

Abstract
Cerebrovascular amyloidosis is caused by amyloid accumulation in walls of blood vessel walls leading to hemorrhagic stroke and cognitive impairment. Transforming growth factor-β1 (TGF-β1) expression levels correlate with the degree of cerebrovascular amyloid deposition in Alzheimer s disease (AD) and TGF-β1 immunoreactivity in such cases is increased along the cerebral blood vessels. Here we show that a nasally administered proteosome-based adjuvant activates macrophages and decreases vascular amyloid in TGF-β1 mice. Animals were nasally treated with a proteosome-based adjuvant on a weekly basis for three months beginning at age 13 months. Using MRI we found that while control animals showed a significant cerebrovascular pathology, proteosome-based adjuvant prevents further brain damage and prevents pathological changes in the blood-brain barrier. Using an object recognition test and Y-maze, we found significant improvement in cognition in the treated group. Our findings support the potential use of a macrophage immuno-modulator as a novel approach to reduce cerebrovascular amyloid, prevent microhemorrhage and improve cognition.
Keywords: cerebrovascular disease, congnitive impairment, intracerebral hemorrhage, macrophage, MRI, vaccine, therapy
1. Introduction
Cerebrovascular amyloidosis is associated with endothelial cell (EC) thinning and loss of mitochondria activity, which leads to blood-brain barrier (BBB) dysfunction (Castellani, et al., 2004,Farfara, et al., 2008,Nicoll, et al., 2004). Vascular amyloidosis may result in intra-parenchymal and subarachnoid bleeding and multiple infarcts, and can present clinically with headache, major hemorrhagic stroke, and cognitive impairment (Greenberg, et al., 2004). Amyloid pathology of cerebrovascular cells has been found in cerebral amyloid angiopathy (CAA) and Alzheimer s Disease (AD). The prevalence of CAA, estimated from autopsy series, is approximately 10% to 40% in the general elderly population. Sporadic CAA increases dramatically with age, increasing in prevalence to more than 50% in people over the age of 90. CAA is found in 80% of AD cases (Nicoll, et al., 2004,Zlokovic, 2005). Although the most common form of cerebrovascular amyloid is the deposition of β–amyloid (Aβ) (1–40), other proteins have been linked to familial forms of CAA such as: APP, cystatin C, BRI, prion protein, gelsolin, and transthyretin (Burgermeister, et al., 2000,Lacombe, et al., 2004). There are no current treatments to reduce amyloid related pathology in CAA (Maia, et al., 2007).
Transforming growth factor-β1 (TGF-β1) is a multifunctional cytokine that is a major regulator of the immune response and has profound effects on vasculogenesis, angiogenesis, and the maintenance of vessel wall integrity. Furthermore, cortical TGF-β1 messenger RNA (mRNA) levels correlate positively with the degree of cerebrovascular amyloid deposition in AD cases, and TGF-β1 immunoreactivity in such cases is elevated along the cerebral blood vessels (Wyss-Coray, et al., 2000). A positive correlation has been reported between TGF-β1 polymorphisms and CAA (Peila, et al., 2007). In mice, transgenic overexpression of TGF-β1 under the control of an astrocyte glial fibrillary acidic protein (GFAP) promoter, causes an age-related (starting at 8 months) deposition of amyloid around cerebral blood vessels and prominent perivascular astrocytosis (Wyss-Coray, et al., 1995), leading to CAA-related vascular alterations and dysfunction.
Several endogenous mechanisms exist for the removal of soluble Aβ from the central nervous system (CNS), including receptor-mediated clearance at the BBB via perivascular macrophages, in light of their localization within perivascular spaces and proximity to vascular amyloid. This process was previously suggested to play a role in regulating the deposition of vascular Aβ (Hawkes and McLaurin, 2009).
We have recently shown that nasal vaccination with a proteosome-based adjuvant (Protollin), comprised of purified outer membrane proteins of Neisseria meningitides and lipopolysaccharide, which is well tolerated in humans, decreased parenchymal Aβ deposition in an AD mouse model (Frenkel, et al., 2008). In this report we treated a mouse model of CAA with Protollin and monitored vascular disease both pathologically and by non-invasive magnetic resonance imaging (MRI). We demonstrate that macrophage activation by Protollin decreases vascular amyloid, reduces brain damage and improves functional outcome in TGF-β1 mice.
2. Materials and methods
2.1. Mice
Heterozygous TGF-β1 mice were obtained from the laboratory of Tony Wyss-Coray (Wyss-Coray, et al., 1995) and maintained on an inbred C57BL/6J genetic background (The Jackson Laboratory, Bar Harbor, ME). We used a total of 16 male and 6 female mice in our studies. No differences were observed between males vs females in terms of CAA development or response to treatment. All animal care and experimental use was in accordance with the Tel Aviv University guidelines and approved by the TAU animal care committee.
2.2. Cell culture
Murine macrophage-like RAW264.7 cells were cultured as described previously (Fujimori, et al., 2001). Briefly, the cells were grown in Dulbecco's modified Eagle's medium supplemented with 10% FCS, 4 mM L-glutamine, 100 units/mL penicillin, 0.1 mg/mL streptomycin, and 12.5 units/mL nystatin. The cell line was maintained at 37°C, 5% CO2 and 95% relative humidity. All of the medium components were obtained from Biological Industries (Beit-Haemek, Israel).
2.3. Nasal Vaccination
Protollin was obtained from GlaxoSmithKline (Laval, Quebec, Canada). Protollin (28.6μg/kg) was given on days 1, 3, and 5 of the first week followed by a weekly boost (Frenkel, et al., 2008). Mice received a weekly boost beginning at age 13 months until age 16 months. Phosphate-buffered saline (PBS) was used as a control. TGF-β1 mice treated with Protollin (n=7) or PBS (n=8) and age-matched wt littermates treated with PBS (n=7) were used, with males and females balanced in all groups. Treated animals exhibited no changes in behavior, as measured by body weight, eating habits, tail tone, or mobility.
2.4. Behavioral tests
Object recognition (ORT) and Y-maze tests were done using young (3 months old) TGF-β1 Tg mice (ORT: n=6; Y-maze: n=5) and age-matched WT mice (ORT: n=7 Y-maze: n=6) and adult (16 months old) immunized TGF-β1 Tg mice with Protollin (ORT: n=5; Y-maze: n=5) or PBS (ORT: n=7; Y-maze: n=5) and age-matched WT mice (n=7).
2.4.1. Object recognition test
Following 3 months of treatment with Protollin or PBS, the mice were tested using a novel object recognition test (ORT). In brief, on the first day of the test, mice were first placed in an apparatus (50×50×20cm) and allowed to explore the testing environment. After a prescribed interval (24 hr), animals were placed on the second day in the same apparatus and allowed to explore an object. On the third day of the test, the animals were placed in the apparatus, containing the familiar object as well as a novel object. Object recognition is distinguished by more time spent interacting with the novel object (Bevins and Besheer, 2006). The exploration time for the familiar or the new object during the test phase (5 min test each day) was recorded. Memory was operationally defined by the discrimination index for the novel object (DI) as the percentage of time the mice spent exploring the novel object as a function of the total amount of time spent exploring both objects during testing (duration spent with novel object/(duration spent with novel object+duration spent with familiar object)×100).
2.4.2. Y-maze test
Behavioral testing was conducted in a Plexiglas Y-maze. Both the start arm (30 cm long) and the two arms forming the Y (both 30 cm long and diverged at a 120° angle from the stem arm) were 8 cm in diameter. Each arm was equipped with a guillotine door which could be operated manually from the experimenters position. The experimental box was decorated with visual cues, which served as distal spatial cues. Each mouse was placed in the steam arm and timed with a stopwatch until they reached one of the arms. The three identical arms were randomly designated: Start (steam) arm, in which the mouse began to explore (always open); Novel arm, which was closed off during the 1st trial, but open in the 2nd trial; and Other arm (always open). The Y-maze test consists of two trials separated by an inter-trial interval (ITI) to assess spatial recognition memory. The first trial (training) lasted 5 min and allowed the mouse to explore only two arms (start and other) of the maze, with access to the third arm (novel arm) blocked off. After 2min ITI, the second trial (retention) was conducted, during which all three arms were accessible; the mouse was returned to the same starting arm and allowed to explore all three arms for 2min. Data is expressed as total time spent in the Novel arm during the 2 min retention period (Akwa, et al., 2001).
