

Abstract
Cellular growth and division are two fundamental processes that are exquisitely sensitive and responsive to environmental fluctuations. One of the most energetically demanding functions of these processes is ribosome biogenesis, the key component to regulating overall protein synthesis and cell growth. Perturbations to ribosome biogenesis have been demonstrated to induce an acute stress response leading to p53 activation through the inhibition of Mdm2 by a number of ribosomal proteins. The energy status of a cell is a highly dynamic variable that naturally contributes to metabolic fluctuations, which can affect both the rates of ribosome biogenesis and p53 function. This, in turn, determines whether a cell is in an anabolic, growth-promoting state or a catabolic, growth-suppressing state. Here the authors integrate the known functions of p53 to postulate how changes in nutrient availability may induce the ribosomal protein–Mdm2-p53 signaling pathway to modulate p53-dependent metabolic regulation.
Keywords: ribosome biogenesis, Mdm2, p53, metabolism
Introduction
The Mdm2-p53 stress response pathway is an important regulator of cellular homeostasis that is generally accepted to respond to a variety of stressors, thus eliciting hallmark effects on cell proliferation, apoptosis, and senescence.1 As such, p53 plays an essential role in monitoring the balance between cellular growth and proliferation.2 Generally, p53 transactivation is determined by stabilization and accumulation of the protein, thus facilitating transcription of downstream target genes. The p53 protein is regulated through an auto-regulatory feedback loop at the posttranslational level by the E3 ubiquitin ligase Mdm2. As the primary negative regulator of p53, Mdm2 plays a key role in determining overall p53 stability and transactivation function.
A number of regulatory factors have been reported to regulate p53 stabilization by binding to and inhibiting the catalytic ubiquitination function of Mdm2 or inducing posttranslational modifications to block the Mdm2-p53 interaction, with the net effect being p53 stabilization. The nucleolar protein ARF (Alternative Reading Frame) is a product of the INK4A/ARF locus that, when induced, binds to Mdm2 to inhibit p53 degradation.3 Protein modification on Mdm2, such as ubiquitination, sumoylation, and phosphorylation,4 work by inhibiting the Mdm2-p53 interaction and thus stabilizing p53 levels. The latest cast of Mdm2 inhibitors comprises a number of ribosomal proteins (RPs), which like ARF, have been consistently demonstrated to bind to Mdm2 and block p53 ubiquitination, thereby inducing p53-directed target gene activation.
The nucleolus is a non-membrane-bound structure of the nucleus where rDNA gene clusters are transcribed and the transcripts processed and modified to produce mature rRNA. The generally accepted paradigm for ribosome biogenesis is the coordinated assembly of equimolar concentrations of ribosomal proteins and ribosomal RNA to generate 60S and 40S ribosome precursors and then mature 80S polysomes that ensure adequate protein synthesis and maintain cellular homeostasis. In humans, this process requires the activity of all 3 RNA polymerases. RNA polymerase I transcribes the 47S precursor rRNA, which is further processed to 18S, 5.8S, and 28S rRNAs. The fourth rRNA, 5S, is transcribed separately by RNA polymerase III in the nucleus, exported to the cytoplasm, and finally imported to the nucleolus for incorporation into the large ribosomal subunit.5 Approximately 79 ribosomal proteins are actively transcribed by RNA polymerase II, exported to the cytosol for translation, and imported to the nucleolus for assembly.6 In addition, approximately 30% of the nucleolar proteome comprises a group of auxiliary factors that assist in the processing of rRNA, assembly of the small and large subunits, and finally export and maturation of the functional ribosome.7-9 The entire process is estimated to use upwards of 60% of cellular resources10 and therefore seems reasonable that it is monitored extensively for quality control.
As a transcription factor, p53 regulates gene expression patterns to alter genetic programs, a paradigm holding true for metabolic regulation as well. To date, there are a number of reports supporting the role of p53 in the regulation of glycolysis, aerobic respiration, and reactive oxygen species (ROS).11 The differential transactivation capabilities of p53 have been found to depend on both the intrinsic DNA binding affinity of p53 to various p53 response elements, as well as the overall protein levels.12 In response to mild cellular stress, p53 may induce high-affinity downstream target genes involved in DNA repair, antioxidant regulation, and metabolism, thereby promoting cell survival through minor corrections that maintain efficient viability. Intermediate to high levels of stress may drive p53 to access lower affinity genes that drive a cell fate toward cell cycle arrest, senescence, and apoptosis—all in an effort to remove cells that retain irreversible genotoxic or cellular damage, which could otherwise be detrimental to an organism. Arguably, the most common stress p53 encounters is the modest day-to-day, or physiological, stress arising from natural metabolic dynamics that represent constitutive effects of cell growth and division.
Perturbations to ribosome biogenesis such as inadequate rRNA transcription, disruption of rRNA processing, or RP imbalances have all been demonstrated to trigger so-called nucleolar stress, resulting, in most cases, in the breakdown of nucleolar structure and activation of the RP-Mdm2-p53 pathway,13 supporting the notion of the nucleolus as a central stress response regulator for p53 activation.14 However, insights into the natural physiological stressors that affect ribosome biogenesis and disrupt nucleolar integrity have been largely elusive, making it difficult to define the RP-Mdm2-p53 pathway as a bona fide intrinsic signaling pathway. Furthermore, the consequences of modulating p53 function through this pathway have, as a result, been primarily undefined. Given the sensitivity of ribosome biogenesis to cellular energy status and the defined roles of p53 in metabolic regulation, we have explored potential avenues of nutrient fluctuation that could integrate the physiological “day-to-day” stress activation of p53 through the RP-Mdm2-p53 pathway to modulate p53-dependent metabolic feedback and maintain overall cellular homeostasis.
The Ribosomal Protein-Mdm2-p53 Pathway
Under normal conditions, p53 is generally kept at relatively low levels through the activity of Mdm2. Mdm2 is thought to negatively regulate p53 through direct binding and masking of the DNA binding domain or by acting as an E3 ubiquitin ligase to mediate the conjugation of ubiquitin to p53 for proteasomal degradation. In regards to these two potential mechanisms, the key to regulating p53 stability is to inhibit the physical interaction of p53 with Mdm2. Previous evidence highlights distinct signaling pathways that respond to specific stressors to inhibit Mdm2. For instance, the tumor suppressor ARF is induced in response to oncogenic stress such as RAS or c-myc overexpression, whereby it binds to the central acidic domain of Mdm2 to inhibit p53 turnover.3,15,16 Additional forms of genotoxic stress resulting from ionizing and ultraviolet radiation, or various chemicals, can activate the ATM-Chk1 or ATM-Chk2 kinase cascades to promote phosphorylation of both Mdm2 and p53, ultimately inhibiting their association.17-19 Additional forms of posttranslational Mdm2 modification such as acetylation, methylation, neddylation, and sumoylation can also work in similar fashion to support p53 stabilization.20-22
The latest proteins implicated in Mdm2 regulation are a subset of ribosomal proteins that, like ARF, have mostly been demonstrated to bind to the central acidic domain of Mdm2 to inhibit the Mdm2-p53 interaction. Initial evidence of direct RP-Mdm2 interactions occurred with the report of RPL5 binding to Mdm2 in a 5S rRNA-RPL5-Mdm2-p53 ribonucleoprotein complex.23 Subsequent reports implicated the large subunit ribosomal proteins RPL5, RPL11, and RPL23 as Mdm2 binding partners that block the E3 ubiquitin ligase function of Mdm2 to promote p53 accumulation.24-29 Additional evidence for the roles of RPS7,30,31 RPL26,32 and RPS333 as Mdm2 binding partners has since been presented, further validating the notion that Mdm2 has multiple RP binding partners.
Despite the common thread of RP binding to Mdm2, there appear to be distinct binding site affinities and mechanisms for each of the ribosomal proteins; findings that may suggest individual responses to nucleolar stress and explain the apparent redundancy of RP-Mdm2 binding partners. For instance, binding analysis has revealed that RPL5 and RPL11 bind to the C4 zinc finger region of Mdm2, but the binding is lost in the naturally occurring Mdm2C305F point mutation variant.34 However, an additional C4 zinc finger variant, Mdm2C305S, is deficient for RPL11 binding but retains the capacity for RPL5 binding,35 suggesting that even slight structural modifications to Mdm2 can have significant implications for RP binding. These studies were taken one step further by generating a mouse knock-in model harboring the Mdm2C305F point mutation, which confirmed in vivo the deficiency of RPL5 and RPL11 binding to the zinc finger region.36 Notably, Mdm2C305F mice exhibited a normal p53 response to DNA damage but an attenuated response when challenged with nucleolar stress. Furthermore, loss of a robust RP-Mdm2-p53 response accelerated Eµ-Myc-induced lymphomagenesis independently of ARF induction, suggesting that the RP-Mdm2-p53 pathway is a genuine failsafe responder to nucleolar stress. It is unclear why RPL23, which binds outside of the zinc finger region, does not compensate for RPL5 and RPL11 deficiency. Given the differences in amino acid sequence preference, RPL5, RPL11, and RPL23 may form a ternary complex on Mdm234,37 or synergize with each other38 to maximize the inhibitory effect on Mdm2. Further studies investigating the importance of individual RPs will be necessary to dissect the relationships, as well as any potential specific growth inhibitory or nucleolar stress responses, to Mdm2.
