addition, p73−/−;p53−/− fibroblasts fail to arrest in S/G2 after DNA damage, and this failure correlates with increased aberrant DNA synthesis and widespread aneuploidy in damaged cells.50 However, other groups have obtained substantially different results,12,51,52 as they detected polyploidy and aneuploidy in p73 null MEFs independently of p53 status. They also described mitotic defects such as lagging chromosomes and aberrant mitotic spindle formation. These defects are dependent on TAp73, as they were detected in oocytes and fibroblasts from TAp73 selective knockout mice.12 Also, the authors showed that TAp73 localizes to the mitotic spindle and interacts with the spindle assembly checkpoint (SAC) proteins, such as Bub1, Bub3, and BubR1.51,52 SAC is a regulatory multiprotein complex that senses the improper attachment of sister chromatids to the mitotic spindle and delays anaphase until all chromosomes are correctly oriented for segregation.53 Consequently, a failure in the SAC results in aberrant mitosis and aneuploidy. Loss of TAp73 impairs SAC function and promotes aneuploidy by directly influencing efficiency and accuracy of mitosis (Fig. 3A, 3B).
Figure 3.
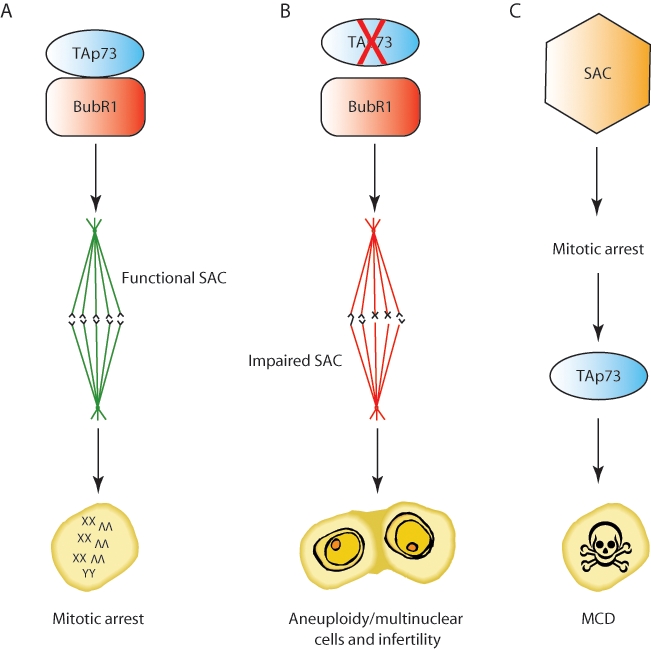
p73 role in mitosis. p73 interaction with BubR1 is shown as a prerequisite for functional SAC activity (A). In the absence of TAp73, a dysfunctional SAC fails to arrest cells in mitosis, leading to aneuploidy, cancer, and infertility (B). TAp73 is also necessary to trigger mitotic cell death upon prolonged mitotic arrest or in the presence of reduced Bub1 levels. This cell death depends on SAC activity, but whether direct interaction between TAp73 and SAC components plays a role is unclear (C). SAC = spindle assembly checkpoint; MCD = mitotic cell death.
Finally, p73 regulates cell death triggered by prolonged arrest in mitosis or by defective activity of the SAC, and this forms a safeguard mechanism that prevents aberrant mitosis and genomic instability. Two different cell death mechanisms have been described, either caspase dependent or independent, and both require p73.54-56 Partial depletion of Bub1, a SAC component, does not impair activation of the SAC and mitotic arrest, and Bub1–partially depleted cells are still able to arrest in mitosis after treatment with spindle inhibitory drugs such as nocodazole and Taxol. Nonetheless, these cells undergo mitotic cell death, showing extensive TUNEL positivity in prophase, prometaphase, and metaphase. This cell death is known as caspase-independent mitotic cell death (CIMD), as caspase activation is dispensable.54 In addition, whereas p53 is not essential for CIMD, depletion of p73 rescues lethality of cells in which CIMD occurs.55 Indeed, spindle drug treatment of Bub1–partially depleted cells triggers c-abl–mediated phosphorylation of p73 and promotes its binding to Bub3. Both events are necessary for p73 to unleash CIMD, but whether Bub3 triggers p73 activation and whether Tyr99 phosphorylation precedes or follows Bub3 binding to p73 are yet to be clearly demonstrated. Intriguingly, overexpression of ΔNp73 inhibits CIMD, through a yet unknown mechanism.55
An alternative p73 and caspase-dependent mitotic cell death has recently been reported by Sabapathy’s group56 to occur in cells with normal Bub1 levels after prolonged engagement of SAC-dependent mitotic arrest. This is, again, p53 independent and is impaired by silencing or knockdown of p73. The mechanism described by Toh and colleagues56 is intriguing: they showed that mitotic cells fail to release cytochrome c from the mitochondria to trigger caspase-9 activation in the absence of transcriptional active p73. Bim is a BH3 only protein required for outer mitochondrial membrane permeabilization and cytochrome c release, and its silencing prevents mitotic cell death.57 In addition, Bim is a p73 but not a p53 transcriptional target.56,58 The authors found that both Bim and p73 expression peak at the G1/S phase of the cell cycle, suggesting that p73 upregulates Bim ahead of mitosis, thus priming cells to die in case of prolonged mitotic arrest (Fig. 3C).
p73 Expression in Human Tumors
As stated above, early studies on the involvement of p73 in tumorigenesis revealed that loss of heterozygosity frequently involved the region containing p73 in many cancers.1 But the issue of p73 expression in human cancers is far more complex. Unlike p53, p73 is rarely mutated in cancers, but its expression is often deregulated (Table 1), and, in passing, it is important for our understanding of oncogenic pathways that we investigate why mutations in p53 are common whereas those in its highly homologous partner, p73, are rare—an area that has received surprisingly little study. p73 dysregulation has been particularly well studied in breast tumors, where one study found that p73 mRNA levels were increased in 40% of patients. This was confirmed by analysis of cell lines derived from breast tumors, where the incidence of increased p73 mRNA levels was even higher.59,60 Furthermore, there was a different pattern of isoform expression between normal tissue, in which the predominant isoforms were α and β, and malignant tissue, in which there was a shift toward other C-terminal splice variants. Increased expression of p73 isoforms has also been detected in other types of tumors. For example, in 70% of ovarian cancers p73 is increased at both mRNA and protein levels. Similar findings have been found in hepatocellular carcinomas, where several studies reported increased p73 protein levels in aggressive tumors, compared with normal liver tissue.61,62 Again, this is also true in neuroblastomas, where neuroblastoma-derived cell lines showed up to a 90-fold increase of p73 transcripts.63 Overexpression of p73 has also been reported in bladder, prostate, and colorectal cancers.64-66 On the contrary, loss of p73 at 1p36 associates frequently with pancreatic malignancies and predicts worst prognosis.67 Interestingly, p73 polymorphisms (G-to-A at exon 2 + 4 and C-to-T at exon 2 + 14) could help in predicting the chemotherapeutic outcome of pancreatic cancer patients, as median survival of p73 wild-type and polymorphic variants was 17.4 and 10.2 months, respectively.68
Table 1.