2.4.3. Rotarod test
Motor coordination and balance were tested with the Rotarod test as previously described (Fowler, et al., 2002). The Rotarod test was performed by placing a mouse on a rotating drum (ENV-576M, Med Associate Inc, Georgia, Vermont) for 5 min habituation, 2 min rest and a test session of no longer than 3 min. The time the animal succeeded in maintaining balance on the rod during the test session was measured. Rotarod was done using adult (16 months old) immunized TGF-β1 Tg mice with Protollin (n=5) or PBS (n=5) and age-matched WT mice (n=7).
2.5. Magnetic resonance imaging
TGF-β1 mice (treated with Protollin or PBS) and age-matched wt littermates (treated with PBS) were analyzed by MRI (5–7 animals in each group) as previously described (Tsenter, et al., 2008). In brief, mice were anesthetized with isoflurane (1–3%) in 1 liter/min oxygen 98% and were scanned in a 7T/30 spectrometer (Bruker, Rheinstetten, Germany) using a 10 mm quadrature surface coil dedicated to mouse heads and 400 mT/m gradient system. The MRI protocol included multi-echo T2-weighted images (RARE, TR/TE 3000/50 ms, RARE factor 8, 4 averages) and contrast enhanced T1 weighted images (SE, TR/TE 600/12 ms, 2 averages) acquired before and 3 and 5 min post ip injection (0.5mmol/kg Gadopentetate Dimeglumine–Gadolinium DPTA) 150 μl, Magnetol, Soreq, Israel) through an ip catheter. In all experiments the field of view (FOV) was 20 × 20 mm2, 12 axial contiguous slices of 1 mm thickness, matrix dimensions of 256 × 128 (zero filled to 256 × 256). Analysis was performed with the Medical Image Analysis (MIA, version 2,4 MATLAB).
2.5.1 T2 weighted analysis
Analysis was performed with the Medical Image Analysis (MIA, version 2,4 MATLAB ®). The area (mm2) of the lateral ventricles from the T2 weighted images were extracted by manual segmentation on selected slices. T2 measurements were carried out using 13 months old immunized TGF-β1 Tg mice (n=10) and following 3 months treatment with Protollin (n=5) or PBS (n=5) and age-matched WT mice (n=7).
2.5.2. Contrast enhanced T1 weighted analysis
The image difference between post-contrast image and pre-contrast provides a contrast enhanced map where enhanced areas suggest areas of vessel permeability. In order to compute the permeability index, regions of interest were manually segmented on 3 consecutive slices of each brain mice from approximately Bregma −0.8 to −2.8. A brain barrier permeability index (I) was calculated by homemade scripts using matlab (The MathWorks, Inc.). (I) was determined using the expression: I = density of enhanced pixels on the brain /(density of enhanced pixels on the brain + density of enhanced pixels on non brain tissue + density of enhanced pixels on noise outside the mice head) (n=5/group).
2.6. ELISA analysis of amyloid peptide from brain and plasma samples
The right hemisphere of each mouse in each treatment group was homogenized with PBS containing protease inhibitor and centrifuged at 40,000g for 40 min to quantify Aβ levels. The supernatant-containing soluble Aβ was stored at −70° C. The pellet containing insoluble Aβ was extracted in 5.0M guanidinium-chloride (pH 8) for 3 h at room temperature. Levels of Aβ (1–40) in brain samples and plasma were assessed by enzyme-linked immunosorbent assay (ELISA) as previously described (DeMattos, et al., 2002) n=5 mice/group.
2.7. Immunohistology and quantitation of amyloid load
Mice were sacrificed (transcardially punctured, and saline-perfused) and their brains rapidly excised and frozen. The brains (left hemisphere) were cut in 14 μm coronal sections at 20° C and used for histological examination. Brain sections (1.44 mm lateral to bregma) were stained with Congo red (Sigma-Aldrich) or ThS (Sigma-Aldrich) and visualized by fluorescence microscopy for quantification of the amount of vascular amyloid depositions. For immunostaining the following antibodies were used: Iba1 (1:1000; Wako); CD11b (1:50; Dako), CD31 (1:75; BD Biosciences). Quantitative analysis of vascular amyloid in the brain was determined by counting the number of vessels per area in 2 consecutive sections (14 μm) per animal, (n=5 mice/group). Vessel radii was determined by using the “Fast Measure Line” tool of the software. The quantification was done in a blinded fashion using Nikon NIS imaging software analysis in an unbiased stereological approach.
2.7.1. Prussian blue staining for microhemorrhages
Staining for deposits was performed on duplicate adjacent coronal sections of the mice containing similar regions located at approximately (1.5 mm lateral to bregma), 14 μm thick, collected from TGF-β1 mice (treated with Protollin or PBS) and age-matched wt littermates (treated with PBS). The sections were stained with Prussian blue working solution (equal parts of freshly made 5% potassium ferrocianide and 5% hydrochloric acid) for 60 min at room temperature, washed in deionized water, and counterstained with Nuclear fast red. Perls's Berlin blue-stained clusters of hemosiderin staining were qualitatively evaluated (presence/absense) from sections throughout the neocortex, hippocampus, and thalamus.
2.8. Real-time PCR analysis
Total RNA was extracted from the right hippocampus, blood samples or peripheral macrophages from 16 months old immunized TGF-β1 Tg mice treated with Protollin (n=7) or PBS (n=6). Reverse transcriptase PCR assays were designed by Applied Biosystems (Foster City, CA) as described previously (Zhang, et al., 2005). The probes for the TaqMan RT–PCRs were: Mm00443258_m1 (TNF-α), Mm00473077_m1 (IDE), Mm00438270_m1 (CCR2), Mm00446211_m1 (SRA), Mm00434455_m1 (CD11b) and Mm00432069_m1 (BDNF). Reactions were performed according to the manufacturer's directions using an Applied Biosystems PRISM 7300 thermal cycler. The mice Actin gene, a housekeeping gene, was used to normalize each sample and each gene. Quantification analysis was performed by the (2−Δ ΔCt method) and statistically analyzed by GraphPad Prism software.
2.9. Western blotting
For protein analysis the hippocampus was dissected out from 16 months old TGF-β1 mice immunized with Protollin (n=4) or PBS (n=6) and from age-matched WT mice (n=3) and homogenized in lysis buffer as previously described (Mitchell, et al., 2007). Proteins were transferred onto a nitrocellulose membrane and probed with a polyclonal anti-synaptophysin antibody (1:25,000, Epitomics) overnight. After incubation with the primary antibody, membranes were incubated for 1 h with goat α rabbit (1:10,000, LI-COR) antibody. Blots were then scanned using the Odyssey Imaging System from LI-COR and normalized to tubulin (1:25,000, Sigma-Aldrich). Relative band intensity was determined using Odyssey software version 1.0 (LI-COR).
2.10. In situ vascular amyloid clearance
10 μm saggital brain sections were prepared from 16-month-old transgenic mice using a cryostat. 6×105 RAW 264 cells were exposed to 0.1 μg/ml Protollin and another set of 6×105 RAW 264 cells were left untreated. In either case, these cells were plated to unfixed brain sections enriched with vascular amyloid and incubated at 37° C for 4 days. Brain sections were then stained with Congo red (Sigma-Aldrich). Quantification of vascular amyloid depositions was done as previously described (Frenkel, et al., 2005). The staining was performed on 6 consecutive sections per animal and repeated 4 times.
2.11. Statistical Analysis
Data from each experiment are expressed as mean± standard error of the mean (SEM.). Two-tailed Student s t-test was performed when two groups were compared. The one-way ANOVA followed by Bonferroni s multiple comparison tests was carried out for multiple samples. Statistical significance was determined at P < 0.05.
3. Results
3.1 Cerebrovascular amyloid deposition results in brain tissue damage in the TGF-β1 animal model
Microscopic examination of brains from 16-month-old TGF-β1 Tg mice showed significant amyloid accumulation around cerebral blood vessels compared to age-matched non-transgenic wild-type (WT) littermates as determined by Congo red staining using different microscopy methods: fluorescence microscopy, bright field microscopy imaging and polarized light microscopy (Fig. 1A) and colocalization with the endothelial cell marker,CD31 (Fig. 1B). To investigate the effect of Protollin on cerebrovascular amyloid, 13-month-old TGF-β1 Tg mice and age-matched non-transgenic littermates mice (WT mice) were nasally administered with Protollin or PBS weekly for 3 months. Disease progression was monitored by non-invasive magnetic resonance imaging (MRI). We used T2 weighted images to assess brain tissue damage as measured by enlargement of the lateral ventricle area. Using T2 weighted analysis (Fig. 1C, D), we found that in 16-month-old TGF-β1 mice there was a 200% (P<0.001) enlargement of the lateral ventricles area as compared with age matched non-transgenic littermates (18.05±1.5 vs. 5.99±0.1 mm2) and 63% compared to 13-month-old TGF-β1 mice (18.05±1.5 vs. 11.05±0.4 mm2). Nasal treatment with Protollin for 3 months (beginning at age 13 months) prevented further enlargement of the lateral ventricles (10.68±1.7 mm2), (F(3,21) = 29.418; P < 0.0001) (Fig. 1C, D).
