

Abstract
The parasitic protozoa Leishmania major produces a peroxidase (L. major peroxidase; LmP) that exhibits activities characteristic of both yeast cytochrome c peroxidase (CCP) and plant cytosolic ascorbate peroxidase (APX). One common feature is a key Trp residue, Trp208 in LmP and Trp191 in CCP, that is situated adjacent to the proximal His heme ligand in CCP, APX, and LmP. In CCP, Trp191 forms a stable cationic radical after reaction with H2O2 to form Compound I; in APX, the radical is located on the porphyrin ring. In order to clarify the role of Trp208 in LmP and to further probe peroxidase structure-function relationships, we have determined the crystal structure of LmP and have studied the role of Trp208 using electron paramagnetic resonance spectroscopy (EPR), mutagenesis, and enzyme kinetics. Both CCP and LmP have an extended section of β structure near Trp191 and Trp208, respectively, which is absent in APX. This region provides stability to the Trp191 radical in CCP. EPR of LmP Compound I exhibits an intense and stable signal similar to CCP Compound I. In the LmP W208F mutant, this signal disappears, indicating that Trp208 forms a stable cationic radical. In LmP conversion of the Cys197 to Thr significantly weakens the Compound I EPR signal and dramatically lowers enzyme activity. These results further support the view that modulation of the local electrostatic environment controls the stability of the Trp radical in peroxidases. Our results also suggest that the biological role of LmP is to function as a cytochrome c peroxidase.
Keywords: Crystal Structure, Enzyme Mechanisms, Enzyme Structure, Peroxidase, Spectroscopy
Introduction
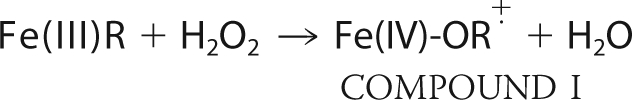
Heme peroxidases are found in many organisms and utilize peroxides to oxidize a variety of physiologically important molecules (1). Because these enzymes are robust and form spectroscopically visible and stable intermediates, heme peroxidases have proven to be a model system to probe structure-function relationships in heme proteins. The general heme peroxidase mechanism is outlined below.
 |
 |
Upon the addition of an equimolar amount of H2O2, the enzyme forms the stable intermediate termed Compound I. H2O2 is heterolytically cleaved, releasing a water molecule while the second peroxide-derived oxygen atom remains coordinated to the heme iron. The iron-linked oxygen atom with only six valence electrons oxidizes the heme iron to Fe(IV) and a nearby organic moiety to a cationic radical. The enzyme returns to the resting state upon two successive electron transfer events from two individual reducing substrate molecules (S in the above scheme), forming the second water that remains in the active site of the resting enzyme. For most heme peroxidases, R is the porphyrin ring. In the case of cytochrome c peroxidase (CCP),2 however, R is an amino acid residue, Trp191, located in the proximal pocket directly adjacent to the proximal His heme ligand (2). CCP is also the only well characterized peroxidase that utilizes another protein, cytochrome c, as a reducing substrate.
These unique mechanistic features of CCP have prompted many investigations probing the relationship between structure and function in heme peroxidases. Structurally, all heme peroxidases have a highly conserved 10-α-helical bundle with sequence identities ranging from ∼16 to 50% (3) and very similar active site architectures. The initial crystal structures and sequence alignments provided reasonable explanations for why CCP forms a Trp radical and not a porphyrin radical. A large majority of heme peroxidases have a Phe at the analogous position of Trp191 in CCP, and because the indole side chain is easier to oxidize than the phenyl side chain, it was postulated that the CCP Trp is preferentially oxidized. This hypothesis was disproved after electron paramagnetic resonance (EPR) experiments illustrated that cytosolic pea ascorbate peroxidase (APX) forms a heme porphyrin radical despite APX maintaining proximal Trp (4).
These unexpected results with APX led to numerous studies that clearly illustrated that stabilization of the Trp191 cation radical is dependent upon the surrounding electrostatic environment (5–10). One obvious difference between CCP and APX is the K+ ion (Ca2+ in other peroxidases) in APX that is located about 8.5 Å from the proximal Trp and is, therefore, in position to electrostatically destabilize the positive charge on the cationic Trp radical. CCP lacks the K+ and instead binds a water molecule at this location. Our group has shown that engineering the APX K+ site into CCP destabilizes the Trp191 radical in a cationic concentration-dependent manner and decreases activity to about ∼0.02% of wild type levels (5–7). Other studies illustrated that the electronegative amino acids unique to CCP, Met230 and Met231, near Trp191 are also important for stabilization of the Trp radical (5, 11, 12).
A recently characterized peroxidase from Leishmania major, LmP, was reported to be a naturally occurring hybrid, a putative “missing link” of heme peroxidases, because it exhibits sequence homology and enzyme activities characteristic of both APX and CCP (13). This peroxidase has been designated LmAPX due to its initial designation as an ascorbate peroxidase, but as will become evident further on, we will refer to the L. major peroxidase as LmP. LmP conserves not only the proximal Trp (Trp208) but also the two Met (positions 248 and 249) residues known to help stabilize the Trp191 radical in CCP (Met230 and Met231), leading us to hypothesize that LmP might be the first well characterized peroxidase to mechanistically mimic CCP (13, 14). We therefore have solved the crystal structure of LmP and employed EPR spectroscopy, enzyme kinetics, and mutagenesis to determine the location of the cationic radical in LmP Compound I.
