

Abstract
Background
Although Toll-like receptor 4 (TLR4) has been implicated in the myocardial injury caused by regional ischemia/reperfusion, its role in the myocardial inflammatory response and in contractile dysfunction after global ischemia/reperfusion is unclear. Cytokines, particularly tumor necrosis factor-α (TNF-α), contribute to the mechanism of myocardial dysfunction after global ischemia/reperfusion. We hypothesized that a TLR4-mediated cytokine cascade modulates myocardial contractile function after global ischemia/reperfusion. This study examined whether TLR4 regulates TNF-α and interleukin (IL)-1β peptide production during global ischemia/reperfusion and whether TLR4 signaling influences postischemic cardiac function through TNF-α and IL-1β.
Methods
Isolated hearts from wild-type mice, two strains of TLR4 mutants, TNF-α knockouts, and IL-1β knockouts underwent global ischemia/reperfusion. Cardiac contractile function was analyzed, and myocardial nuclear factor-κB activity and TNF-α and IL-1β levels were measured.
Results
In wild-type hearts, global ischemia/reperfusion induced nuclear factor-κB activation and the production of TNF-α and IL-1β peptides. In TLR4-mutant hearts, these changes were significantly reduced and postischemic functional recovery was improved. Application of TNF-α and IL-1β to TLR4-mutant hearts abrogated this improvement in postischemic functional recovery. Postischemic functional recovery also improved in TNF-α knockout and IL-1β knockout hearts, as well as in wild-type hearts treated with TNF-binding protein or IL-1 receptor antagonist.
Conclusions
This study demonstrates that TLR4 signaling contributes to cardiac dysfunction after global ischemia/reperfusion. TLR4 signaling mediates the production of TNF-α and IL-1β peptides, and these two cytokines link TLR4 signaling to postischemic cardiac dysfunction.
Global myocardial ischemia/reperfusion, which is often obligatory during cardiac surgery, induces an inflammatory response in the heart characterized by cytokine production [1]. Previous studies showed that proinflammatory cytokines, particularly tumor necrosis factor-α (TNF-α) and interleukin-1β (IL-1β), depress myocardial contractility [2] and contribute to cardiac dysfunction [3]. Thus, preservation of cardiac function after global ischemia/reperfusion requires regulation of the myocardial inflammatory response.
Toll-like receptor 4 (TLR4) signaling has been implicated in the cardiac dysfunction induced by hemorrhagic shock [4] and has also been linked to the production of proinflammatory mediators after ischemia/reperfusion in the liver and brain [5, 6]. In addition, TLR4 signaling is involved in the myocardial expression of cytokine (IL-1β and IL-6) messenger RNA (mRNA) [7, 8] and influences the size of myocardial infarcts after regional ischemia/reperfusion [7–10]. Together, these studies suggest that TLR4 might also play a role in cardiac dysfunction after global ischemia/reperfusion, which induces an inflammatory response without severe tissue injury [11, 12].
Increased levels of TNF-α have been found in human myocardial tissue after surgery with cardiopulmonary bypass [1, 13], and animal models of global ischemia/reperfusion have implicated TNF-α in the myocardial inflammatory response [14–17]. Neutralization of TNF-α improved cardiac functional recovery in several studies [11, 12], but gene target knockout of both the p55 and the p75 TNF-α receptors resulted in myocardial apoptosis and an enlargement of infarct size after regional ischemia/reperfusion [18]. Thus, the role of TNF-α in cardiac function after ischemia/reperfusion remains to be determined. Pomerantz and colleagues [19] have shown that human myocardial tissue expresses elevated levels of IL-1β after ischemia/reperfusion, and we reported earlier that TNF-α and IL-1β can both depress myocardial contractility in vitro [2]. Although two studies found that TLR4 plays a role in the myocardial expression of IL-1β and IL-6 mRNA after regional ischemia/reperfusion [7, 8], the effect of TLR4 signaling on the production of TNF-α and IL-1β peptides remains to be determined. Nor is it clear whether TNF-α and IL-1β mediate the effect of TLR4 on postischemic cardiac dysfunction.
We hypothesized that TLR4 signaling modulates post-ischemic cardiac function by regulating the production of TNF-α and IL-1β. This study examined (1) the effect of TLR4 mutation (defect or deletion) on cardiac function after global ischemia/reperfusion, (2) the influence of TLR4 signaling on the production of TNF-α and IL-1β peptides during global ischemia/reperfusion, and (3) the role of TNF-α and IL-1β in mediating the effect of TLR4 on cardiac function.