Fig. 1.

Nasal Protollin reduces brain damage in TGF-β1 mice. (A) Congo red staining in the brains of 16-month-old TGF-β1 Tg mice (A, iii–vi) and age-matched non-transgenic littermates (WT) (A, i–ii). Vascular amyloid (Congo red) is apparent in the cortex (A,iii, scale bar: 100 μm ) and hippocampus (A,iv, scale bar: 100 μm) of a 16-month-old TGF-β1 Tg mice. Fluorescence image (v) and brightfield microscopy image (vi), after Congo red staining. Scale bar: 50 μm. (B) Colocalization (right) of vascular amyloid (Congo red, left) and endothelial cells (EC) (CD31 marker, green, middle) in the brain of 16-month-old TGF-β1 Tg mice compared to WT mice. Scale bar: 10 μm. (C) T2-weighted representative images and (D) analysis of the lateral ventricle area (mm2) of TGF-β1 mice brain with (n=7 mice) and without Protollin treatment (n=5 mice). Arrows point to lateral ventricles (**P<0.01, *** P<0.001).
3.2 Nasal vaccination with a proetosome based adjuvant reduces cerebrovascular amyloid and prevents microhemorrhage
The severity of cerebrovascular amyloidosis has been associated with increased microhemorrhages both in human studies and in animal models of AD (Hawkes and McLaurin, 2009). Perls blue staining of 16-month-old TGF-β1 Tg brain tissue revealed vessels outlined by hemosiderin, representing extravasation of blood, which is associated with CAA (Fig. 2A). To further assess the effect of nasal Protollin, contrast enhanced (CE) T1 weighted images, measuring gadolinium (Gd-DTPA) penetration through the blood-brain barrier (BBB), were used for microhemorrhage detection and to detect increased BBB permeability. We found a 12% (P<0.01) increase in BBB permeability in 16-month-old TGF-β1 mice compared to WT (Fig. 2B), as revealed by a higher intensity signal. After Protollin treatment, the BBB permeability index (signal intensities on T1-weighted MRI) was similar in Tg and wt mice. Histology confirmed the presence of brain tissue microhemorrhage in Tg mice. These results point to a general pattern of vascular abnormalities (leakiness of BBB to endogenous blood proteins) in TGF-β1 mice tissue and demonstrates that nasal Protollin treatment reduces pathological changes in the BBB.
Fig. 2.
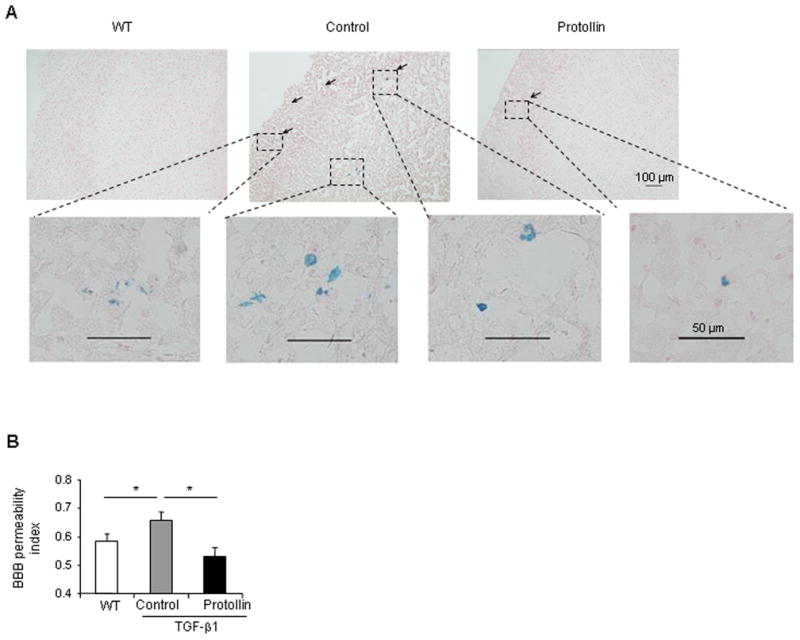
Protollin treatment prevents microhemorrhage incidents in TGF-β1 mice. (A) Representative Perls Berlin Blue stain of the brains of a 16-month old TGF-β1 mice shows hemosiderin, indicative of the local extravasion of blood (arrows) (upper middle, original magnification ×10) and lower panels (original magnification ×40) compared to Protollin treated mice (uper right) and WT mice (uper left)(n=4). The sections are counterstained with nuclear fast red. (B) Brain permeability index as quantified from T1 weighted analysis in control mice compared to the WT and Protollin treated mice (*P<0.01, n=5).
We also found that vascular amyloid deposition was prominent in the leptomeningeal and parenchyma vessels of 16-month-old TGF-β1 mice compared to WT mice as measured by Congo red staining (Fig. 3A). Protollin treatment significantly (P<0.03) reduced by 47% the incidence of cerebrovascular amyloid (8.2±1.8 affected vessels per brain section in Protollin group vs. 15.4±2.7 in PBS treated group) (Fig. 3B, upper panel). Protollin treatment showed a higher effect in vessels with radii of 20–35μm (2.2± 0.6 vs. 3.67±0.5; P=0.05) and greater than 35μm (0.6± 0.2 vs. 1.6± 0.4 affected vessels per brain section; P<0.04) compared to PBS treatment (Fig. 3B, lower panel).
Fig. 3.
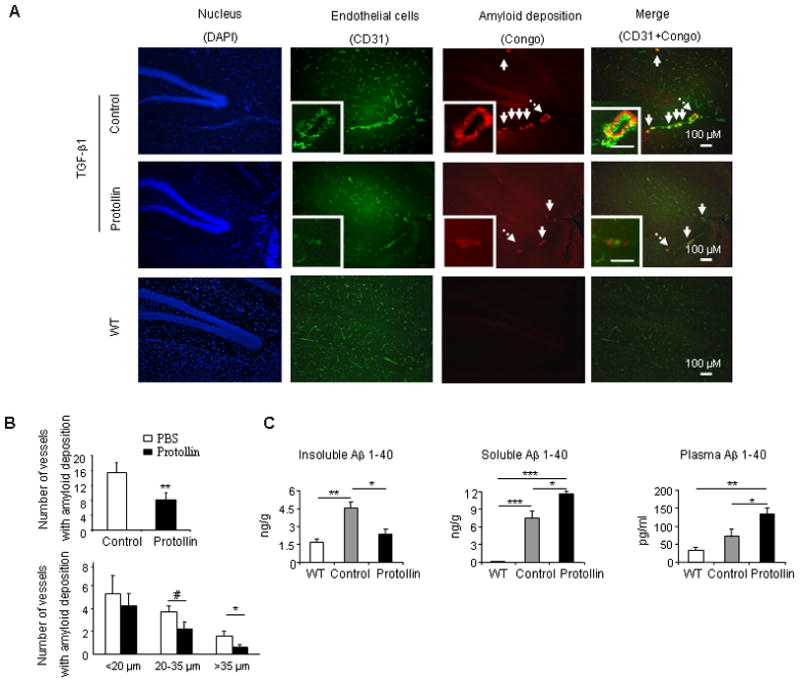
TGF-β1 mice treated with Protollin showed reduction in vascular amyloid. (A) Double-label immunofluorescent staining of vascular amyloid (Congo red) and endothelial cells (CD31) in brain sections of control mice compared to Protollin treated mice and WT mice. Scale bar: 100 μm. (B) Quantified analysis of the number of vessels containing amyloid deposition in brain section of control and Protollin treated group (upper panel) and according to the size of affected vessels (lower panel). Comparisons were made using unpaired t tests (**P<0.03, #P=0.05, *P<0.04; n=5 mice/ group). (C) Brain insoluble, soluble and plasma mouse Aβ (1–40) levels were measured by ELISA (*P<0.05, **P<0.01, ***P<0.001; n=5 mice/ group).
