

Abstract
BACKGROUND
Patients with osteosarcoma (OS) lung metastases have poor response to the salvage therapy. Understanding the mechanisms involved in metastatic process of OS may lead to new effective therapeutic approaches. We previously found that upregulation of Fas receptor by transfecting OS cells with fas plasmid inhibited their metastases growth in the lungs in vivo.
METHODS
In this study we treated OS cells with HDAC inhibitor, SNDX-275 and studied its cytotoxicity and effect on Fas signaling in vitro and in vivo.
RESULTS
We found that subtoxic doses of SNDX-275 can activate the Fas pathway in OS cells by increasing expression of Fas mRNA, which was not always followed by the increased levels of its receptor expression on the cell surface. Treatment of cells with the combination of SNDX-275 and FasL had a stronger cytotoxic effect on tested OS cells than either agent alone. Inhibition of the Fas pathway in cells by inhibition of the FADD molecule eliminated this combination effect, indicating that activity of FADD is important for the efficacy of this agent in a FasL-expressing environment to which lungs belong. Intranasal administration of SNDX-275 in mice with OS lung metastases showed that it may inhibit metastatic growth at a dose of 0.13 mg/kg, which is ~200-fold lower than the therapeutically effective oral dose reported previously.
CONCLUSION
These findings indicate that SNDX-275 can activate Fas signaling in OS cells in vitro and in vivo and that administration of SDNX-275 via inhalation is feasible for treatment of OS metastases and warrants its further investigation.
Introduction
The most common and almost exclusive site for OS metastasis is the lungs. Metastatic OS is difficult to treat. The overall survival rated for these patients range from 20% to 25%. Over the past 25 years, alterations of chemotherapy regimens have not significantly improved their survival rates (1), indicating the need for new therapeutic approaches for OS lung metastases treatment.
Formation of metastases is a complicated process. Evaluation of the key mechanisms that control metastatic process should lead to the creation of novel therapeutic targets. We previously found that the Fas pathway plays an important role in OS metastasis growth in the lungs. Lungs belong to few organs in our body that constitutively express Fas ligand (FasL). When tumor cells that express Fas receptor on their surface migrate to the lungs, their Fas binds with FasL, which is expressed in the lung environment. This event further leads to trimerization of the receptor and recruitment of Fas-associated death domain (FADD) adaptor molecule. Assembly of these molecules all together is called death-inducing signaling complex (DISC). Formation of DISC ultimately results in proteolytic cleavage of caspases and apoptosis (2). Using LM7 and DLM8 models of OS lung metastases, we demonstrated that due to this mechanism only Fas-negative cells were able to survive and form lung metastases, whereas Fas-positive OS cells were rapidly cleared from the lung (3). These findings were further confirmed by analysis of Fas expression in OS lung metastases from patients. In most cases OS lung metastases had negligible levels of Fas. Elevation of the Fas expression was observed in those patients who received chemotherapy prior to lung metastases resection (4). This finding indicates that Fas expression could be triggered by treatment and, that Fas may contribute to the therapeutic effect. Indeed, we made similar observations in LM7 and DLM8 animal models with OS lung metastases that received aerosol chemotherapy (5, 6). Inhibition of the Fas-mediated signaling significantly corrupted tumor response to the treatment (6). These results indicated, that the Fas signaling is a critical mechanism of the treatment of OS lung metastases. Therefore, identification of agents that can enhance Fas receptor expression in OS tumor cells and/or activate downstream Fas signaling may lead to the discovery of novel therapeutic approach for patients with relapsed OS in the lungs.
Histone deacetylase inhibitors (HDACi) is the novel class of promising anticancer agents approved for clinical use. HDACi promote acetylation of histones, which is required for promotion of gene transcription. Several classes of HDACi have been identified, but their effect on OS cells regarding the Fas pathway is not well understood. The most detailed studies were performed by Imai et al. demonstrating that treatment of sarcoma cells with FK901228 HDACi was able to induce apoptosis in tumor cells upregulating FasL expression. However, they did not observe changes in Fas receptor expression (7).
In the study described herein, we studied the effect of another HDACi, SNDX-275, which specifically inhibits class I HDACs (8), on the Fas signaling in different OS cells. We used low doses of SNDX-275 that alone had little effect of the OS cell growth in vitro. The purpose of this approach was to evaluate the ability of SNDX-275 to sensitize tumor cells to apoptosis in a FasL-expressing environment without inducing a direct killing effect by itself, and to determine the mechanism of this sensitization. We found that in one OS cell line, DLM8, SNDX-275 activated the Fas pathway by direct upregulation of Fas mRNA and its receptor expression on cell surface, whereas in the other cell line, LM7 we did not observe the elevated levels of Fas receptor on cell surface, despite increased mRNA levels. Treatment of both cell lines with the combination of SNDX-275 and FasL showed increased cytotoxic effect than either agent alone. Corruption of the Fas pathway in both tumor cell lines by inhibiting Fas adapter molecule FADD, reversed the killing effect of SNDX-275 and FasL combination indicating on important role of this molecule in the Fas pathway activation by SNDX-275. Based on these results we concluded that SNDX-275 is able to activate Fas signaling in OS cells and induce their cell death in FasL-expressing environment without having a direct cytotoxic effect by itself.
To date there have been few therapeutic studies using SNDX-275 in solid tumors, including sarcomas (9-12). However, these studies were performed on the subcutaneous xenograft models and high levels of FasL expression were not reported in subcutaneous tissues. In addition, OS metastasize almost exclusively to the lungs. Therefore, in this present study we verified the efficacy of SNDX-275 treatment using a more relevant experimental mouse model with established OS metastases in the lungs (13, 14). To minimize systemic exposure and deliver the drug directly in the lungs we used an aerosol delivery approach. We have previously shown that aerosol administration is feasible for delivering anticancer drugs directly to the lungs (5, 15, 16) and have developed a unique technology of outpatient aerosol anticancer therapy (17). Here, we treated mice intranasally with SNDX-275 at dose, which was ~ 200-fold lower than the previously reported therapeutically effective oral dose (9, 10) and showed that this dose was able to inhibit OS lung metastases growth in mice.