Expression of p73 Isoforms in Human Tumors
| Tumor type | p73 Isoforms | References |
|---|---|---|
| Breast cancer | ΔEx2; ΔEx2/3; ΔNp73↑ | 59, 60, 72 |
| Ovarian cancer | ΔEx2↑ | 75 |
| HCC | ΔNp73↑ | 61, 62, 76 |
| Thyroid carcinoma | ΔNp73↑ | 70 |
| Prostate carcinoma | ΔNp73↑ | 64 |
| Pancreatic cancer | p73↓ | 67 |
| Cervical cancer | ΔNp73↑ | 73 |
| HNSSC | ΔNp73↑ | 74 |
| Colorectal cancer | ΔNp73↑ | 66 |
| AML | γ, δ, ϵ, θ↑ | 69 |
| CML | γ, δ, ϵ, θ↑ | 69 |
| B-CLL | ΔNp73↑ | 71 |
| Neuroblastoma | ΔNp73↑;p73↓ | 63, 78, 79, 80 |
| Glioma | ΔNp73↑ | 70 |
| RMS | ΔNp73↑ | 90 |
AML = acute myeloid leukemia; B-CLL = B-cell chronic leukemia; CML = chronic myeloid leukemia; HCC = hepatocellular carcinoma; HNSSC = head and neck squamous cell carcinoma; RMS = rhabdomyosarcoma; ↑ = upregulated; ↓ = downregulated/lost.
In acute myeloid leukemia (AML) and chronic myeloid leukemia (CML), expression of γ, δ, ϵ, and θ isoforms is also strongly upregulated.69 Although of potential interest, most of these studies have focused on mRNA levels, and only a few have studied protein expression, perhaps reflecting the unsatisfactory nature of the available antibodies; moreover, no distinction was made between TA and δN isoforms. Indeed, there is increasing evidence that p73 effects on tumor progression do not rely on total p73 levels but rather on the ratio between TA and δN expression. In support of this hypothesis, the δEx2/3 isoform has been identified for the first time in a neuroblastoma cell line1 and, furthermore, has been detected at high levels in around 50% of breast cancer-derived cell lines.15 The relative overexpression of this isoform confers resistance to apoptosis induced by chemotherapeutic drugs, and δEx2/3 has been shown to inhibit the activity of both p53 and TAp73α.59 In addition, δN isoforms have a longer half-life than TAp73, implying that even if the transcription of N-terminal isoforms is identical, δN predominates due to its higher stability.14 For these reasons, overexpression of δNp73 in tumors could be an important mechanism in blocking the proapoptotic functions of TAp73 and p53. Thus, in most thyroid carcinomas70 and B-cell chronic leukemias,71 both TA and δN isoforms are overexpressed. In colon and breast cancers, also, there are reports of tumors upregulating expression of both isoforms but with a bias toward higher levels of δNp73.72
Moreover, in many cases, δNp73 expression correlates with prognosis. Indeed, many reports have highlighted that increased levels of δNp73, without a concomitant increase of TA, are a significant risk factor for reduced survival. This has been shown in neuroblastoma, prostate, and cervical cancers. In the latter case, high levels of δNp73 were also associated with resistance to radiotherapy.73 Finally, similar findings have been reported in head and neck squamous cell carcinomas with a significant correlation between δNp73 levels and poor prognosis.74 However, N-terminal truncated p73 isoforms are not all equivalent, and tumors expressing isoforms from the P1 promoter (δEx2/3 or δEx2) are usually more aggressive than those overexpressing the isoforms generated from the P2 promoter (δNp73). For example, in ovarian tumors, the δEx2 isoform has been detected in malignant tumors but is absent in noninvasive tumors or normal tissues.75 Prostate carcinomas, gliomas, and hepatocellular carcinomas are others for which the presence of δNp73 correlates with increased aggressiveness.76
Zooming In: p73 in Neuroblastoma
Given the complexity and, sometimes, confusion around the field of p73 and cancer, in the last part of this review we will focus in more detail on the particular contribution of p73 to neuroblastoma development and outcome.
Neuroblastoma (NB) is the most common extracranial tumor in childhood. Epidemiological studies show that NB represents more than 7% of malignancies in patients younger than 15 years.77
Clinically, NB is a malignant embryonic tumor of the sympathetic lineage of neuronal crest cells, and for this reason it can arise anywhere in the sympathetic nervous system.
Genetically, the most common aberrations in NB are genomic amplification of MYCN (which occurs in about 20% of primary tumors), deletion of the short arm of chromosome 1 (1p36 deletion is present in 25%-35% of NB), and the duplication of chromosome 17q. All these chromosomal alterations are associated with poor patient survival. The human Trp73 gene maps within the region1p36, which led to the idea that p73 could have a role in the development of this tumor (Figure 4).
Figure 4.
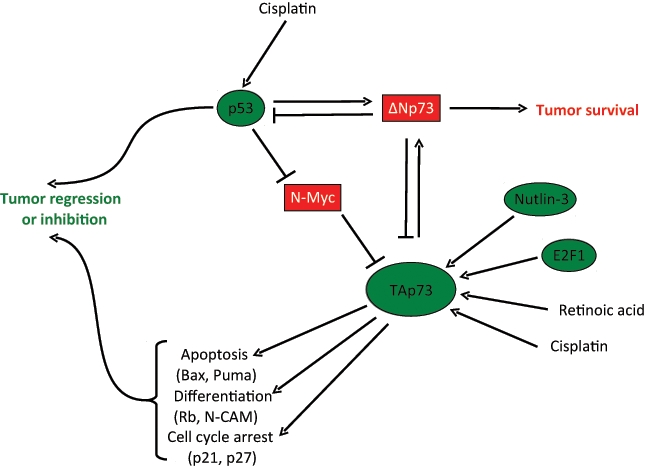
p73 role in neuroblastoma. TAp73 plays a tumor suppressive role by inducing target genes (some of which are reported) that trigger cell cycle arrest, apoptosis, and differentiation. These activities are promoted by chemotherapeutic drugs, such as cisplatin or nutlin 3 or retinoic acid, and counteracted by the dominant negative function of ΔNp73. Two other important players are the transcription factors N-myc and E2F1, which upregulate and repress TAp73, respectively.