EXPERIMENTAL PROCEDURES
Cloning and Site-directed Mutagenesis
LmP was expressed without the hydrophobic N-terminal tail, as Δ34LmP. The construct was generously supplied by Dr. Subrata Adak (13). Δ34LmP was inserted into the pPAL7 (Bio-Rad) vector using the Infusion® Dry-Down PCR cloning kit. The primers were designed to conserve the SpeI and NotI restriction sites in the pPAL7 vector and therefore code for two extra amino acids on the N terminus of Δ34LmP, Thr and Ser. The oligonucleotides were synthesized by Operon®: forward primer (SpeI), 5′-G CTC TTC AAA GCT TTG ACT AGT GAG GAG CCG CCG TTC-3′ and reverse primer (NotI), 5′ CGG GCT TAT GCG GCC GCT TAG CTC TCC GAA GCG-3′. Successful insertion was first verified with double digests using SpeI and NotI, followed by sequencing. All mutants in this paper were prepared using the Stratagene® Lightning site-directed mutagenesis kit; and all mutagenic oligonucleotides, forward and reverse complement, were also obtained through Operon®. The forward mutagenic primers for Δ34LmP C197T and Δ34LmP W208F were 5′-GCG CAC ACA TGC GGT GAG ACC CAC ATC GAA TTC TCC GGC-3′ and 5′-GGC TAC CAT GGG CCG TTC ACA CAC GAC AAG AAC-3. CCPK1 is a CCP mutant that has been engineered to bind K+ at the same location as the K+ site in APX and LmP (7). CCPK1/T180C was prepared in the same manner, using the CCPK1 mutagenic construct from our earlier studies (7). The forward mutagenic oligonucleotide was 5′-GGG GCT CAC ACC CTG GGC AAG TGC CAC TTG AAG AAC TCT GGA TAC G-3′. Mutagenesis was confirmed with sequencing.
Protein Expression and Purification
The pPAL7/LmP plasmid was transformed into Escherichia coli BL21(DE3) cells and plated onto minimal media agar with ampicillin (100 μg/ml). One colony was picked and grown in LB containing 0.5% glucose, to repress leaky expression, and ampicillin (100 μg/ml) overnight at 37 °C and 220 rpm agitation. Expression cultures of TB media with ampicillin (100 μg/ml) were inoculated with the overnight culture (1:100). The cells were grown at 37 °C with agitation of 220 rpm in a New Brunswick® Scientific C25KC incubator. When the culture reached an A600 of 0.8, the temperature was dropped to 25 °C, and agitation was decreased to 100 rpm. Induction with isopropyl 1-thio-β-d-galactopyranoside was found to be unnecessary because it caused a greater level of insoluble protein. The cells were harvested by centrifugation after 18 h of growth and stored at −80 °C. The cells were thawed and resuspended by stirring for 1 h at 4 °C in 50 mm sodium phosphate, pH 7.0, 10% glycerol, 150 mm sodium acetate, pH 6.0, 0.5 mm PMSF, 2 mm l-ascorbic acid, 25 μm magnesium acetate, 100 μg/ml lysozyme, 0.2 μg/ml DNase, and 0.2 μg/ml leupeptin. Sonication on ice for 2 min ensured efficient lysis. The soluble fraction was isolated by centrifugation at 37,500 × g, 4 °C for 1 h; the lysate was loaded onto a 10-ml Profinity eXact (Bio-Rad) gravity column that had been pre-equilibrated with 100 mm sodium phosphate, pH 7.0, 10% glycerol at a rate of 2 ml/min at 4 °C. The column then was washed with 10 column volumes of 100 mm sodium phosphate, pH 7.0, 10% glycerol, followed by 10 column volumes of 300 mm sodium phosphate, pH 7.0, 10% glycerol. The column was incubated overnight at 4 °C with 1 column volume of 100 mm sodium phosphate, pH 7.0, 10% glycerol, 100 mm sodium fluoride. The protein was eluted with the same buffer the following day at room temperature. The eluted protein was then concentrated in a 10,000 molecular weight cut-off Amicon® concentrator at 4 °C and then loaded onto a Superdex 75 16/60 column that had been pre-equilibrated with 50 mm potassium phosphate, 5% glycerol at a flow rate of 0.3 ml/min at 4 °C. The resulting chromatogram exhibited three peaks. Active LmP elutes in the second peak as determined by the spectroscopic formation of Compound I upon equimolar addition of hydrogen peroxide. Sample homogeneity was determined by SDS-PAGE. Fractions with an Rz (A408/A280) of ≥1.2 were pooled and concentrated to at least 30 mg/ml. The C197T and W208F mutants were expressed and purified in the same manner. The protein concentration of LmP was calculated with the molar extinction coefficient, ϵ280 = 74.3 mm−1 cm−1, determined using the Pierce® modified Lowry protein assay kit. The same kit was used to determine the molar extinction coefficients of the mutants, W208F ϵ280 = 70.4 mm−1 cm−1 and C197T ϵ280 = 69.2 mm−1 cm−1. The heme content of LmP and both mutants was between 95.6 and 99.7%. A slightly modified hemochromagen assay (15), 20% pyridine and 0.2 m NaOH, was used to determine heme content and these Soret molar extinction coefficients: LmP ϵ408 = 113.6 mm−1 cm−1, C197T ϵ408 = 104.3 mm−1 cm−1 and W208F ϵ410 = 108 mm−1 cm−1. Purified protein was flash frozen in liquid nitrogen and stored at −80 °C.
