

Abstract
Identification and study of genetic variation in recently admixed populations not only provides insight into historical population events but also is a powerful approach for mapping disease loci. We studied a population (OG-W-IP) that is of African-Indian origin and has resided in the western part of India for 500 years; members of this population are believed to be descendants of the Bantu-speaking population of Africa. We have carried out this study by using a set of 18,534 autosomal markers common between Indian, CEPH-HGDP, and HapMap populations. Principal-components analysis clearly revealed that the African-Indian population derives its ancestry from Bantu-speaking west-African as well as Indo-European-speaking north and northwest Indian population(s). STRUCTURE and ADMIXTURE analyses show that, overall, the OG-W-IPs derive 58.7% of their genomic ancestry from their African past and have very little inter-individual ancestry variation (8.4%). The extent of linkage disequilibrium also reveals that the admixture event has been recent. Functional annotation of genes encompassing the ancestry-informative markers that are closer in allele frequency to the Indian ancestral population revealed significant enrichment of biological processes, such as ion-channel activity, and cadherins. We briefly examine the implications of determining the genetic diversity of this population, which could provide opportunities for studies involving admixture mapping.
Introduction
The Indian population represents a substantial fraction of global diversity and has been shaped by multiple waves of migration and local admixture events.1,2–5 Admixed populations offer special opportunities for mapping disease loci6,7,8 as well as studying signatures of selection.9,10,11 An admixture event between populations leads to an extended linkage disequilibrium (LD), which could greatly facilitate the mapping of human disease loci.12 The power of gene mapping by admixture linkage disequilibrium (MALD) primarily depends upon two factors: (1) the extent and strength of LD and (2) the systematic difference of the genotype and phenotype in the ancestral populations.6,12,13 Typical large admixed populations such as the African Americans and the Latinos in the United States have been traditionally used for MALD.6,7,8,11,14 Recently, admixture in Asian populations such as the Uygurs in China has also been reported.15,16 Reich et al. have proposed that populations in India have arisen out of extremely ancient admixture events and that, because of this antiquity, the extent of LD in these admixed populations is small5. Furthermore, in populations within India the difference in allele frequency in the ancestral populations is small, and thus they might not furnish any distinct advantage in terms of MALD.5 In this study take a detailed look at a population that behaves as a distinct out-group when included in a study comprising populations sampled from different parts of India.3 This population, known as the Siddi, has been given a nomenclature of OG-W-IP1 by convention of the Indian Genome Variation Consortium (IGVC) because it is an out-group (OG) isolated population (IP) from the western (W) part of India. OG, reckoned to be the “lost tribe of Africa,” is one of the major nonnative tribal communities of Gujarat, and they have adapted to the local language and the religious practices of the place. It has been said that African slaves are the ancestors of this tribal community and they came to India during the 12th–15th century with the Arab merchants.17 It is also argued that the Portuguese merchants brought the African slaves to the west coast of India, possibly to Karnataka and Maharashtra. They eventually expanded and migrated northward. Apart from being located in Gujarat, this community resides in some parts of Karnataka, Goa, and Maharashtra.17,18 Interestingly, the region in Gujarat, where this tribe resides, is extremely saline, and most of the salt that is exported from India is produced in this area. The anthropological and other evidences linking the OG to their African ancestors has been weak and limited primarily to musical instruments, folklores, and traditions.17 Few genetic studies3,19 have attempted to decipher the ancestry of this population.
Our study not only provides a window to their past but also evaluates them as a resourceful population for disease variant mapping. We demonstrate that this population derives its ancestry from both Africa and India. We also demonstrate that, because of differences in the allele frequency of important genic SNPs, this admixture between African and Indian populations makes the Siddi a wonderful potential candidate population for admixture mapping. The LD structure in this particular population is large and extended, indicating recent population admixture. As indigenous and migrant populations from two different continents intermated and subsequently formed the admixed population, there were novel opportunities for natural selection to occur. Annotation of genes and analysis of associated functional enrichments revealed a significant representation of ion-channel genes, especially those related to potassium transport and cadherins.
Material and Methods
Subjects
The analysis was carried with three population datasets. The first dataset consisted of a subset of 26 reference populations based on our previous study of IGVC3 comprising 509 samples of Austro-Asiatic (AA), Tibeto-Burman (TB), Dravidian (DR), and Indo-European (IE) linguistic origins from the north (N), east (E), west (W), south (S), northeast (NE), and central (C) parts of India and one out-group population of African origin (OG). These populations represent diverse linguistic groups residing in different geographical regions and encompassing the genetic spectrum of India. The details of population identification, sample collection and DNA isolation are described elsewhere.3 For naming of populations we have followed a convention where each population was represented by their linguistic affiliation followed by geographical location and ethnicity (Table S1). The second dataset comprised 210 samples from four population of the International HapMap Project (60 CEU [Utah residents with ancestry from northern and western Europe], 60 YRI (Yoruba in Ibadan, Nigeria), 45 CHB (Han Chinese in Beijing), and 45 JPT (Japanese in Tokyo).20 The third dataset consisted of 52 populations comprising 1043 CEPH-HGDP samples (Centre d'Étude du Polymorphisme Humain [CEPH]-obtained samples from the Human Genome Diversity Panel [HGDP]).21
Genotype Datasets
We used the following genotype data on the three population sets: (1) 509 IGVC samples generated with Affymetrix 50k Xba1 240 GeneChip Human Mapping array (Affymetrix, Santa Clara, CA, USA) as a part of the IGVC project, 22 (2) 1043 samples from the CEPH-HGDP Human Genome Diversity Panel generated on an Illumina Human Hap650K Beadchip, 21 and (3) genotypes of 210 samples from the International HapMap Project.20 A common set of 18,534 SNPs that met all the standard QC criteria were merged from the three datasets (IGVC, HapMap, CEPH-HGDP) and used for further analysis. The SNPs that showed deviation from Hardy-Weinberg equilibrium within the population were excluded from data analysis. We ensured that all the genotype data were from the same strand prior to merging the data. The physical positions of the SNPs were retrieved from Homo sapiens NCBI Build 36. The average spacing between adjacent markers was 166.7 kb, and the minimum and maximum spacing was 17 bp and 31.5 Mb, respectively.