Ribosome Biogenesis and Nucleolar Stress
As mentioned, ribosome biogenesis is a highly complex activity requiring the coordinated responses of all 3 RNA polymerases to synthesize new rRNA, transcribe individual RPs and cofactors, and finally assemble all components in the nucleolus to manufacture ribosomes. Mdm2 is a nucleo-cytoplasmic shuttling protein, whereas RPs are translated in the cytosol and shuttled to the nucleolus, where they are incorporated into nascent subunits of ribosomes for cytoplasmic export and maturation. So when and where do individual RPs interact with Mdm2 to facilitate p53 stabilization? It is conceivable that RPs may interact with Mdm2 upon nuclear import in transit to the nucleolus. If so, elevated rates of RP translation would enhance association of RPs with Mdm2, thereby increasing p53 in response to an overabundance of RPs. Conversely, degradation or breakdown of cytosolic polysomes could enhance the levels of freely available nuclear RPs and trigger a p53 response.27,29,35 Given that the nucleolus contains no physical membrane, RPs could freely shuttle between the nucleolus and nucleus to interact with Mdm2,39 or vice versa, Mdm2, potentially bound to the nucleolar protein ARF,40,41 could transiently shuttle to the nucleolus to bind to RPs. Finally, the most favored possibility posits that the nucleolus sequesters free ribosomal proteins until disruption of ribosome biogenesis triggers breakdown of the nucleolus, thereby releasing a free pool of ribosomal proteins into the nuclear space to bind to and inhibit Mdm2. This so-called nucleolar stress response is supported by a plethora of observations in the literature and can be broken down into 3 components: disruption of rRNA transcription, perturbation to rRNA processing, and RP imbalances.
Disruption of rRNA Synthesis Activates p53
In the context of ribosome biogenesis, nucleolar stress specifically refers to the perturbation to the dynamics and flow of ribosome synthesis.13 Disruptions to rRNA transcription and processing, as well as imbalances in ribosomal proteins and processing factors, in many cases, have been reported to induce the breakdown of nucleolar structure and activate a p53 stress response. Experimental techniques designed to mimic these imbalances include inhibition of precursor rRNA synthesis through administration of low doses of actinomycin D,26,29 an antineoplastic antibiotic compound that, at low concentrations (<10nM), specifically disrupts ribosome biogenesis by intercalating into the GC-rich regions of rDNA to inhibit PolI-mediated transcription of nascent 47S rRNA.42,43 Additional commonly used chemotherapeutic compounds such as 5-flourouracil (5-FU), a uracil analogue antimetabolite that functions by misincorporation into nascent RNA to block complete RNA synthesis,44 and mycophenolic acid (MPA), an agent that selectively inhibits inosine monophosphate dehydrogenase to deplete the guanine nucleotide pool and disrupt pre-ribosomal RNA synthesis,45 have also been used to induce nucleolar stress responses to demonstrate p53 stabilization through RPL5- and RPL11-directed inhibition of Mdm2.46,47
Genetic models that lead to suppression of rRNA transcription have produced a number of fairly consistent observations in regards to p53 activation. One applicable example is deletion of the RNA PolI transcription cofactor TIF-1A in mouse embryonic fibroblasts, where loss of TIF-1A disrupts nucleolar integrity and correlates to elevated p53 with activation of apoptosis.48 Other murine cell types, such as neural progenitors and hippocampal neurons, have also been demonstrated to induce a p53 response in the absence of TIF-1A,49 further supporting the notion that depletion of the rRNA precursor can trigger a p53-mediated nucleolar stress response. Disruption of rDNA transcription, as well as processing, by ablation of BAP28, a component of the PolI machinery and the U3 small nucleolar RNA-containing RNP complex, respectively, triggers a p53-dependent apoptotic phenotype in the developing nervous system of zebrafish that can subsequently be rescued by deletion of p53.50 SL1 complexes are recruited to the rDNA promoters to facilitate PolI-mediated transcription. Activation of the tumor suppressor PTEN can block SL1 recruitment,51 thereby reducing rRNA production and potentially contributing to a p53 response. Likewise, the tumor suppressor ARF, already established as a robust negative regulator of Mdm2, can inhibit phosphorylation of upstream binding factor (UBF)52 to effectively inhibit rRNA synthesis and induce p53 independently of Mdm2 binding.
Disruption of rRNA Processing Activates p53
Ribosomal RNA is initially transcribed as a long 47S precursor that must be efficiently processed by a series of nucleolar proteins to 18S, 5.8S, and 28S rRNAs to be incorporated into ribosomal subunits. Infidelity in rRNA processing can lead to accumulation of unprocessed intermediate transcripts that can retard subunit assembly, thereby triggering a nucleolar stress event. One example of this comes from the study of the rRNA processing factor Bop1, which is necessary for 47S cleavage at the internal transcribed spacers ITS1 and ITS2, as well as the 3′ external spacer.53 Expression of a dominant negative form of Bop1 in mouse cells inhibits production of 28S and 5.8S rRNA and, as a result, blocks cell cycle progression in a p53-dependent manner.54 In a similar vein, mutation or depletion of the WD40 repeat protein WDR12, which forms a complex with Bop1 and Pes1 and is involved in rRNA processing, also induces p53 to block cell proliferation.55 The tumor suppressor ARF can delay rRNA processing,56 in part through mediating degradation of nucleophosmin NPM/B23, a factor essential for a number of processing events.57,58
Additional work in animal models further validates the concept of disrupted rRNA processing triggering p53-dependent effects. For instance, Wrd36 is required for 18S processing in zebra-fish and, when deficient, leads to p53 activation.59 Furthermore, inactivation of RNA binding motif protein 19 (Rpm19) in the mouse triggers apoptosis in the morula stage of the developing embryo, subsequently terminating further development.60 Collectively, these data support the notion that inappropriate accumulation of mature rRNA, via the disruption of appropriate rRNA precursor processing, is sufficient to signal a stress response to p53 that may derive from the RP-Mdm2 pathway.
Ribosomal Protein Imbalances Activate p53
A consistent supply of ribosomal proteins is essential for maintaining the fidelity of nascent ribosome synthesis, so naturally an imbalance in the stoichiometric ratio of RPs might be considered to hinder the flow of subunit assembly to induce nucleolar stress. Using cell culture models, depletion of the small 40S protein RPS9 was shown to negatively affect cell proliferation by activating p53-mediated cell cycle arrest.61 Moreover, knockdown of HIP/RPL29 was shown to have similar effects in colon cancer cells.62 For the most part, in the absence of nucleolar stress, transient decreases in the reported RP binding partners RPL11,26 RPL5,27 RPS7,31 and RPS3,33 which would be thought to contribute to an RP imbalance, had no effect on p53 stabilization in cell culture. This may reflect the importance of each RP in promoting Mdm2 inhibition, leading to the speculation that the RPs may work in a cooperative fashion to fully enable the p53 stress response. Conversely, knockdown of RPL23, another Mdm2 binding protein, did in fact activate p53.29 These apparent discrepancies regarding RP depletion may be reconciled by acknowledging the variability in RNAi knockdown and the cell types studied, both of which could have an impact on the p53-dependent phenotypes.