3.3 Nasal Protollin reduces brain Aβ (1–40) levels
It was shown that large parts of the amyloid in the TGF-β1 mice is contained in the extracellular matrix and that basement membrane proteins will also trap endogenous proteins such as Aβ (1–40) leading to CAA (Wyss-Coray, et al., 2000). While we did not detect Aβ (1–40), by immunohistology, we were able to detect and quantify it using ELISA. These findings are consistent with previous reports (Lacombe et al., 2004). We thus investigated the levels of endogenous Aβ (1–40), as an indication of cerebrovascular amyloid, by ELISA (Frenkel, et al., 2005) and by ANOVA found a difference among groups in soluble (F(2,12)=50.056, P<0.0001), insoluble (F(2,10) =12.9, P=0.0017) and plasma (F(2,12) =10.75, P=0.0021) endogenous Aβ (1–40) levels. Brains of 16-month-old TGF-β1 mice (Fig. 3C) showed a significant increase in levels of both soluble and insoluble mouse endogenous Aβ (1–40) compared to WT. It was previously suggested that low-density lipoprotein receptor–related protein-1 (LRP-1) binds the Aβ, at the abluminal side of the BBB, and initiates Aβ clearance from brain to blood via transcytosis across the BBB to the periphery (Sagare, et al., 2007). We thus measured LRP-1 and found that in TGF-β1 mice there was a significant reduction of LRP-1 expression (Supplemental figure 1) as compared to WT mice. .Nasal Protollin significantly (P<0.05) reduced the insoluble Aβ (1–40) fraction (2.34±0.4 vs. 4.5±0.6 ng/gr) in the brain, and increased the soluble Aβ (1–40) fraction (11.7±0.4 vs. 7.4±1.38 ng/gr, P<0.05) in the brain and plasma (135±15.4 vs. 72±20 pg/ml, P<0.05). Our data suggest that Protollin treatment leads to a progressive shift of insoluble brain Aβ (1–40) to soluble, reducing CAA and demonstrating the important role of a peripheral mechanism for amyloid clearance.
3.4 Nasal Protollin improved cognitive impairment associated with cerebrovascular amyloidosis
Vascular dementia (VaD) is the second most common cause of dementia in the elderly, after Alzheimer s disease (AD) and is described as a multifaceted cognitive decline resulting from cerebrovascular injury to brain regions associated with memory (Lifshitz and Frenkel, 2009). We investigated cognition of aged TGF-β1 mice to determine whether cerebrovascular amyloid in those mice can lead to cognitive impairment and whether Protollin treatment can ameliorate memory dysfunction. To evaluate cognitive function, two behavioral tests were used: 1) object recognition test (ORT) to investigate visual recognition memory; and 2) Y-maze test to investigate spatial perception. We found that 3-month-old mice, prior to signs of cerebrovascular amyloid deposition (data not shown), show no observable difference in cognition between TGF-β1 Tg mice and age matched WT littermates (83.04±8.57 vs. 88.96±5.25 %, respectively) as measured by ORT and (54.64±6.77 vs. 54.87±8.82 sec, respectively) by Y-maze test. However, at 16 months of age, when cerebrovascular amyloid is well established (Fig. 1A), there is significant (P<0.05) cognitive impairment in TGF-β1 Tg mice compared to age matched WT mice (42.2±7.58 vs. 75.35±9.11 %, respectively) as measured by the ORT (ANOVA; F(4,24)=7.395, p=0.0005) and Y-maze (P<0.04) (34.68±6.6 vs. 48.36±3.32 sec, respectively) (Fig. 4A, B). Furthermore, amelioration of CAA like pathology following Protollin treatment (Fig. 3A) was associated with improved memory in TGF-β1 Tg mice as observed by ORT (83.6±4.12 vs. 42.2±7.58 % , P<0.01) and Y-maze (49.58±3.85 vs 34.68±6.6 sec, P<0.05,) (Fig. 4A, B). In order to establish that the impairment of TGF-β1 mice in ORT and Y-maze test was linked to cognitive impairment and not motor dysfunction, we used a Rotarod test. No motor disabilities were observed in TGF-β1 Tg mice as compared to the WT or Protollin treated groups (Fig. 4C).
Fig. 4.
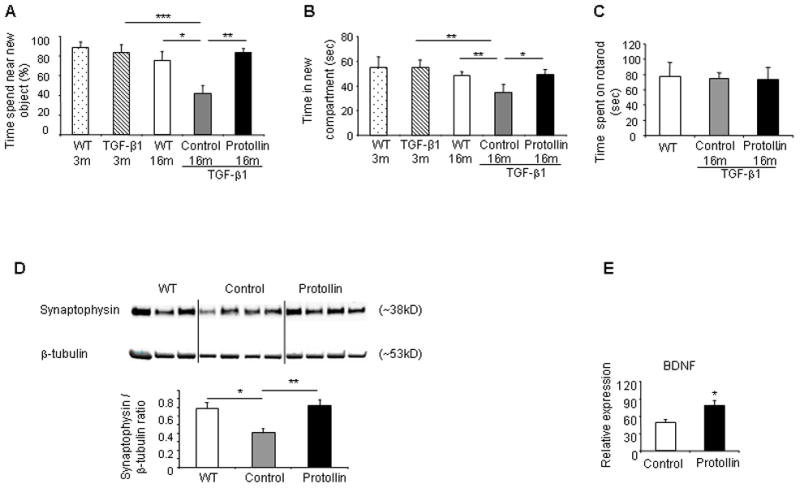
Protollin treatment improved cognitive impairment associated with cerebrovascular amyloidosis and induced elevation in synatophysin. (A) Object recognition test in Protollin treated TGF-β1 Tg mice vs. control (*P<0.05, **P<0.01, ***P<0.006). Object recognition was done using young (3 months old) TGF-β1 Tg mice (n=6), age-matched WT mice (n=7) and adult (16 months old) immunized TGF-β1 Tg mice with Protollin (n=5) or PBS (n=7) and age-matched WT mice (n=7).
(B) Y-maze test in Protollin-treated TGF-β1 Tg mice vs. control mice (*P<0.05, **P<0.04). Y-maze tests was done using young (3 months old) TGF-β1 Tg mice (n=5) and age-matched WT mice (n=6) and adult (16 months old) immunized TGF-β1 Tg mice with Protollin (n=5) or PBS (n=5) and age-matched WT mice (n=7).
(C) Locomotor behavior (Rotarod test) showed no significant difference between the groups (n=5–7 mice/group). Rotarod was done using adult (16 months old) immunized TGF-β1 Tg mice with protollin (n=5) or PBS (n=5) and age-matched WT mice (n=7).
(D) Representative Western blot and densitometric analysis demonstrating protein expression of hippocampal synaptophysin (*P<0.05, **P<0.01, n=3–6 mice/group). (E) BDNF expression in the hippocampal region of Protollin treated mice vs control (*P<0.05) from 16 months old TGF-β1 mice immunized with Protollin (n=4) or PBS (n=6) and from age-matched WT mice (n=3). Values are mean ± SEM of the relative expression of the RNA compared to ACTB RNA levels.
3.5 Nasal Protollin increases hippocampal expression of marker proteins for synaptic plasticity
Previous studies showed that a reduction in synaptophysin immunoreactivity may serve as a marker for synaptic loss in the hippocampus and correlates with cognitive decline in an animal model (Buttini, et al., 2005). We found a 40% reduction in synaptophysin in the hippocampus of 16-month-old TGF-β1 Tg mice compared to WT mice by Western blot (P<0.05) (Fig. 4D). Protollin treatment resulted in an 80% (P<0.01) increase in synaptophysin expression levels in TGF-β1 mice vs. PBS treated Tg mice (Anova; F(2,10)=9.428, P=0.005) (Fig. 4D, lower panel). Other reports suggest a reduction in brain-derived neurotrophic factor (BDNF) protein in the hippocampus in Alzheimer s disease (Connor, et al., 1997) and that administration of BDNF may prevent cell death and ameliorate behavioral deficits in several animal models of AD (Nagahara, et al., 2009). Furthermore, it was reported that BDNF has an important role in angiogenesis (Kermani and Hempstead, 2007). We found a 45% increase in BDNF (P<0.05) in the hippocampus region of Protollin treated mice vs. control group (Fig. 4E). BDNF is known to modulate the levels of the vesicle proteins such as synaptophysin at the hippocampus synapses (Lu and Chow, 1999). BDNF, which is known to modulate the levels of the vesicle proteins such as synaptophysin at the hippocampus synapses(Lu and Chow, 1999), was positively correlated (+0.7) with synaptophysin values.