Statistical Analysis
We performed principal-components analysis (PCA) by using EIGENSOFT 3.0.23,24 We used a model-based clustering algorithm, STRUCTURE,25–27 for estimation of individual and population ancestries. For STRUCTURE analysis, we assumed two and three clusters (K = 2, 3) with 20,000 burnin period and 20,000 iterations. We also used ADMIXTURE28 software to infer individual ancestry proportions and validate our STRUCTURE results. We calculated analysis of molecular variance (AMOVA) as in Excoffier et al.,29 by using the software package ARLEQUIN.30 We computed FST and Reynold's distance31 to estimate the extent of genetic differentiation between populations. We calculated FST and its significance for all 18,534 markers by using the Weir and Hill32 method in ARLEQUIN30 with 10,000 permutations, which adjusts for sample size variation across populations. For estimation of pairwise LD33 (r2), we used PLINK34 for all the SNPs on one chromosome separately for one population at a time. The average LD per 200,000 bases was plotted.
Functional Annotation of Ancestry-Informative Markers
From our entire SNP dataset we defined a set of ancestry-informative markers (AIMS) for the ancestral populations of OG. These are markers that occur at polymorphic loci and which differ substantially in terms of allele frequencies between the two ancestral populations. We defined two hypothetical populations that can serve as putative ancestors to OG (see details in the next section). The putative African ancestry comes from a hypothetical population consisting of 32 individuals (11 belonging to Bantu [Kenya], 21 to Yoruba), and the non-African ancestor population consists of 86 individuals from four different IE-speaking groups (24 from IE-N-LP10, 23 from IE-N-LP18, 20 from IE-W-LP2, and 19 from IE-W-LP4).
We collated a set of 3396 SNPs that have an FST value > 0.1 between the two ancestral populations (Table S2). We mapped these ancestry-informative markers (AIMs) to variants reported to be associated with diseases in the Genetic Association Database (GAD) by using the SNPnexus tool. We also explored whether OG had specific functional enrichment of genes that could be attributed to either of their ancestors. For this we computed the closeness of OG to either of the ancestors by comparing the allele frequency of AIMs in OG with the Indian and African ancestral populations. We excluded from analysis all those AIMs whose frequencies were similar to their expected frequency, i.e., within a cutoff of 5% of the weighted average (weights used are the approximate ancestry estimates of 0.59 for African and 0.41 from Indian populations) of the ancestral allele frequencies. The AIMs were now binned into two groups, one close to African ancestors in terms of allele frequency and one close to the Indian ancestors, and their functional gene classification and functional annotation clustering were performed with DAVID bioinformatics resources 2008.35 We used the most stringent criteria for classification of genes, and a cutoff > 1.5 was set. Functional-annotation clustering was also carried out at the highest stringency. The results with >3-fold enrichments at ≤1% FDR have been represented. (Table 5 and Table S3).
Table 5.
Functional Annotation of Genes Encompassing the Ancestry-Informative Markers that Are Closer in Allele Frequency to the Indian Ancestral Population
| Term | Count | p Value | n-Fold Enrichment | Bonferroni | Benjamini | FDR |
|---|---|---|---|---|---|---|
| Enrichment Score: 4.324 | ||||||
| GO:0007409∼axonogenesis | 16 | 9.43 × 10−6 | 4.063 | 0.015 | 0.015 | 0.016 |
| GO:0048667∼cell morphogenesis involved in neuron differentiation | 16 | 2.44 × 10−5 | 3.752 | 0.039 | 0.008 | 0.041 |
| GO:0048812∼neuron projection morphogenesis | 16 | 3.04 × 10−5 | 3.682 | 0.048 | 0.008 | 0.051 |
| GO:0000904∼cell morphogenesis involved in differentiation | 16 | 1.42 × 10−4 | 3.214 | 0.207 | 0.023 | 0.238 |
| GO:0031175∼neuron projection development | 16 | 2.41 × 10−4 | 3.063 | 0.325 | 0.030 | 0.403 |
| Enrichment Score: 3.240 | ||||||
| potassium transport | 10 | 2.95 × 10−4 | 4.672 | 0.097 | 0.011 | 0.403 |
| potassium | 10 | 5.79 × 10−4 | 4.264 | 0.182 | 0.012 | 0.788 |
| GO:0030955∼potassium ion binding | 10 | 0.001112 | 3.871 | 0.437 | 0.062 | 1.596 |
Results
Identification of Putative Populations of OG Ancestors
Initial PCA with 26 IGV populations revealed that the OG population was distant from all TB and IE isolated populations of northern and northeastern regions as well as AA and DR isolated populations of IGV along both the principal components (Figure 1). We excluded these distant IGV populations and carried out the next level of PCA with the remaining 18 IGV populations, including OG. In the search for the possible African ancestor(s) to OG, we included all African populations from the CEPH-HGDP panel (except for Mozambites, who are highly admixed between Africans and Middle Easterners21). We also included all the CEPH-HGDP panel populations from Pakistan because of their geographical proximity to OG. Because history states that the OG was brought into India by Portuguese traders,36 we also included the CEU population from HapMap. This combined analysis was carried out on a set of 18,534 markers that were common and typed in all the studies (IGV, CEPH-HGDP, and HapMap) (see details in the Material and Methods).
Figure 1.
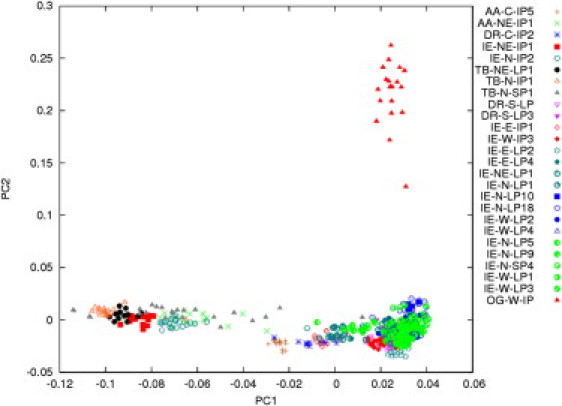
PCA of 26 Indian Populations Showing the Siddis as an OG
The second eigenvector explains the separation of the OG population from other Indian populations. The first eigenvector explains the variation in the rest of the 25 population groups. Along the first eigenvector, the populations on the left are primarily Tibeto-Burman (TB)-speaking populations, who separate from the Indo-European (IE) and Dravidian (DR) speaking populations. The populations are coded by linguistic lineage (AA, Austro-Asiatic; IE, Indo-European; DR, Dravidian; and TB, Tibeto-Burman) followed by geographical location (N, north; NE, northeast; W, west; E, east; S, south; and C, central) and ethnic category (LP, castes; Sp, religious groups; and IP, tribes).