Animal models have been a valuable tool to gain more precise insight into the nature of ribosomal protein deficiencies and the p53 response. Forward genetic screens in zebrafish identified 11 different ribosomal proteins that, under haploinsufficient conditions, greatly increased the rates of spontaneous malignant peripheral sheath tumors (MPSTs),63 a phenotype that was found to be, in a number of cases, dependent on the loss of p53 synthesis.64,65 Further analysis investigating RP mutants and contribution to MPST found a subset of RPs that also contributed to growth impairment.66 Despite the expectation of increased p53 activation in these RP-deficient models, the observations of decreased protein synthesis and growth impairment may in fact reflect a partial p53-dependent phenotype given that p53 itself plays a negative feedback role in suppressing ribosome biogenesis, a process that would be expected to attenuate production of new ribosomes, followed by decreased protein synthesis and therefore growth impairment. Deficiencies of RPs in other zebrafish studies have directly validated a p53 response, as is the case for RPS19, a gene commonly mutated in Diamond-Blackfan anemia, as well as RPS8, RPS11, and RPS18, where the occurrence of hematopoietic and developmental abnormalities could be rescued by concomitant loss of p53.67 Finally, loss of RPL11 was also demonstrated to be embryonic lethal due to developmental abnormalities, and the abnormality can be rescued, at least in part, by depletion of p53.68
Taken together, there are fairly clear connections between RP levels and p53 function in zebrafish, but murine models have perhaps provided the majority of evidence to support a p53 response to nucleolar stress. For instance, T cell– specific homozygous deletion of RPS6, a small subunit protein that acts through the mTOR pathway as the downstream effector of S6 kinase, resulted in impairment of T cell development in a manner dependent on p53.69 Moreover, haploinsufficiency of Rps6 in frog oocytes was embryonic lethal at day E5.5 owing to p53-dependent apoptosis.70 One key mediator of the RP-Mdm2-p53 pathway in RPS6 knockouts was demonstrated in the liver, where RPL11 inhibited Mdm2 to activate p53. Interestingly, RPL11 was able to activate p53 function in the absence of nucleolar breakdown,71 suggesting that more subtle perturbations to ribosome synthesis may leave the nucleolus intact and functional but modulate the activity of p53 to account for dynamic, yet transient, fluctuations in ribosome biogenesis. Given the ubiquitous necessity of ribosome biogenesis across cell types, it is surprising to find a range of seemingly limited phenotypes associated with RP imbalances. Mutations in RPS19 and RPS20 are reported to reduce overall body size and activate p53-dependent expression of Kit ligand, resulting in excessive pigmentation and a dark skin phenotype (aka epidermal melanocytosis).72 RPL24 ablation, one of the first RP mutant mice to be characterized, also displays skin pigmentation defects but produces congenital malformations of the eye and skeleton73,74—phenotypes that are likely dependent on elevation of basal p53 levels.75 Finally, RPL22 was found to block αβ T cell but not γδ T cell lineage progression by inducing a p53-dependent cell cycle arrest.76 These observations reflect the variability of phenotypes observed when ribosomal proteins are deficient, but the common theme among all RP imbalances is the activation of p53. Therefore, it seems likely that the RP-Mdm2-p53 pathway is functional in vivo and may represent a common surveillance system that responds to a wide variety of ribosome biogenesis perturbations.
Metabolic Fluctuations Alter Ribosome Biogenesis
Nucleolar integrity is influenced by a number of factors, including, but not limited to, genotoxic, osmotic, and oncogenic stress, as well as hypoxia and viral infection (reviewed in Boulon et al.77). As stated, ribosome biogenesis is a highly coordinated function requiring all 3 classes of RNA polymerase to appropriately maintain the balance between rRNA transcription and processing to production of ribosomal proteins. Given the intense energetic demand for producing new ribosomes, it should come as no surprise that fluctuations in cellular energy status have a profound impact on the overall capacity for ribosome biogenesis. The most obvious contributor to cellular energetics is nutrient availability, which may constitute a common physiological stressor requiring constant and dynamic monitoring to accommodate conditions where fluctuations in nutrient load lead to adaptations that couple the energetic requirements of ribosome biogenesis to overall cellular energy levels.
One key regulator of the link between nutrient availability and ribosome biogenesis is the mammalian target of rapamycin (mTOR) pathway. mTOR has a well-established role in sensing nutrient deprivation to initiate signaling cascades resulting in downregulation of ribosome biogenesis with reductions in protein synthesis, ultimately leading to reduced cell growth and proliferation.78 The mechanisms surrounding these series of events are likely numerous and beyond the scope of this review. However, mTOR does play a key role in regulating ribosome biogenesis, in part through coordinating the activities of RNA polymerases PolI, PolII, and PolIII (reviewed in Mayer and Grummt79). The impact on ribosome biogenesis by mTOR occurs primarily by modulating the rates of rRNA synthesis or ribosomal protein translation. Observations regarding PolI-mediated transcription of precursor rRNA have demonstrated how mTOR works on multiple levels to affect the activity,80 subcellular localization,81 and cofactor recruitment of PolI to rDNA genes.82,83 Likewise, 5S rRNA synthesis has also been implicated as a target of mTOR, whereby cofactor recruitment of PolIII to nuclear 5S rDNA genes is affected by mTOR-dependent phosphorylation status.84 Finally, mTOR has been reported to regulate both the transcription and translation of PolII-dependent ribosomal protein mRNAs.85 Collectively, these studies support the notion that nutrient deprivation–triggered suppression of mTOR can induce downregulation of ribosome biogenesis by specifically influencing the rates of polymerase-dependent rRNA synthesis and RP production. Given the role of mTOR in sensing nutrient availability and the previously stated imbalances in rRNA transcription, processing, and RP protein availability directly affecting nucleolar integrity, it stands to reason that specific nutrient signals acting through mTOR to mediate changes to specific aspects of ribosome biogenesis may contribute to nucleolar stress and activate the RP-Mdm2-p53 pathway.
p53 and Energy Metabolism
To carry out normal metabolic functions to maintain cell growth and proliferative homeostasis, eukaryotic cells must meet energetic demands through ATP production. The primary carbon source for ATP production is glucose, but lipids and amino acids can also be catabolized to provide fuel for energy production. ATP synthesis can occur through the more “ancient” process of glycolysis or by aerobic respiration, which occurs in the mitochondria. Glycolysis and mitochondrial respiration are both used simultaneously to yield the net energy necessary for eukaryotic cell function, but the balance between the two processes is tightly regulated and adaptable to varying metabolic conditions. Although the yield of ATP from glycolysis may be low, it is a rapid process that may become the preferred method of ATP production in contexts where high demand for energy is necessary such as contraction of muscle fibers or increased biosynthesis of proteins and cell structures. Glycolysis may also be more relied on during conditions of mitochondrial dysfunction, where oxygen concentrations are low or the mitochondria are otherwise impaired.
Research over the past decade has begun to emphasize the role of p53 in regulating cell metabolism under low to moderate stress conditions. Since a cell may be constantly undergoing metabolic perturbations due to constantly changing physiological conditions, a more accurate representation of p53 in this context is a metabolic stress response regulator, altering cellular conditions during nonlethal or low-stress conditions. Metabolic stress can come in a variety of forms, including increased or decreased ATP demands, carbon source availability, fluctuating oxygen concentrations, growth factor signaling, and any other number of common “day-to-day” stresses a cell might encounter. Stabilization of p53 has been demonstrated to decrease glycolysis and enhance aerobic respiration, whereas loss of the gene corresponds to decreased mitochondrial biogenesis, lowered oxygen consumption, and increased rates of glycolysis. Interestingly, in the presence of wild-type p53, net ATP production remains stable but is skewed in favor of mitochondrial respiration. However, when p53 function is lost, ATP is primarily derived through glycolytic energy production.86 In part, this observation has led to some insight regarding the genetic switches accounting for the Warburg effect. This is the observation that, even under conditions of high oxygen availability, most cancer cells shift ATP production from oxidative phosphorylation in the mitochondria to the less efficient (in terms of total energy produced from glucose) process of glycolysis. Given that p53 is mutated or inactivated in the majority of human cancer, it is reasonable to suspect that p53 plays a role in governing the switch from aerobic to anaerobic metabolism.