3.6 Nasal Protollin activates macrophags to clear cerebrovascular amyloid deposition
It has been suggested that vascular amyloid deposits may result from impaired clearance of Aβ (1–40) by macrophages along perivascular spaces (Hawkes and McLaurin, 2009). In addition, other matrix proteins are likely to be cleared by macrophages. We observed an increase of perivascular macrophages in Protollin treated mice (Fig. 5A, B), in association with a reduction in cerebrovascular amyloid. Amyloid pahagocytosis by macrophage was seen by colocalization (yellow) of macrophage (red) with vascular amyloid (ThS, green) in Protollin treated mice (Fig 5B). Furthermore, we found significantly increased expression of monocyte activation markers, such as TNFα (by 227%, P<0.02) and CD11b (by 158%, P<0.05), in the blood of Protollin treated mice, by Real-Time PCR analysis (Fig. 5C). Analysis of isolated peritoneal macrophages from Protollin treated mice as compared to the control group showed a significant increase (Fig. 5D) in the levels of macrophage activation markers: a 58% (P<0.04) increase in the expression of CD11b, and a 122% (P<0.02) increase in the TNF-α expression. Furthermore we found a significant increase in macrophage markers that were previously described to be important for amyloid clearance (Farfara, et al., 2008): - a 266% (P<0.02) increase in scavenger receptor A (SRA) expression, a 498% (P<0.02) increase in insulin-degrading enzyme (IDE) involved in Aβ degradation and a 120% (P<0.05) increase in chemokine receptor CCR2 which has been recently linked to macrophage migration toward amyloid deposits (El Khoury, et al., 2007).
Fig. 5.
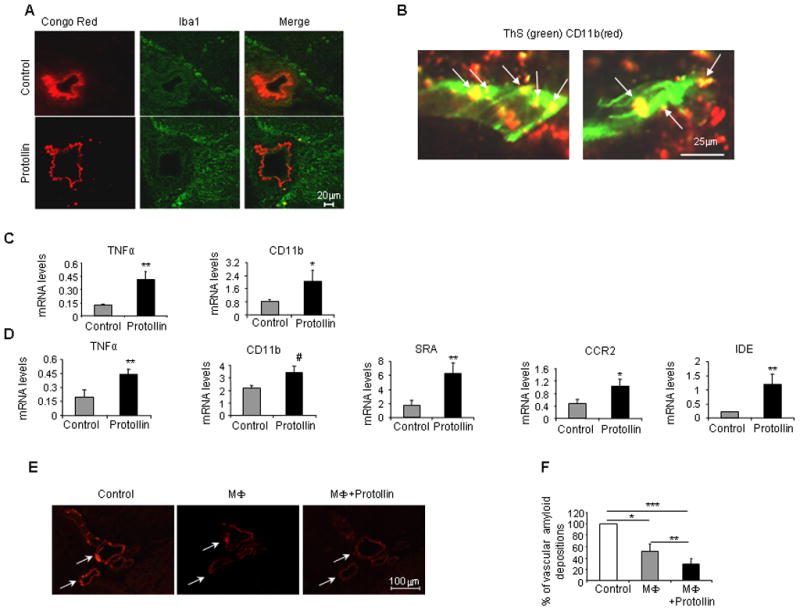
Cerebrovascular amyloid clearance in Protollin-treated mice is mediated by activated macrophages. (A) Activated perivascular macrophage cells (Iba1, positive) colocalized with a reduction of vascular amyloid deposition (Congo red) in the brains of Protollin-treated mice vs. control mice. Scale bar: 20 μm. (B) Confocal image of colocalization of CD11b positive cells (red) with vascular amyloid (ThS green) in Protollin treated mice. Scale bar: 50 μm. (C) Elevation of monocyte mRNA expression markers TNF-α (**P<0.02) and CD11b (*P<0.05) in the blood of Protollin-treated mice compared to the control mice (n=6 mice/group). (D) Elevation of macrophage activation markers from Protollin-treated mice compared to control: SRA (**P<0.02), TNFα (**P<0.02), IDE (**P<0.02), CD11b (#P<0.04), CCR2 (*P<0.05 from 16 months old immunized TGF-β1 Tg mice treated with Protollin (n=7) or PBS (n=6). (E) Representative figures and (F) analysis of reduction of cerebrovascular amyloid (Congo red) following incubation for 4 days at 37° C with Macrophage cell line (RAW264.7) with (right panel) and without (middle panel) 0.1 μg/ml Protollin ( *P<0.05, **P<0.0003, ***P<0.0001, n = 7).
3.7 Protollin activates macrophage to degrade vascular amyloid in situ
To directly determine the effect of Protollin on phagocytosis of vascular amyloid deposition by macrophages, we established an in situ assay as previously described (Wyss-Coray, et al., 2003). In brief (Fig. 5E), a macrophage cell line (RAW 264.7) was plated on unfixed insoluble vascular amyloid rich brain sections from transgenic mice with or without 0.1 μg/ml Protollin for 4 days (Fig. 5E, F). We found that Protollin-activated macrophages significantly decreased amyloid deposition by 71% (P<0.0001) compared to control (only medium), and 43% (P<0.0003) as compared to macrophages alone (Fig. 5F).
4. Discussion
In the present study, we investigated a novel immunotherapeutic approach to reduce amyloid deposition in TGF-β1 mice. This study demonstrates that chronic elevation of TGF-β1 production of astrocytes in 16-month-old transgenic mice leads to pathological signs: vascular and meningeal amyloid deposition, brain tissues damage and cognitive impairment and this was improved by nasal Protollin treatment. Treated animals exhibited no changes in behavior or toxicity as measured by body weight, eating habits, tail tone, or mobility.
We demonstrated a decrease in the congophilic material in the TGF-β1 animal model of cerebral amyloid angiopathy. “Amyloid” refers to any protein that becomes fibrillar, insoluble, and precipitates upon adopting a beta-pleated sheet configuration instead of the alpha-helical one (Lacombe, et al., 2004). Over twenty proteins qualify as amyloids by the criterion of thioflavin-S and congo red staining. Other than Aβ, other amyloids include fibronectin, laminin and cystatin C, which are prone to misfold and deposit as congophilic material in human brain vessels and lead to several forms of CAA (Lacombe, et al., 2004,Wyss-Coray, et al., 1995,Wyss-Coray, et al., 2000) .Because the increase in insoluble Aβ 1–40 in the brains of aged TGF- β 1 mice we measured using ELISA was not detected by immunohistology, it is likely that other congophilic proteins mask Aβ staining and that the congophilic material in the TGF-β1 model we studied contains these proteins in addition to the Aβ we identified by ELISA . Indeed, it was previously shown that astroglial overexpression of TGF-β1 consistently induced a strong upmodulation of the extracellular matrix proteins such as laminin and fibronectin in the CNS (Buckwalter and Wyss-Coray, 2004,Wyss-Coray, et al., 1995). Furthermore, blood vessels in brains of TGF-β1 mice also showed increase in fibronectin and laminin immunoreactivity (Wyss-Coray, et al., 1995). Thus our results demonstrate a decrease in congophilic material that appears to also contain Aβ 1–40.
Patients with CAA have a higher risk of developing hemorrhages and ischemic stroke (Olichney, et al., 1995). We monitored vascular disease progression in TGF-β1 mice using an MRI approach as is done in humans, which demonstrates the clinical application of our approach. BBB permeability increases with advancing age in patients with Alzheimer s disease and vascular dementia (Farrall and Wardlaw, 2007). The focal increase in the signal intensity change on the contrast-enhanced T1-weighted images, caused by leakage of the contrast agent (Gd-DTPA) into the surrounding tissue, was used in our study to detect the increase of BBB permeability (Liu, et al., 2008). In TGF-β1 mice, we found a significant increase in the passage of Gd-DTPA across the BBB, compared to wt mice. Protollin treatment prevented pathological BBB permeability in TGF-β1 mice and reduced BBB permeability to similar levels as in wt mice. This was correlated with a reduction in microhemorrhages as detected by Perls blue stain. Blood vessel diseases which mainly affect the elderly population, often result in typical T2 MRI findings that include region-specific volume loss, enlargement of the lateral ventricles, and patchy areas of abnormal signal intensity within the white matter and basal ganglia (Tsenter, et al., 2008). Homozygote TGF-β1 mice with high levels of cerebral TGF-β1 expression developed a severe hydrocephalus that results in enlargement of brain ventricles (Wyss-Coray, et al., 1995). Nevertheless, heterozygous TGF-β1 mice, with lower levels of TGF-β1 such as those used here, do not develop such alterations at young age (Wyss-Coray, et al., 1995). However, we found that 16 months TGF-β1 mice (Fig. 1C,D) had significant enlargement of lateral ventricles compared to wt littermates. These results are consistent with increased amyloid deposition in cerebral blood vessels. Indeed, Protollin treatment that reduced vascular amyloid also prevented pathological enlargement of the lateral ventricles.