The PCA along the first principal component (PC1) separated all African from the non-African populations and explained 6.5% of the entire variation, whereas the second principal component (PC2) led to the separation of the various non-African groups (Figure 2). Of note, the separation of the non-African population groups is achieved prior to the separation of the African populations, even though the latter are known to be extremely diverse in terms of genetic variation. We observed a distinct gradient of decreasing genetic similarity (representing a cline) of Indian populations with the west- and central-Asian gene pools as we looked eastward or southward from the northwestern corridor along the PC2. Figure 2 also reveals that the OG population lies on a direct line between north and northwest Indian populations and the Africans, revealing varying levels of admixture between these two broad groups. The distance from the HapMap CEU along the PC2 reduces the possibility that the OGs derive their non-African ancestry from Portuguese traders. We estimated the European (Portuguese) ancestry among the OGs by using STRUCTURE and ADMIXTURE, and the estimate obtained via both the methods was about 0.03.
Figure 2.
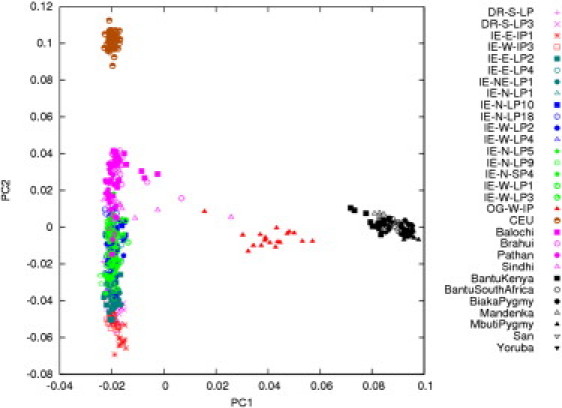
Genetic Relatedness of the OGs with Populations of the Indian Subcontinent and Africa
PCA of 17 Indian populations (IE speakers and DR speakers), seven African populations, and four Pakistani populations from HGDP, CEU from HapMap, and the OGs clearly shows that the OGs are admixed between Indians and Africans and that there is no contribution from CEU.
To further narrow down on the possible ancestors of OG in India and Africa, we carried out PCAs of OG with African populations as well as Dravidian and Indo-European-speaking Indian populations that are close to OG (Figure 3), i.e., we left out the Pakistani populations and the CEU. As before, PC1 separated the Africans and the Indians (7.95%), and PC2 separated the various African groups. The two pygmy populations (Biaka, Mbuti) and the San of South Africa were well separated from the other African groups, whereas a greater genetic affinity appeared to exist between the Mandenka of west Africa, the Yoruba of central west Africa, and the Bantu speakers, who derive from Kenya in east Africa. It is also clear in Figure 3 that the OGs lies between the Indians and the Bantu Kenyans, reflecting the varying levels of admixture between these two continental groups. The next step was the identification of the non-African population(s) who might be considered ancestral to the OG. On the basis of the distance matrix of EIGENSTRAT(Figures 2 and 3) and the Reynold's distances between the populations, we reduced our search of ancestral populations to the following: IE-N-LP10, IE-N-LP18, IE-W-LP2, and IE-W-LP4. Although these might not be the exact population(s) that has contributed to the gene pool of the OG, the genetic distance between population(s) that might be the actual ancestors to OG should not be genetically very different from that of our shortlisted choice of four.
Figure 3.
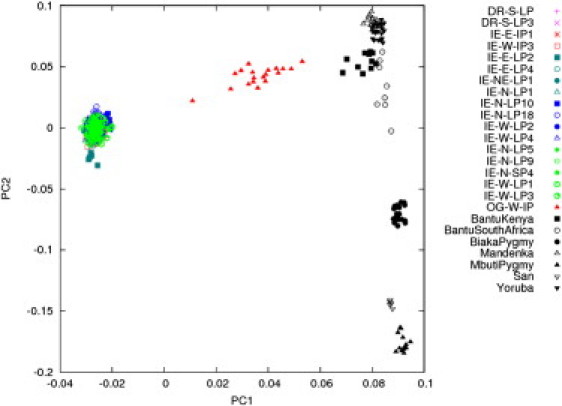
Genetic Relatedness of OGs with Populations in India and Africa
PCA of 17 Indian populations (IE speakers and DR speakers) and seven African populations from HGDP shows that the OGs are admixed from Indians, west Africans, and the Bantu Kenyans.
The PCA with the four Indo-European-speaking Indian populations along with OG and the two African populations show the separation of the Indo-Europeans and Africans along the PC1 (8.67%) (Figure 4). The variation between the Indo-European populations is more than that between the two African populations, resulting in a clearer separation of the Indo-Europeans along PC2 (1.44%). The variation along PC2 separates seven individuals of the IE-N-LP18 from the rest of the samples. We have seen that these individuals are separate and distinct from the rest of the group in all subsequent analyses.
Figure 4.
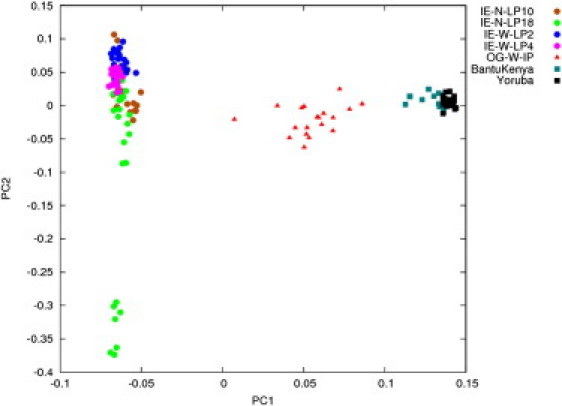
Genetic Composition of the Most Likely Ancestors of OGs in the Indian and African Population
PCA of four Indian IE-speaking populations and two African populations that were considered ancestors of the OGs shows separation of Indian and Africans populations along PC1 and a clear separation of Indo-Europeans along PC2.
Variability between the Ancestral Populations
Table 1 shows FST estimates for the OG population and various other populations contributing to the admixture. It is to be noted that the FST estimates between the African and the IE speaking Indian populations are extremely high (>0.1), whereas the OG lies in between (<0.05). The large pairwise FST values between the Indian and the African populations are indicative of the large genetic separation between the populations. We performed an Analysis of Molecular Variance Analysis or AMOVA29,30 with 3 groups. The four Indian populations constituted one group, the two African populations constituted the second group while the OG was the third group. The AMOVA results confirmed what we observed in Table 1; the percentage of variation among groups was 7.92 with a permutation p-value of 0.01 (Table 2).
Table 1.