A number of p53 target genes have been identified that provide a partial explanation for the observation of p53 as a metabolic switch (Figure 1). One of the first metabolic genes to be identified as a p53-regulated target is phosphoglycerate mutase (PGM),87 a glycolytic enzyme that catalyzes the reversible conversion of 3-phosphoglycerate to 2-phosphoglycerate through a 2,3 bisphosphoglycerate intermediate. PGM is actually repressed by p53, and in doing so, glycolytic function is decreased. However, since PGM is not critical for regulation of glycolysis, it only created an initial framework for explaining the role of p53 in shifting metabolic tides. A second gene identified as a p53-inducible target gene is synthesis of cytochrome c oxidase 2 (SCO2).86 SCO2 regulates the cytochrome c oxidase (COX) complex of the electron transport chain where the majority of oxygen is consumed during oxidative phosphorylation. Targeted disruption of one Sco2 allele in mice was sufficient to recapitulate the altered distribution of ATP production observed in p53 null mice, where glycolysis is favored over respiration. Expression of SCO2 in p53 null HCT116 cell lines restored the oxygen consumption levels seen in p53 wild-type mice, indicating that SCO2 was critical in driving ATP production in the mitochondria. Another p53-inducible target gene is Tp53-induced glycolysis and apoptosis regulator (TIGAR), a gene that inhibits glycolysis by lowering fructose-2,6-bisphosphate levels.88 This favors the accumulation of fructose-1,6-bisphosphate, which is effectively shunted into the pentose phosphate pathway to produce NADPH nucleotides. An additional biological effect of TIGAR expression is to lower detrimental levels of ROS in the cell and to enhance resistance to apoptotic stimuli. Another gene identified as a p53-regulated metabolic target is guanidinoacetate methyltransferase (GAMT), an enzyme critical for creatine biosynthesis.89 GAMT catalyzes the conversion of guanidinoacetate to creatine from glycine, arginine, or methionine substrates. Creatine is produced from these amino acids primarily in the kidneys and liver, where it is secreted into peripheral blood circulation for utilization by muscle cells for energy production. Specifically, once in muscle cells, creatine enhances ATP recycling by using a phosphocreatine intermediate to convert ADP back to the usable ATP. On an additional note, under glucose-deprived conditions, GAMT enhances fatty acid oxidation (FAO), thereby enhancing this alternative fuel source for maintenance of energy production. The latest gene identified in the p53-regulated metabolic suite is glutaminase 2 (GLS2), a mitochondrial enzyme that promotes the conversion of the amino acid glutamine to glutamate.90,91 Through de-amination, glutamate is converted to α-ketoglutarate, where it can be shunted into the TCA cycle to promote mitochondrial respiration. Positive regulation of both GAMT and GLS2 highlights the implications of p53 in promoting lipid and amino acid catabolism, respectively; both are mitochondrial functions that are upregulated when glucose-derived pyruvate becomes limiting and the cell is forced to switch to alternative carbon sources. Taken together, these genes provide partial mechanistic insight into the function of p53 in surveying metabolic conditions and driving a shift toward aerobic respiration. Moreover, this evidence highlights how modulation of p53 levels may be imperative to regulating the cellular response to daily fluctuations in nutrient load and availability. In doing so, p53 would need to transactivate a host of target genes to adapt to changing metabolic conditions but do so largely at the expense of activating the more extreme functions of apoptosis and senescence: functions that need not be mutually exclusive from metabolic regulation but may be appropriately attenuated in the face of mild physiological stress.
Figure 1.
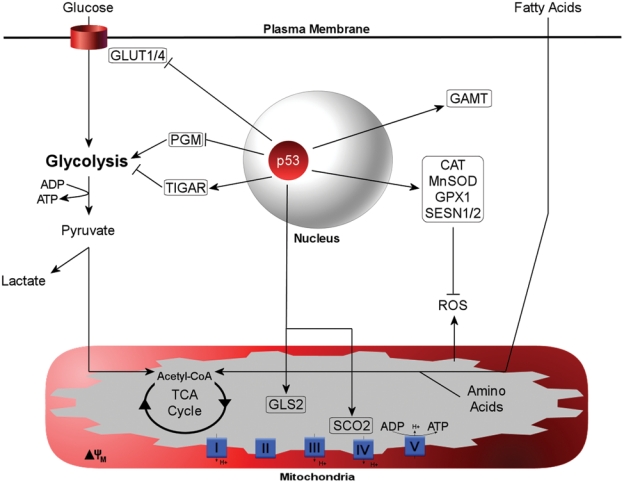
Metabolic target genes of p53. Reported target genes for p53 are boxed. The functional outcomes of the p53 metabolic program include inhibition of glucose import, attenuation of glycolysis, upregulation of creatine levels for increased ATP, enhanced mitochondrial respiration correlating to increased amino acid and fatty acid oxidation, and upregulation of the antioxidant system.
Additional metabolic targets of p53 include a number of antioxidant genes that serve to maintain homeostasis of ROS. Several antioxidants have been reported to be downstream transcriptional targets of p53 and include catalase (CAT), manganese superoxide dismutase 2 (SOD2), glutathione peroxidase (GPX1), and sestrins 1 and 2 (SESN1/2).92 Furthermore, GLS2, by increasing glutamate levels, enhances glutathione production, which can act as an additional buffer to protect against ROS-induced oxidative damage and apoptosis.90,91 Basal p53 function has been reported to be essential for ROS regulation under conditions of low stress.93 Since mitochondria, specifically the electron transport chain, are the primary intracellular source or ROS, it is reasonable that a p53-mediated increase in oxidative phosphorylation may be coupled with p53-induced antioxidant genes to offset ROS generated from enhanced mitochondrial function.
Based on the target genes that have thus far been characterized, the functional outcome of the p53 metabolic response includes inhibition of glucose import, attenuation of glycolysis, promotion of ATP production through increased creatine levels, and enhancement of mitochondrial function where amino acid and lipid oxidation is increased (Figure 1). These functions are consistent with p53 inhibiting anabolic cell growth and promoting catabolic energy-sparing pathways. Although it is not precisely clear under what conditions such a program would be implemented, changes in nutrient availability are a logical place to look and have received some attention. The majority of evidence surrounding nutrient sensing and p53 function is in the context of glucose availability, the primary preferred carbon source for energy acquisition. Interestingly, glucose starvation has been demonstrated in a cell culture model to induce a transient rise in p53 Ser15 phosphorylation, a marker for p53 transactivation potential.94 Complete glucose deprivation promoted maximal phosphorylation of p53 at 45 minutes and returned to baseline levels within 3 hours, signifying a rapid, yet transient, response to glucose depletion. Amp-activated protein kinase (AMPK) plays a key role in monitoring energy status by sensing and responding to fluctuating AMP/ATP ratios, generally stimulating catabolic responses to generate ATP during periods of energy depletion or increased demand.95 The effects of glucose deprivation on p53 phosphorylation are generally correlated with and may be dependent on AMPK activation. When cells were starved for glucose, p53 induced G1/S cell cycle arrest that was shown to be dependent on AMPK function and could be reversed upon glucose addition.96 Furthermore, the effects of glucose deprivation are further supported in a study analyzing p53 levels and function in TSC–/– mouse embryonic fibroblasts (MEFs), where the constitutively high levels of mTOR activity promote p53 translation and therefore greater basal protein levels. Glucose deprivation in TSC–/– MEFs induced p53 Ser15 phosphorylation and accumulation, an effect correlating to enhanced p53-dependent apoptosis.97 Finally, glucose deprivation was demonstrated to activate AMPKα, the catalytic subunit of the AMPK complex, and promote p53 accumulation. Although the investigators in this study were unable to detect Ser15 or Ser20 phosphorylation, Ser46 was phosphorylated and subsequently associated with enhanced transactivation of p53.98 Collectively, there is a decent body of evidence supporting p53 activation in response to glucose deprivation. When combined with the evidence surrounding the known cadre of p53 metabolic target genes, it seems that p53 could act as a promoter of “energy scavenging” during periods of acute glucose starvation. Given that glucose is the primary carbon source of the cell, when glucose becomes a limiting nutrient, we can surmise that transitory p53 activation promotes an acute metabolic shift to bridge the energy gap as a cell transitions from conventional glucose oxidation to alternative substrates, such as amino acids and lipids, through activation of catabolic pathways. In line with the well-established role of p53 in promoting survival under low to moderate stress, this more subtle “burst” of p53 activity may actually promote conservation of cellular resources and energy by shifting carbon source utilization to those derived from catabolic processes. Furthermore, these effects can be reconciled with the known functions of p53, whereby temporary stalling of cell cycle progression and inhibition of cell growth could provide a window of opportunity to restructure nutrient partitioning and utilization to allow for sufficient energetic recovery that is permissive to anabolic growth and proliferation.
Perspectives: Does Nutrient Stress Regulate the Ribosomal Protein-Mdm2-p53 Pathway?
Decades of research have solidified the critical functions of p53 in stress surveillance, particularly as they pertain to cell cycle regulation and apoptosis. As such, p53 has garnered a reputation as a first responder to significant deleterious genotoxic and cytotoxic stress to elicit repair processes that either preserve cell viability or initiate cell death in times of irreversible damage. Although certainly still true, it seems difficult to believe that the origin of p53 was to function solely in this context. A recent review on the history of p53 traces an evolutionary function back over one billion years to the preservation of germline and stem cell integrity,99 suggesting that p53 has a remarkable track record for monitoring cellular homeostasis. Here we aimed to extend the well-established role of p53 as an overall stress response regulator to include the monitoring of nutrient stress and utilization through the RP-Mdm2-p53 pathway.