Expression Studies
Although the Trp73 gene is rarely mutated in tumors, (indeed only 2 mutations [P405R and P425L] in the TP73 gene have been identified in tumors.63,78,79 Trp73 silencing has been reported in NB, leukemia (acute lymphoblastic), and Burkitt’s and non-Hodgkin lymphomas. The lack of expression of p73 can be due to several mechanisms, such as by the loss of 1 allele at 1p36 and by methylation or mutations in the promoter region of the apoptotic TAp73 that would result in the loss of p73 apoptotic activity. Already in 2002, Casciano and co-workers80 reported that δNp73 expression, but not that of TAp73, correlates with poor overall and progression-free survival in patients with NB. In particular, the investigators found high levels of expression of δNp73 by RT-PCR in 30% of 52 primary NB. In addition, expression of δNp73 is a negative prognostic marker independent of age, primary tumor site, stage, and MYCN amplification.
Hypothesis Concerning p73 Molecular Mechanisms in NB
In a majority of undifferentiated NB, wild-type p53 accumulates in the cytoplasm and is therefore unable to function as a transcription factor.81 In addition, the overexpression of the δNp73 isoforms described above could functionally block any residual nuclear p53 activity.82 δNp73 could also interact with TAp73 to inhibit its transcriptional role both in terms of apoptosis and in the neuronal differentiation actions of TAp73.83
Crosstalk between MYCN and p73 could also be involved. Whereas the data on c-MYC are controversial, with some studies showing enhancement and others reduction or no effect on transcriptional activity of p73,84-86 the overexpression of TAp73 clearly downregulates MYCN.86 This might allow NB cells to escape from the growth inhibiting properties of p73. TAp73 is also able to reduce MYCN expression and thus induce differentiation,83 suggesting an antagonistic role of the 2 transcription factors on NB cell proliferation and differentiation.
Epigenetic modifications, particularly by hyper- or hypomethylation, are critical events in cell transformation. Methylation of the TP73 promoter has been described,87 and the internal P2 promoter that controls the transcription of δNp73 is demethylated in NB cell lines and primary tumors, indicating a possible mechanism for the differential regulation of TAp73 and δNp73.
Therapeutic Approaches Targeting the p73 Pathway
Conventional therapies used in the treatment of NB include surgery, chemotherapy, and radiotherapy. Focusing on the molecular mechanism depicted in Figure 4, we will now discuss in particular the possibility of targeting the p73 pathway in therapy of NB. Assuming that upregulation of TAp73 could be of therapeutic benefit, several direct or indirect mechanisms for increasing its expression are possible. Like p53, TAp73 is upregulated by chemotherapeutic agents such as cisplatin, doxorubicin, and etoposide or by γ-irradiation.88-90 This occurs at both transcriptional and posttranscriptional levels, and there is evidence that drugs and irradiation increase p73 expression also in NB cell lines.91
p73 may also be a therapeutic target in chemoresistant tumors with inactive p53.92 Thus, in a p53 null and doxorubicin-resistant NB cell line, nutlin-3 treatment increased TAp73 expression in a E2F-dependent manner, resulting in an increase in the ability of doxorubicin to block cell proliferation and induce apoptosis. Knockdown of TAp73 largely abrogated this sensitization, suggesting that apoptosis induced by nutlin-3/doxorubicin is TAp73 dependent.
A number of experimental and clinical studies show that retinoids inhibit or reverse the carcinogenic process of some tissues, including hematological malignancies as well as premalignant and malignant lesions of the oral cavity, head and neck, breast, skin, and liver. Retinoic acid (RA) is used as standard therapy of NB.93 RA acts as a cell growth inhibitor and induces cell differentiation through the regulation of the expression of several target genes, including p73.83 Indeed, treatment of NB cell lines with RA increases endogenous levels of TAp73, and ectopic expression of TAp73 induces neuronal terminal differentiation. Moreover, TAp73 regulates the N-CAM promoter, and TAp73 overexpression has been reported to reduce N-MYC and increase expression of pRB, mimicking the RA effect on these 2 genes.94,95 These findings suggest that the therapeutic effects of RA are at least in part mediated by p73.
p73 and Rhabdomyosarcoma: The mTOR Connection
Rhabdomyosarcoma (RMS) is the most common childhood malignancy, and p73 has been implicated in the cell cycle withdrawal triggered by myogenic differentiation.96 Induction of differentiation induces the expression of TAp73β in myogenic cells, which in turn triggers the expression of the cell cycle inhibitor p57KIP2, which acts by inhibiting the cyclin/CDK complexes and promoting hypophosphorylation of retinoblastoma (RB) protein. p57 is already known to be a p73 target gene.97 As may be expected, ΔNp73-overexpressing myoblasts show no upregulation of p57 and do not accumulate hypophosphorylated active RB. A potential oncogenic role for ΔNp73 in RMS is suggested by the observation that ΔNp73 cooperates with typical RMS oncogenes (such as Pax3) in eliciting myoblast malignant transformation in vivo. Indeed, injection into nude mice of myoblasts transduced with Pax3 does not result in tumor formation, due to induction of muscle differentiation and cell cycle arrest; but co-expression of ΔNp73 results in the formation of undifferentiated, highly proliferative tumors. In addition, p73 depletion in the human RMS cell line Rh30 impairs xenograft tumor growth and triggers expression of p57. p73 is undetectable in normal muscle but is abundant in RMS tumor samples, where both TAp73 and ΔNp73 are present, suggesting that in RMS, as in other tumors, the ratio between the N-terminal isoforms determines the phenotype.96
Recent findings have added more complexity to this picture by revealing a role for the mTOR-p73 axis in the genetic signature of RMS.98 mTOR is a serine/threonine kinase that regulates cell size and proliferation and negatively affects p73. Indeed, treatment of cancer cells with the widely used mTOR inhibitor rapamycin increases p73 protein levels and triggers its activity, through a yet unidentified mechanism.99 Whole genome ChIP and microarray analysis of the Rh30 cell line showed that inhibition of mTOR by rapamycin increased by 9% p73 occupancy of genomic sites (corresponding to a 5% increase in regulated genes). Cluster analysis of this data creates a mTOR-p73 gene signature in which expression of 154 genes is increased and that of 156 genes decreased by p73 in a mTOR-dependent manner. Twenty-nine percent of these genes are putatively direct p73 targets, as deduced by ChIP-on ChIP data.