Extent of Genetic Differentiation Estimated by Pairwise FST, × 1000, between the Populations
| IE-N-LP10 | IE-N-LP18 | IE-W-LP2 | IE-W-LP4 | OG | Bantu | |
|---|---|---|---|---|---|---|
| IE-N-LP10 | ||||||
| IE-N-LP18 | 8 | |||||
| IE-W-LP2 | 10 | 15 | ||||
| IE-W-LP4 | 8 | 11 | 12 | |||
| OG | 43 | 47 | 49 | 42 | ||
| Bantu | 115 | 119 | 121 | 113 | 34 | |
| Yoruba | 128 | 132 | 135 | 126 | 41 | 9 |
The pairwise FST values between the OG and the other populations are given in bold.
Table 2.
Extent of Genetic Differentiation Estimated by AMOVA
| Sources of Variation | Degrees of Freedom | Sum of Squares | Variance Components | Percentage of Variation |
|---|---|---|---|---|
| Among groups | 2 | 40,816.413 | 226.08412 Va | 7.92 |
| Among populations within groups | 4 | 14,701.267 | 28.20762 Vb | 0.99 |
| Among individuals within populations | 131 | 335,986.892 | −35.59279 Vc | −1.25 |
| Within individuals | 138 | 363,764 | 2635.97101 Vd | 92.34 |
| Total | 275 | 755,268.572 | 2854.66996 |
Individual Ancestry Proportions
We carried out STRUCTURE25–27 and ADMIXTURE28 analysis to estimate the individual ancestry (IA) of the OGs under the assumption that they were admixed between Africans (Yoruba and Bantu Kenya) and Indians (IE-N-LP10, IE-N-LP18, IE-W-LP2, and IE-W-LP4). The amount of African ancestry as estimated by STRUCTURE was 58.7% ± 8.4%, and there was a range of 40% in the OG individuals (Figure S1). STRUCTURE models LD arising out of admixture between two genetically distinct populations with different allele-frequency spectra but does not model the existing LD in the ancestral populations. It therefore performs best in dealing with genotypes that are not under strong LD. The SNP density that we have in our dataset is hence unlikely to affect the STRUCTURE results. Also, the ancestral populations we are dealing with here are very old and do not have much background LD. However, because our results are highly contingent upon the STRUCTURE findings, we validated our findings by using a reduced set of informative markers. From our set of markers, we chose those that had FST values ≥ 0.1 among ancestral Africans and Indians. By using the above-mentioned cutoff, we got a set of 3396 SNPs. We ran STRUCTURE with this set of SNPs and thus reduced the chance of background LD to minimum. The overall findings with the reduced set of SNPs and our findings with the complete set of 18534 SNPs were highly concordant (Figure 5, Table 3, and Table 4). However, it is important to note that when we increase the number of clusters in the STRUCTURE run to more than 2, neither the African populations nor the Indian populations separate. Rather, seven individuals of the population IE-N-LP18 separate as a population and the likelihood of the data is also reduced.
Figure 5.
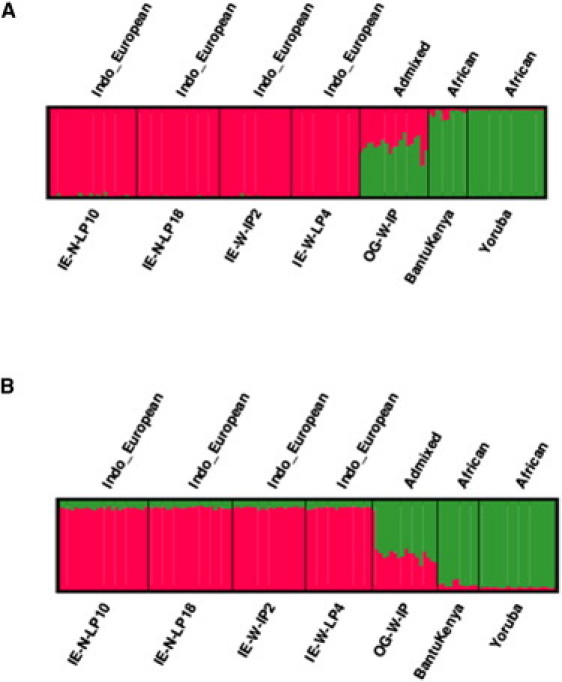
Summary Plot of Individual Admixture Proportions in OGs from Indo-European and African Ancestral Populations
Each individual is represented by a vertical line broken into two colored segments. Red lines indicate Indo-European ancestral proportions, and green lines indicate African ancestral proportions. The relative proportion of each ancestor in OGs and also in the ancestral populations is represented with length proportional to each of the inferred clusters.
(A) Analysis of admixture via the program STRUCTURE under the assumption of two ancestral populations and including data on all 18,534 SNPs.
(B) Analysis of admixture via the program STRUCTURE under the assumption of two ancestral populations and including data on 3396 AIMs (FST ≥ 0.1).
Table 3.
Proportion of Membership of Each Predefined Population in Each of the Two Clusters as Determined with 18,534 Markers
| Given Population | Inferred Cluster 1 | Inferred Cluster 2 | Number of Individuals |
|---|---|---|---|
| IE-N-LP10 | 0.009 | 0.991 | 24 |
| IE-N-LP18 | 0.002 | 0.998 | 23 |
| IE-W-LP2 | 0.003 | 0.997 | 20 |
| IE-W-LP4 | 0.001 | 0.999 | 19 |
| OG-W-IP | 0.587 | 0.413 | 19 |
| BANTUKENYA | 0.96 | 0.04 | 11 |
| YORUBA | 1 | 0 | 21 |
Table 4.
Proportion of Membership of Each Predefined Population in Each of the Two Clusters as Determined with 3396 Markers
| Given Population | Inferred Cluster 1 | Inferred Cluster 2 | Number of Individuals |
|---|---|---|---|
| IE-N-LP10 | 0.077 | 0.923 | 24 |
| IE-N-LP18 | 0.069 | 0.931 | 23 |
| IE-W-LP2 | 0.068 | 0.932 | 20 |
| IE-W-LP4 | 0.068 | 0.932 | 19 |
| OG-W-IP | 0.585 | 0.415 | 19 |
| BANTUKENYA | 0.937 | 0.063 | 11 |
| YORUBA | 0.972 | 0.028 | 21 |
Linkage Disequilibrium
It is known that, irrespective of the existence of LD in the ancestral parental populations, an admixed population derived from them will have an elevated LD for a period of time. The LD in the OG population is observed to be much higher than the Indian and/or ancestral African population (Figure 6). The pairwise LD analysis for the ancestral populations (r2 threshold > 0.2) resulted in 360 marker pairs in the Indian population and 329 marker pairs in the African population. In contrast, in OG there were 1100 marker pairs with r2 > 0.2. The average r2 drops below 0.05 for Africans and Indians within 250 Kbp, whereas the average LD is steady and above 0.1 for the OG even at distances larger than 800 Kbp. The extremely long-ranged LD among OG, compared to the African or the Indian populations, indicates that the admixture event is relatively recent.