Material and Methods
Animals
Male mice, TLR4-defective (C3H/HeJ), TLR4-deleted (C57BL/10ScNJ), TNF-α knockout, and wild-type controls (C57BL/10ScSn for TLR4-deleted and B6 for TNF-α knockout) were purchased from Jackson Laboratory (Bar Harbor, ME). Male C3H/HeN mice (wild-type controls for TLR4-defective) were purchased from Charles River Company (Wilmington, MA). Breeding pairs of IL-1β knockout and wild-type controls (B6.129) were generous gifts from Dr Fantuzzi [20]. We confirmed previously that TNF-α knockout mice lack a myocardial TNF-α response to endotoxin [21]. In preliminary experiments, we also confirmed that IL-1β knockout mice lack a myocardial IL-1β response to endotoxin.
The mice (weight, 22 to 26 g) were acclimated in a 12-hour light/12-hour dark room and maintained on a standard pellet diet. They received humane care in compliance with the Guide for the Care and Use of Laboratory Animals (National Research Council, revised 1996). All experiments were approved by the Animal Care and Research Committee of the University of Colorado Denver.
Isolated Heart Perfusion
Isolated hearts were perfused by the Langendorff technique as described previously [4]. Mice were anesthetized with intraperitoneal sodium pentobarbital (50 mg/kg) and heparinized with intraperitoneal sodium heparin (300 U). Their hearts were rapidly excised into oxygenated ice-cold Krebs-Henseleit solution (pH 7.4) and were retrograde-perfused in non-recirculating mode at a constant pressure of 70 mm Hg. All hearts were perfused within 2 minutes after excision.
An ultrathin latex balloon was inserted into the left ventricle. Approximately 60 μL of water was injected into the balloon to achieve a left ventricular end-diastolic pressure (LVEDP) of 8 to 20 mm Hg at the beginning of equilibration, and the balloon volume was not altered thereafter. Mean LVEDP was approximately 10 mm Hg at the onset of ischemia. Hearts were paced at 450 beats/min.
After 20 minutes of equilibration, hearts underwent 20 minutes of normothermic global ischemia followed by 60 minutes of reperfusion. During ischemia, hearts were placed in a normal saline-filled organ bath chamber without pacing, and chamber temperature was maintained at 37°C. Left ventricular developed pressure (LVDP), dP/dt, and LVEDP were continuously recorded with a computerized pressure amplifier/digitizer.
To determine the role of TLR4 signaling in postischemic cardiac dysfunction and the myocardial inflammatory response, four groups of hearts (8 TLR4-defective and 8 wild-type controls, 6 TLR4-deleted and 6 wild-type controls) underwent ischemia/reperfusion. LVDP, +dP/dt max, and LVEDP were recorded before and during ischemia/reperfusion. Ventricular tissue specimens were prepared for analysis of nuclear factor (NF)-κB DNA-binding activity and cytokine peptide levels. An additional 6 TLR4-defective hearts and 6 wild-type controls underwent 20 minutes of ischemia, 20 minutes of ischemia with 60 minutes of reperfusion, or perfusion only. Ventricular tissue specimens were prepared for analysis of NF-κB p65 phosphorylation.
To determine the role of cytokines in mediating the effect of TLR4 signaling on postischemic cardiac function, three groups of 6 hearts each (TNF-α knockout, IL-1β knockout, and wild-type controls) underwent ischemia/reperfusion. In addition, two groups of 6 hearts each from wild-type mice were treated with TNF-α binding protein (TNF-BP, 1.0 μg/mL) or IL-1 receptor antagonist (IL-1RA, 1.0 μg/mL) during reperfusion. We previously found that TNF-BP at this concentration abolishes endotoxemic cardiac dysfunction in rats [22]. Finally, 6 TLR4-defective hearts were treated with murine TNF-α and IL-1β (0.1 ng/mL each) during reperfusion.