Generally speaking, nutrient load and composition have a significant bearing on the anabolic (energy-consuming) versus catabolic (energy-sparing) state of the cell. At the organismal level, nutrient starvation induces a milieu of hormonal changes that drive a shift toward alternative forms of fuel in an effort to conserve energy and preserve basic cellular functions. If no immediate forms of fuel are readily available, meaning glucose in particular, catabolic pathways are activated to mobilize alternative energy substrates from stored depots or generate glucose from noncarbohydrate sources. For example, additional glucose can be derived from the oxidation of liver and muscle glycogen, amino acids can be generated from the peptides of muscle breakdown, or, conversely, lipids can be mobilized from adipocytes to provide energy rich fatty acids in times of low glucose availability. These 3 primary micronutrients—glucose, amino acids, and lipids—are in a dynamic flux dependent on cellular supply and demand. The cellular decision regarding how best to partition and use these resources is likely based on a delicate balance of feedback mechanisms that respond to reprogram a cell according to a current metabolic climate. We propose that the RP-Mdm2-p53 pathway may contribute to the highly dynamic process of nutrient partitioning and metabolic reprogramming, thereby constituting a nutrient stress surveillance system.
Thus far, we have briefly characterized three major concepts: 1) Nutrient availability regulates the rate of ribosome biogenesis and can contribute to nucleolar stress, 2) the RP-Mdm2-p53 pathway monitors nucleolar stress to elicit a p53 response, and 3) p53 governs a suite of genes involved in energy metabolism. Given that nutrient availability plays a key role in regulating ribosome biogenesis, it is expected that the RP-Mmd2-p53 pathway may function in some capacity to mediate the nucleolar stress response to nutrient supply and, in turn, modulate the metabolic program of p53 to appropriately compensate for the nutrient changes. For instance, during anabolic phases when glucose levels are abundant, the activity of Mdm2 would keep p53 levels relatively low, amino acid and fatty acid oxidation would be largely suppressed by the high availability of glucose, and glycolysis-derived pyruvate would supply sufficient acetyl-CoA to the mitochondria for aerobic respiration. In turn, there would be sufficient energy available to facilitate ribosome biogenesis and support protein synthesis to drive cell growth and proliferation. In contrast, under glucose-deprived conditions, the activation of AMPK and resulting decrease in mTOR activity could induce nucleolar stress to activate the RP-Mdm2-p53 pathway, inhibiting Mdm2 by RP binding and contributing to p53 stabilization. During this period, p53 would elicit a transcriptional response that would suppress glucose metabolism and shift the balance toward energy-scavenging catabolic pathways such as amino acid and fatty acid oxidation to support mitochondrial respiration. In addition, these effects may coincide with reduction in cell growth and p53-mediated stalling of the cell cycle to inhibit premature proliferation. Indeed, in this context, p53 could act as a switch during acute metabolic perturbations when a shift in nutrient partitioning is essential and, by doing so, create a cellular environment conducive to utilization of readily available carbon sources (Figure 2).
Figure 2.
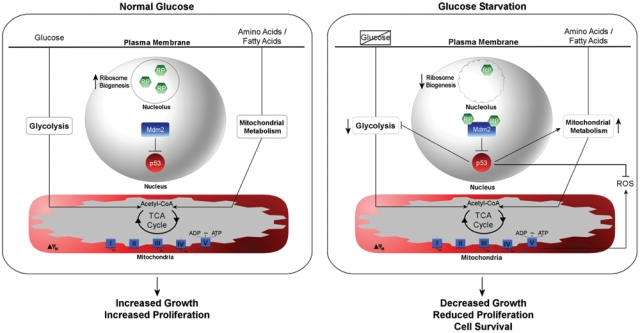
Hypothetical model for nutrient partitioning through the RP-Mdm2-p53 pathway. (Left panel) During normal periods of growth when glucose levels are adequate, ribosomal proteins (RPs) are sequestered in the nucleolus to promote ribosome biogenesis, and p53 levels are maintained at low levels by Mdm2. Glucose utilization is high and amino acid or fatty acid catabolism may supplement energetic requirements as necessary for anabolic growth and division. (Right panel) Glucose deprivation, through suppression of ribosome biogenesis, may induce a nucleolar stress response (squiggled circle), prompting RP inhibition of Mdm2 to stabilize p53. p53 transactivation may downregulate glycolytic rates and promote mitochondrial metabolism through enhanced oxidation of amino acids and fatty acids. In addition, enhanced reactive oxygen species (ROS) levels may be neutralized by p53-induced antioxidants. The functional outcome of glucose deprivation could be to promote cell survival by using alternative carbon sources while simultaneously attenuating energetically demanding growth and proliferation.
Based on the current proposed model, if p53 is suppressing glycolysis under conditions where glucose is limiting, then why would it also be enhancing aerobic respiration if the available pool of acetyl-CoA derived from glycolysis were suppressed? Alternative carbon sources are provided through catabolic pathways that meet the unmet mitochondrial demand for TCA cycle intermediates to sustain and even enhance respiration. Under conditions of glucose deprivation, FAO provides an abundant source of energy-rich fatty acids that are readily oxidized to acetyl-CoA to feed into the TCA cycle to maintain mitochondrial respiration and adequate energy production. By suppressing glycolysis but simultaneously enhancing mitochondrial respiration, is p53 creating an environment favorable to mitochondrial beta-oxidation of fatty acids? A glimpse into this possibility was presented from evidence illustrating how GAMT could support FAO.89 In addition, p53 has been reported being induced in adipocytes of obese ob/ob mice,100 resulting in upregulation of proinflammatory cytokines and decreased insulin sensitivity but also in suppression of lipogenic genes.101 Taken together, p53 may contribute to lipolysis in adipocytes, promoting liberation of fatty acids for utilization not only by adipocytes but also by peripheral tissues such as the liver and muscle, where p53 may promote mitochondrial FAO. It is of interest to note that previous observations regarding p53-mediated enhancement of mitochondrial function have used oxygen consumption as a marker for activity. Enhanced oxygen consumption is also observed when mitochondrial membrane potential is uncoupled from oxidative phosphorylation and mitochondrial uncoupling is associated with enhanced FAO.102 Treatment of mitochondria with chemical uncouplers such as 2,4-dinitrophenol or FCCP has been shown to correlate with enhanced rates of FAO.103 Perhaps even more telling is the fact that cells use a natural mechanism via uncoupling proteins to promote depletion of mitochondrial membrane potential, a process that coincides with enhanced FAO.104 For instance, inhibition of biological uncoupling proteins, such as UCP2, blunts the rate of lipid oxidation in the mitochondria,105 indicating that mitochondrial uncoupling may be a natural process associated with FAO. Higher rates of mitochondrial FAO would coincide with increased levels of oxygen consumption while still maintaining adequate ATP production from oxidative phosphorylation to meet cellular demands. The p53-dependent switch from glycolysis to aerobic respiration may actually reflect nutrient partitioning effects of p53, whereby fatty acid oxidation and amino acid catabolism are enhanced in the mitochondria when p53 is stabilized, leading to the observed enhanced oxygen consumption. Much experimentation is necessary to determine if p53 influences utilization of particular carbon sources and how this, in turn, affects energy synthesis from glycolysis or mitochondrial respiration.Furthermore, p53 induces a host of antioxidant genes, which, in this scenario, would help to buffer the enhanced levels of ROS produced from the mitochondrial uncoupling effects of increased FAO. The research investigating whether p53 might play any role in promoting lipid metabolism is essentially untouched but certainly an area ripe for exploration. The use of animal models to study p53-dependent effects on metabolism will better reflect the intricate hormonal makeup affecting nutrient load, partitioning, and tissue utilization that are not readily produced in cell culture models.