Materials and Methods
Cell Lines and Reagents
The mouse osteosarcoma Dunn LM8 (DLM8) cell line was provided by Dr. Akira Myoui (University of Osaka, Osaka, Japan) (14). The human LM7 OS lung metastatic cell line was derived from SAOS-2 cell line by repeated intravenous recycling through the lungs of nude mice in our laboratory (13). Cells had been MAP tested and were mycoplasma-negative. Cell lines were validated by STR DNA fingerprinting using the AmpFℓSTR Identifier kit according to manufacturer instructions (Applied Biosystems cat 4322288). The STR profiles were compared to known ATCC fingerprints (ATCC.org), to the Cell Line Integrated Molecular Authentication database (CLIMA) version 0.1.200808 (http://bioinformatics.istge.it/clima/) (Nucleic Acids Research 37:D925-D932 PMCID: PMC2686526) and to the MD Anderson fingerprint database. The STR profiles matched known DNA fingerprints or were unique. Authenticity of cells was determined on 12/11/2010 by Characterized Cell Line core at U.T. M.D. Anderson Cancer Center. All cell lines were maintained in the complete Dulbecco’s modified Eagle’s medium (DMEM) (Whittaker Bioproducts Inc. Walkersville, MD) supplemented with 10% heat-inactivated bovine serum (Intergen, Purchase, NJ). Recombinant soluble superFasL was purchased from Alexis Biochemicals, Inc. (Farmingdale, NY). SNDX-275 agent was a kind gift from Syndax Pharmaceuticals Inc. (Waltham, MA). PE-labeled anti-Fas antibodies and isotype-match controls were purchased from BD Biosciences (San Diego, CA). Anti-human FADD monoclonal antibody was purchased from Upstate (Lake Placid, NY).
Flow cytometry
For OS cells for Fas, one million cells were suspended in FACS buffer (PBS, containing 2% fetal calf serum and 0.1% sodium azide) and incubated with either 1.0 mg/ml PE-conjugated mouse anti-human Fas antibody (clone DX2) or hamster anti-mouse Fas monoclonal antibody (Pharmingen, San Diego, CA) for human and mouse cells, respectively. PE-conjugated isotype-matched control IgG antibodies were used as negative controls. Samples were analyzed using a FACScan (Becton Dickinson, Mountain View, CA). Experiments for all cell lines were performed simultaneously.
Cytotoxicity Assay
The sensitivity of OS cells to soluble FasL (sFasL) (Alexis Biochemicals, San Diego, CA) was determined by 3-(4,5-dimethylthiazol-2yl)2,5-diphenyltetrazolium bromide (MTT) assay as described previously (3). Briefly, 3,000 cells/well were grown in 96-well plates and treated with SNDX-275 for the time specified in the text followed by the addition of 10 ng/ml soluble FasL for 24 h; wells treated with a single agent or without cells served as controls. MTT reagent was added to each well at a concentration of 0.08 mg/ml for 2-4 h. Cells were then lysed with 0.1 ml of dimethylsulfoxide. Cytotoxicity was quantified using a microtiter plate-reader at 570 nm. Experiments for all cell lines were performed simultaneously.
Chromatin immuno-precipitation assay (ChIP)
Cells were cross-linked with 1% formaldehyde added directly to cell culture medium for 10 min at room temperature. The cell monolayers were washed twice with ice-cold 1x PBS and collected into dithiothreitol solution (100 mM Tris-HCl, pH 9.4) and 10 mM dithiothreitol) followed by incubation at 30°C for 10 min. Cells were washed sequentially with ice-cold PBS, buffer I (0.25 M Triton X-100, 1 mM EDTA, 0.5 mM EGTA, 10 mM HEPES, pH 6.5) and buffer II (200 mM NaCl, 1 mM EDTA, 10 mM HEPES, pH 6.5), containing protease and phosphatase inhibitors, and then lysed in lysis buffer (1% sodium dodecyl sulfate, 10 mM EDTA, 50 mM Tris-HCl, pH 8.1 plus protease inhibitors and 1 mM phenylmethylsulfonylfluoride) for 10 min on ice. Sonication was performed using a Branson-450 Sonifier with microtip in 7-sec bursts followed by 1 min of cooling on ice for a total sonication time of 21 sec per sample resulted in DNA fragment sizes of 0.7–1.5 kb. Then samples were centrifuged at 14,000 for 10 min at 4°C. Supernatants were diluted 5-fold in ChIP dilution buffer (1% Triton X-100, 2 mM EDTA, 150 mM NaCl, 20 mM Tris-HCl, pH 8.1 plus protease and phosphatase inhibitors) and precleared for 30 min at 4°C with 20 μl preimmune serum and 80 μl salmon sperm DNA/protein A/G agarose slurry. Ten percent of total supernatant was saved as a total input control and processed with the eluted immunoprecipitates beginning with the cross-linking reversal step. Five micrograms of ChIP-grade specific rabbit polyclonal anti-acetyl H3 antibody (Abcam, Cambridge, MA) was added to the chromatin solutions and samples were incubated at 4°C overnight with rotation. Immunocomplexes were collected with 60 μl of the salmon sperm DNA/protein A/G agarose beads for 1 h at 4°C with rotation. Beads were then washed consecutively for 10 min each with low salt wash buffer (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 150 mM NaCl, 20 mM Tris-HCl, pH 8.1), high salt wash buffer (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 500 mM NaCl, 20 mM Tris-HCl, pH 8.1), and LiCl wash buffer (0.25 mM LiCl, 1% Nonidet P-40, 1% deoxycholate, 1 mM EDTA, 10 mM Tris-HCl, pH 8.1) and twice in 1x TE buffer. Final wash with TE buffer was divided in half and the pellet was frozen at −80°C until use. Half of the complex was further examined for fas promoter PCR analysis and the other half for the global chromatin acetylation status on histone H3 by Western blot.
For PCR analysis reverse formaldehyde cross-link was performed by eluting the pellet twice in 100 μl freshly made elution buffer (1% SDS/0.1 M NaHCO3) during 15 min. One μl 10 mg/ml RNase and 5 M NaCl to a final concentration of 0.3 M was added to the eluate and the sample was incubated at 64°C overnight. DNA was recovered using the QiaQuick spin columns (Qiagen, Valencia, CA) and eluted in 80 μl of 10 mM Tris (pH 8.0). Recovered DNA was then quantified by PCR (see below).
For Western blot analysis the chromatin-DNA complex pellet was dissociated by adding 25 μl of 1× Laemmli buffer and boiling for 10 min. The beads were pelleted by centrifugation and discarded, supernatants were loaded onto 15% SDS-PAGE gel. The proteins from the gel were transferred to nitrocellulose membrane and stained with a ChIP grade monoclonal mouse anti-acetyl K9 Histone 3 antibody from Abcam, followed by HRP-labeled secondary anti-mouse IgG antibody exposure.
Plasmids and establishment of FADD-dominant negative (FDN) stable clones
The plasmid constructs for human FDN was a kind gift from Drs. Rokhlin and Taghiyev (Univ. of Iowa, IO) (18) and mouse FDN constructs were kindly provided by Dr. Winoto (Univ. of California, Berkley, CA) (19). Both cell lines were transfected with plasmids using the Fugene6 reagent (Roche Applied Biosystems, Indianapolis, IN).