Immunoblotting
Myocardial tissue was homogenized in phosphate-buffered saline containing 0.5% Triton X-100 and a protease inhibitor cocktail. Size fraction of crude protein (20 μg) was performed by electrophoresis as previously described [23]. After transfer, the membrane was incubated in phosphate-buffered saline containing 5% nonfat dry milk to block nonspecific binding. The membrane was then incubated for 60 minutes with an antibody against phosphorylated NF-κB p65, total NF-κB p65, or TLR4 (purchased from AbD Serotec, Oxford, United Kingdom) at 1:1000 to 1:2000 dilution with phosphate-buffered saline containing 0.05% Tween 20 and 5% dry milk.
After thorough washes, the membrane was treated with peroxidase-labeled secondary antibody (1:5000 dilution with phosphate-buffered saline containing 0.05% Tween 20 and 5% dry milk) for 45 minutes. Protein bands were developed using enhanced chemiluminescence technique. Densitometry was performed using a computerized densitometer (Molecular Dynamics, Sunnyvale, CA).
Nuclear Factor-κB Activity Assay
A transcription factor assay kit (Active Motif, Carlsbad, CA) was used to measure NF-κB p65 DNA-binding activity in myocardial homogenate. This assay is based on the specific binding of the active form of NF-κB to a consensus oligonucleotide attached to the plate and is reported to be 10-fold more sensitive than electrophoretic mobility shift assays [24].
Cytokine Measurements
Cytokine levels in myocardial homogenate were analyzed using enzyme-linked immunosorbent kits (R & D Systems, Minneapolis, MN) as previously reported [21]. The detection limits are 5.1 pg/mL for TNF-α and 3.0 pg/mL for IL-1β.
Statistics
Data are expressed as means ± standard error of the mean. Statistical analysis was performed using Stat-View software (Abacus Concepts, Calabasas, CA). The statistical significance of differences between groups was determined by analysis of variance with a post hoc Bonferroni/Dunn test and was accepted within a 95% confidence limit.
Results
Effect of TLR4 Mutation
ON POSTISCHEMIC CARDIAC FUNCTION
During equilibration, we found no difference in mean LVDP among TLR4-defective and TLR4-deleted hearts and their respective controls. LVDP declined to zero in all groups during ischemia, but TLR4-mutant hearts regained contractile function more quickly during reperfusion. At 40, 50 and 60 minutes of reperfusion, LVDP and +dP/dt max were significantly higher (p < 0.05) in TLR4-defective and TLR4-deleted hearts than in wild-type controls (Fig 1A and B). LVEDP recovery was significantly improved during reperfusion in TLR4 mutants (Fig 1C).
Fig 1.

Effect of Toll-like receptor 4 (TLR4) mutation on postischemic cardiac function. Hearts from 8 TLR4-defective (TLR4-def, black squares) and 6 TLR4-deleted (TLR4-del, black circles) mice and their respective wild-type (WT) controls (WT-def, n = 8, white squares; WT-del, n = 6, white circles) underwent 20 minutes of global ischemia followed by 60 minutes of reperfusion. TLR4-mutant hearts displayed improved recovery of (A) left ventricular developed pressure (LVDP), (B) +dP/dt max, and (C) left ventricular end-diastolic pressure (LVEDP) compared with wild-type controls. Data are expressed as mean ± SEM; *p < 0.05 vs WT-def; #p < 0.05 vs WT-del.
ON NUCLEAR FACTOR-κB ACTIVATION
In wild-type hearts, levels of phosphorylated NF-κB p65 increased with ischemia or ischemia/reperfusion (Fig 2A). In TLR4-defective hearts, although levels of total NF-κB p65 were comparable, levels of phosphorylated NF-κB p65 were unchanged during ischemia or ischemia/reperfusion (Fig 2A and B). NF-κB DNA-binding activity was four times greater after ischemia/reperfusion in wild-type hearts but was significantly attenuated in TLR4-defective or TLR4-deleted hearts (p < 0.05, Table 1).
Fig 2.