Figure 6.
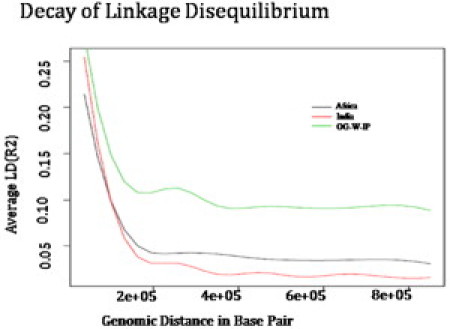
Extent of Linkage Disequilibrium among the Ancestor Populations and the Admixed Population
The figure shows the long-range LD present among the OGs compared to the ancestral Indian and African populations.
Gene Ontology Analysis of Ancestry-Informative Markers
We also wanted to see whether there were some biological processes that were selectively enriched in the admixed populations from either of the ancestors. Considering the SNPs that have an FST value ≥0.1 between the two ancestral populations, we selected 3396 of the 18,534 SNPs for functional analysis. Of these, 1218 SNPs were filtered out because their frequencies in the OG population were within 5% of the expected frequency, which is the ancestry proportionate weighted average of the allele frequencies of the two ancestral populations. The remaining SNPs were classified into two groups of 1240 and 938 SNPs on the basis of their closeness, in terms of allele frequency, to the Indian and African ancestral populations, respectively. Analysis of gene classes in these groups revealed significant enrichment of cadherins, potassium channels, membrane proteins, and solute carriers as well as protein kinases from the group close to IE and kinases and immune-related genes from the group close to African ancestry. Further functional annotation clustering (FAC) revealed significant enrichment of processes related to axonogenesis and potassium transport in genes from the group for which the frequency of SNPs is close to that of the Indian ancestral population (Table 5). However, FAC did not reveal any specific enrichment of the processes contributed by the other group.
Discussion
In this study we have dissected the ancestry and genetic structure of an Afro-Indian population residing in Gujarat by using a set of 18,534 genome-wide markers. Although it is well acknowledged that the OG population has an African origin, their specific ancestry and time of spread in mainland India has been enigmatic. We tried to elucidate the genetic structure of this population by using a set of populations from the CEPH-HGDP panel, HapMap, and 26 diverse Indian populations. In an earlier study involving 55 Indian populations and a set of 405 SNPs, we observed that the OG population was distinct from other Indian populations.3 Extending the analysis to genome-wide markers in a subset of these populations further substantiated our earlier observations (Figure 1). This is also supported by the recent observation of Reich et al.5
A prior analysis that used 377 autosomal short tandem repeat (STR) loci to examine genetic structure among the African populations included in the HGDP was able to define distinct genetic clusters for the Biaka, Mbuti, and San; however, the study lacked the power to differentiate between the Mandenka, Yoruba, and Bantu groups.37 In contrast, greater resolution of African ethnic groups, particularly for the Mandenka and Yoruba, was possible in multiple recent studies11,38,39, Our study suggests that the African slaves brought into India were unlikely to be of diverse origin; PCA revealed that they are closer to Bantu Kenya and YRI. This is unlike the multiple migration and massive slave trade that happened in the new world. Hence, although it is of interest to compare African admixture estimates to descriptions of proportional representation of various African groups during the Middle Passage and during slave trade in post-Columbian America, it is unlikely that the situation will be replicated in the Indian context. However, in the absence of data from population groups representative of southeastern and other parts of southern Africa, their genetic representation in OG remains a possibility.
It is important to note that considerable migration has occurred among African ethnic groups over the past three millennia or more. For example, the two Bantu groups included in our analysis originated from a more central African location (Nigeria-Cameroon) several millennia ago, making precise geographic localization of African ancestry difficult.39,40 This difficulty is also reflected in the close genetic relationships among the various western, west-central, and southwest African groups, who also show considerable overlap in terms of mtDNA haplotypes vis-à-vis the autosomal genome.38 Recent large-scale studies of the African genetic diversity also substantiate the closeness of the Bantu and Yorubans and have only limited representation of southern and southeastern population groups.38 Previous genetic study of the OG population have suffered from similar limitations and have rarely tried to address this population's ancestry.19 This is because researchers either lacked data on reference population or had very little genetic data, often from a single marker and a few loci. Our results are based on an examination of the entire autosomal genome and, therefore, provide a more robust picture of the admixed African ancestry of individual African Indians than have prior analyses, which focused on only a single locus (mtDNA or Y chromosome).
Our exploratory analysis looking for the non-African component of the ancestry of OG started in an agnostic fashion. We included six north Indian, IE-speaking populations, five IE-speaking populations from west India, and three IE-speaking populations from east India. We also included two Dravidian populations and one IE population from northeast. From the most comprehensive study of the human genetic diversity of Indian populations (at least in terms of the number of populations represented),3 we only excluded the Tibeto-Burman (TB)-speaking populations of India. The TB, who primarily inhabit the northeastern parts of India, are genetically close to east Asians and comprise a distinct genetic pool.1,3 They do not overlap the domain of the OG geographically, linguistically, or otherwise. Our analysis reclaims, as shown by Reich et al.5 and other groups,1,2,3 a distinct gradient of decreasing genetic similarity (representing a cline) of Indian populations with the west- and central-Asian gene pools as we move eastward or southward from the northwestern corridor (Figure 3). It also shows that if we include CEU, along with Pakistani, IE, and DR populations from all over India, the genetic variation is more than that observed in the HGDP African populations. However, when we narrow down our still agnostic search to include only the Indian populations, the intra-African variation is much larger. The oral history states that African ancestors to the OG population were brought into India by the Portuguese traders. It is possible that a very small fraction of the non-African ancestry is actually derived from the Portuguese traders. Using both ADMIXTURE and STRUCTURE, we estimated the CEU ancestry to be around 3%. Given the marker density in our dataset, it is difficult to determine whether that small proportion is real or an artifact.
Prior studies of African populations suggest close genetic kinship among various west, west-central, and southwest African ethnic groups.38 It is to be noted here that identification of exact ancestors to admixed populations is a problem that is impossible to address. This applies especially for a population such as the OG, which has remained small and has undergone random genetic drift and possible selection. We do not claim here that the four Indian and two African populations that we introduce as possible ancestors to OG are their exact ancestors. We rather claim that the differences between the two ancestral populations contributing in OG admixture are genetically so diverse that our choice is a good approximation for all practical purposes. The ancestry estimates are also largely independent of the number of markers used, and whether we use the entire genome or ancestry-informative markers, our estimates are pretty robust to the choice.