Western Blot Assay
Cell lysates were processed by Western blot analysis as described previously (3). Briefly 0.02 mg of proteins were separated on 12% SDS-PAGE gel and blotted to the nitrocellulose membrane (Amersham Biosciences Inc., Pittsburg, PA). The membranes were blocked with 5% non-fat dry milk in PBS containing 0.1% Tween-20 and then incubated with an anti-FADD antibody (Upstate) according to the manufacturer’s instructions.
RT-PCR
Total RNA from cells was isolated using Trizol reagent (Life Technologies, Inc, Gaitherburg, MD). Three micrograms of RNA was reverse-transcribed using a Reverse Transcription System (Promega Co., Madison, WI). The 2.5 μL aliguot of resulting cDNA was used for PCR amplification with Taq-polymerase (Promega Co., Madison, WI) with specific primers for Fas and FADD (see primers and annealing temperatures in Table 1). Primers for beta-actin were used as loading controls.
Table 1.
Primers sequences and annealing conditions for the Fas signaling molecules
| Gene target |
Annealing T (°C) |
Forward primer | Reverse primer |
|---|---|---|---|
| Fas | 59 | GGCTATAGATCACCTTCATGTA | GCAGTTAACTCAGGGACCAAG |
| FADD | 50 | AGGCATCTACACAGCCTGGACTTT | CCGCTGCCTTGGCAATTCTGTTAT |
| beta-actin | 49-60 | ATCATGTTTGAGACCTTCAACA3 | CATCTCTTGCTCGAAGTCCA |
Cycling conditions were 94°C for 60s, annealing for 60s, and primer extension at 72°C for 30s.
Animal study
Nu/nu mice and C3H mice were purchased from the National Cancer Institute (Bethesda, MD) and housed in standard cages with food and water provided ad libitum. All animal experiments were performed with the approval of the Institutional Animal Care and Use Committee. Mice were injected with human LM7 cells via the tail vein. After that, mice were randomly divided into 2 groups (6 mice/group). Both groups received treatment intranasally under isoflurane anesthesia. Inhalation-based treatment was initiated 32 days after tumor cell inoculation when micrometastases were established in lungs. Mice received 0.13 mg/kg SNDX-275 or PBS (as a control) 3 times weekly for 3 weeks. Mice are nose-obligatory breathers; thus, substances administered through the nose will go into the lungs. We observed a slight toxicity in mice after 3 weeks, most likely due to frequent anesthesia because the control mice showed similar signs of toxicity as those that received SNDX-275 treatment; therefore, the frequency of treatment was decreased to twice a week at the same dose of drug and treatment was continued for another 2 weeks. At the end of treatment, mice were euthanized, their lungs were resected and weighed, and visible lung metastases were counted.
Immunohistochemistry
Lung tissues from mice were fixed in formalin and embedded in paraffin. Tissue sections were then deparafinized in xylene and rehydrated. Antigen retrieval was performed by boiling slides with tissues in a microwave for 5 min in 0.01 M sodium citrate buffer, pH 6.0. To block exogenous peroxidases, tissues were blocked with 3% hydrogen peroxide for 12 min. Nonspecific binding was then blocked with PBS containing 10% normal horse serum and 1% normal goat serum (protein blocking buffer). The primary antibody, polyclonal rabbit anti-human Fas (C-20) or FADD (H-181) antibodies from Santa Cruz Biotechnology, Inc (Santa Cruz, CA) at 1:50 dilution in protein blocking buffer were applied over the tissue and left overnight at 4°C. The secondary goat anti-rabbit antibody labeled with horse-radish peroxidase (Jackson Laboratory, Bar Harbor, Maine) was then applied for 2 hr at ambient temperature. The slides were finally developed with 3,30-diaminobenzidine as a substrate and lightly counterstained with hematoxylin. Negative controls were prepared by omitting the primary antibodies. Liver tissue was used as a positive control for Fas as described previously (4) and for FADD as verified by antibody provider. Tissues were examined under the conventional light microscope (Leica) and representative tumor lesions were photographed at magnification 100x.
Statistical Analysis
The unpaired Student’s t-test was used to determined significance of differences between experimental groups. Mann-Whitney rank-sum test was used for data evaluation of in vivo experiments. P value less than 0.05 was considered statistically significant.
Results
SNDX-275 Sensitizes DLM8 Mouse OS cells to FasL by Upregulating Fas Receptor Expression
Mouse OS cells DLM8 were treated with a low dose of SNDX-275 (3 μM) for 24, 48 and 72 h. Flow cytometry analysis of Fas expression on the cell surface of DLM8 cells showed that it increased over time (Fig. 1A). Elevation of Fas expression was also observed with the dose increase of this agent: in the untreated population ~ 25% of DLM8 cells were Fas-positive, treatment of cells with 1 and 3 μM of SNDX-275 increased this number to 40% and 98%, respectively. Cytotoxicity studies revealed that this concentration of SNDX-275 was not toxic in DLM8 cells (Fig. 1B).
Fig. 1. Treatment with SNDX-275 increases Fas expression on the surface of DLM8 cells and increases its sensitivity to FasL.
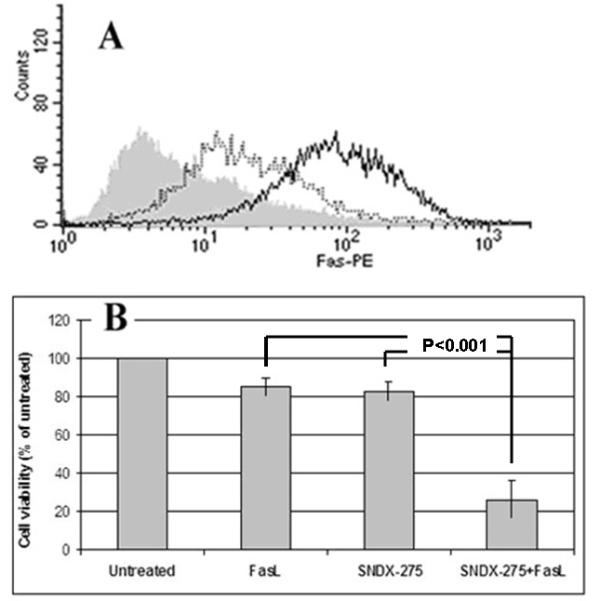
(A) murine DLM8 cells were treated with 3 μM SNDX-275 for 24 h (dashed line), or 48 h (solid line), or left untreated (grey shading). Fas expression on the cell surface was detected using flow cytometry. (B) DLM8 cells were pretreated with 3 μM SNDX-275 for 24 h and then treated with 10 ng/ml soluble FasL for another 24 h. Untreated cells and cells treated with either agent were used as controls. The cell viability was detected by MTT assay.
To examine the biological activity of overexpressed Fas, we pretreated DLM8 cells with the same dose of SNDX-275 for 48 h and then added 10 ng/ml FasL for another 24 h. The cytotoxic effect was determined by MTT assay. Combination of SNDX-275 with FasL showed an increased cytotoxic effect than either agent alone in DLM8 cells (P<0.001, Student’s t-test; Fig. 1B).