There 2 main types of RMS: alveolar and embryonic. The former is more severe and affects older children compared with embryonic RMS. From the array data above, the identified signature segregates and exclusively defines the alveolar subtype of RMS and is not relevant to embryonic tumors. Indeed, alveolar tumors express genes upregulated by p73 and show reduced levels of p73-repressed genes. More strikingly, 17 selected genes from the mTOR-p73 signature predict patients’ survival, as tumors with the highest score for the selected targets were associated with reduced survival.98
In summary, 2 main axes are acting in RMS: a p73-p57KIP2 axis and a mTOR-p73 axis. How, and indeed whether, these 2 pathways are interrelated is yet to be established. Indeed, the effect of mTOR on p73 remains poorly defined and the ΔNp73 status (i.e., the TA vs. ΔN ratio) in the alveolar RMS Rh30 cells used in the array study has not been investigated. Nonetheless, the available data arguably prove an involvement of p73 in RMS and pose a challenge for its further investigation and possible therapeutic exploitation.
p73 as a Target for Cancer Therapy
Apart from the specific cancers, such as NB and RMS discussed in more detail above, can p73 be exploited as a target for cancer therapy in general? In theory, chemotherapeutic compounds able to trigger the activity of the proapototic p73 isoforms could be potentially beneficial and improve patients’ response to chemotherapy and overall survival.
Already, there are several possibilities here. Drugs developed to improve the apoptotic activity of p53 are indeed also able to act on p73. There are 3 major candidate compounds: Nutlins, RETRA, and PRIMA.100
Nutlins are small molecules able to disrupt the interaction between p53 and MDM2, thus stabilizing the former and promoting cell cycle arrest and apoptosis.101 MDM2, and its human counterpart HDM2, bind to TAp73 and inhibit its transcriptional activity (although this is somewhat controversial), without promoting its degradation. Two pioneering studies92,102 showed that Nutlins disrupt the HDM2/p73 interaction in cancer cell lines, potentiating TAp73 transcriptional activity, and Nutlin-3a treatment increases doxorubicin-mediated cell death of the p53-null neuroblastoma cell line LA155N, an effect that is dependent on induction of TAp73 proapoptotic activity.
RETRA is a compound discovered in high-throughput screening aimed at identifying small molecules capable of reactivating the normal function of mutant p53 (mt-p53).103 Administration of RETRA to cancer cells with mt-p53 triggers a strong apoptotic response with accumulation of p21 and PUMA. Importantly, further studies suggest that RETRA, rather than promoting p53 activity, releases mt-p53 inhibition of p73. Although more studies are needed to confirm this, these early data nonetheless strongly implicate p73 as a targetable tumor suppressor.
The quest for molecules able to reactivate the transcriptional activity of mt-p53 proteins also led to the isolation of PRIMA.104,105 PRIMA binds the mt-p53 core domain and promotes p53-dependent apoptosis by rescuing the transcriptional activity of mutant p53. A recent study has extended these findings to p73,106 showing that PRIMA can promote transcriptional and apoptotic activity by mutant p73. Thus, PRIMA could also be useful for reactivation of p73 in the small subset of human tumors that harbor p73 mutations.
Alternative approaches for therapeutic development could be focused on inhibiting the ubiquitin E3 ligases involved in the degradation of p73. One of these is the HECT-containing ligase Itch.107 The HECT family has intrinsic catalytic activity, so it is should be possible to design a specific inhibitor directed at the Cys active site or its catalytic pocket. Alternatively, the hinge region of Itch, required for catalysis, could be used to generate allosteric inhibitors. Although this approach seems promising, several issues remain unresolved. First, Itch degrades both TAp73 and ΔNp73, leaving the question of the balance of the isoforms unresolved. Second, alternative degradation pathways have been described involving calpain,108 caspases,109 or the cullin-dependent F-Box protein FBX045.110 However, the Itch pathway seems to be the most relevant, being regulated by JNK,111 Wwox,112 Yes-associated protein (YAP),113 and hippo.114 More recently, pathways specific for the ΔNp73 isoform have been identified, the antizyme system115 and the E3 ligase PIR2,116 although the development of their specific inhibitors is theoretically significantly more complicated than that of Itch. Thus, although highly promising, this area is still preliminary.
Concluding Remarks
Although the phenotype of the total p73 knockout mouse is developmental without evidence of enhanced tumor susceptibility, TAp73 can clearly act as a tumor suppressor. Thus, in the majority of cancers in which p53 is mutated or otherwise inactive, exploiting TAp73 as a proapoptotic p53 substitute is an attractive therapeutic option. However, any such strategy must take into account the inhibitory effects of both mt-p53 and ΔNp73 on TAp73 activity, and a combination of approaches that activate TAp73 while removing or reducing these inhibitory actions would seem the way forward for exploiting the apoptotic effects of TAp73 in cancer.
Finally, it is important to remember that the complexity of the p53-family also determines its multiple mechanisms of action, both transcriptional dependent and independent. In this regard, we would like to conclude this review with the recent suggestion that p73 may act to control micro-RNA biogenesis.117,118 If true, this opens up a whole new area of investigation in the role of p73 in cancer. Another chapter to be written in the future?