CCP, CCPK1, and CCPK1/T180C were expressed in E. coli BL21(DE3) cells and purified as described previously by Fishel et al. (12) and Meharenna et al. (39). However, after the heme incorporation, both the CCP and CCPK1 were eluted from the DEAE column using a gradient of 20–500 mm potassium phosphate pH 6, whereas the CCPK1/T180C was eluted with 500 mm potassium phosphate, pH 6.0.
Steady-state Activity Assays
Spectrophotometric steady-state activity assays using both reduced horse heart cytochrome c and l-ascorbic acid as substrates were performed at room temperature using a Cary 3E UV-visible spectrophotometer. The assays were carried out in 50 mm potassium phosphate buffer, which had been filtered with a 0.2-μm sterile filter unit. The pH for CCP and the CCP mutants was 6.0, whereas the pH for LmP and its mutants was 7.0. Attempts to dialyze wild type and mutant LmP against a pH 6.0 buffer resulted in protein precipitating out of solution. Horse heart cytochrome c (Sigma) was dissolved in 50 mm potassium phosphate, pH 7.0, 10 mm sodium dithionite and incubated on ice for 30 min. Dithionite was removed by gel filtration over a disposable PD-10 column pre-equilibrated with 50 mm potassium phosphate, pH 7.0. The concentration of reduced horse heart cytochrome c was determined using the molar extinction coefficient, ϵ550 = 27.6 mm−1 cm−1, and the steady-state oxidation of reduced horse heart cytochrome c was calculated using Δϵ550 = 19.6 mm−1 cm−1 (16–18). The final reaction conditions were as follows: LmP, 29 pm; CCP, 38 pm; C197T, 47 nm; W208F, 79 nm; CCPK1, 47 nm; and CCPK1/T180C, 39 nm with 40 μm reduced horse heart cytochrome and 0.180 mm hydrogen peroxide that had been standardized as described by Fowler and Bright (19). The reaction was initiated by the addition of hydrogen peroxide and monitored for 30 s. l-Ascorbic acid was dissolved in water, and the concentration was determined using ϵ265 = 14.5 mm−1 cm−1 (20). Final reaction conditions for ascorbate oxidation activity assays were as follows: LmP, 585 nm; CCP, 766 nm; C197T, 621 nm; W208F, 600 nm; CCPK1, 627 nm; and CCPK1/T180C, 521 nm with 0.1 mm ascorbate and 0.3 mm hydrogen peroxide. The addition of hydrogen peroxide initiated the reactions, which were monitored at 290 nm (ϵ290 = 2.8 mm−1cm−1) over 1 min (21, 22). All LmP enzyme concentrations were determined using the Soret molar extinction coefficients reported in this paper, and the molar extinction coefficient used for CCP was ϵ408 = 93 mm−1 cm−1, and that used for CCPK1 and CCPK1/T180C was ϵ408 = 96 mm−1 cm−1 (7, 17).
EPR Spectroscopy
CCP, CCPK1, and CCPK1/T180C were dialyzed against 50 mm potassium phosphate, pH 6.0, and LmP C197T and W208F were dialyzed against 50 mm potassium phosphate, pH 7.0, which had been filtered sterile, at 4 °C, overnight. Enzyme concentrations were determined using the Soret molar extinction coefficients. Compound I EPR samples were prepared by combining equal volumes of 0.3 mm enzyme with 0.36 mm of hydrogen peroxide to total 0.3 ml. The sample was immediately transferred to an EPR tube and flash frozen in liquid nitrogen; this process took no more than 3 min for each sample. After the initial EPR spectra were collected at 7 K, each sample was concurrently removed from a cryo-Dewar and allowed to sit at room temperature for 30 min. The samples were simultaneously refrozen in liquid nitrogen and again subjected to EPR at 7 K. All spectra were standardized against a 1,1 diphenyl-2-picrylhydrazyl, g = 2.0036 (23, 24). EPR settings were as follows: microwave frequency, 9.387 GHz; modulation amplitude, 0.40 G; modulation frequency, 100 kHz; field sweep rate, 23.84 G/s; microwave power, 0.638 milliwatt; receiver gain, 1.0 × 104; and time constant, 5.120 ms.
Crystallization of LmP and C197T
The precrystallization test (Hampton) was used to determine the most promising range of protein concentrations, 5–7 mg/ml, for the initial screenings of LmP. Various commercial crystallization kits were used to set up hanging drop vapor diffusion trays with a high throughput nanoliter dispensing robot, the Mosquito®. LmP crystallized in a variety of conditions, the precipitant usually being PEG 3350. All crystallization reagents were filtered sterile (0.2 μm). Diffraction quality crystals were grown overnight using sitting drop vapor diffusion trays at room temperature. An equal volume, usually 2 μl, of 6 mg/ml LmP in 50 mm potassium phosphate, pH 7.0, 5% glycerol was combined with the crystallization solution, 0.2 m potassium chloride, 16% PEG 3350, and 2.5–5% glycerol.