SNDX-275 Promotes Fas Expression in DLM8 Cells by Increasing Histone Acetylation of the Fas Gene Promoter and mRNA Expression
HDACi activate transcription by histone acetylation. To verify that SNDX-275 enhances histone acetylation in DLM8 cells we performed a ChIP assay. We examined the acetylation status of global histone 3 and on the fas promoter. The ChIP assay followed by the Western blot analysis of the protein lysate confirmed that overall histone 3 acetylation of chromatin protein was increased in DLM8 cells following treatment with SNDX-275 (Fig.2A).
Fig. 2. Treatment with SNDX-275 increases acetylation of global H3 histones and on the fas gene promoter in DLM8 cells.
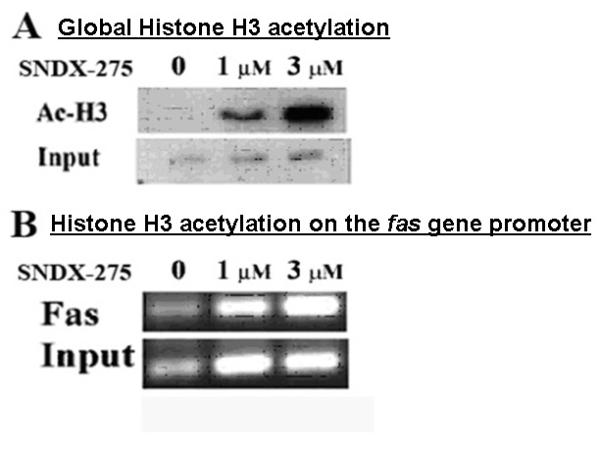
DLM8 cells were treated with 1 and 3 μM SNDX-275 for 24 h and processed using ChIP analysis for expression of (A) global acetylation of Histone H3 in DLM8 cells was analyzed by Western blot method and (B) acetylation of H3 histones on the fas promoter was determined by PCR assay.
ChIP assay followed by the PCR with a specific primers for fas promoter was also used to determine the level of fas gene promoter acetylation after treatment with SNDX-275 in these cells. As shown in Fig. 2B, acetylation of fas promoter did not significantly increase at a low SNDX-275 concentration (1μM) but increased after exposure to 3 μM SNDX-275. The following RT-PCR analysis demonstrated the elevation of mRNA expression in DLM8 cells after SNDX-275 treatment in dose and time-dependent manner (Fig. 3).
Fig. 3. Treatment with SNDX-275 stimulate expression of Fas mRNA.
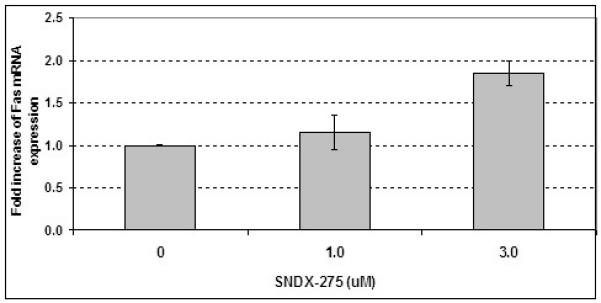
DLM8 cells were treated with 1 and 3 μM SNDX-275 for 24 h, total RNA was extracted and analyzed by quantitative RT-PCR.
Inactivation of FADD Molecule in DLM8 Cells Reverses Cytotoxic Effect of SNDX-275 and FasL Combination
To verify the significance of the Fas signaling in response to the SNDX-275 treatment for DLM8 cells in the environment that constitutively express FasL, we inhibited the pathway by stable transfection of cells with FDN plasmid (Fig. 4A ). This transformation completely reversed the combination effect of SNDX-275 and FasL treatment (Fig. 4B).
Fig. 4. Transfection of DLM8 cells with the FDN plasmid induces expression of the inactive (truncated) form of FADD and reverses SNDX-275 induced sensitization of DLM8 cells to FasL.
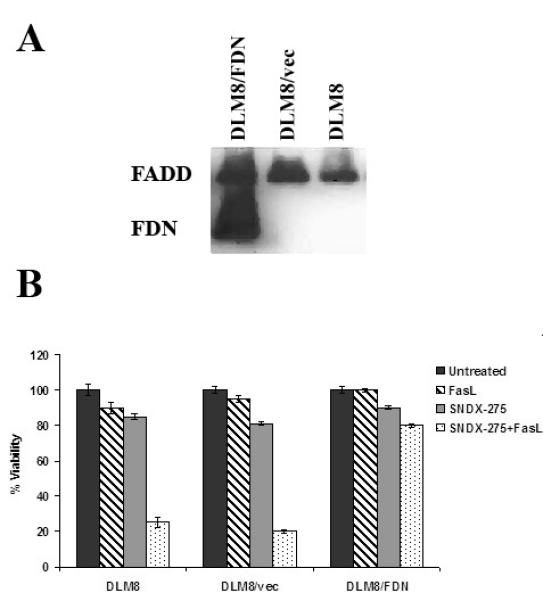
(A) DLM8 cells were transfected with FDN plasmid (DLM8/FDN cells) or an empty control vector (DLM8/vec). Cell lysates were processed using Western blot analysis for the expression of full-length FADD protein and its truncated form, FDN, using an anti-FADD antibody. (B) DLM8, DLM8/vec and DLM8/FDN cells were pretreated with 3 μM SNDX-275 for 48 h and then treated with 10 ng/ml soluble FasL for another 24 h. Untreated cells and cells treated with either agent were used as controls. The cells’ viability was detected by MTT assay.
SNDX-275 Stimulates fas Gene Transcription in LM7 Cells Without Changing the Expression of Fas Receptor on the Cell Surface and Sensitizes LM7 Cells to FasL Treatment
Real-time PCR analysis revealed that 2 μM SNDX-275 was able to increase expression of Fas mRNA in LM7 cells (Fig. 5). However, flow cytometry analysis did not reveal upregulation of the Fas receptor expression on the cell surface of LM7 cells after treatment with SNDX-275 alone (Fig. 6A). The combination effect of SNDX-275 and FasL observed in DLM8 cells, also occurred in LM7 cells (P<0.001 Student’s t-test, Fig. 6B).
Fig. 5. SNDX-275 increases expression of Fas mRNA in LM7 cells.
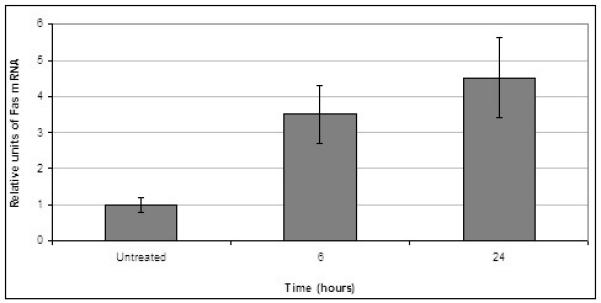
LM7 cells were treated with 2 μM SNDX-275 for 6 and 24 h, total RNA was extracted and analyzed by quantitative RT-PCR.