The development of dementia in several neurodegenerative diseases has been shown to correlate with impairment of the cerebrovascular system and in these cases has been defined as vascular dementia. Cognitive impairment has been observed in both familial (Grabowski, et al., 2001) and sporadic (Greenberg, et al., 1993) instances of severe CAA. These observations suggest that CAA can cause clinically important vascular dysfunction(Greenberg, et al., 2004). Here we have shown that accumulation of amyloid in the cerebrovascular vessels of 16-month-old TGF-β1 mice is associated with a deficiency in memory and learning in these animals and that Protollin treatment improves the impairment of recognition memory.
Synaptophysin is a specific component of the membrane of presynaptic vesicles, and appears to be important for the biogenesis of synaptic vesicles, vesicle budding, and endocytosis (Daly, et al., 2000,Tartaglia, et al., 2001). Cognitive impairment has been observed in both familial (Grabowski, et al., 2001) and sporadic (Greenberg, et al., 1993) instances of severe CAA. It was previously demonstrated (Calhoun, et al., 1999) that expression of a mutation in APP, that leads to significant deposition of amyloid β (Aβ) in the cerebral vasculature (CAA) in mice, was associated with local neuron loss and synaptic abnormalities as shown by dystrophic staining of synaptophysin. In the present study, we found a marked decrease in hippocampal synaptophysin protein expression in 16-month-old TGF-β1 mice as compare to wt littermates. We postulate that the marked reduction in synaptophysin in TGF-β1 mice may correlate with the increase in CAA appearance, a reduction in brain volume (T2 MRI) and oxidative stress due to microhemorrhage incidents. Our results support previous reports of reduced synaptophysin expression in AD brains (Heffernan, et al., 1998). We found that Protollin administration leads to an increase in hippocampal synaptophysin that is important to hippocampal synaptic plasticity and cognition. Another factor that may regulate neuroplasticity in multiple brain regions is BDNF (Gomez-Pinilla, et al., 2002). BDNF is known to improve memory and also regulate synaptic transmission and plasticity in the CNS (Suzuki, et al., 2004). Furthermore, BDNF is decreased in the brains, serum, and CSF of patients with mild cognitive impairment and AD and may even correlate with Mini Mental State Examination scores (Laske, et al., 2006). In Protollin-treated animals, BDNF level increased in the hippocampus and was positively correlated with synaptophysin values. Indeed, it was previously shown that BDNF rapidly (within 2 h) upregulates levels of synaptophysin in cerebellar granule cells (Coffey, et al., 1997) and modulates the levels of the vesicle proteins (synaptobrevin and synaptophysin) at the hippocampus synapses (Lu and Chow, 1999).
Abnormal deposition of Aβ (1–40) that occurs in amyloid angiopathy (Leblanc, et al., 1992) and certain related disorders including hereditary cerebral hemorrhage amyloidosis-Dutch type (HCHWA-D) (Bornebroek, et al., 1996) leads to the degeneration of vascular smooth muscle cells of the large penetrating vessels as well as the cerebral capillaries that represent the BBB (Prior, et al., 1996) and results in cerebrovascular hemorrhage (Winkler, et al., 2001).
Previous reports (Koudinov, et al., 2001,Lacombe, et al., 2004) demonstrated that insoluble diffuse endogenous Aβ can be detected (verified by immunohistochemistry, Congo Red fluorescence and ELISA) in aged non-transgenic mice. In this work we analyzed endogenous Aβ 1–40 using ELISA both in transgenic and non transgenic littermates. We found by ELISA that we could detect insoluble but not soluble Aβ in the brain of aged normal mice. However, we did find soluble Aβ in the plasma of aged normal mice. We hypothesized that the inability to detect soluble Aβ in a normal mouse was related to transport of Aβ from the brain to the periphery. To investigate this we measured the levels of LRP-1 in brain capillaries of WT vs. TGF-β1 mice. LRP-1 is important for the clearance of Aβ from brain to blood via transcytosis across the BBB to the periphery. Furthermore, clearance of Aβ is induced by circulating lipoprotein receptors (Sagare, et al., 2007). We found that in TGF-β1 mice there was a significant reduction of LRP-1 expression (Supplemental figure 1) as compared to WT mice. These results suggest that in normal mice, LRP-1 might be responsible for the rapid transport of soluble Aβ 1–40 to the plasma whereas this mechanism is defective in TGF-β1 mice.
A progressive shift of Aβ from soluble to insoluble forms is evident in brains of patients with AD (Wang, et al., 1999). In our study, we found that an increase in endogenous Aβ (1–40) in 16-month-old TGF-β1 mice, correlated to the presence of vascular and meningeal Congo red reactivity. A decrease of insoluble Aβ (1–40) and an increase of brain and plasma soluble Aβ (1–40) following Protollin treatment, raises a possibility that the insoluble Aβ is converted to a soluble form by Aβ degrading enzymes and then transferred to the plasma. Conversion of Aβ from insoluble to soluble forms by Protollin may facilitate the removal of Aβ from the brain and explain the improved performance of the TGF-β1 mice when cognitively tested.
Recently, a physiological role for macrophages was reported in the regulation and clearing of cerebrovascular amyloid in AD mice (Hawkes and McLaurin, 2009, Weiss, et al., 2010). We showed that activated macrophage cells were present around amyloid depositions in Protollin treated mouse brains and this activation was associated with a reduction in CAA like pathology. In addition, we found a significantly increased expression of monocyte activation markers, such as TNFα and CD11b, in the blood of Protollin-treated mice. These results are in agreement with those of an earlier studies (Frenkel, et al., 2005,Frenkel, et al., 2008) showing that nasal Protollin, in an AD mouse model, activates microglia or macrophage cells to clear amyloid.
It was previously suggested that a defect in IDE activity contributes to the accumulation of amyloid in the cortical microvasculature (Morelli, et al., 2004). Furthermore, IDE activity was significantly reduced in CAA as compared with age-matched controls (Morelli, et al., 2004). We found an upregulation of IDE in the peritoneal macrophages after nasal Protollin. Furthermore, we found an increase of CCR2 in peritoneal macrophages after nasal Protollin. CCR2 is a chemokine receptor expressed on microglia, which mediates the accumulation of mononuclear phagocytes at sites of inflammation. CCR2 is also required for the migration of CD11b+ cells into the brain.
In summary, our study demonstrates that activation of macrophages by nasal Protollin is a novel therapeutic approach to reduce microhemorrhage stroke and improve cognition in a model of cerebral amyloid angiopathy.