Footnotes
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
References
- 1. Kaghad M, Bonnet H, Yang A, et al. Monoallelically expressed gene related to p53 at 1p36, a region frequently deleted in neuroblastoma and other human cancers. Cell. 1997;90:809-19 [DOI] [PubMed] [Google Scholar]
- 2. Dötsch V, Bernassola F, Coutandin D, Candi E, Melino G. p63 and p73 the ancestors of p53. Cold Spring Harb Perspect Biol. 2010;2:a004887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Yang A, Walker N, Bronson R, et al. p73-deficient mice have neurological, pheromonal and inflammatory defects but lack spontaneous tumors. Nature. 2000;404:99-103 [DOI] [PubMed] [Google Scholar]
- 4. Melino G, De Laurenzi V, Vousden KH. p73: Friend or foe in tumorigenesis. Nat Rev Cancer. 2002;2:605-15 [DOI] [PubMed] [Google Scholar]
- 5. Murray-Zmijewski F, Lane DP, Bourdon JC. p53/p63/p73 isoforms: an orchestra of isoforms to harmonise cell differentiation and response to stress. Cell Death Differ. 2006;13:962-72 [DOI] [PubMed] [Google Scholar]
- 6. Ishimoto O, Kawahara C, Enjo K, Obinata M, Nukiwa T, Ikawa S. Possible oncogenic potential of DeltaNp73: a newly identified isoform of human p73. Cancer Res. 2002;62:636-41 [PubMed] [Google Scholar]
- 7. Ou HD, Löhr F, Vogel V, Mäntele W, Dötsch V. Structural evolution of C-terminal domains in the p53 family. EMBO J. 2007;26:3463-73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Deutsch GB, Zielonka EM, Coutandin D, et al. DNA damage in oocytes induces a switch of the quality control factor TAp63α from dimer to tetramer. Cell. 2011;144:566-76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Serber Z, Lai HC, Yang A, et al. A C-terminal inhibitory domain controls the activity of p63 by an intramolecular mechanism. Mol Cell Biol. 2002;22:8601-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Rossi M, Sayan AE, Terrinoni A, Melino G, Knight RA. Mechanism of induction of apoptosis by p73 and its relevance to neuroblastoma biology. Ann N Y Acad Sci. 2004;1028:143-9 [DOI] [PubMed] [Google Scholar]
- 11. Ramadan S, Terrinoni A, Catani MV, et al. p73 induces apoptosis by different mechanisms. Biochem Biophys Res Commun. 2005;331:713-7 [DOI] [PubMed] [Google Scholar]
- 12. Tomasini R, Tsuchihara K, Wilhelm M, et al. TAp73 knockout shows genomic instability with infertility and tumor suppressor functions. Genes Dev. 2008;22:2677-91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zaika AI, Slade N, Erster SH, et al. DeltaNp73, a dominant-negative inhibitor of wild-type p53 and TAp73, is up-regulated in human tumors. J Exp Med. 2002;196:765-80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Stiewe T, Theseling CC, Pützer BM. Transactivation-deficient Delta TA-p73 inhibits p53 by direct competition for DNA binding: implications for tumorigenesis. J Biol Chem. 2002;277:14177-85 [DOI] [PubMed] [Google Scholar]
- 15. Fillippovich I, Sorokina N, Gatei M, et al. Transactivation-deficient p73alpha (p73Deltaexon2) inhibits apoptosis and competes with p53. Oncogene. 2001;20:514-22 [DOI] [PubMed] [Google Scholar]
- 16. Joerger AC, Rajagopalan S, Natan E, Veprintsev DB, Robinson CV, Fersht AR. Structural evolution of p53, p63, and p73: implication for heterotetramer formation. Proc Natl Acad Sci U S A. 2009;106:17705-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Grob TJ, Novak U, Maisse C, et al. Human delta Np73 regulates a dominant negative feedback loop for TAp73 and p53. Cell Death Differ. 2001;8:1213-23 [DOI] [PubMed] [Google Scholar]
- 18. Nakagawa T, Takahashi M, Ozaki T, et al. Autoinhibitory regulation of p73 by Delta Np73 to modulate cell survival and death through a p73-specific target element within the Delta Np73 promoter. Mol Cell Biol. 2002;22:2575-85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Harvey M, McArthur MJ, Montgomery CA, Jr, Butel JS, Bradley A, Donehower LA. Spontaneous and carcinogen-induced tumorigenesis in p53-deficient mice. Nat Genet. 1993;5:225-9 [DOI] [PubMed] [Google Scholar]
- 20. Flores ER, Sengupta S, Miller JB, et al. Tumor predisposition in mice mutant for p63 and p73: evidence for broader tumor suppressor functions for the p53 family. Cancer Cell. 2005;7:363-73 [DOI] [PubMed] [Google Scholar]
- 21. Wilhelm MT, Rufini A, Wetzel MK, et al. Isoform-specific p73 knockout mice reveal a novel role for delta Np73 in the DNA damage response pathway. Genes Dev. 2010;24:549-60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Petrenko O, Zaika A, Moll UM. deltaNp73 facilitates cell immortalization and cooperates with oncogenic Ras in cellular transformation in vivo. Mol Cell Biol. 2003;23:5540-55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Beitzinger M, Hofmann L, Oswald C, et al. p73 poses a barrier to malignant transformation by limiting anchorage-independent growth. EMBO J. 2008;27:792-803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Nemajerova A, Petrenko O, Trümper L, Palacios G, Moll UM. Loss of p73 promotes dissemination of Myc-induced B cell lymphomas in mice. J Clin Invest. 2010;120:2070-80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Polager S, Ginsberg D. E2F—at the crossroads of life and death. Trends Cell Biol. 2008;18:528-35 [DOI] [PubMed] [Google Scholar]
- 26. Stiewe T, Pützer BM. Role of the p53-homologue p73 in E2F1-induced apoptosis. Nat Genet. 2000;26:464-9 [DOI] [PubMed] [Google Scholar]
- 27. Irwin M, Marin MC, Phillips AC, et al. Role for the p53 homologue p73 in E2F-1-induced apoptosis. Nature. 2000;407:645-8 [DOI] [PubMed] [Google Scholar]
- 28. Seelan RS, Irwin M, van der Stoop P, Qian C, Kaelin WG, Jr, Liu W. The human p73 promoter: characterization and identification of functional E2F binding sites. Neoplasia. 2002;4:195-203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Irwin MS, Kondo K, Marin MC, Cheng LS, Hahn WC, Kaelin WG., Jr Chemosensitivity linked to p73 function. Cancer Cell. 2003;3:403-10 [DOI] [PubMed] [Google Scholar]