Fig. 6. Treatment with SNDX-275 increases the sensitivity of LM7 cells to FasL without increasing Fas receptor expression on the cell surface.
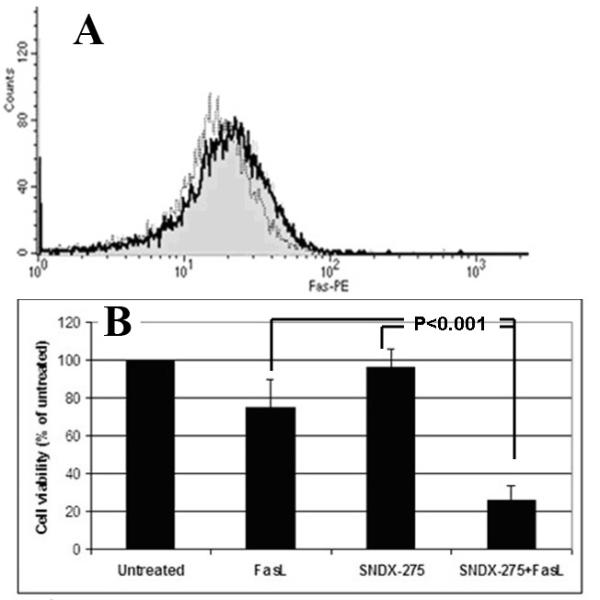
(A) LM7 cells were treated with 2̣ μM of SNDX-275 and Fas expression was detected at 48 h (dashed line), 72 hr (solid line). Untreated cells were used as control (grey shading). Fas expression on the cell surface was detected by flow cytometry. (B) LM7 cells were pretreated with 2 μM SNDX-275 for 48 h and then treated with 10 ng/ml soluble FasL for another 24 h. Untreated cells and cells treated with either agent were used as controls. The cell viability was detected by MTT assay.
SNDX-275 Activates Fas Signaling in LM7 Cells Via FADD
To determine whether the combined effect of SNDX-275 and FasL in LM7 cells can be modulated by downstream Fas signaling, we studied changes in mRNA expression for the most upstream Fas adapter molecule FADD. RT-PCR and Western blot analysis showed increased levels of FADD in the cytoplasm of these cells (Figs. 7 and 8).
Fig. 7. Treatment with SNDX-275 changes the pattern of mRNA expression of the molecules downstream from Fas signaling in favor of apoptosis in LM7 cells.
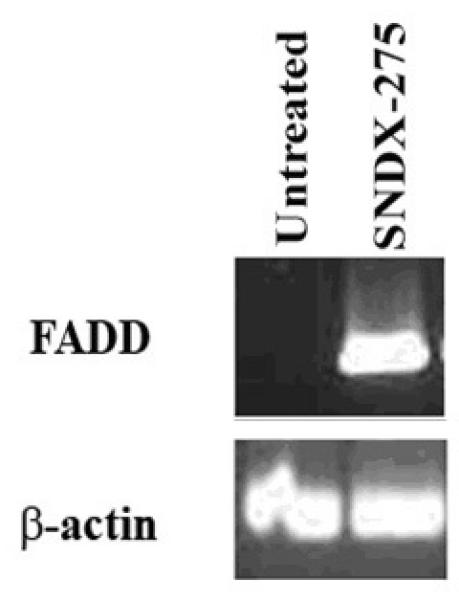
Cells were treated with 2̣ μM SNDX-275 for 24 h. mRNA was extracted from the cells and examined using RT-PCR assay to determine the levels of FADD expression.
Fig. 8. SNDX-275 increases expression of FADD protein in LM7 cells.
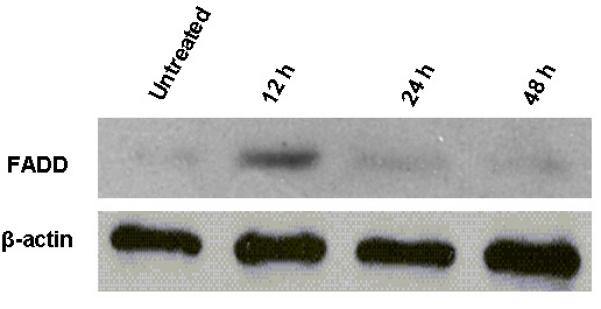
Cells were treated with 2̣ μM SNDX-275 for different periods of time. Total protein lysates were analyzed for FADD expression by Western blot using anti-FADD antibody.
To further verify the significance of FADD in activation of Fas signaling in LM7 cells in response to SNDX-275 treatment, we transfected them with FDN plasmid to inhibit FADD activity (Fig. 9A), as was done in DLM8 cells. Then LM7 cells stably transfected with FDN plasmid (LM7/FDN) cells, LM7 cells transfected with a control vector (LM7/Vec), and parental LM7 cells were treated with SNDX-275 and FasL either alone and in combination. In LM7 and LM7/vec cells combination of SNDX-275 and FasL showed significantly increased cytotoxic effect when compared with either agent alone, whereas this effect of combined treatment was inhibited in LM7/FDN cells (Fig. 9B).
Fig. 9. Transfection of LM7 cells with the FDN plasmid induces expression of the inactive (truncated) form of FADD and reverses SNDX-275 induced sensitization of LM7 cells to FasL.
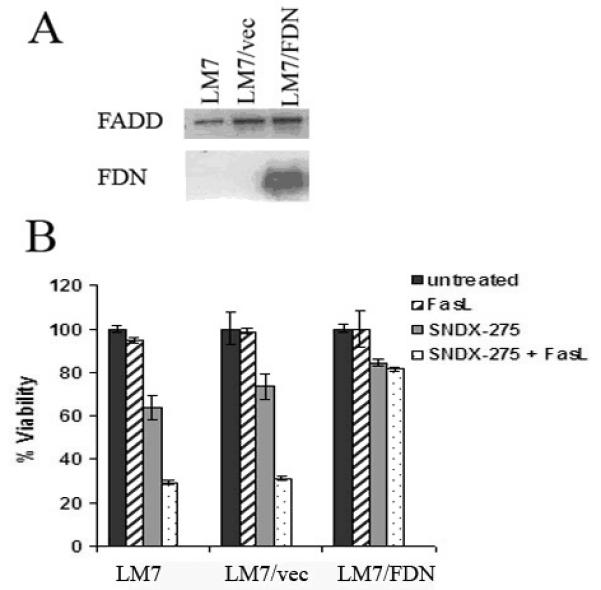
(A) LM7 cells were transfected with FDN plasmid (LM7/FDN cells) or an empty control vector (LM7/vec). Cell lysates were processed using Western blot analysis for the expression of full-length FADD protein and its truncated form, FDN, using an anti-human FADD antibody. (B) LM7, LM7/vec and LM7/FDN cells were pretreated with 2 μM SNDX-275 for 48 h and then treated with 10 ng/ml soluble FasL for another 24 h. Untreated cells and cells treated with either agent were used as controls. The cell viability was detected by MTT assay.