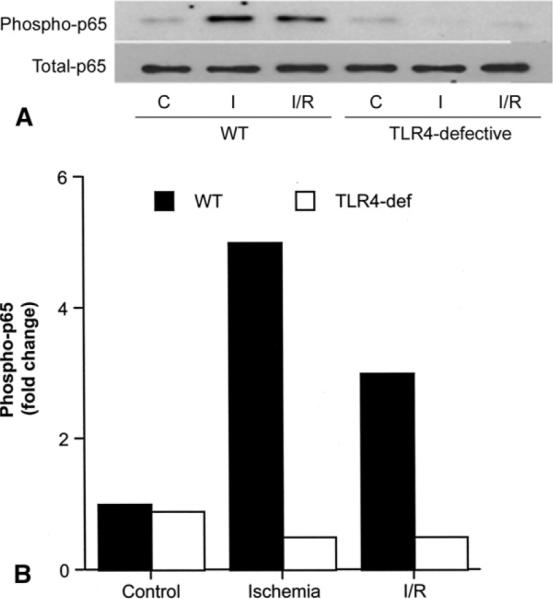
Effect of Toll-like receptor 4 (TLR4) mutation on nuclear factor (NF)-κB phosphorylation. (A) A representative gel shows results after hearts of wild-type (WT) and TLR4-defective (TLR4-def) mice underwent either 40 minutes of perfusion (C–control), 20 minutes of ischemia (I), or 20 minutes of ischemia followed by 60 minutes of reperfusion (I/R). (B) Densitometry data of two separate experiments show that levels of phosphorylated NF-κB p65 increased after ischemia and ischemia/reperfusion in WT hearts (black bars), but not in TLR4-defective hearts (white bars).
Table 1.
Myocardial Nuclear Factor-κB Activity and Cytokine Levels
| NF-κB DNA-binding, OD/mg Protein (n = 6)a |
TNF-α, pg/mg Protein (n = 5)a |
IL-1β, pg/mg Protein (n = 5)a |
||||
|---|---|---|---|---|---|---|
| Mouse Type | Control | I/R | Control | I/R | Control | I/R |
| Wild-type | ||||||
| Defective | 0.08 ± 0.02 | 0.31 ± 0.05b | 3.18 ± 0.60 | 28.4 ± 3.86b | 1.40 ± 0.72 | 22.4 ± 0.82b |
| Deleted | 0.07 ± 0.01 | 0.29 ± 0.04b | 4.16 ± 0.70 | 26.6 ± 3.22b | 2.60 ± 0.80 | 21.5 ± 2.65b |
| TLR4 mutants | ||||||
| Defective | 0.09 ± 0.02 | 0.14 ± 0.04c | 3.84 ± 0.57 | 7.44 ± 1.17c | 1.80 ± 0.55 | 5.18 ± 0.98c |
| Deleted | 0.10 ± 0.01 | 0.16 ± 0.03d | 3.23 ± 1.55 | 6.84 ± 1.36d | 2.24 ± 1.48 | 5.44 ± 1.76d |
Data are expressed as mean ± standard error of the mean.
p < 0.05 vs control.
p < 0.05 vs wild-type defective I/R.
p < 0.05 vs wild-type deleted I/R.
Control = 100 minutes of perfusion; IL = interleukin; I/R = ischemia (20 min)/reperfusion (60 min); NF = nuclear factor; TLR4 = Toll-type receptor 4; TNF = tumor necrosis factor.
ON POSTISCHEMIC MYOCARDIAL CYTOKINE LEVELS
In wild-type hearts, TNF-α levels after ischemia/reperfusionwere six to eight times greater than perfusion controls (p < 0.05) and IL-1β levels were six to fifteen times greater than perfusion controls (p < 0.01). In TLR4-mutant hearts, neither TNF-α nor IL-1β was significantly elevated above perfusion control levels after ischemia/reperfusion (Table 1).
Role of TNF-α and IL-1β in Postischemic Cardiac Dysfunction
First, we examined postischemic cardiac function in TNF-α and IL-1β knockouts. Immunoblotting demonstrated that myocardial TLR4 levels in TNF-α and IL-1β knockouts are comparable with wild-type hearts (Fig 3A). LVDP and +dP/dt max were not significantly different in knockouts and wild-type controls before ischemia. Throughout reperfusion, LVDP and +dP/dt max were greater in TNF-α and IL-1β knockouts than in wild-type controls, and significantly greater (p < 0.05) in the late phase of reperfusion (Fig 3B and C). In addition, LVDP and +dP/dt max were significantly greater (p < 0.05) in TNF-α knockouts than in IL-1β knockouts after 50 and 60 minutes of reperfusion (Fig 3B and C). Similarly, LVEDP recovery was significantly greater in TNF-α knockouts than in IL-1β knockouts (data not shown).
Fig 3.