The FST estimates between the African and the IE-speaking Indian populations are extremely high (>0.1). The large pairwise FST values between the Indian and the African populations are indicative of the large genetic separation between the populations. They are also indicative of the fact that there is expected to be a large number of ancestry-informative markers, which ensures a relatively easy and efficient study design for MALD. We observed 3396 (>18%) of 18,534 SNPs to have FST values larger than or equal to 0.1 between the Indian and the African ancestral populations, indicative of the large genetic separation between the populations. This as well as the distribution of the AIMs on genes associated with different diseases as listed in GAD (Table S2 and Figure S2) also indicates that the OG population is likely to be extremely informative in MALD.
The extent of LD is contingent upon the allele-frequency difference between the ancestral populations as well as the number of generations that have elapsed after the admixture event took place, and it rapidly decreases with each passing generation.13,41,42 The long-range LD that we observe among the OG is indicative of very recent admixture. Because there is little prior study of the African diaspora in the ocean of Indian population diversity, it is difficult to state when the African ancestors to OG started settling there. However, it is likely that there was limited mate exchange with native populations until recent times, probably because of sociological constraints. This is also evident from individual estimates of Indian ancestry; such estimates show that the Indian contribution to the admixture is approximately 40%. Some historians argue that in the 19th century, Zanzibar emerged as the hub for the distribution of African slaves to Arabia, southern Persia, and probably western India. Even after the nominal abolition of the slave trade by the British, a small number of male and female African slaves continued to be shipped to the western coasts of south Asia, especially to Makran and Gujarat, where they were mostly employed as servants and bodyguards at the courts of local rulers. The long-range LD is also possible if the 19th century Zanzibar residents are the African ancestors to the OG. The genetic similarity of the African ancestors of the OG to the current west Africans could still be due to the wide spread of Bantu speakers throughout Africa and their genetic homogeneity.39
Migrations of Africans into mainland India brought individuals from different continents into close physical proximity and resulted in inter-mating between migrant and indigenous populations. This meant sudden confluence of geographically diverged genomes with novel environmental challenges. These unprecedented events brought together genomes that had evolved independently and optimized to different continents and conditions for tens of thousands of years and presented new environmental challenges for the indigenous and migrant populations, as well as their offspring. These circumstances provided novel opportunities for natural selection to occur and perhaps resulted in large deviations from the genome-wide ancestry distribution at specific locations.9,10 As we have already shown, the OG is a relatively recent admixed population, and so we did not expect to see very large deviations from genome-wide ancestry distribution at specific locations. However, we wanted to examine whether the OG have retained any enriched biological processes from either of the ancestors. Our search for functional enrichments was directed at the AIMs that were associated with genes and whose frequency in OG was close to either of the ancestral populations. We observed a significant enrichment of processes related to ion-channel activity and cadherin genes; the genotypic spectrum in these enriched processes was close to that of the IE ancestors (Figure 7). Selection in ion-channel genes among populations of African ancestry has been a long-term global enigma.43 However, the fact that the population resides in an extremely saline region of the country and has shown deviations in these genes was intriguing and made it compelling to speculate that this finding is biologically relevant. This is especially interesting in the light of the fact that a recent GWAS study of hypertension and blood pressure in African Americans implicated a similar family of genes related to ion channels, cadherins, and calmodulins.44
Figure 7.
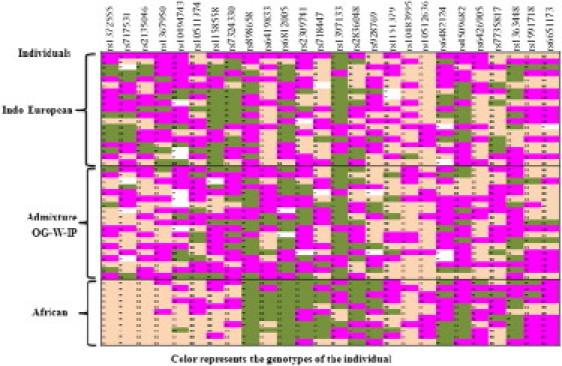
Genotype Distribution of AIMs Related to Ion-Channel Activity and Cadherin Genes in OGs and the Ancestral Populations
The genotypes of the various AIMs depicted in the columns are represented for each individual in a row. Heterozygous genotypes are represented in pink, and the two homozygous genotypes are represented in green and cream for each of the markers. The genotypes of 19 individuals of OG and representative Indian (IE-N-LP4) and African (YRI) ancestral populations are depicted. The genotypes of OGs are markedly similar to those of the Indian IE ancestor than the African ancestor.
Ramana et al. studied the variation in the Y chromosome among the OG19 and found that there is considerable infusion of Y chromosomes from different Indian caste populations into the gene pool of OG. Although the African Indian population was sampled from a different geographical location, it probably shares a common history with the population we sampled. Despite the Y-chromosomal variation, there is little chance that the maternal founding gene pool for the OG population is large. The population also lives in isolated small endogamous groups, which is the likely cause of the deep-rooted founder effect. The population history hence resembles, in terms of forces shaping genetic architecture, the European Roma population and the Ashkenazi Jews, who have long served as a model population for identification and mapping of founder mutations and diseases.45,46
The overarching goal of identifying an admixed population lies in the potential of the population for mapping disease-causing mutations. Admixture mapping is based on the hypothesis that differences in disease rates between populations are due in part to frequency differences in disease-causing genetic variants. In admixed populations, these genetic variants occur more often on chromosome segments inherited from the ancestral population with the higher disease-variant frequency.13 Thus, the chances of successfully mapping disease-causing variants are vastly improved if the divergence between the ancestral populations is large. Admixture mapping also takes advantage of long-range haplotypes that are generated by gene flow among recently admixed ethnic groups. The chances of successfully mapping disease-causing variants further increase if there is a large difference in the prevalence of the disease between the ancestral populations. The extent of LD also ensures that the admixture event is recent. We speculate that the divergence of the two ancestral populations and the recent admixture makes OG a highly potent population for admixture mapping.
Acknowledgments
The project was supported by funding from the Council of Scientific and Industrial Research (CMM-0016 and SIP0006 to M.M.). Collection of endogamous population samples across India was coordinated under the Indian Genomics Variation Consortium. The genotype was done at The Centre for Genomic Application (a collaboration between the Institute of Genomics and Integrative Biology and the Institute of Molecular Medicine). A Department of Science and Technology Inspire Fellowship to A.N. and a Senior Research Fellowship to P.J. from the Council of Scientific and Industrial Research are duly acknowledged.