Much effort has gone into defining how nutrient deprivation influences cellular responses to this common metabolic stress. However, nutrient stress works on both sides of the spectrum, begging the question, does the RP-Mdm2-p53 pathway facilitate the proposed role of p53 as a metabolic stress response regulator when it comes to nutrient overload? Given the emerging preponderance of obesity as a worldwide epidemic, it seems just as imperative to determine the cellular stress responses associated with nutrient abundance. Physiologically, obesity is associated with enhanced levels of circulating glucose and fatty acids, and this surplus of nutrients may be constantly stimulating anabolic processes. In the context of the model proposed here, this likely means enhanced rates of growth factor signaling, upregulation of ribosome biogenesis, and, as a result, enhanced protein synthesis that may further drive cell growth and proliferation. Just as p53 has a significant role in regulating unrestricted proliferation when it comes to cancer, the paradigm may be essentially the same when it comes to nutrient overload and unrestricted anabolism. Proto-oncogenes that normally stimulate anabolic processes are mutated in cancer to become constitutively active, but in an overfed state, the surplus of nutrients may supply a constant stimulus to activate many of the same signaling pathways. Henceforth, p53 may have a vital role in suppressing overt nutrient-stimulated signaling by restricting the rates of cell growth and proliferation. Some insight into this potential paradigm comes from work with the Mdm2C305F zinc finger mutation, which was demonstrated to lose binding to ribosomal proteins L5 and L11, thereby circumventing the nucleolar stress response to p53.34 As stated earlier, the mouse model harboring a “knock-in” allele of the Mdm2C305F mutation was shown to respond normally to DNA damage but was deficient in response to various forms of nucleolar stress. In addition, the Mdm2C305F mouse was crossed with a transgenic model constitutively expressing the oncogene Eµ-Myc, which, in part, functions to significantly increase the rates of ribosome biogenesis. Interestingly, mice with a normal Mdm2 allele exhibited a p53 stress response to the upregulated levels of ribosome biogenesis, whereas the response was greatly attenuated in the Mdm2C305F mutant.36 These results indicate that upregulated ribosome biogenesis may constitute just as a significant a stressor as decreased ribosome biogenesis, highlighting the RP-Mdm2-p53 axis as a critical responder to nucleolar stress. Therefore, this observation could be extended to more physiological stress, specifically as it pertains to nutrient abundance and induction of nucleolar stress. It has yet to be determined if nutrient excess contributes to an RP-Mdm2-p53 metabolic stress response and, in turn, how p53 may affect nutrient partitioning in this context. Mouse models such as the Mdm2C305F mutant, as well as conditional and tissue-specific knockouts of some of the key ribosomal protein players such as RPL5, RPL11, and RPL23, will provide valuable tools to further our understanding of the RP-Mdm2-p53 pathway as a potential metabolic stress response pathway.
When put into a collective perspective, the aforementioned observations support the notion that energy homeostasis may be regulated through a balance between nutrient availability and utilization. Dynamic fluctuations in substrate availability may be constantly monitored to drive transcriptional responses that induce cellular reprogramming to most efficiently use the forms of energy that are available at any given time. Although not a seemingly major player in energy sensing, the evidence suggests it is certainly plausible for p53 to have a contributing role as a molecular metabolic switch to direct nutrient utilization in an effort to promote cell survival. Future research oriented toward clarifying the role of the RP-Mdm2-p53 pathway as a metabolic stress response system and any role it may play in governing potential p53 modulation of nutrient partitioning and energy production provides an exciting opportunity to expand upon the myriad aspects of p53 in regulating cellular homeostasis.
Acknowledgments
We thank Paula L. Miliani de Marval and Everardo Macias for sharing unpublished information. We apologize for not being able to cite all of the relevant papers due to limited space.
Footnotes
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Y.Z. is support by grants from the Leukemia & Lymphoma Society, the American Cancer Society, and the National Institutes of Health.
References
- 1. Levine AJ, Feng Z, Mak TW, You H, Jin S. Co-ordination and communication between the p53 and IGF-1-AKT-TOR signal transduction pathways. Genes Dev. 2006;20:267-75 [DOI] [PubMed] [Google Scholar]
- 2. Vogelstein B, Lane D, Levine AJ. Surfing the p53 network. Nature. 2000;408:307-10 [DOI] [PubMed] [Google Scholar]
- 3. Sharpless NE. INK4a/ARF: a multifunctional tumor suppressor locus. Mutat Res. 2005;576:22-38 [DOI] [PubMed] [Google Scholar]
- 4. Meek DW, Knippschild U. Posttransiational modification of MDM2. Mol Cancer Res. 2003;1:1017-26 [PubMed] [Google Scholar]
- 5. Szymanski M, Barciszewska MZ, Erdmann VA, Barciszewski J. 5 S rRNA: structure and interactions. Biochem J. 2003;371:641-51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lempiainen H, Shore D. Growth control and ribosome biogenesis. Curr Opin Cell Biol. 2009;21:855-63 Epub Sep 30, 2009 [DOI] [PubMed] [Google Scholar]
- 7. Kressler D, Linder P, de La Cruz J. Protein trans-acting factors involved in ribosome biogenesis in Saccharomyces cerevisiae. Mol Cell Biol. 1999;19:7897-912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Venema J, Tollervey D. Ribosome synthesis in Saccharomyces cerevisiae. Annu Rev Genet. 1999;33:261-311 [DOI] [PubMed] [Google Scholar]
- 9. Boisvert F-M, van Koningsbruggen S, Navascues J, Lamond AI. The multifunctional nucleolus. Nat Rev Mol Cell Biol. 2007;8:574-85 [DOI] [PubMed] [Google Scholar]
- 10. Warner JR. The economics of ribosome biosynthesis in yeast. Trends Biochem Sci. 1999;24:437-40 [DOI] [PubMed] [Google Scholar]
- 11. Gottlieb E, Vousden KH. p53 regulation of metabolic pathways. Cold Spring Harb Perspect Biol. 2009;2:a001040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Inga A, Storici F, Darden TA, Resnick MA. Differential transactivation by the p53 transcription factor is highly dependent on p53 level and promoter target sequence. Mol Cell Biol. 2002;22:8612-25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Deisenroth C, Zhang Y. Ribosome biogenesis surveillance: probing the ribosomal protein-Mdm2-p53 pathway. Oncogene. 2010;29:4253-60 [DOI] [PubMed] [Google Scholar]
- 14. Rubbi CP, Milner J. Disruption of the nucleolus mediates stabilization of p53 in response to DNA damage and other stresses. Embo J. 2003;22:6068-77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zhang Y, Xiong Y. Control of p53 ubiquitination and nuclear export by MDM2 and ARF. Cell Growth Differ. 2001;12:175-86 [PubMed] [Google Scholar]
- 16. Sherr CJ. Divorcing ARF and p53: an unsettled case. Nat Rev Cancer. 2006;6:663-73 [DOI] [PubMed] [Google Scholar]
- 17. Blattner C, Hay T, Meek DW, Lane DP. Hypophosphorylation of Mdm2 augments p53 stability. Mol Cell Biol. 2002;22:6170-82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hay TJ, Meek DW. Multiple sites of in vivo phosphorylation in the MDM2 oncoprotein cluster within two important functional domains. FEBS Lett. 2000;478:183-6 [DOI] [PubMed] [Google Scholar]
- 19. Maya R, Balass M, Kim ST, et al. ATM-dependent phosphorylation of Mdm2 on serine 395: role in p53 activation by DNA damage. Genes Dev. 2001;15:1067-77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Brooks CL, Gu W. Ubiquitination, phosphorylation and acetylation: the molecular basis for p53 regulation. Curr Opin Cell Biol. 2003;15:164-71 [DOI] [PubMed] [Google Scholar]
- 21. Melchior F, Hengst L. SUMO-1 and p53. Cell Cycle. 2002;1:245-9 [PubMed] [Google Scholar]
- 22. Prives C, Manley JL. Why is p53 acetylated? Cell. 2001;107:815-8 [DOI] [PubMed] [Google Scholar]
- 23. Marechal V, Elenbaas B, Piette J, Nicolas JC, Levine AJ. The ribosomal L5 protein is associated with mdm-2 and mdm-2-p53 complexes. Mol Cell Biol. 1994;14:7414-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lohrum MA, Ludwig RL, Kubbutat MH, Hanlon M, Vousden KH. Regulation of HDM2 activity by the ribosomal protein L11. Cancer Cell. 2003;3:577-87 [DOI] [PubMed] [Google Scholar]