In an attempt to obtain the ascorbate-bound structure, we soaked these crystals in cryo (well solution plus 20% glycerol) containing 0.5 and 1 mm ascorbic acid. The crystals, however, began to melt immediately upon introduction of this substrate. Nevertheless, LmP co-crystallized in the presence of ascorbic acid; 2 μl of 9 mg/ml LmP was combined with an equal volume of 0.1 m HEPES, pH 7.4, 3.0% tacsimate, pH 7.0, 5 mm l-ascorbic acid, 20% PEG 3350, and 5–12.5% glycerol. These crystals were grown using sitting drop vapor diffusion at 4 °C and would not grow without l-ascorbic acid. C197T crystals were obtained by using hanging drop trays and combining an equal volume of 7 mg/ml C197T with the reservoir solution of 0.1–0.12 m potassium chloride, 7.5–11.5 mm calcium chloride, 12% glycerol, and 18% PEG 3350. All cryoprotectants were identical to the specific crystallization solutions and included a final concentration of 20% glycerol.
Structural Determination and Refinement
LmP crystals diffracted to 1.76 Å resolution and belong to space group P212121 with two molecules per asymmetric unit. Data for LmP were collected at SSRL beamline 7-1. The LmP structure was solved by combining phase information from molecular replacement and single wavelength anomalous dispersion (SAD) at the iron absorption edge. The SAD data were collected at 1.7 Å x-ray wavelength using an inverse beam protocol. High resolution data were also collected with a different crystal at 0.97 Å wavelength in two scans; in the low resolution scan, 100 frames were collected with 5-s exposure and 1° oscillation per frame, and in the high resolution scan, 200 frames were collected with 25-s exposure and 0.5° oscillation per frame. The data for each scan were indexed and integrated separately and scaled together using HKL-2000 software (25). SAD data were also processed using HKL-2000. Molecular replacement calculations were carried out with Phaser (26) through the CCP4i graphic interface (27) using cytochrome c peroxidase (Protein Data Bank entry 2CYP) as a search model. Phaser was also used for the SAD phasing calculation. Phases were combined using SigmaA followed by density modification using DM in the CCP4 suite. The resulting electron density was in excellent quality. The structure was then inspected and some surface regions manually rebuilt using COOT (28, 29). Refinement was completed with REFMAC5 (30).
LmP crystals grown in the presence of ascorbate diffracted to 1.98 Å and were also in the P212121 space group with two molecules per asymmetric unit. Data were indexed, integrated, and scaled in the aforementioned manner; molecular replacement was done with Phaser using substrate-free LmP as the search model. The resulting maps showed no electron density for ascorbate. C197T crystals diffracted to 2.38 Å and were also in the P212121 space group. Two data sets from two separate crystals were collected at SSRL beamline 7-1; the first data set was 200 frames exposed for 25 s/frame, and the second data set was 200 frames exposed for 12 s/frame, both with 0.5° oscillations. Data were indexed and integrated separately and scaled together with HKL-2000. For the initial density calculation, all cations were removed from the LmP model, and Cys197 was mutated into Ala to prevent any bias. After three rounds of refinements, the density continued to clearly exhibit the conserved cation binding sites (density visible up to 13 σ in Fo − Fc maps) and the Thr density at position 197. Crystallographic data collection and refinement statistics are provided in Table 1, where all Rfree values are calculated with 5% of the data excluded from refinements.
TABLE 1.
Crystallographic data collection and refinement statistics
| Data set | LmP native SAD | LmP native HiRes (PDB code 3RIV) | LmP C197T (PDB code 3RIW) |
|---|---|---|---|
| Space group | P212121 | P212121 | P212121 |
| Unit cell dimensions (a, b, c) (Å) | 46.535, 77.048, 155.641 | 46.600, 77.101, 155.734 | 45.819, 77.766, 160.580 |
| Resolution range (Å) | 50–2.58 | 50–1.76 | 50–2.38 |
| Radiation source | SSRL 7-1 | SSRL 7-1 | SSRL 7-1 |
| Wavelength (Å) | 1.7 | 0.97 | 1.09 |
| Total observations | 141,780 | 278,239 | 186,582 |
| Unique reflections (highest shell) | 33,860 (1,622) | 56,061 (2,779) | 23,951 (1,191) |
| Completeness (%) (highest shell) | 99.9 (97.7) | 99.9 (100) | 100 (99.9) |
| Rsym (highest shell) | 0.06 (0.18) | 0.074 (0.399) | 0.097 (0.584) |
| 〈I/σ〉 (highest shell) | 25.05 (6.01) | 33.22 (2.82) | 17.15 (3.08) |
| Wilson B factor | 27.1 | 35.5 | |
| Reflections used in refinement | 53,055 | 22,558 | |
| Resolution range (Å) in refinement | 38.5–1.76 | 35.5–2.38 | |
| Rwork | 0.161 | 0.181 | |
| Rfree | 0.209 | 0.242 |
RESULTS
Crystal Structure of LmP and C197T
The structure of LmP has been solved to 1.76 Å resolution and exhibits the characteristic peroxidase 10 α-helical bundle fold (Fig. 1). The triple β-strand unique to CCP is present in LmP. This structural element is not present in APX. The electron density of LmP shows two cations, a potassium that is visible up to 20 σ in the Fo − Fc difference map and a calcium visible up to 26 σ in the same map (Fig. 2). Given the coordinating amino acids (Table 2) and the electron density, the distal cation is probably a Ca2+, whereas the proximal cation is probably a K+. These cations are conserved among the majority of heme peroxidases except CCP, which has no cations, and APX, which conserves only the proximal Na+/K+. The presence or absence of the K+ ion is critically important for stabilization of the Trp191 cationic radical in CCP Compound I. The mechanism of stabilization is electrostatic. When Trp191 is converted to a Gly, thus creating a proximal pocket cavity, the cavity can be filled with small molecules with a strong preference for positively charged imidazoles (8) and monovalent cations (9). In addition, computational studies indicate that the Trp191 local environment is designed to stabilize a positive charge (10). Our laboratory has engineered the APX K+ binding site into CCP to give the mutant termed CCPK1. The presence of this positive charge so close to Trp191 destabilized the Trp191 cation radical in Compound I, as observed through EPR and significantly reduced cytochrome c peroxidase activity (6, 7). We also showed that Met230 and Met231 are important for stabilization of the cationic Trp191 radical (11). Like CCP, changing the proximal Trp to Phe in LmP greatly decreased the cytochrome c peroxidase activity (31), suggesting that LmP might also form a Trp radical. This would be inconsistent with the presence of the proximal K+ site in LmP. Therefore, it was important to determine if LmP forms a stable Trp208 radical using EPR spectroscopy.