Intranasal Treatment of Established LM7 OS Lung Metastases in Mice with SNDX-275 Inhibits Metastatic Growth
To verify that treatment with SNDX-275 in vivo can affect OS metastases growth in lungs, which constitutively express FasL, we injected nu/nu mice with LM7 cells intravenously. On day 32 after the injection, pulmonary micrometastases formed (as confirmed using microscopic analysis), and we began administering SNDX-275 to the mice intranasally as described in Materials and Methods. Because solubility of the drug in water is low (0.016 mg/ml) and the volume that can be administered intranasally is limited to 0.02 ml, we were able to administer only 0.13 mg/kg of SNDX-275. Control mice received PBS intranasally on the same schedule. Mice receiving SNDX-275 treatment showed no evidence of pulmonary obstruction or other general morbidity signs, such as body weight loss or the lack of activity. Histopathological analysis did not reveal any difference between control group and the treated group. At the end of treatment, we evaluated the tumor burdens in the animals’ lungs and performed immunohistochemical analysis. The mean number of visible lung metastases was 56±43 in the control group and 18±12 in the treated group (p=0.04, Mann-Whitney test). During immunohistochemical examination we compared the lung metastases of medium size, as larger tumors have a higher background of apoptosis and necrosis. Immunohistochemical staining of the lungs did not show any significant difference in Fas expression in the LM7 metastases obtained from SNDX-275 treated mice and those obtained from control mice (Fig. 10A), but FADD expression levels were higher in metastases obtained from SNDX-275-treated mice than in those obtained from control mice (Fig. 10B). All metastatic lesions in mice from the SNDX-275 treated group were positive for FADD. However the pattern of staining within tumors was not even – some areas within the same tumors were positive for FADD and some not. The intensity of staining also varied from low (mouse #3) to high as shown in the lesion from mouse #2. Lesion in mouse #1 had an intermediate level of FADD expression. None of the metastases in the PBS treated group were positive for FADD.
Fig. 10. The effect of treatment with SNDX-275 on Fas and FADD expression in LM7 lung metastases in vivo.

Lungs were obtained from mice that received SNDX-275 or PBS treatment, and then fixed and stained with (A) anti-Fas and (B) anti-FADD antibodies followed with the secondary antibody as described in Materials and Methods. Representative tumor lesions in the lungs from different mice are shown. Brown staining indicates on the expression of Fas or FADD protein in tissue. Magnification 100x.
Discussion
Currently, there is sufficient evidence indicating that downregulation of Fas receptor may be associated with a poor prognosis and metastasis formation in patients with different types of cancer (20-22). In our previous studies with OS, we found that levels of Fas expression in OS lung metastases from patients and animals were increased following the treatment (4-6). This indicates that the fas gene is present in OS tumor cells, but its expression is inhibited in OS metastatic lesions and may be reactivated by chemotherapy. Therefore, detection of the mechanisms responsible for epigenetic silencing of the fas gene and identification of the agents capable of increasing its expression in tumor cells will have important therapeutic significance.
Several mechanisms are responsible for epigenetic gene-expression regulation. Our studies of the fas gene methylation status and the use of demethylation agents revealed that methylation mechanisms can not change fas gene expression in OS cells (23). Another mechanism that plays a key role in regulation of gene transcription is chromatin histone acetylation. Several studies have shown the ability of HDACi to stimulate fas gene transcription in different types of cancer cells (24-26). However, little is known about these inhibitors effect on the Fas pathway in OS. Imai and colleagues were the first to describe the ability of pan-HDACi FR901228 to induce OS tumor regression via Fas signaling by inducing expression of FasL in vitro (7). Watanabe et al. later described the ability of FR901228 to sensitize OS tumor cells to death receptor-mediated apoptosis by suppression of cFLIP expression (27). In the present study, we observed that transcription of the fas gene can be stimulated in OS cells by treatment with the HDACi SNDX-275. Treatment of DLM8 cells, which express low levels of Fas, with a subtoxic dose of SNDX-275 enhanced expression of the Fas mRNA and its protein on the tumor-cell surface. This elevation of Fas receptor expression increased the sensitivity of DLM8 cells to FasL treatment. ChIP analysis revealed an increased acetylation status of the fas gene promoter in these cells, indicating that chromatin histone acetylation is the mechanism responsible for upregulation of the fas gene expression in these cells. Because SNDX-275 works by inhibiting class I HDACs (8), inactivation of only these HDACs was sufficient for induction of Fas expression in OS cells and increased their sensitivity to FasL treatment.
Interestingly, in LM7 cells, which also have low levels of Fas receptor expression, treatment with SNDX-275 was followed by elevated levels of Fas, but it was not followed by the elevation of Fas receptor expression on their cell surface. Despite the fact that Fas receptor expression on tumor cell surface was not increased, treatment of LM7 cells with a combination of SNDX-275 and FasL showed an increased cytotoxic effect when compared with treatments by either agent alone. We believe that the lack of correlation between Fas mRNA and its protein expression on cell surface may be caused by some post-translational dysregulation. Alterations of miRNA levels, proteasome activity or receptor trafficking all of which regulate different post-translation events, by HDACi (28-33) may be responsible for this incongruity.
Despite the fact that Fas receptor expression on LM7 tumor cell surface was not increased, treatment of LM7 cells with a combination of SNDX-275 and FasL showed an increased cytotoxic effect when compared with treatments by either agent alone. This indicates that in LM7 cells the levels of Fas receptor molecules was sufficient to induce apoptosis, but there was a discrepancy in the expression of some downstream signaling molecule. RT-PCR analysis revealed that treatment of LM7 cells with SNDX-275 increased expression of FADD mRNA, which is the most upstream adaptor molecule to Fas receptor and is an essential component of the DISC formation (2). Inhibition of FADD activity in these cells reversed cytotoxic effect of the combination of SNDX-275 with FasL. Similarly, inhibition of the FADD activity in DLM8 cells reversed the cytotoxic effect of this combination.
Taken together, our in vitro data indicated that SNDX-275 can activate Fas signaling by stimulating transcription of the fas gene and increased expression of the Fas mRNA and protein. However, in some cells elevation of Fas mRNA was not followed by upregulation of Fas receptor expression on the cell surface and this did not interfere with their increased sensitivity to SNDX-275 and FasL combination treatment. FADD mRNA expression in these cells was elevated in response to SNDX-275 treatment and its inhibition reversed the cytotoxic effect of SMDX-275 and FasL combination. These findings indicate on an important role of FADD as a mediator of the Fas signaling in OS cells in response to SNDX-275 treatment.