- 30. Urist M, Tanaka T, Poyurovsky MV, Prives C. p73 induction after DNA damage is regulated by checkpoint kinases Chk1 and Chk2. Genes Dev. 2004;18:3041-54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Marabese M, Vikhanskaya F, Rainelli C, Sakai T, Broggini M. DNA damage induces transcriptional activation of p73 by removing C-EBPalpha repression on E2F1. Nucleic Acids Res. 2003;31:6624-32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Rödicker F, Stiewe T, Zimmermann S, Pützer BM. Therapeutic efficacy of E2F1 in pancreatic cancer correlates with TP73 induction. Cancer Res. 2001;61:7052-5 [PubMed] [Google Scholar]
- 33. Pediconi N, Ianari A, Costanzo A, et al. Differential regulation of E2F1 apoptotic target genes in response to DNA damage. Nat Cell Biol. 2003;5:552-8 [DOI] [PubMed] [Google Scholar]
- 34. Fontemaggi G, Gurtner A, Strano S, et al. The transcriptional repressor ZEB regulates p73 expression at the crossroad between proliferation and differentiation. Mol Cell Biol. 2001;21:8461-70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ibrahim N, He L, Leong CO, et al. BRCA1-associated epigenetic regulation of p73 mediates an effector pathway for chemosensitivity in ovarian carcinoma. Cancer Res. 2010;70:7155-65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Agami R, Blandino G, Oren M, Shaul Y. Interaction of c-Abl and p73alpha and their collaboration to induce apoptosis. Nature. 1999;399:809-13 [DOI] [PubMed] [Google Scholar]
- 37. Gong JG, Costanzo A, Yang HQ, et al. The tyrosine kinase c-Abl regulates p73 in apoptotic response to cisplatin-induced DNA damage. Nature. 1999;399:806-9 [DOI] [PubMed] [Google Scholar]
- 38. Yuan ZM, Shioya H, Ishiko T, et al. p73 is regulated by tyrosine kinase c-Abl in the apoptotic response to DNA damage. Nature. 1999;399:814-7 [DOI] [PubMed] [Google Scholar]
- 39. Tsai KK, Yuan ZM. c-Abl stabilizes p73 by a phosphorylation-augmented interaction. Cancer Res. 2003;63:3418-24 [PubMed] [Google Scholar]
- 40. Sanchez-Prieto R, Sanchez-Arevalo VJ, Servitja JM, Gutkind JS. Regulation of p73 by c-Abl through the p38 MAP kinase pathway. Oncogene. 2002;21:974-9 [DOI] [PubMed] [Google Scholar]
- 41. Gaiddon C, Lokshin M, Gross I, et al. Cyclin-dependent kinases phosphorylate p73 at threonine 86 in a cell cycle-dependent manner and negatively regulate p73. J Biol Chem. 2003;278:27421-31 [DOI] [PubMed] [Google Scholar]
- 42. Di Como CJ, Gaiddon C, Prives C. p73 function is inhibited by tumor-derived p53 mutants in mammalian cells. Mol Cell Biol. 1999;19:1438-49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Marin MC, Jost CA, Brooks LA, et al. A common polymorphism acts as an intragenic modifier of mutant p53 behaviour. Nat Genet. 2000;25:47-54 [DOI] [PubMed] [Google Scholar]
- 44. Bergamaschi D, Gasco M, Hiller L, et al. p53 polymorphism influences response in cancer chemotherapy via modulation of p73-dependent apoptosis. Cancer Cell. 2003;3:387-402 [DOI] [PubMed] [Google Scholar]
- 45. Bruno T, Desantis A, Bossi G, et al. Che-1 promotes tumor cell survival by sustaining mutant p53 transcription and inhibiting DNA damage response activation. Cancer Cell. 2010;18:122-34 [DOI] [PubMed] [Google Scholar]
- 46. Bruno T, De Nicola F, Iezzi S, et al. Che-1 phosphorylation by ATM/ATR and Chk2 kinases activates p53 transcription and the G2/M checkpoint. Cancer Cell. 2006;10:473-86 [DOI] [PubMed] [Google Scholar]
- 47. Robinson RA, Lu X, Jones EY, Siebold C. Biochemical and structural studies of ASPP proteins reveal differential binding to p53, p63, and p73. Structure. 2008;16:259-68 [DOI] [PubMed] [Google Scholar]
- 48. Patel S, George R, Autore F, Fraternali F, Ladbury JE, Nikolova PV. Molecular interactions of ASPP1 and ASPP2 with the p53 protein family and the apoptotic promoters PUMA and Bax. Nucleic Acids Res. 2008;36:5139-51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Merlo P, Fulco M, Costanzo A, et al. A role of p73 in mitotic exit. J Biol Chem. 2005;280:30354-60 [DOI] [PubMed] [Google Scholar]
- 50. Talos F, Nemajerova A, Flores ER, Petrenko O, Moll UM. p73 suppresses polyploidy and aneuploidy in the absence of functional p53. Mol Cell. 2007;27:647-59 [DOI] [PubMed] [Google Scholar]
- 51. Tomasini R, Tsuchihara K, Tsuda C, et al. TAp73 regulates the spindle assembly checkpoint by modulating BubR1 activity. Proc Natl Acad Sci U S A. 2009;106:797-802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Vernole P, Neale MH, Barcaroli D, et al. TAp73alpha binds the kinetochore proteins Bub1 and Bub3 resulting in polyploidy. Cell Cycle. 2009;8:421-9 [DOI] [PubMed] [Google Scholar]
- 53. Musacchio A, Salmon ED. The spindle-assembly checkpoint in space and time. Nat Rev Mol Cell Biol. 2007;8:379-93 [DOI] [PubMed] [Google Scholar]
- 54. Niikura Y, Dixit A, Scott R, Perkins G, Kitagawa K. BUB1 mediation of caspase-independent mitotic death determines cell fate. J Cell Biol. 2007;178:283-96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Niikura Y, Ogi H, Kikuchi K, Kitagawa K. BUB3 that dissociates from BUB1 activates caspase-independent mitotic death (CIMD). Cell Death Differ. 2010;17:1011-24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Toh WH, Nam SY, Sabapathy K. An essential role for p73 in regulating mitotic cell death. Cell Death Differ. 2010;17:787-800 [DOI] [PubMed] [Google Scholar]
- 57. Piñon JD, Labi V, Egle A, Villunger A. Bim and Bmf in tissue homeostasis and malignant disease. Oncogene. 2008;27(Suppl 1):S41-52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Busuttil V, Droin N, McCormick L, et al. NF-kappaB inhibits T-cell activation-induced, p73-dependent cell death by induction of MDM2. Proc Natl Acad Sci U S A. 2010;107:18061-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Domínguez G, García JM, Peña C, et al. DeltaTAp73 upregulation correlates with poor prognosis in human tumors: putative in vivo network involving p73 isoforms, p53, and E2F-1. J Clin Oncol. 2006;24:805-15 [DOI] [PubMed] [Google Scholar]
- 60. Zaika AI, Kovalev S, Marchenko ND, Moll UM. Overexpression of the wild type p73 gene in breast cancer tissues and cell lines. Cancer Res. 1999;59:3257-63 [PubMed] [Google Scholar]