- 25. Zhang Y, Wolf GW, Bhat K, et al. Ribosomal protein L11 negatively regulates oncoprotein MDM2 and mediates a p53-dependent ribosomal-stress checkpoint pathway. Mol Cell Biol. 2003;23:8902-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bhat KP, Itahana K, Jin A, Zhang Y. Essential role of ribosomal protein L11 in mediating growth inhibition-induced p53 activation. EMBO J. 2004;23:2402-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Dai MS, Lu H. Inhibition of MDM2-mediated p53 ubiquitination and degradation by ribosomal protein L5. J Biol Chem. 2004;279:44475-82 [DOI] [PubMed] [Google Scholar]
- 28. Dai MS, Zeng SX, Jin Y, Sun XX, David L, Lu H. Ribosomal protein L23 activates p53 by inhibiting MDM2 function in response to ribosomal perturbation but not to translation inhibition. Mol Cell Biol. 2004;24:7654-68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Jin A, Itahana K, O’Keefe K, Zhang Y. Inhibition of HDM2 and activation of p53 by ribosomal protein L23. Mol Cell Biol. 2004;24:7669-80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Chen D, Zhang Z, Li M, et al. Ribosomal protein S7 as a novel modulator of p53-MDM2 interaction: binding to MDM2, stabilization of p53 protein, and activation of p53 function. Oncogene. 2007;26:5029-37 [DOI] [PubMed] [Google Scholar]
- 31. Zhu Y, Poyurovsky MV, Li Y, et al. Ribosomal protein S7 is both a regulator and a substrate of MDM2. Mol Cell. 2009;35:316-26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ofir-Rosenfeld Y, Boggs K, Michael D, Kastan MB, Oren M. Mdm2 regulates p53 mRNA translation through inhibitory interactions with ribosomal protein L26. Mol Cell. 2008;32:180-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Yadavilli S, Mayo LD, Higgins M, Lain S, Hegde V, Deutsch WA. Ribosomal protein S3: a multi-functional protein that interacts with both p53 and MDM2 through its KH domain. DNA Repair (Amst). 2009;8:1215-24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lindstrom MS, Jin A, Deisenroth C, White Wolf G, Zhang Y. Cancer-associated mutations in the MDM2 zinc finger domain disrupt ribosomal protein interaction and attenuate MDM2-induced p53 degradation. Mol Cell Biol. 2007;27:1056-68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Gilkes DM, Chen L, Chen J. MDMX regulation of p53 response to ribosomal stress. EMBO J. 2006;25:5614-25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Macias E, Jin A, Deisenroth C, et al. An ARF-independent c-MYC-activated tumor suppression pathway mediated by ribosomal protein-Mdm2 interaction. Cancer Cell. 2010;18:231-43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Dai MS, Shi D, Jin Y, et al. Regulation of the MDM2-p53 pathway by ribosomal protein L11 involves a post-ubiquitination mechanism. J Biol Chem. 2006;281:24304-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Horn HF, Vousden KH. Cooperation between the ribosomal proteins L5 and L11 in the p53 pathway. Oncogene. 2008;27:5774-84 [DOI] [PubMed] [Google Scholar]
- 39. Chen D, Huang S. Nucleolar components involved in ribosome biogenesis cycle between the nucleolus and nucleoplasm in interphase cells. J Cell Biol. 2001;153:169-76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Tao W, Levine AJ. P19(ARF) stabilizes p53 by blocking nucleo-cytoplasmic shuttling of Mdm2. Proc Natl Acad Sci U S A. 1999;96:6937-41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Weber JD, Taylor LJ, Roussel MF, Sherr CJ, Bar-Sagi D. Nucleolar Arf sequesters Mdm2 and activates p53. Nat Cell Biol. 1999;1:20-6 [DOI] [PubMed] [Google Scholar]
- 42. Perry RP. Selective effects of actinomycin D on the intracellular distribution of RNA synthesis in tissue culture cells. Exp Cell Res. 1963;29:400-6 [Google Scholar]
- 43. Sobell HM, Jain SC, Sakore TD, Nordman CE. Stereochemistry of actinomycin–DNA binding. Nat New Biol. 1971;231:200-5 [DOI] [PubMed] [Google Scholar]
- 44. Longley DB, Harkin DP, Johnston PG. 5-fluorouracil: mechanisms of action and clinical strategies. Nat Rev Cancer. 2003;3:330-8 [DOI] [PubMed] [Google Scholar]
- 45. Huang M, Ji Y, Itahana K, Zhang Y, Mitchell B. Guanine nucleotide depletion inhibits pre-ribosomal RNA synthesis and causes nucleolar disruption. Leuk Res. 2008;32:131-41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Sun XX, Dai MS, Lu H. 5-Fluorouracil activation of p53 involves an MDM2-ribosomal protein interaction. J Biol Chem. 2007;282:8052-9 [DOI] [PubMed] [Google Scholar]
- 47. Sun XX, Dai MS, Lu H. Mycophenolic acid activation of p53 requires ribosomal proteins L5 and L11. J Biol Chem. 2008;283:12387-92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Yuan X, Zhou Y, Casanova E, et al. Genetic inactivation of the transcription factor TIF-IA leads to nucleolar disruption, cell cycle arrest, and p53-mediated apoptosis. Mol Cell. 2005;19:77-87 [DOI] [PubMed] [Google Scholar]
- 49. Parlato R, Kreiner G, Erdmann G, et al. Activation of an endogenous suicide response after perturbation of rRNA synthesis leads to neurodegeneration in mice. J Neurosci. 2008;28:12759-64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Azuma M, Toyama R, Laver E, Dawid IB. Perturbation of rRNA synthesis in the bap28 mutation leads to apoptosis mediated by p53 in the zebrafish central nervous system. J Biol Chem. 2006;281:13309-16 [DOI] [PubMed] [Google Scholar]
- 51. Zhang C, Comai L, Johnson DL. PTEN represses RNA Polymerase I transcription by disrupting the SL1 complex. Mol Cell Biol. 2005;25:6899-911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Ayrault O, Andrique L, Fauvin D, Eymin B, Gazzeri S, Seite P. Human tumor suppressor p14ARF negatively regulates rRNA transcription and inhibits UBF1 transcription factor phosphorylation. Oncogene. 2006;25:7577-86 [DOI] [PubMed] [Google Scholar]
- 53. Strezoska Z, Pestov DG, Lau LF. Functional inactivation of the mouse nucleolar protein Bop1 inhibits multiple steps in pre-rRNA processing and blocks cell cycle progression. J Biol Chem. 2002;277:29617-25 [DOI] [PubMed] [Google Scholar]
- 54. Pestov DG, Strezoska Z, Lau LF. Evidence of p53-dependent cross-talk between ribosome biogenesis and the cell cycle: effects of nucleolar protein Bop1 on G(1)/S transition. Mol Cell Biol. 2001;21:4246-55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Holzel M, Rohrmoser M, Schlee M, et al. Mammalian WDR12 is a novel member of the Pes1-Bop1 complex and is required for ribosome biogenesis and cell proliferation. J Cell Biol. 2005;170:367-78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Sugimoto M, Kuo ML, Roussel MF, Sherr CJ. Nucleolar Arf tumor suppressor inhibits ribosomal RNA processing. Mol Cell. 2003;11:415-24 [DOI] [PubMed] [Google Scholar]
- 57. Itahana K, Bhat KP, Jin A, et al. Tumor suppressor ARF degrades B23, a nucleolar protein involved in ribosome biogenesis and cell proliferation. Mol Cell. 2003;12:1151-64 [DOI] [PubMed] [Google Scholar]
- 58. Bertwistle D, Sugimoto M, Sherr CJ. Physical and functional interactions of the Arf tumor suppressor protein with nucleophosmin/B23. Mol Cell Biol. 2004;24:985-96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Skarie JM, Link BA. The primary open-angle glaucoma gene WDR36 functions in ribosomal RNA processing and interacts with the p53 stress-response pathway. Hum Mol Genet. 2008;17:2474-85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Zhang J, Tomasini AJ, Mayer AN. RBM19 is essential for preimplantation development in the mouse. BMC Dev Biol. 2008;8:115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Lindstrom MS, Zhang Y. Ribosomal protein S9 is a novel B23/NPM-binding protein required for normal cell proliferation. J Biol Chem. 2008;283:15568-76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Liu JJ, Huang BH, Zhang J, Carson DD, Hooi SC. Repression of HIP/RPL29 expression induces differentiation in colon cancer cells. J Cell Physiol. 2006;207:287-92 [DOI] [PubMed] [Google Scholar]