To further confirm the feasibility of SNDX-275 in treatment of OS lung metastases, we treated mice with established OS lung metastases with this drug. To minimize systemic exposure of SNDX-275 and to verify the feasibility of topical drug delivery approach to the lungs via inhalation route, mice received SNDX-275 intranasally. Of note is that the intranasal dose we used in our study (0.1 mg/kg) was significantly lower than the oral dose (24.5 mg/kg) reported to have therapeutic potential in studies of different types of s.c. tumor xenograft models (9, 10). Intranasal administration of SNDX-275 for 5 weeks was well tolerated and significantly inhibited LM7 OS metastasis growth in the lungs, indicating that local administration of the drug to the lungs by inhalation facilitates delivery of a therapeutic dose. The discrepancy in the inhalation and systemic therapeutic doses could be explained by the specificity of FasL expression in the pulmonary environment and ability of SNDX-275 to activate Fas-induced apoptosis at the subtoxic doses as we determined here in vitro. Indeed, immunohistochemical analysis of LM7 OS metastases obtained from mice showed that Fas expression did not change significantly after SNDX-275 treatment but levels of FADD expression in these mice were higher than in the control group. These in vivo findings correlate with our in vitro data and indicate that activation of the downstream FADD molecule in the Fas signaling may be important for the therapeutic effect of this agent. We realize that intranasal administration can not be used in humans and does not allow equal distribution of SNDX-275 in the lungs and deep penetration of the agent in the respiratory zone, where lung metastases frequently form. In addition, low solubility of the drug in water did now allow us to use higher doses, which could provide stronger antitumor effect. Therefore, in our future studies we will develop liposome aerosol formulation for this agent, which will allow to bypass the limitations of intranasal administration and poor water-solubility, and will provide topical non-invasive method of drug delivery to the lungs. We also plan to investigate whether this agent is capable of inducing other mechanisms besides Fas, since it is recognized that these agents can target multiple mechanisms in cancer cells. Investigation of this drug effect on normal lung tissues also warrants further studies.
In summary, we identified that inhibition of HDACs by SNDX-275 at subtoxic doses is sufficient to activate the Fas pathway in OS cells and trigger apoptosis induction in the FasL-expressing environment. Topical intranasal administration of SNDX-275 to the lungs of mice with established OS pulmonary metastases inhibited metastatic growth and was followed by changes in FADD expression downstream of the Fas receptor indicating that the Fas pathway may play an important role in the response to this treatment. Currently, SNDX-275 is in clinical trials in adults and our studies indicate that its application as an aerosol therapy should be considered for the treatment of pediatric OS lung metastases as well.
Acknowledgements
We would like to thank the American Legion Auxiliary Organization for the Fellowship Award to Mrs. Krithi Rao-Bindal. We would also like to thank Mrs. Barbara Liddle and Kristine Ash for their assistance in the preparation of this manuscript.
Grant support: National Cancer Institute grant R01 CA42992 (Kleinerman); Legends of Friendswood Award, Amschwand Sarcoma Cancer Foundation and in part by Liddy’s Shriver Sarcoma Initiative (Koshkina).
References
- 1.Anderson P, Kopp L, Anderson N, et al. Novel bone cancer drugs: investigational agents and control paradigms for primary bone sarcomas (Ewing’s sarcoma and osteosarcoma) Expert Opin Investig Drugs. 2008;17(11):1703–15. doi: 10.1517/13543784.17.11.1703. [DOI] [PubMed] [Google Scholar]
- 2.Sartorius U, Schmitz I, Krammer PH. Molecular mechanisms of death-receptor-mediated apoptosis. Chembiochem. 2001;2(1):20–9. doi: 10.1002/1439-7633(20010105)2:1<20::AID-CBIC20>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 3.Koshkina NV, Khanna C, Mendoza A, Guan H, DeLauter L, Kleinerman ES. Fas-negative osteosarcoma tumor cells are selected during metastasis to the lungs: the role of the Fas pathway in the metastatic process of osteosarcoma. Mol Cancer Res. 2007;5(10):991–9. doi: 10.1158/1541-7786.MCR-07-0007. [DOI] [PubMed] [Google Scholar]
- 4.Gordon N, Arndt CA, Hawkins DS, et al. Fas expression in lung metastasis from osteosarcoma patients. J Pediatr Hematol Oncol. 2005;27(11):611–5. doi: 10.1097/01.mph.0000188112.42576.df. [DOI] [PubMed] [Google Scholar]
- 5.Koshkina NV, Kleinerman ES. Aerosol gemcitabine inhibits the growth of primary osteosarcoma and osteosarcoma lung metastases. Int J Cancer. 2005;116(3):458–63. doi: 10.1002/ijc.21011. [DOI] [PubMed] [Google Scholar]
- 6.Gordon N, Koshkina NV, Jia SF, et al. Corruption of the Fas pathway delays the pulmonary clearance of murine osteosarcoma cells, enhances their metastatic potential, and reduces the effect of aerosol gemcitabine. Clin Cancer Res. 2007;13(15 Pt 1):4503–10. doi: 10.1158/1078-0432.CCR-07-0313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Imai T, Adachi S, Nishijo K, et al. FR901228 induces tumor regression associated with induction of Fas ligand and activation of Fas signaling in human osteosarcoma cells. Oncogene. 2003;22(58):9231–42. doi: 10.1038/sj.onc.1207184. [DOI] [PubMed] [Google Scholar]
- 8.Khan N, Jeffers M, Kumar S, et al. Determination of the class and isoform selectivity of small-molecule histone deacetylase inhibitors. Biochem J. 2008;409(2):581–9. doi: 10.1042/BJ20070779. [DOI] [PubMed] [Google Scholar]
- 9.Saito A, Yamashita T, Mariko Y, et al. A synthetic inhibitor of histone deacetylase, MS-27-275, with marked in vivo antitumor activity against human tumors. Proc Natl Acad Sci U S A. 1999;96(8):4592–7. doi: 10.1073/pnas.96.8.4592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jaboin J, Wild J, Hamidi H, et al. MS-27-275, an inhibitor of histone deacetylase, has marked in vitro and in vivo antitumor activity against pediatric solid tumors. Cancer Res. 2002;62(21):6108–15. [PubMed] [Google Scholar]