FIGURE 1.

Crystal structures of the three heme peroxidases that conserve the proximal Trp: CCP, APX, and LmP. The triple β-strand feature unique to CCP and LmP is colored in yellow. The loop between the A and B helices is colored in cyan. The Trp pocket, underneath the heme prosthetic group, for each enzyme is unique. LmP has the potassium ion that CCP lacks and an extra sulfur-containing residue, Cys197 (C197). The large cyan spheres represent K+ or Ca2+. Figs. 1–3 were prepared with PyMOL (available on the World Wide Web).
FIGURE 2.

Calcium and potassium binding coordination in LmP. A, calcium Fo − Fc density contoured at 13 σ and the coordinating amino acids. B, potassium Fo − Fc density also contoured at 13 σ and the coordinating amino acids. Note the proximity of Trp208, ∼8.5 Å from the potassium.
TABLE 2.
Amino acid coordination of potassium and calcium binding sites in LmP and C197T
| Amino acid | Contributing group | Distance in LmP | Distance in C197T |
|---|---|---|---|
| Å | Å | ||
| Potassium binding site | |||
| Thr193 | (OH) | 2.8 | 2.9 |
| Thr193 | bb(CO)a | 2.7 | 2.6 |
| Thr209 | (OH) | 2.9 | 2.8 |
| Asp211 | bb(CO) | 2.8 | 2.7 |
| Gly214 | bb(CO) | 2.7 | 2.9 |
| Ser218 | (OH) | 3 | 3.1 |
| Calcium binding site | |||
| Glu69 | (COO−) | 2.7 | 3.1 |
| Glu69 | bb(CO) | 2.7 | 3.0 |
| Ser72 | (OH) | 2.8 | 2.9 |
| Ser81 | bb(CO) | 2.9 | 3.0 |
| Ser86 | (OH) | 2.5 | 2.8 |
| Glu92 | (COO−) | 2.8 | 2.7 |
a bb, backbone.
EPR Spectroscopy
CCP exhibits a characteristic EPR spectra of g = 2.006, and LmP exhibits a very intense signal centered at g = 2.000, consistent with Trp radical formation (Fig. 3). Note that the overall shapes of the wild type signals are different. The CCP Trp191 EPR radical signal results in part from weak coupling between Trp191 and the Fe4+=O center (32), whereas various conformational states result in a distribution of coupling from antiferromagnetic to ferromagnetic. In other words, the precise shape of the spectrum is quite sensitive to subtle differences in the local environment, so it is not too surprising that the LmP radical signal differs from that of CCP. To ensure that the radical derives from oxidation of Trp208, the mutant W208F was prepared. This mutant previously has been shown to exhibit diminished cytochrome c peroxidase activity (31). As expected, the Compound I EPR signal of W208F is essentially lost (Fig. 3). Therefore, like CCP, LmP requires the proximal Trp208 for cytochrome c peroxidase activity and is the site of radical formation in Compound I.
FIGURE 3.

A, relative initial Compound I EPR spectra of LmP and mutants, W208F and C197T. Both mutations significantly decrease the initial signal of the spectra. B, relative initial Compound I EPR spectra of CCP, CCPK1, and CCPK1/T180C. Note the restoration in signal of CCPK1/T180C compared with the complete loss of signal in CCPK1. C–F, immediate (dashed line) Compound I EPR spectra versus the same sample allowed to sit at room temperature for 30 min and then refrozen (solid line). C, LmP; D, C197T; E, W208F; F, CCPK1/T180C.
What remains a puzzle, however, is why LmP forms a stable Trp208 radical despite the K+ bound about 8 Å away. Based on our previous studies, a cation this close should greatly diminish the stability of the Trp208 cationic radical. A close examination of the CCP and LmP active sites reveals one possible difference that may enhance the stability of the LmP Trp208 radical even with a K+ nearby. As shown in Fig. 1, where CCP has Thr180 LmP has Cys197, whose sulfur atom directly contacts Trp208. The electronegative sulfur or even a partially/fully deprotonated sulfur might provide additional electrostatic stability to the Trp208 cation radical. We therefore prepared the C197T mutant in LmP and the T180C mutant in CCPK1, the engineered version of CCP that contains the K+ site, to give CCPK1/T180C. Fig. 4 shows the electron density of the mutant in this region. Outside the site of mutation, there is no significant differences between the mutant and wild type structures. The C197T LmP mutant exhibits a significantly decreased EPR signal at g = 2.001 (Fig. 3). In fact, this mutation produces the same decrease in signal as the W208F mutant. Of all of the mutants, however, CCPK1 exhibits the weakest Compound I EPR spectra (Fig. 3). The EPR signal of CCPK1/T180C exhibits a stronger signal at g = 2.004, indicating some restabilization but not nearly to the level of wild type CCP. Cys197 is, therefore, an essential residue for the Trp208 cation radical.