- 61. Moll UM, Erster S, Zaika A. p53, p63 and p73—solos, alliances and feuds among family members. Biochim Biophys Acta. 2001;1552:47-59 [DOI] [PubMed] [Google Scholar]
- 62. Tannapfel A, Wasner M, Krause K, et al. Expression of p73 and its relation to histopathology and prognosis in hepatocellular carcinoma. J Natl Cancer Inst. 1999;91:1154-8 [DOI] [PubMed] [Google Scholar]
- 63. Kovalev S, Marchenko N, Swendeman S, LaQuaglia M, Moll UM. Expression level, allelic origin, and mutation analysis of the p73 gene in neuroblastoma tumors and cell lines. Cell Growth Differ. 1998;9:897-903 [PubMed] [Google Scholar]
- 64. Yokomizo A, Mai M, Bostwick DG, et al. Mutation and expression analysis of the p73 gene in prostate cancer. Prostate. 1999;39:94-100 [DOI] [PubMed] [Google Scholar]
- 65. Yokomizo A, Mai M, Tindall DJ, et al. Overexpression of the wild type p73 gene in human bladder cancer. Oncogene. 1999;18:1629-33 [DOI] [PubMed] [Google Scholar]
- 66. Sunahara M, Ichimiya S, Nimura Y, et al. Mutational analysis of the p73 gene localized at chromosome 1p36.3 in colorectal carcinomas. Int J Oncol. 1998;13:319-23 [PubMed] [Google Scholar]
- 67. Loukopoulos P, Shibata T, Katoh H, et al. Genome-wide array-based comparative genomic hybridization analysis of pancreatic adenocarcinoma: identification of genetic indicators that predict patient outcome. Cancer Sci. 2007;98:392-400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Chen J, Li D, Killary AM, Sen S, et al. Polymorphisms of p16, p27, p73, and MDM2 modulate response and survival of pancreatic cancer patients treated with preoperative chemoradiation. Ann Surg Oncol. 2009;16:431-9 [DOI] [PubMed] [Google Scholar]
- 69. Tschan MP, Grob TJ, Peters UR, et al. Enhanced p73 expression during differentiation and complex p73 isoforms in myeloid leukemia. Biochem Biophys Res Commun. 2000;277:62-5 [DOI] [PubMed] [Google Scholar]
- 70. Malaguarnera R, Vella V, Vigneri R, Frasca F. p53 family proteins in thyroid cancer. Endocr Relat Cancer. 2007;14:43-60 [DOI] [PubMed] [Google Scholar]
- 71. Leupin N, Lüthi A, Novak U, et al. p73 status in B-cell chronic lymphocytic leukaemia. Leuk Lymphoma. 2004;45:1205-7 [DOI] [PubMed] [Google Scholar]
- 72. Díaz R, González-Sancho JM, Soldevilla B, et al. Differential regulation of TP73 isoforms by 1α,25-dihydroxyvitamin D3 and survivin in human colon and breast carcinomas. Genes Chromosomes Cancer. 2010;49:1135-42 [DOI] [PubMed] [Google Scholar]
- 73. Buhlmann S, Pützer BM. ΔNp73 a matter of cancer: mechanisms and clinical implications. Biochim Biophys Acta. 2008;1785:207-16 [DOI] [PubMed] [Google Scholar]
- 74. Faridoni-Laurens L, Tourpin S, Alsafadi S, et al. Involvement of N-terminally truncated variants of p73, deltaTAp73, in head and neck squamous cell cancer: a comparison with p53 mutations. Cell Cycle. 2008;7:1587-96 [DOI] [PubMed] [Google Scholar]
- 75. Ng SW, Yiu GK, Liu Y, et al. Analysis of p73 in human borderline and invasive ovarian tumor. Oncogene. 2000;19:1885-90 [DOI] [PubMed] [Google Scholar]
- 76. Buhlmann S, Racek T, Schwarz A, Schaefer S, Pützer BM. Molecular mechanism of p73-mediated regulation of hepatitis B virus core promoter/enhancer II: implications for hepatocarcinogenesis. J Mol Biol. 2008;378:20-30 [DOI] [PubMed] [Google Scholar]
- 77. Maris JM. Neuroblastoma. Lancet. 2007;369:2106-20 [DOI] [PubMed] [Google Scholar]
- 78. Douc-Rasy S, Barrois M, Echeynne M, et al. DeltaN-p73alpha accumulates in human neuroblastic tumors. Am J Pathol. 2002;160:631-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Ichimiya S, Nimura Y, Kageyama H, et al. p73 at chromosome 1p36.3 is lost in advanced stage neuroblastoma but its mutation is infrequent. Oncogene. 1999;18:1061-6 [DOI] [PubMed] [Google Scholar]
- 80. Casciano I, Mazzocco K, Boni L, et al. Expression of DeltaNp73 is a molecular marker for adverse outcome in neuroblastoma patients. Cell Death Differ. 2002;9:246-51 [DOI] [PubMed] [Google Scholar]
- 81. Moll UM, LaQuaglia M, Bénard J, Riou G. Wild-type p53 protein undergoes cytoplasmic sequestration in undifferentiated neuroblastomas but not in differentiated tumors. Proc Natl Acad Sci U S A. 1995;92:4407-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Pozniak CD, Radinovic S, Yang A, McKeon F, Kaplan DR, Miller FD. An anti-apoptotic role for the p53 family member, p73, during developmental neuron death. Science. 2000;289:304-6 [DOI] [PubMed] [Google Scholar]
- 83. De Laurenzi V, Raschellá G, Barcaroli D, et al. Induction of neuronal differentiation by p73 in a neuroblastoma cell line. J Biol Chem. 2000;275:15226-31 [DOI] [PubMed] [Google Scholar]
- 84. Zaika A, Irwin M, Sansome C, Moll UM. Oncogenes induce and activate endogenous p73 protein. J Biol Chem. 2001;276:11310-6 [DOI] [PubMed] [Google Scholar]
- 85. Watanabe K, Ozaki T, Nakagawa T, et al. Physical interaction of p73 with c-Myc and MM1, a c-Myc-binding protein, and modulation of the p73 function. J Biol Chem. 2002;277:15113-23 [DOI] [PubMed] [Google Scholar]
- 86. Horvilleur E, Bauer M, Goldschneider D, et al. p73alpha isoforms drive opposite transcriptional and post-transcriptional regulation of MYCN expression in neuroblastoma cells. Nucleic Acids Res. 2008;36:4222-32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Casciano I, Banelli B, Croce M, et al. Role of methylation in the control of DeltaNp73 expression in neuroblastoma. Cell Death Differ. 2002;9:343-5 [DOI] [PubMed] [Google Scholar]
- 88. Costanzo A, Merlo P, Pediconi N, et al. DNA damage-dependent acetylation of p73 dictates the selective activation of apoptotic target genes. Mol Cell. 2002;9:175-86 [DOI] [PubMed] [Google Scholar]
- 89. Lin KW, Nam SY, Toh WH, Dulloo I, Sabapathy K. Multiple stress signals induce p73beta accumulation. Neoplasia. 2004;6:546-57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Müller M, Schilling T, Sayan AE, et al. TAp73/Delta Np73 influences apoptotic response, chemosensitivity and prognosis in hepatocellular carcinoma. Cell Death Differ. 2005;12:1564-77 [DOI] [PubMed] [Google Scholar]