Supplementary Material
{kind=link}
Acknowledgments
This work is supported by grants from the HFSP organization and the Dana Foundation (to D.F.), NIH (NIA AG027437 to H.L.W.), the United States-Israel Bi-National Science Foundation (to D.F and H.L.W.). There is no conflict of Interest.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Akwa Y, Ladurelle N, Covey DF, Baulieu EE. The synthetic enantiomer of pregnenolone sulfate is very active on memory in rats and mice, even more so than its physiological neurosteroid counterpart: distinct mechanisms? Proc Natl Acad Sci U S A. 2001;98(24):14033–7. doi: 10.1073/pnas.241503698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bevins RA, Besheer J. Object recognition in rats and mice: a one-trial non-matching-to-sample learning task to study 'recognition memory'. Nat Protoc. 2006;1(3):1306–11. doi: 10.1038/nprot.2006.205. [DOI] [PubMed] [Google Scholar]
- Bornebroek M, Haan J, Maat-Schieman ML, Van Duinen SG, Roos RA. Hereditary cerebral hemorrhage with amyloidosis-Dutch type (HCHWA-D): I--A review of clinical, radiologic and genetic aspects. Brain Pathol. 1996;6(2):111–4. doi: 10.1111/j.1750-3639.1996.tb00793.x. [DOI] [PubMed] [Google Scholar]
- Buckwalter MS, Wyss-Coray T. Modelling neuroinflammatory phenotypes in vivo. J Neuroinflammation. 2004;1(1):10. doi: 10.1186/1742-2094-1-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgermeister P, Calhoun ME, Winkler DT, Jucker M. Mechanisms of cerebrovascular amyloid deposition. Lessons from mouse models. Ann N Y Acad Sci. 2000;903:307–16. doi: 10.1111/j.1749-6632.2000.tb06381.x. [DOI] [PubMed] [Google Scholar]
- Buttini M, Masliah E, Barbour R, Grajeda H, Motter R, Johnson-Wood K, Khan K, Seubert P, Freedman S, Schenk D, Games D. Beta-amyloid immunotherapy prevents synaptic degeneration in a mouse model of Alzheimer's disease. J Neurosci. 2005;25(40):9096–101. doi: 10.1523/JNEUROSCI.1697-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calhoun ME, Burgermeister P, Phinney AL, Stalder M, Tolnay M, Wiederhold KH, Abramowski D, Sturchler-Pierrat C, Sommer B, Staufenbiel M, Jucker M. Neuronal overexpression of mutant amyloid precursor protein results in prominent deposition of cerebrovascular amyloid. Proc Natl Acad Sci U S A. 1999;96(24):14088–93. doi: 10.1073/pnas.96.24.14088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castellani RJ, Smith MA, Perry G, Friedland RP. Cerebral amyloid angiopathy: major contributor or decorative response to Alzheimer's disease pathogenesis. Neurobiol Aging. 2004;25(5):599–602. doi: 10.1016/j.neurobiolaging.2003.12.019. [DOI] [PubMed] [Google Scholar]
- Coffey ET, Akerman KE, Courtney MJ. Brain derived neurotrophic factor induces a rapid upregulation of synaptophysin and tau proteins via the neurotrophin receptor TrkB in rat cerebellar granule cells. Neurosci Lett. 1997;227(3):177–80. doi: 10.1016/s0304-3940(97)00335-2. [DOI] [PubMed] [Google Scholar]
- Connor B, Young D, Yan Q, Faull RL, Synek B, Dragunow M. Brain-derived neurotrophic factor is reduced in Alzheimer's disease. Brain Res Mol Brain Res. 1997;49(1–2):71–81. doi: 10.1016/s0169-328x(97)00125-3. [DOI] [PubMed] [Google Scholar]
- Daly C, Sugimori M, Moreira JE, Ziff EB, Llinas R. Synaptophysin regulates clathrin-independent endocytosis of synaptic vesicles. Proc Natl Acad Sci U S A. 2000;97(11):6120–5. doi: 10.1073/pnas.97.11.6120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeMattos RB, Bales KR, Cummins DJ, Paul SM, Holtzman DM. Brain to plasma amyloid-beta efflux: a measure of brain amyloid burden in a mouse model of Alzheimer's disease. Science. 2002;295(5563):2264–7. doi: 10.1126/science.1067568. [DOI] [PubMed] [Google Scholar]
- El Khoury J, Toft M, Hickman SE, Means TK, Terada K, Geula C, Luster AD. Ccr2 deficiency impairs microglial accumulation and accelerates progression of Alzheimer-like disease. Nat Med. 2007;13(4):432–8. doi: 10.1038/nm1555. [DOI] [PubMed] [Google Scholar]
- Farfara D, Lifshitz V, Frenkel D. Neuroprotective and neurotoxic properties of glial cells in the pathogenesis of Alzheimer's disease. J Cell Mol Med. 2008;12(3):762–80. doi: 10.1111/j.1582-4934.2008.00314.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrall AJ, Wardlaw JM. Blood-brain barrier: Ageing and microvascular disease - systematic review and meta-analysis. Neurobiol Aging. 2007 doi: 10.1016/j.neurobiolaging.2007.07.015. [DOI] [PubMed] [Google Scholar]
- Fowler SC, Zarcone TJ, Vorontsova E, Chen R. Motor and associative deficits in D2 dopamine receptor knockout mice. Int J Dev Neurosci. 2002;20(3–5):309–21. doi: 10.1016/s0736-5748(02)00009-6. [DOI] [PubMed] [Google Scholar]
- Frenkel D, Maron R, Burt DS, Weiner HL. Nasal vaccination with a proteosome-based adjuvant and glatiramer acetate clears beta-amyloid in a mouse model of Alzheimer disease. J Clin Invest. 2005;115(9):2423–33. doi: 10.1172/JCI23241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frenkel D, Puckett L, Petrovic S, Xia W, Chen G, Vega J, Dembinsky-Vaknin A, Shen J, Plante M, Burt DS, Weiner HL. A nasal proteosome adjuvant activates microglia and prevents amyloid deposition. Ann Neurol. 2008;63(5):591–601. doi: 10.1002/ana.21340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujimori Y, Maeda S, Saeki M, Morisaki I, Kamisaki Y. Inhibition by nifedipine of adherence- and activated macrophage-induced death of human gingival fibroblasts. Eur J Pharmacol. 2001;415(1):95–103. doi: 10.1016/s0014-2999(01)00810-x. [DOI] [PubMed] [Google Scholar]
- Gomez-Pinilla F, Ying Z, Roy RR, Molteni R, Edgerton VR. Voluntary exercise induces a BDNF-mediated mechanism that promotes neuroplasticity. J Neurophysiol. 2002;88(5):2187–95. doi: 10.1152/jn.00152.2002. [DOI] [PubMed] [Google Scholar]
- Grabowski TJ, Cho HS, Vonsattel JP, Rebeck GW, Greenberg SM. Novel amyloid precursor protein mutation in an Iowa family with dementia and severe cerebral amyloid angiopathy. Ann Neurol. 2001;49(6):697–705. doi: 10.1002/ana.1009. [DOI] [PubMed] [Google Scholar]
- Greenberg SM, Gurol ME, Rosand J, Smith EE. Amyloid angiopathy-related vascular cognitive impairment. Stroke. 2004;35(11 Suppl 1):2616–9. doi: 10.1161/01.STR.0000143224.36527.44. [DOI] [PubMed] [Google Scholar]
- Greenberg SM, Vonsattel JP, Stakes JW, Gruber M, Finklestein SP. The clinical spectrum of cerebral amyloid angiopathy: presentations without lobar hemorrhage. Neurology. 1993;43(10):2073–9. doi: 10.1212/wnl.43.10.2073. [DOI] [PubMed] [Google Scholar]
- Hawkes CA, McLaurin J. Selective targeting of perivascular macrophages for clearance of beta-amyloid in cerebral amyloid angiopathy. Proc Natl Acad Sci U S A. 2009;106(4):1261–6. doi: 10.1073/pnas.0805453106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heffernan JM, Eastwood SL, Nagy Z, Sanders MW, McDonald B, Harrison PJ. Temporal cortex synaptophysin mRNA is reduced in Alzheimer's disease and is negatively correlated with the severity of dementia. Exp Neurol. 1998;150(2):235–9. doi: 10.1006/exnr.1997.6772. [DOI] [PubMed] [Google Scholar]
- Kermani P, Hempstead B. Brain-derived neurotrophic factor: a newly described mediator of angiogenesis. Trends Cardiovasc Med. 2007;17(4):140–3. doi: 10.1016/j.tcm.2007.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koudinov AR, Berezov TT, Koudinova NV. The levels of soluble amyloid beta in different high density lipoprotein subfractions distinguish Alzheimer's and normal aging cerebrospinal fluid: implication for brain cholesterol pathology? Neurosci Lett. 2001;314(3):115–8. doi: 10.1016/s0304-3940(01)02263-7. [DOI] [PubMed] [Google Scholar]