FIGURE 4.

2Fo − Fc map contoured at 1.0 σ of the C197T mutant.
We also examined the relative stability of the Trp radical in both wild type and mutant proteins by subtracting the g = 2.00 maxima of the sample thawed for 30 min from the initially rapidly frozen samples (Fig. 3). The stability of the Trp radical in LmP is very high, with only a 20% loss of signal intensity after 30 min at room temperature. In addition, the UV-visible spectra of wild type LmP Compound I and the mutants show little change after 30 min at room temperature (supplemental Fig. S1), indicating that the Fe(IV)=O center is also quite stable. The stability of CCP Compound I is very similar (33). In sharp contrast, LmP C197T exhibits a complete loss of EPR signal after 30 min at room temperature. The W208F mutant is nearly as unstable as the C197T mutant in that it exhibits a 96% signal reduction. CCP exhibits a 78% reduction in signal. Interestingly, the CCPK/T180C refrozen signal exhibits only a 64% reduction, suggesting that the introduction of the Cys residue contributes more to longer term radical stability. Where the radical is situated in the mutants remains unknown. However, mutagenesis studies of CCP where the Trp191 Compound I radical no longer forms indicates that tyrosine(s) serves as electron donor(s) in forming Compound I and that the weak EPR signal in the CCP mutants is due to residual partial spin remaining on an unstable Tyr radical (5, 11, 34). The similarity in the EPR signals of the LmP mutants suggests that Tyr is the most likely alternate electron donor which is much less stable than the Trp radical in wild type LmP.
Steady-state Activity Assays
The cytochrome c peroxidase activity assays were carried out using one substrate (reduced horse heart cytochrome c) and at one ionic strength (50 mm). We observe a much higher cytochrome c peroxidase activity for LmP (Table 2) than reported previously (31). This could be due to the differences in the expression vector. The pTrcHis vector (Invitrogen) used in the previous studies codes for an extra 21-amino acid N-terminal tail. Such a long, flexible, and seemingly unstructured region could interfere with substrate interactions. Furthermore, the highly purified samples we prepared for crystallization exhibited a higher Rz, at least 1.2, compared with an Rz of ∼1 previously obtained (13). Both factors could explain why we observe a higher cytochrome c peroxidase activity for LmP.
The wild type enzymes, CCP and LmP, had much higher activities than the mutant enzymes. As expected, both W208F and C197T exhibited a significantly decreased ability to oxidize cytochrome c. These data are consistent with the decreased EPR signals of both mutants. We initially thought that introduction of Cys into the CCPK1 mutant might restore cytochrome c peroxidase activity by providing increased stability to the Trp191 radical; this does not occur although the conversion of Thr180 to Cys in CCPK1 does enhance the Compound I EPR signal. Nevertheless, the EPR spectra is still much lower than wild type CCP and exhibits a completely different profile. Therefore, it is not surprising that the CCPK/T180C exhibits very low activity.
Table 2 also shows that the LmP activity using ascorbate as the substrate is quite low. We attempted to solve the LmP-ascorbate structure by either soaking in high concentrations of ascorbate or by co-crystallization. Co-crystallization in the presence of 5 mm ascorbate did yield reasonable crystals, allowing us to collect a 1.98 Å data set, but there was no hint of ascorbate binding. Perhaps this is not too surprising because the anticipated binding site for ascorbate in LmP has some important differences from APX. First, the loop connecting the A and B helices where ascorbate binds in APX is one residue longer in LmP. Second, Arg172, which in APX hydrogen-bonds with ascorbate (35) and is known to be essential for APX activity (36), is Phe201 in LmP. Simple modeling studies show that Phe201 and the longer loop would present severe steric problems for ascorbate binding in this region and further indicate that ascorbate interacts very weakly or not at all at this site.
DISCUSSION
As expected from sequence alignments, LmP is a hybrid peroxidase that shares structural features of the various classes of heme peroxidases. Like class 3 peroxidases, such as horseradish peroxidase, LmP contains two cation binding sites. In class 3 peroxidases, both cations are Ca2+, but in LmP, like in plant APX, the proximal cation is monovalent, probably K+. Unique to yeast CCP is a long insertion between the A and B helices that provides contacts with cytochrome c in the CCP-cytochrome c complex (37). This region is much shorter in APX and forms part of the ascorbate binding pocket. LmP more closely resembles APX in this region, although the loop is one residue longer in LmP. LmP also lacks the key Arg172 residue in APX required for binding ascorbate (35, 37). The analogous residue in LmP is Phe201. Simple modeling indicates that ascorbate cannot bind to LmP the same as in APX due to steric clashes both with the Phe201 and the longer loop connecting the A and B helices, which may account for the fairly poor ascorbate peroxidase activity of LmP, 9.2 min−1 compared with ∼250 s−1 for plant APX (38). Similar mutations have been made in yeast CCP that also improve ascorbate peroxidase activity (39, 40). As expected, the mutants' decrease in cytochrome c peroxidation correlates with an increased ascorbate peroxidase activity (Table 3). The final segment of structure unique to LmP and CCP is the triple β-strand segment near the Trp radical. This region very likely provides additional protection of the Trp radical in Compound I.