- 91. Million K, Horvilleur E, Goldschneider D, et al. Differential regulation of p73 variants in response to cisplatin treatment in SH-SY5Y neuroblastoma cells. Int J Oncol. 2006;29:147-54 [PubMed] [Google Scholar]
- 92. Peirce SK, Findley HW. The MDM2 antagonist nutlin-3 sensitizes p53-null neuroblastoma cells to doxorubicin via E2F1 and TAp73. Int J Oncol. 2009;34:1395-402 [PubMed] [Google Scholar]
- 93. Wagner LM, Danks MK. J Cell Biochem. New therapeutic targets for the treatment of high-risk neuroblastoma. 2009;107:46-57 [DOI] [PubMed] [Google Scholar]
- 94. Thiele CJ, Reynolds CP, Israel MA. Decreased expression of N-myc precedes retinoic acid-induced morphological differentiation of human neuroblastoma. Nature. 1985;313:404-6 [DOI] [PubMed] [Google Scholar]
- 95. Kranenburg O, Scharnhorst V, Van der Eb AJ, Zantema A. Inhibition of cyclin-dependent kinase activity triggers neuronal differentiation of mouse neuroblastoma cells. J Cell Biol. 1995;131:227-34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Cam H, Griesmann H, Beitzinger M, et al. p53 family members in myogenic differentiation and rhabdomyosarcoma development. Cancer Cell. 2006;10:281-93 [DOI] [PubMed] [Google Scholar]
- 97. Blint E, Phillips AC, Kozlov S, Stewart CL, Vousden KH. Induction of p57(KIP2) expression by p73beta. Proc Natl Acad Sci U S A. 2002;99:3529-34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Rosenbluth JM, Mays DJ, Jiang A, Shyr Y, Pietenpol JA. Differential regulation of the p73 cistrome by mammalian target of rapamycin reveals transcriptional programs of mesenchymal differentiation and tumorigenesis. Proc Natl Acad Sci U S A. 2011;108:2076-81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Rosenbluth JM, Mays DJ, Pino MF, Tang LJ, Pietenpol JA. A gene signature-based approach identifies mTOR as a regulator of p73. Mol Cell Biol. 2008;28:5951-64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Zawacka-Pankau J, Kostecka A, Sznarkowska A, Hedström E, Kawiak A. p73 tumor suppressor protein: a close relative of p53 not only in structure but also in anti-cancer approach? Cell Cycle. 2010;9:720-8 [DOI] [PubMed] [Google Scholar]
- 101. Vassilev LT, Vu BT, Graves B, et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science. 2004;303:844-8 [DOI] [PubMed] [Google Scholar]
- 102. Lau LM, Nugent JK, Zhao X, Irwin MS. HDM2 antagonist Nutlin-3 disrupts p73-HDM2 binding and enhances p73 function. Oncogene. 2008;27:997-1003 [DOI] [PubMed] [Google Scholar]
- 103. Kravchenko JE, Ilyinskaya GV, Komarov PG, et al. Small-molecule RETRA suppresses mutant p53-bearing cancer cells through a p73-dependent salvage pathway. Proc Natl Acad Sci U S A. 2008;105:6302-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Bykov VJ, Issaeva N, Shilov A, et al. Restoration of the tumor suppressor function to mutant p53 by a low-molecular-weight compound. Nat Med. 2002;8:282-8 [DOI] [PubMed] [Google Scholar]
- 105. Bykov VJ, Issaeva N, Selivanova G, Wiman KG. Mutant p53-dependent growth suppression distinguishes PRIMA-1 from known anticancer drugs: a statistical analysis of information in the National Cancer Institute database. Carcinogenesis. 2002;23:2011-8 [DOI] [PubMed] [Google Scholar]
- 106. Rökaeus N, Shen J, Eckhardt I, Bykov VJ, Wiman KG, Wilhelm MT. PRIMA-1(MET)/APR-246 targets mutant forms of p53 family members p63 and p73. Oncogene. 2010;29:6442-51 [DOI] [PubMed] [Google Scholar]
- 107. Rossi M, De Laurenzi V, Munarriz E, et al. The ubiquitin-protein ligase Itch regulates p73 stability. EMBO J. 2005;24:836-48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Munarriz E, Bano D, Sayan AE, Rossi M, Melino G, Nicotera P. Calpain cleavage regulates the protein stability of p73. Biochem Biophys Res Commun. 2005;333:954-60 [DOI] [PubMed] [Google Scholar]
- 109. Sayan AE, Sayan BS, Gogvadze V, et al. p73 and caspase-cleaved p73 fragments localize to mitochondria and augment TRAIL-induced apoptosis. Oncogene. 2008;27:4363-72 [DOI] [PubMed] [Google Scholar]
- 110. Peschiaroli A, Scialpi F, Bernassola F, Pagano M, Melino G. The F-box protein FBXO45 promotes the proteasome-dependent degradation of p73. Oncogene. 2009;28:3157-66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Chang L, Kamata H, Solinas G, et al. The E3 ubiquitin ligase itch couples JNK activation to TNFalpha-induced cell death by inducing c-FLIP(L) turnover. Cell. 2006;124:601-13 [DOI] [PubMed] [Google Scholar]
- 112. Aqeilan RI, Pekarsky Y, Herrero JJ, et al. Functional association between Wwox tumor suppressor protein and p73, a p53 homolog. Proc Natl Acad Sci U S A. 2004;101:4401-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Basu S, Totty NF, Irwin MS, Sudol M, Downward J. Akt phosphorylates the Yes-associated protein, YAP, to induce interaction with 14-3-3 and attenuation of p73-mediated apoptosis. Mol Cell. 2003;11:11-23 [DOI] [PubMed] [Google Scholar]
- 114. Salah Z, Melino G, Aqeilan RI. Negative regulation of the hippo pathway by E3 ubiquitin ligase ITCH is sufficient to promote tumorigenicity. Cancer Res. 2011;71:2010-20 [DOI] [PubMed] [Google Scholar]
- 115. Dulloo I, Gopalan G, Melino G, Sabapathy K. The antiapoptotic DeltaNp73 is degraded in a c-Jun-dependent manner upon genotoxic stress through the antizyme-mediated pathway. Proc Natl Acad Sci U S A. 2010;107:4902-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Sayan BS, Yang AL, Conforti F, et al. Differential control of TAp73 and DeltaNp73 protein stability by the ring finger ubiquitin ligase PIR2. Proc Natl Acad Sci U S A. 2010;107:12877-82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Boominathan L. The tumor suppressors p53, p63, and p73 are regulators of microRNA processing complex. PLoS One. 2010;5:e10615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Boominathan L. The guardians of the genome (p53, TA-p73, and TA-p63) are regulators of tumor suppressor miRNAs network. Cancer Metastasis Rev. 2010;29:613-39 [DOI] [PubMed] [Google Scholar]