- Lacombe P, Mathews PM, Schmidt SD, Breidert T, Heneka MT, Landreth GE, Feinstein DL, Galea E. Effect of anti-inflammatory agents on transforming growth factor beta over-expressing mouse brains: a model revised. J Neuroinflammation. 2004;1(1):11. doi: 10.1186/1742-2094-1-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laske C, Stransky E, Leyhe T, Eschweiler GW, Wittorf A, Richartz E, Bartels M, Buchkremer G, Schott K. Stage-dependent BDNF serum concentrations in Alzheimer's disease. J Neural Transm. 2006;113(9):1217–24. doi: 10.1007/s00702-005-0397-y. [DOI] [PubMed] [Google Scholar]
- Leblanc R, Haddad G, Robitaille Y. Cerebral hemorrhage from amyloid angiopathy and coronary thrombolysis. Neurosurgery. 1992;31(3):586–90. doi: 10.1227/00006123-199209000-00025. [DOI] [PubMed] [Google Scholar]
- Lifshitz V, Frenkel D. Animal Models for Cerebrovascular Impairment and its Relevance in Vascular Dementia. In: Landow Melanie L., editor. Cognitive Impairment: Causes, Diagnosis and Treatments. 2009. [Google Scholar]
- Liu HL, Wai YY, Chen WS, Chen JC, Hsu PH, Wu XY, Huang WC, Yen TC, Wang JJ. Hemorrhage detection during focused-ultrasound induced blood-brain-barrier opening by using susceptibility-weighted magnetic resonance imaging. Ultrasound Med Biol. 2008;34(4):598–606. doi: 10.1016/j.ultrasmedbio.2008.01.011. [DOI] [PubMed] [Google Scholar]
- Lu B, Chow A. Neurotrophins and hippocampal synaptic transmission and plasticity. J Neurosci Res. 1999;58(1):76–87. [PubMed] [Google Scholar]
- Maia LF, Mackenzie IR, Feldman HH. Clinical phenotypes of Cerebral Amyloid Angiopathy. J Neurol Sci. 2007;257(1–2):23–30. doi: 10.1016/j.jns.2007.01.054. [DOI] [PubMed] [Google Scholar]
- Mitchell AN, Jayakumar L, Koleilat I, Qian J, Sheehan C, Bhoiwala D, Hushmendy SF, Heuring JM, Crawford DR. Brain expression of the calcineurin inhibitor RCAN1 (Adapt78) Arch Biochem Biophys. 2007;467(2):185–92. doi: 10.1016/j.abb.2007.08.030. [DOI] [PubMed] [Google Scholar]
- Morelli L, Llovera RE, Mathov I, Lue LF, Frangione B, Ghiso J, Castano EM. Insulin-degrading enzyme in brain microvessels: proteolysis of amyloid {beta} vasculotropic variants and reduced activity in cerebral amyloid angiopathy. J Biol Chem. 2004;279(53):56004–13. doi: 10.1074/jbc.M407283200. [DOI] [PubMed] [Google Scholar]
- Nagahara AH, Merrill DA, Coppola G, Tsukada S, Schroeder BE, Shaked GM, Wang L, Blesch A, Kim A, Conner JM, Rockenstein E, Chao MV, Koo EH, Geschwind D, Masliah E, Chiba AA, Tuszynski MH. Neuroprotective effects of brain-derived neurotrophic factor in rodent and primate models of Alzheimer's disease. Nat Med. 2009;15(3):331–7. doi: 10.1038/nm.1912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicoll JA, Yamada M, Frackowiak J, Mazur-Kolecka B, Weller RO. Cerebral amyloid angiopathy plays a direct role in the pathogenesis of Alzheimer's disease. Pro-CAA position statement. Neurobiol Aging. 2004;25(5):589–97. doi: 10.1016/j.neurobiolaging.2004.02.003. discussion 603–4. [DOI] [PubMed] [Google Scholar]
- Olichney JM, Hansen LA, Hofstetter CR, Grundman M, Katzman R, Thal LJ. Cerebral infarction in Alzheimer's disease is associated with severe amyloid angiopathy and hypertension. Arch Neurol. 1995;52(7):702–8. doi: 10.1001/archneur.1995.00540310076019. [DOI] [PubMed] [Google Scholar]
- Peila R, Yucesoy B, White LR, Johnson V, Kashon ML, Wu K, Petrovitch H, Luster M, Launer LJ. A TGF-beta1 polymorphism association with dementia and neuropathologies: the HAAS. Neurobiol Aging. 2007;28(9):1367–73. doi: 10.1016/j.neurobiolaging.2006.06.004. [DOI] [PubMed] [Google Scholar]
- Prior R, D'Urso D, Frank R, Prikulis I, Pavlakovic G. Loss of vessel wall viability in cerebral amyloid angiopathy. Neuroreport. 1996;7(2):562–4. doi: 10.1097/00001756-199601310-00044. [DOI] [PubMed] [Google Scholar]
- Sagare A, Deane R, Bell RD, Johnson B, Hamm K, Pendu R, Marky A, Lenting PJ, Wu Z, Zarcone T, Goate A, Mayo K, Perlmutter D, Coma M, Zhong Z, Zlokovic BV. Clearance of amyloid-beta by circulating lipoprotein receptors. Nat Med. 2007;13(9):1029–31. doi: 10.1038/nm1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki S, Numakawa T, Shimazu K, Koshimizu H, Hara T, Hatanaka H, Mei L, Lu B, Kojima M. BDNF-induced recruitment of TrkB receptor into neuronal lipid rafts: roles in synaptic modulation. J Cell Biol. 2004;167(6):1205–15. doi: 10.1083/jcb.200404106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tartaglia N, Du J, Tyler WJ, Neale E, Pozzo-Miller L, Lu B. Protein synthesis-dependent and -independent regulation of hippocampal synapses by brain-derived neurotrophic factor. J Biol Chem. 2001;276(40):37585–93. doi: 10.1074/jbc.M101683200. [DOI] [PubMed] [Google Scholar]
- Tsenter J, Beni-Adani L, Assaf Y, Alexandrovich AG, Trembovler V, Shohami E. Dynamic changes in the recovery after traumatic brain injury in mice: effect of injury severity on T2-weighted MRI abnormalities, and motor and cognitive functions. J Neurotrauma. 2008;25(4):324–33. doi: 10.1089/neu.2007.0452. [DOI] [PubMed] [Google Scholar]
- Wang J, Dickson DW, Trojanowski JQ, Lee VM. The levels of soluble versus insoluble brain Abeta distinguish Alzheimer's disease from normal and pathologic aging. Exp Neurol. 1999;158(2):328–37. doi: 10.1006/exnr.1999.7085. [DOI] [PubMed] [Google Scholar]
- Weiss R, Lifshitz V, Frenkel D. TGF-β1 (2010) affects endothelial cell interaction with macrophage and Th1 T-cells leading to the development of cerebrovascular amyloidosis. Brain Behav Immun. doi: 10.1016/j.bbi.2010.11.012. [DOI] [PubMed] [Google Scholar]
- Winkler DT, Bondolfi L, Herzig MC, Jann L, Calhoun ME, Wiederhold KH, Tolnay M, Staufenbiel M, Jucker M. Spontaneous hemorrhagic stroke in a mouse model of cerebral amyloid angiopathy. J Neurosci. 2001;21(5):1619–27. doi: 10.1523/JNEUROSCI.21-05-01619.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyss-Coray T, Feng L, Masliah E, Ruppe MD, Lee HS, Toggas SM, Rockenstein EM, Mucke L. Increased central nervous system production of extracellular matrix components and development of hydrocephalus in transgenic mice overexpressing transforming growth factor-beta 1. Am J Pathol. 1995;147(1):53–67. [PMC free article] [PubMed] [Google Scholar]
- Wyss-Coray T, Lin C, Sanan DA, Mucke L, Masliah E. Chronic overproduction of transforming growth factor-beta1 by astrocytes promotes Alzheimer's disease-like microvascular degeneration in transgenic mice. Am J Pathol. 2000;156(1):139–50. doi: 10.1016/s0002-9440(10)64713-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyss-Coray T, Loike JD, Brionne TC, Lu E, Anankov R, Yan F, Silverstein SC, Husemann J. Adult mouse astrocytes degrade amyloid-beta in vitro and in situ. Nat Med. 2003;9(4):453–7. doi: 10.1038/nm838. [DOI] [PubMed] [Google Scholar]
- Zhang C, Shen JK, Lam TT, Zeng HY, Chiang SK, Yang F, Tso MO. Activation of microglia and chemokines in light-induced retinal degeneration. Mol Vis. 2005;11:887–95. [PubMed] [Google Scholar]
- Zlokovic BV. Neurovascular mechanisms of Alzheimer's neurodegeneration. Trends Neurosci. 2005;28(4):202–8. doi: 10.1016/j.tins.2005.02.001. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.