TABLE 3.
Turnover numbers from peroxidase activity assays for wild type and mutant enzymes
| Enzyme | kcat |
|
|---|---|---|
| Cytochrome c | Ascorbate | |
| s−1 | min−1 | |
| LmP | 594.4 ± 63 | 9.2 ± 0.3 |
| LmP C197T | 4.72 ± 0.1 | 15.4 ± 0.9 |
| LmP W208F | 0.25 ± 0.02 | 4.9 ± 0.2 |
| CCP | 1,846.8 ± 127.5 | 4.3 ± 0.3 |
| CCPK1 | 2.3 ± 0.03 | 7.7 ± 0.5 |
| CCPK1/T180C | 3.37 ± 0.05 | 11.5 ± 0.7 |
Although LmP forms the Trp radical like CCP, the shape of the spectrum is very different. A complete understanding of the CCP Trp191 radical required isotope substitution (2), EPR, and electron double resonance spectroscopy (32). Although these types of investigations are beyond the scope of the present study, the lessons learned from CCP can shed light on the LmP Compound I EPR spectrum. The shape of the EPR spectrum clearly indicates more than one, probably two, species that are in fast exchange on the time scale of the EPR experiment. The CCP Trp191 radical also has two species that are weakly coupled to the spin = 1.0 Fe4+=O center, one of which is the dominant antiferromagnetically and the other a minor ferromagnetically coupled species. These two species arise from very subtle differences in conformational states. Minor changes in the active site can cause major differences in Compound I EPR properties. For example, conversion of Asp235 in CCP (Fig. 1) to Glu gives rise to a much narrower EPR signal of Compound I (41) and has been attributed to a near elimination of one of the conformational states observed in wild type CCP (32). The LmP Compound I EPR spectrum more closely resembles the CCP D235E EPR spectrum than wild type CCP, indicating that LmP favors one of the two states observed in CCP. The His-Asp-Trp hydrogen-bonded network is thought to play a key role in the Trp radical EPR properties (32), suggesting, perhaps, that there may be subtle differences between CCP and LmP in this hydrogen-bonding network. Of course, the major structural difference between LmP and CCP is that LmP has Cys197 where CCP has Thr180. This could affect the hydrogen-bonding network or, by an as yet to be determined mechanism, favor one EPR conformer/state over another. Full dissection of this problem and determination of the precise effects of Cys197 on the LmP EPR Compound I properties are readily possible but will require the analysis of mutants of CCP and LmP that both retain activity and form a stable Trp radical.
Taken together, these structural and functional results clearly show that LmP is indeed a structural hybrid of different peroxidases but is not truly a functional hybrid. The observations that our preparations of the Lm peroxidase form a stable Trp radical and that cytochrome c is a much better substrate than ascorbate suggest that the biological function of the Lm enzyme is to serve as a CCP. Moreover, it has recently been shown that LmP is localized to the inner mitochondrial membrane of promastisgotes (42) similar to yeast CCP. It thus is tempting to rename LmP to LmCCP. However, what remains to be demonstrated is whether or not the putative native substrate, cytochrome c from L. major (Lm cytochrome c), is a good redox partner. All studies for the Lm peroxidase so far have used horse heart cytochrome c as a substrate. Until such data become available, we suggest that the L. major enzyme be referred to as simply Lm peroxidase or LmP.
Supplementary Material
Acknowledgment
We thank Dr. Subrata Adak for the LmP expression system and for valuable advice and discussions.
This work was supported, in whole or in part, by National Institutes of Health Grant GM42614.

The on-line version of this article (available at http://www.jbc.org) contains supplemental Fig. S1.
The atomic coordinates and structure factors (codes 3RIV and 3RIW) have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ (http://www.rcsb.org/).
- CCP
- cytochrome c peroxidase
- APX
- ascorbate peroxidase
- SAD
- single wavelength anomalous dispersion
- LmP
- L. major peroxidase.