Contributor Information
Analabha Basu, Email: ab1@nibmg.ac.in.
Mitali Mukerji, Email: mitali@igib.res.in.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Arlequin, http://cmpg.unibe.ch/software/arlequin3/
EIGENSOFT, http://genepath.med.harvard.edu/∼reich/Software.htm
HapMap, http://hapmap.ncbi.nlm.nih.gov/downloads/index.html.en
IGVBrowser, http://igvbrowser.igib.res.in
SNPnexus, http://www.snp-nexus.org/
References
- 1.Basu A., Mukherjee N., Roy S., Sengupta S., Banerjee S., Chakraborty M., Dey B., Roy M., Roy B., Bhattacharyya N.P. Ethnic India: A genomic view, with special reference to peopling and structure. Genome Res. 2003;13:2277–2290. doi: 10.1101/gr.1413403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sengupta S., Zhivotovsky L.A., King R., Mehdi S.Q., Edmonds C.A., Chow C.E., Lin A.A., Mitra M., Sil S.K., Ramesh A. Polarity and temporality of high-resolution y-chromosome distributions in India identify both indigenous and exogenous expansions and reveal minor genetic influence of Central Asian pastoralists. Am. J. Hum. Genet. 2006;78:202–221. doi: 10.1086/499411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Indian Genome Variation Consortium Genetic landscape of the people of India: a canvas for disease gene exploration. J. Genet. 2008;87:3–20. doi: 10.1007/s12041-008-0002-x. [DOI] [PubMed] [Google Scholar]
- 4.Abdulla M.A., Ahmed I., Assawamakin A., Bhak J., Brahmachari S.K., Calacal G.C., Chaurasia A., Chen C.H., Chen J., Chen Y.T., HUGO Pan-Asian SNP Consortium. Indian Genome Variation Consortium Mapping human genetic diversity in Asia. Science. 2009;326:1541–1545. doi: 10.1126/science.1177074. [DOI] [PubMed] [Google Scholar]
- 5.Reich D., Thangaraj K., Patterson N., Price A.L., Singh L. Reconstructing Indian population history. Nature. 2009;461:489–494. doi: 10.1038/nature08365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhu X., Luke A., Cooper R.S., Quertermous T., Hanis C., Mosley T., Gu C.C., Tang H., Rao D.C., Risch N. Admixture mapping for hypertension loci with genome-scan markers. Nat. Genet. 2005;37:177–181. doi: 10.1038/ng1510. [DOI] [PubMed] [Google Scholar]
- 7.Basu A., Tang H., Arnett D., Gu C.C., Mosley T., Kardia S., Luke A., Tayo B., Cooper R., Zhu X. Admixture mapping of quantitative trait loci for BMI in African Americans: Evidence for loci on chromosomes 3q, 5q, and 15q. Obesity (Silver Spring) 2009;17:1226–1231. doi: 10.1038/oby.2009.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Basu A., Tang H., Lewis C.E., North K., Curb J.D., Quertermous T., Mosley T.H., Boerwinkle E., Zhu X., Risch N.J. Admixture mapping of quantitative trait loci for blood lipids in African-Americans. Hum. Mol. Genet. 2009;18:2091–2098. doi: 10.1093/hmg/ddp122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tang H., Choudhry S., Mei R., Morgan M., Rodriguez-Cintron W., Burchard E.G., Risch N.J. Recent genetic selection in the ancestral admixture of Puerto Ricans. Am. J. Hum. Genet. 2007;81:626–633. doi: 10.1086/520769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Basu A., Tang H., Zhu X., Gu C.C., Hanis C., Boerwinkle E., Risch N. Genome-wide distribution of ancestry in Mexican Americans. Hum. Genet. 2008;124:207–214. doi: 10.1007/s00439-008-0541-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zakharia F., Basu A., Absher D., Assimes T.L., Go A.S., Hlatky M.A., Iribarren C., Knowles J.W., Li J., Narasimhan B. Characterizing the admixed African ancestry of African Americans. Genome Biol. 2009;10:R141. doi: 10.1186/gb-2009-10-12-r141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Smith M.W., O'Brien S.J. Mapping by admixture linkage disequilibrium: Advances, limitations and guidelines. Nat. Rev. Genet. 2005;6:623–632. doi: 10.1038/nrg1657. [DOI] [PubMed] [Google Scholar]
- 13.Winkler C.A., Nelson G.W., Smith M.W. Admixture mapping comes of age. Annu. Rev. Genomics Hum. Genet. 2010;11:65–89. doi: 10.1146/annurev-genom-082509-141523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Freedman M.L., Haiman C.A., Patterson N., McDonald G.J., Tandon A., Waliszewska A., Penney K., Steen R.G., Ardlie K., John E.M. Admixture mapping identifies 8q24 as a prostate cancer risk locus in African-American men. Proc. Natl. Acad. Sci. USA. 2006;103:14068–14073. doi: 10.1073/pnas.0605832103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xu S., Huang W., Qian J., Jin L. Analysis of genomic admixture in Uyghur and its implication in mapping strategy. Am. J. Hum. Genet. 2008;82:883–894. doi: 10.1016/j.ajhg.2008.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xu S., Jin L. A genome-wide analysis of admixture in Uyghurs and a high-density admixture map for disease-gene discovery. Am. J. Hum. Genet. 2008;83:322–336. doi: 10.1016/j.ajhg.2008.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Singh K.S. Popular Prakashan Pvt Ltd.; Mumbai, India: 2002. People of India, Gujarat: Anthropological survey of India. pp. 1295–1297. [Google Scholar]
- 18.Gauniyal M., Chahal S.M., Kshatriya G.K. Genetic affinities of the Siddis of South India: An emigrant population of East Africa. Hum. Biol. 2008;80:251–270. doi: 10.3378/1534-6617-80.3.251. [DOI] [PubMed] [Google Scholar]
- 19.Ramana G.V., Su B., Jin L., Singh L., Wang N., Underhill P., Chakraborty R. Y-chromosome SNP haplotypes suggest evidence of gene flow among caste, tribe, and the migrant Siddi populations of Andhra Pradesh, South India. Eur. J. Hum. Genet. 2001;9:695–700. doi: 10.1038/sj.ejhg.5200708. [DOI] [PubMed] [Google Scholar]
- 20.International HapMap Consortium The International HapMap Project. Nature. 2003;426:789–796. doi: 10.1038/nature02168. [DOI] [PubMed] [Google Scholar]
- 21.Li J.Z., Absher D.M., Tang H., Southwick A.M., Casto A.M., Ramachandran S., Cann H.M., Barsh G.S., Feldman M., Cavalli-Sforza L.L. Worldwide human relationships inferred from genome-wide patterns of variation. Science. 2008;319:1100–1104. doi: 10.1126/science.1153717. [DOI] [PubMed] [Google Scholar]