- 11.Bracker TU, Sommer A, Fichtner I, Faus H, Haendler B, Hess-Stumpp H. Efficacy of MS-275, a selective inhibitor of class I histone deacetylases, in human colon cancer models. Int J Oncol. 2009;35(4):909–20. doi: 10.3892/ijo_00000406. [DOI] [PubMed] [Google Scholar]
- 12.Khandelwal A, Gediya L, Njar V. MS-275 synergistically enhances the growth inhibitory effects of RAMBA VN/66-1 in hormone-insensitive PC-3 prostate cancer cells and tumours. Br J Cancer. 2008;98(7):1234–43. doi: 10.1038/sj.bjc.6604295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jia SF, Worth LL, Kleinerman ES. A nude mouse model of human osteosarcoma lung metastases for evaluating new therapeutic strategies. Clin Exp Metastasis. 1999;17(6):501–6. doi: 10.1023/a:1006623001465. [DOI] [PubMed] [Google Scholar]
- 14.Asai T, Ueda T, Itoh K, et al. Establishment and characterization of a murine osteosarcoma cell line (LM8) with high metastatic potential to the lung. Int J Cancer. 1998;76(3):418–22. doi: 10.1002/(sici)1097-0215(19980504)76:3<418::aid-ijc21>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- 15.Koshkina NV, Kleinerman ES, Waidrep C, et al. 9-Nitrocamptothecin liposome aerosol treatment of melanoma and osteosarcoma lung metastases in mice. Clin Cancer Res. 2000;6(7):2876–80. [PubMed] [Google Scholar]
- 16.Koshkina NV, Waldrep JC, Roberts LE, Golunski E, Melton S, Knight V. Paclitaxel liposome aerosol treatment induces inhibition of pulmonary metastases in murine renal carcinoma model. Clin Cancer Res. 2001;7(10):3258–62. [PubMed] [Google Scholar]
- 17.Verschraegen CF, Gilbert BE, Loyer E, et al. Clinical evaluation of the delivery and safety of aerosolized liposomal 9-nitro-20(s)-camptothecin in patients with advanced pulmonary malignancies. Clin Cancer Res. 2004;10(7):2319–26. doi: 10.1158/1078-0432.ccr-0929-3. [DOI] [PubMed] [Google Scholar]
- 18.Guseva NV, Taghiyev AF, Rokhlin OW, Cohen MB. Contribution of death receptor and mitochondrial pathways to Fas-mediated apoptosis in the prostatic carcinoma cell line PC3. Prostate. 2002;51(4):231–40. doi: 10.1002/pros.10095. [DOI] [PubMed] [Google Scholar]
- 19.Zhang J, Winoto A. A mouse Fas-associated protein with homology to the human Mort1/FADD protein is essential for Fas-induced apoptosis. Mol Cell Biol. 1996;16(6):2756–63. doi: 10.1128/mcb.16.6.2756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bebenek M, Dus D, Kozlak J. Fas/Fas-ligand expressions in primary breast cancer are significant predictors of its skeletal spread. Anticancer Res. 2007;27(1A):215–8. [PubMed] [Google Scholar]
- 21.Onodera H, Mori A, Nagayama S, et al. Fas/CD95 signaling rather than angiogenesis or proliferative activity is a useful prognostic factor in patients with resected liver metastases from colorectal cancer. Int J Colorectal Dis. 2005;20(6):477–84. doi: 10.1007/s00384-004-0708-z. [DOI] [PubMed] [Google Scholar]
- 22.Asensio C, Zapata A, Garcia-Ahijado J, Gil B, Salvadores P, Schneider J. Fas expression is associated with a better prognosis in laryngeal squamous cell carcinoma. Anticancer Res. 2007;27(6B):4083–6. [PubMed] [Google Scholar]
- 23.Huang G, Koshkina NV, Kleinerman ES. Fas expression in metastatic osteosarcoma cells is not regulated by CpG island methylation. Oncol Res. 2009;18(1):31–9. doi: 10.3727/096504009789745638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Escaffit F, Vaute O, Chevillard-Briet M, et al. Cleavage and cytoplasmic relocalization of histone deacetylase 3 are important for apoptosis progression. Mol Cell Biol. 2007;27(2):554–67. doi: 10.1128/MCB.00869-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kim HR, Kim EJ, Yang SH, et al. Trichostatin A induces apoptosis in lung cancer cells via simultaneous activation of the death receptor-mediated and mitochondrial pathway? Exp Mol Med. 2006;38(6):616–24. doi: 10.1038/emm.2006.73. [DOI] [PubMed] [Google Scholar]
- 26.Wood TE, Dalili S, Simpson CD, et al. Selective inhibition of histone deacetylases sensitizes malignant cells to death receptor ligands. Mol Cancer Ther. 9(1):246–56. doi: 10.1158/1535-7163.MCT-09-0495. [DOI] [PubMed] [Google Scholar]
- 27.Watanabe K, Okamoto K, Yonehara S. Sensitization of osteosarcoma cells to death receptor-mediated apoptosis by HDAC inhibitors through downregulation of cellular FLIP. Cell Death Differ. 2005;12(1):10–8. doi: 10.1038/sj.cdd.4401507. [DOI] [PubMed] [Google Scholar]
- 28.Zhou Q, Agoston AT, Atadja P, Nelson WG, Davidson NE. Inhibition of histone deacetylases promotes ubiquitin-dependent proteasomal degradation of DNA methyltransferase 1 in human breast cancer cells. Mol Cancer Res. 2008;6(5):873–83. doi: 10.1158/1541-7786.MCR-07-0330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Scott GK, Mattie MD, Berger CE, Benz SC, Benz CC. Rapid alteration of microRNA levels by histone deacetylase inhibition. Cancer Res. 2006;66(3):1277–81. doi: 10.1158/0008-5472.CAN-05-3632. [DOI] [PubMed] [Google Scholar]
- 30.Bandres E, Agirre X, Bitarte N, et al. Epigenetic regulation of microRNA expression in colorectal cancer. Int J Cancer. 2009;125(11):2737–43. doi: 10.1002/ijc.24638. [DOI] [PubMed] [Google Scholar]
- 31.Kong X, Lin Z, Liang D, Fath D, Sang N, Caro J. Histone deacetylase inhibitors induce VHL and ubiquitin-independent proteasomal degradation of hypoxia-inducible factor 1alpha. Mol Cell Biol. 2006;26(6):2019–28. doi: 10.1128/MCB.26.6.2019-2028.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jung ID, Lee JS, Jeong YI, et al. Apicidin, the histone deacetylase inhibitor, suppresses Th1 polarization of murine bone marrow-derived dendritic cells. Int J Immunopathol Pharmacol. 2009;22(2):501–15. doi: 10.1177/039463200902200227. [DOI] [PubMed] [Google Scholar]
- 33.Watanabe M, Adachi S, Matsubara H, et al. Induction of autophagy in malignant rhabdoid tumor cells by the histone deacetylase inhibitor FK228 through AIF translocation. Int J Cancer. 2009;124(1):55–67. doi: 10.1002/ijc.23897. [DOI] [PubMed] [Google Scholar]