REFERENCES
- 1. Dunford H. B. (2010) in Peroxidases and Catalases: Biochemistry, Biophysics, Biotechnology, and Physiology, 2nd Ed., Wiley & Sons, Inc., New York [Google Scholar]
- 2. Sivaraja M., Goodin D. B., Smith M., Hoffman B. M. (1989) Science 245, 738–740 [DOI] [PubMed] [Google Scholar]
- 3. Gajhede M., Schuller D. J., Henriksen A., Smith A. T., Poulos T. L. (1997) Nat. Struct. Biol. 4, 1032–1038 [DOI] [PubMed] [Google Scholar]
- 4. Patterson W. R., Poulos T. L., Goodin D. B. (1995) Biochemistry 34, 4342–4345 [DOI] [PubMed] [Google Scholar]
- 5. Barrows T. P., Bhaskar B., Poulos T. L. (2004) Biochemistry 43, 8826–8834 [DOI] [PubMed] [Google Scholar]
- 6. Bonagura C. A., Sundaramoorthy M., Bhaskar B., Poulos T. L. (1999) Biochemistry 38, 5538–5545 [DOI] [PubMed] [Google Scholar]
- 7. Bonagura C. A., Sundaramoorthy M., Pappa H. S., Patterson W. R., Poulos T. L. (1996) Biochemistry 35, 6107–6115 [DOI] [PubMed] [Google Scholar]
- 8. Fitzgerald M. M., Churchill M. J., McRee D. E., Goodin D. B. (1994) Biochemistry 33, 3807–3818 [PubMed] [Google Scholar]
- 9. Miller M. A., Han G. W., Kraut J. (1994) Proc. Natl. Acad. Sci. U.S.A. 91, 11118–11122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Jensen G. M., Bunte S. W., Warshel A., Goodin D. B. (1998) J. Phys. Chem. B 102, 8221–8228 [Google Scholar]
- 11. Barrows T. P., Poulos T. L. (2005) Biochemistry 44, 14062–14068 [DOI] [PubMed] [Google Scholar]
- 12. Fishel L. A., Farnum M. F., Mauro J. M., Miller M. A., Kraut J., Liu Y. J., Tan X. L., Scholes C. P. (1991) Biochemistry 30, 1986–1996 [DOI] [PubMed] [Google Scholar]
- 13. Adak S., Datta A. K. (2005) Biochem. J. 390, 465–474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Yadav R. K., Dolai S., Pal S., Adak S. (2010) Arch. Biochem. Biophys. 495, 129–135 [DOI] [PubMed] [Google Scholar]
- 15. Berry E. A., Trumpower B. L. (1987) Anal. Biochem. 161, 1–15 [DOI] [PubMed] [Google Scholar]
- 16. Margoliash E. (1952) Nature 170, 1014–1015 [DOI] [PubMed] [Google Scholar]
- 17. Yonetani T. (1965) J. Biol. Chem. 240, 4509–4514 [PubMed] [Google Scholar]
- 18. Yonetani T., Ray G. S. (1965) J. Biol. Chem. 240, 4503–4508 [PubMed] [Google Scholar]
- 19. Fowler R. M., Bright H. A. (1935) J. Res. Natl. Bur. Stand. 15, 493–501 [Google Scholar]
- 20. Buettner G. R. (1988) J. Biochem. Biophys. Methods 16, 27–40 [DOI] [PubMed] [Google Scholar]
- 21. Nakano Y., Asada K. (1981) Plant Cell Physiol. 22, 867–880 [Google Scholar]
- 22. Nakano Y., Asada K. (1987) Plant Cell Physiol. 28, 131–140 [Google Scholar]
- 23. Yordanov N. D. (1996) Appl. Magn. Reson. 10, 339–350 [Google Scholar]
- 24. Yordanov N. D., Christova A. (1994) Appl. Magn. Reson. 6, 341–345 [Google Scholar]
- 25. Otwinowski Z., Minor W. (1997) Macromol. Crystallogr. Pt. A 276, 307–326 [DOI] [PubMed] [Google Scholar]
- 26. McCoy A. J. (2007) Acta. Crystallogr. D Biol. Crystallogr. 63, 32–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. (1994) Acta Crystallogr. D Biol. Crystallogr. 50, 760–763 [DOI] [PubMed] [Google Scholar]
- 28. Emsley P., Cowtan K. (2004) Acta Crystallogr. D 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 29. Emsley P., Lohkamp B., Scott W. G., Cowtan K. (2010) Acta Crystallogr. D 66, 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Murshudov G. N., Vagin A. A., Dodson E. J. (1997) Acta Crystallogr. D 53, 240–255 [DOI] [PubMed] [Google Scholar]
- 31. Yadav R. K., Dolai S., Pal S., Adak S. (2008) Biochim. Biophys. Acta 1784, 863–871 [DOI] [PubMed] [Google Scholar]
- 32. Houseman A. L., Doan P. E., Goodin D. B., Hoffman B. M. (1993) Biochemistry 32, 4430–4443 [DOI] [PubMed] [Google Scholar]
- 33. Yonetani T. (1970) Adv Enzymol. Relat. Areas Mol. Biol. 33, 309–335 [DOI] [PubMed] [Google Scholar]
- 34. Musah R. A., Goodin D. B. (1997) Biochemistry 36, 11665–11674 [DOI] [PubMed] [Google Scholar]
- 35. Sharp K. H., Mewies M., Moody P. C., Raven E. L. (2003) Nat. Struct. Biol. 10, 303–307 [DOI] [PubMed] [Google Scholar]
- 36. Bursey E. H., Poulos T. L. (2000) Biochemistry 39, 7374–7379 [DOI] [PubMed] [Google Scholar]
- 37. Pelletier H., Kraut J. (1992) Science 258, 1748–1755 [DOI] [PubMed] [Google Scholar]
- 38. Lad L., Mewies M., Raven E. L. (2002) Biochemistry 41, 13774–13781 [DOI] [PubMed] [Google Scholar]
- 39. Meharenna Y. T., Oertel P., Bhaskar B., Poulos T. L. (2008) Biochemistry 47, 10324–10332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Murphy E. J., Metcalfe C. L., Basran J., Moody P. C., Raven E. L. (2008) Biochemistry 47, 13933–13941 [DOI] [PubMed] [Google Scholar]
- 41. Goodin D. B., McRee D. E. (1993) Biochemistry 32, 3313–3324 [PubMed] [Google Scholar]
- 42. Dolai S., Yadav R. K., Pal S., Adak S. (2009) Eukaryotic Cell 8, 1721–1731 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.