- 22.Narang A., Roy R.D., Chaurasia A., Mukhopadhyay A., Mukerji M. Indian Genome Variation Consortium, and Dash, D. (2010). IGVBrowser—A genomic variation resource from diverse Indian populations. Database (Oxford) 2010;2010:baq022. doi: 10.1093/database/baq022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Patterson N., Price A.L., Reich D. Population structure and eigenanalysis. PLoS Genet. 2006;2:e190. doi: 10.1371/journal.pgen.0020190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Price A.L., Patterson N.J., Plenge R.M., Weinblatt M.E., Shadick N.A., Reich D. Principal components analysis corrects for stratification in genome-wide association studies. Nat. Genet. 2006;38:904–909. doi: 10.1038/ng1847. [DOI] [PubMed] [Google Scholar]
- 25.Pritchard J.K., Stephens M., Donnelly P. Inference of population structure using multilocus genotype data. Genetics. 2000;155:945–959. doi: 10.1093/genetics/155.2.945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Falush D., Stephens M., Pritchard J.K. Inference of population structure using multilocus genotype data: Linked loci and correlated allele frequencies. Genetics. 2003;164:1567–1587. doi: 10.1093/genetics/164.4.1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Falush D., Stephens M., Pritchard J.K. Inference of population structure using multilocus genotype data: Dominant markers and null alleles. Mol. Ecol. Notes. 2007;7:574–578. doi: 10.1111/j.1471-8286.2007.01758.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Alexander D.H., Novembre J., Lange K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009;19:1655–1664. doi: 10.1101/gr.094052.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Excoffier L., Smouse P.E., Quattro J.M. Analysis of molecular variance inferred from metric distances among DNA haplotypes: Application to human mitochondrial DNA restriction data. Genetics. 1992;131:479–491. doi: 10.1093/genetics/131.2.479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Excoffier L., Laval G., Schneider S. Arlequin (version 3.0): An integrated software package for population genetics data analysis. Evol. Bioinform. Online. 2005;1:47–50. [PMC free article] [PubMed] [Google Scholar]
- 31.Reynolds J., Weir B.S., Cockerham C.C. Estimation of the coancestry coefficient: Basis for a short-term genetic distance. Genetics. 1983;105:767–779. doi: 10.1093/genetics/105.3.767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Weir B.S., Hill W.G. Estimating F-statistics. Annu. Rev. Genet. 2002;36:721–750. doi: 10.1146/annurev.genet.36.050802.093940. [DOI] [PubMed] [Google Scholar]
- 33.Hill W.G. Estimation of linkage disequilibrium in randomly mating populations. Heredity. 1974;33:229–239. doi: 10.1038/hdy.1974.89. [DOI] [PubMed] [Google Scholar]
- 34.Purcell S., Neale B., Todd-Brown K., Thomas L., Ferreira M.A., Bender D., Maller J., Sklar P., de Bakker P.I., Daly M.J., Sham P.C. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dennis G., Jr., Sherman B.T., Hosack D.A., Yang J., Gao W., Lane H.C., Lempicki R.A. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol. 2003;4:3. [PubMed] [Google Scholar]
- 36.Mohanty P.K. Gyan Publication; Delhi: 2006. Encyclopaedia of Scheduled Tribes in India. pp. 81. [Google Scholar]
- 37.Rosenberg N.A., Pritchard J.K., Weber J.L., Cann H.M., Kidd K.K., Zhivotovsky L.A., Feldman M.W. Genetic structure of human populations. Science. 2002;298:2381–2385. doi: 10.1126/science.1078311. [DOI] [PubMed] [Google Scholar]
- 38.Tishkoff S.A., Reed F.A., Friedlaender F.R., Ehret C., Ranciaro A., Froment A., Hirbo J.B., Awomoyi A.A., Bodo J.M., Doumbo O. The genetic structure and history of Africans and African Americans. Science. 2009;324:1035–1044. doi: 10.1126/science.1172257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jallow M., Teo Y.Y., Small K.S., Rockett K.A., Deloukas P., Clark T.G., Kivinen K., Bojang K.A., Conway D.J., Pinder M., Wellcome Trust Case Control Consortium. Malaria Genomic Epidemiology Network Genome-wide and fine-resolution association analysis of malaria in West Africa. Nat. Genet. 2009;41:657–665. doi: 10.1038/ng.388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vansina J. Valleys of the Niger. J. Afr. Hist. 1995;36:491–495. [Google Scholar]
- 41.Chakraborty R., Weiss K.M. Admixture as a tool for finding linked genes and detecting that difference from allelic association between loci. Proc. Natl. Acad. Sci. USA. 1988;85:9119–9123. doi: 10.1073/pnas.85.23.9119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Stephens J.C., Briscoe D., O'Brien S.J. Mapping by admixture linkage disequilibrium in human populations: Limits and guidelines. Am. J. Hum. Genet. 1994;55:809–824. [PMC free article] [PubMed] [Google Scholar]
- 43.Genovese G., Friedman D.J., Ross M.D., Lecordier L., Uzureau P., Freedman B.I., Bowden D.W., Langefeld C.D., Oleksyk T.K., Uscinski Knob A.L. Association of trypanolytic ApoL1 variants with kidney disease in African Americans. Science. 2010;329:841–845. doi: 10.1126/science.1193032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Adeyemo A., Gerry N., Chen G., Herbert A., Doumatey A., Huang H., Zhou J., Lashley K., Chen Y., Christman M., Rotimi C. A genome-wide association study of hypertension and blood pressure in African Americans. PLoS Genet. 2009;5:e1000564. doi: 10.1371/journal.pgen.1000564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Risch N., de Leon D., Ozelius L., Kramer P., Almasy L., Singer B., Fahn S., Breakefield X., Bressman S. Genetic analysis of idiopathic torsion dystonia in Ashkenazi Jews and their recent descent from a small founder population. Nat. Genet. 1995;9:152–159. doi: 10.1038/ng0295-152. [DOI] [PubMed] [Google Scholar]
- 46.Kalaydjieva L., Gresham D., Calafell F. Genetic studies of the Roma (Gypsies): A review. BMC Med. Genet. 2001;2:5. doi: 10.1186/1471-2350-2-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.