- 63. Amsterdam A, Sadler KC, Lai K, et al. Many ribosomal protein genes are cancer genes in zebrafish. PLoS Biol. 2004;2:E139 Epub May 11, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Berghmans S, Murphey RD, Wienholds E, et al. tp53 mutant zebrafish develop malignant peripheral nerve sheath tumors. Proc Natl Acad Sci U S A. 2005;102:407-12 Epub Jan 3, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. MacInnes AW, Amsterdam A, Whittaker CA, Hopkins N, Lees JA. Loss of p53 synthesis in zebrafish tumors with ribosomal protein gene mutations. Proc Natl Acad Sci U S A. 2008;105:10408-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Lai K, Amsterdam A, Farrington S, Bronson RT, Hopkins N, Lees JA. Many ribosomal protein mutations are associated with growth impairment and tumor predisposition in zebrafish. Dev Dyn. 2009;238:76-85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Danilova N, Sakamoto KM, Lin S. Ribosomal protein S19 deficiency in zebrafish leads to developmental abnormalities and defective erythropoiesis through activation of p53 protein family. Blood. 2008;112:5228-37 [DOI] [PubMed] [Google Scholar]
- 68. Chakraborty A, Uechi T, Higa S, Torihara H, Kenmochi N. Loss of ribosomal protein L11 affects zebrafish embryonic development through a p53-dependent apoptotic response. PLoS One. 2009;4:e4152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Sulic S, Panic L, Barkic M, Mercep M, Uzelac M, Volarevic S. Inactivation of S6 ribosomal protein gene in T lymphocytes activates a p53-dependent checkpoint response. Genes Dev. 2005;19:3070-82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Panic L, Tamarut S, Sticker-Jantscheff M, et al. Ribosomal protein S6 gene haploinsufficiency is associated with activation of a p53-dependent checkpoint during gastrulation. Mol Cell Biol. 2006;26:8880-91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Fumagalli S, Di Cara A, Neb-Gulati A, et al. Absence of nucleolar disruption after impairment of 40S ribosome biogenesis reveals an rpL11-translation-dependent mechanism of p53 induction. Nat Cell Biol. 2009;11:501-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. McGowan KA, Li JZ, Park CY, et al. Ribosomal mutations cause p53-mediated dark skin and pleiotropic effects. Nat Genet. 2008;40:963-70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Oliver ER, Saunders TL, Tarle SA, Glaser T. Ribosomal protein L24 defect in belly spot and tail (Bst), a mouse Minute. Development. 2004;131:3907-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Tang Q, Rice DS, Goldowitz D. Disrupted retinal development in the embryonic belly spot and tail mutant mouse. Dev Biol. 1999;207:239-55 [DOI] [PubMed] [Google Scholar]
- 75. Barkic M, Crnomarkovic S, Grabusic K, et al. The p53 tumor suppressor causes congenital malformations in Rpl24-deficient mice and promotes their survival. Mol Cell Biol. 2009;29:2489-504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Anderson SJ, Lauritsen JP, Hartman MG, et al. Ablation of ribosomal protein L22 selectively impairs alphabeta T cell development by activation of a p53-dependent checkpoint. Immunity. 2007;26:759-72 [DOI] [PubMed] [Google Scholar]
- 77. Boulon S, Westman BJ, Hutten S, Boisvert FM, Lamond AI. The nucleolus under stress. Mol Cell. 2010;40:216-27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Zoncu R, Efeyan A, Sabatini DM. mTOR: from growth signal integration to cancer, diabetes and ageing. Nat Rev Mol Cell Biol. 2010;12:21-35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Mayer C, Grummt I. Ribosome biogenesis and cell growth: mTOR coordinates transcription by all three classes of nuclear RNA polymerases. Oncogene. 2006;25:6384-91 [DOI] [PubMed] [Google Scholar]
- 80. Mahajan PB. Modulation of transcription of rRNA genes by rapamycin. Int J Immunopharmacol. 1994;16:711-21 [DOI] [PubMed] [Google Scholar]
- 81. Li H, Tsang CK, Watkins M, Bertram PG, Zheng XF. Nutrient regulates Tor1 nuclear localization and association with rDNA promoter. Nature. 2006;442:1058-61 [DOI] [PubMed] [Google Scholar]
- 82. Hannan KM, Brandenburger Y, Jenkins A, et al. mTOR-dependent regulation of ribosomal gene transcription requires S6K1 and is mediated by phosphorylation of the carboxy-terminal activation domain of the nucleolar transcription factor UBF. Mol Cell Biol. 2003;23:8862-77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Claypool JA, French SL, Johzuka K, et al. Tor pathway regulates Rrn3p-dependent recruitment of yeast RNA polymerase I to the promoter but does not participate in alteration of the number of active genes. Mol Biol Cell. 2004;15:946-56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Zaragoza D, Ghavidel A, Heitman J, Schultz MC. Rapamycin induces the G0 program of transcriptional repression in yeast by interfering with the TOR signaling pathway. Mol Cell Biol. 1998;18:4463-70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Powers T, Walter P. Regulation of ribosome biogenesis by the rapamycin-sensitive TOR-signaling pathway in Saccharomyces cerevisiae. Mol Biol Cell. 1999;10:987-1000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Matoba S, Kang JG, Patino WD, et al. p53 regulates mitochondrial respiration. Science. 2006;312:1650-3 [DOI] [PubMed] [Google Scholar]
- 87. Kondoh H, Lleonart ME, Gil J, et al. Glycolytic enzymes can modulate cellular life span. Cancer Res. 2005;65:177-85 [PubMed] [Google Scholar]
- 88. Bensaad K, Tsuruta A, Selak MA, et al. TIGAR, a p53-inducible regulator of glycolysis and apoptosis. Cell. 2006;126:107-20 [DOI] [PubMed] [Google Scholar]
- 89. Ide T, Brown-Endres L, Chu K, et al. GAMT, a p53-inducible modulator of apoptosis, is critical for the adaptive response to nutrient stress. Mol Cell. 2009;36:379-92 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 90. Hu W, Zhang C, Wu R, Sun Y, Levine A, Feng Z. Glutaminase 2, a novel p53 target gene regulating energy metabolism and antioxidant function. Proc Natl Acad Sci U S A. 2010;107:7455-60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Suzuki S, Tanaka T, Poyurovsky MV, et al. Phosphate-activated glutaminase (GLS2), a p53-inducible regulator of glutamine metabolism and reactive oxygen species. Proc Natl Acad Sci U S A. 2010;107:7461-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Olovnikov IA, Kravchenko JE, Chumakov PM. Homeostatic functions of the p53 tumor suppressor: regulation of energy metabolism and antioxidant defense. Semin Cancer Biol. 2009;19:32-41 Epub. Dec 3, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Sablina AA, Budanov AV, Ilyinskaya GV, Agapova LS, Kravchenko JE, Chumakov PM. The antioxidant function of the p53 tumor suppressor. Nat Med. 2005;11:1306-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Feng Z, Zhang H, Levine AJ, Jin S. The coordinate regulation of the p53 and mTOR pathways in cells. Proc Natl Acad Sci U S A. 2005;102:8204-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Canto C, Auwerx J. AMP-activated protein kinase and its downstream transcriptional pathways. Cell Mol Life Sci. 2010;67:3407-23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Jones RG, Plas DR, Kubek S, et al. AMP-activated protein kinase induces a p53-dependent metabolic checkpoint. Mol Cell. 2005;18:283-93 [DOI] [PubMed] [Google Scholar]
- 97. Lee CH, Inoki K, Karbowniczek M, et al. Constitutive mTOR activation in TSC mutants sensitizes cells to energy starvation and genomic damage via p53. EMBO J. 2007;26:4812-23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Okoshi R, Ozaki T, Yamamoto H, et al. Activation of AMP-activated protein kinase induces p53-dependent apoptotic cell death in response to energetic stress. J Biol Chem. 2008;283:3979-87 [DOI] [PubMed] [Google Scholar]
- 99. Belyi VA, Ak P, Markert E, et al. The origins and evolution of the p53 family of genes. Cold Spring Harb Perspect Biol. 2009;2:a001198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Yahagi N, Shimano H, Matsuzaka T, et al. p53 Activation in adipocytes of obese mice. J Biol Chem. 2003;278:25395-400 [DOI] [PubMed] [Google Scholar]
- 101. Minamino T, Orimo M, Shimizu I, et al. A crucial role for adipose tissue p53 in the regulation of insulin resistance. Nat Med. 2009;15:1082-7 [DOI] [PubMed] [Google Scholar]
- 102. Harper JA, Dickinson K, Brand MD. Mitochondrial uncoupling as a target for drug development for the treatment of obesity. Obes Rev. 2001;2:255-65 [DOI] [PubMed] [Google Scholar]
- 103. Modriansky M, Gabrielova E. Uncouple my heart: the benefits of inefficiency. J Bioenerg Biomembr. 2009;41:133-6 [DOI] [PubMed] [Google Scholar]
- 104. Ledesma A, de Lacoba MG, Rial E. The mitochondrial uncoupling proteins. Genome Biol. 2002;3:REVIEWS3015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Sheets AR, Fulop P, Derdak Z, et al. Uncoupling protein-2 modulates the lipid metabolic response to fasting in mice. Am J Physiol Gastrointest Liver Physiol. 2008;294:G1017-24 Epub Feb 21, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]