Role of tumor necrosis factor (TNF)-α and interleukin (IL)-1β in postischemic cardiac dysfunction. (A) Immunoblotting demonstrates that TNF-α knockout (TNF KO), IL-1β knockout (IL-1 KO) and wild-type (WT) hearts have comparable Toll-like receptor 4 (TLR4) levels. (B, C) Hearts isolated from TNF KO (black circles), IL-1 KO (black squares) and WT (white circles, a combined group of TNF-α WT and IL-1β WT) underwent 20 minutes of global ischemia followed by 60 minutes reperfusion. TNF KO and IL-1 KO hearts had higher left ventricular developed pressure (LVDP) and +dP/dt max after I/R than hearts from wild-type controls. Data are expressed as mean ± SEM. n = 6 in each group; *p < 0.05 vs WT; #p < 0.05 vs IL-1 KO. (D) Wild-type hearts underwent 20 minutes of global ischemia followed by 60 minutes reperfusion, and were treated with TNF-binding protein (TNF-BP, 1.0 μg/mL) or IL-1 receptor antagonist (IL-1 RA, 1.0 μg/mL) during reperfusion. The percentage recovery of LVDP in hearts treated with TNF-BP and IL-1 RA was greater than in untreated hearts and was similar to knockouts. Data are expressed as mean ± standard error of the mean; n = 6 in each group; *p < 0.05 vs WT.
To confirm these results, we applied TNF-BP and IL-1RA to wild-type hearts during reperfusion and calculated the percentage recovery of LVDP (LVDP after 60 minutes reperfusion/baseline LVDP × 100%). Treatment with TNF-BP or IL-1RA improved the percentage recovery of LVDP to levels comparable with those in knockout hearts (Fig 3D). The percentage recovery of LVDP was greater in TNF-BP–treated hearts than in IL-1RA–treated hearts, but this difference was not statistically significant.
Effect of TNF-α and IL-1β on Postischemic Cardiac Function in TLR4-Defective Hearts
To evaluate the role of TNF-α and IL-1β in the modulation of postischemic cardiac function by TLR4, we applied TNF-α and IL-1β to TLR4-defective hearts during reperfusion. The addition of these two cytokines to TLR4-defective hearts reduced LVDP and +dP/dt max to levels similar to those in wild-type hearts, abrogating the beneficial effects of defective TLR4 on postischemic cardiac functional recovery (Fig 4A and B).
Fig 4.

Effect of tumor necrosis factor (TNF)-α and interleukin (IL)-1β on postischemic cardiac functional recovery in Toll-like receptor 4 (TLR4)-defective hearts. TLR4-defective (TLR4-def) hearts underwent 20 minutes of global ischemia followed by 60 minutes of reperfusion, and were treated with TNF-α and IL-1β (0.1 ng/mL each) during reperfusion. Postischemic (A) left ventricular developed pressure (LVDP) and (B) +dP/dt max were similar in TLR4-defective hearts treated with TNF-α and IL-1β (TLR4-def + Cytok, black circles) and wild-type (WT, white squares) hearts, and significantly lower than in untreated TLR4-defective hearts (white circles). Data are expressed as mean ± SEM; n = 6 in each group; *p < 0.05 vs WT; #p < 0.05 vs TLR4-def.
Comment
It is well known that global ischemia/reperfusion causes cardiac dysfunction in patients undergoing cardiac operations [25, 26]; however, the underlying mechanism is not fully understood. In the present study, we evaluated the role of TLR4 in cardiac dysfunction after global ischemia/reperfusion in isolated mouse hearts. Our results show that cardiac functional parameters, including LVDP, +dP/dt max, and LVEDP, recovered faster and to a greater extent after global ischemia/reperfusion in TLR4-defective and TLR4-deleted hearts. Previous studies on models of regional myocardial ischemia/reperfusion showed that TLR4 mutation or inhibition reduces infarct size [7–10]. Our study demonstrates that TLR4 also plays a critical role in cardiac dysfunction after global myocardial ischemia/reperfusion.
Studies have reported that TLR4 mutation or inhibition reduces myocardial cytokine (IL-1β and IL-6) mRNA levels after regional ischemia/reperfusion [7, 8]. We evaluated the influence of TLR4 mutation on myocardial NF-κB activity and levels of TNF-α and IL-1β peptides after global ischemia/reperfusion. We observed that NF-κB p65 phosphorylation was absent in TLR4-mutant hearts and NF-κB DNA-binding activity was markedly reduced. These changes correlated with the abolishment of TNF-α and IL-1β peptide production. Therefore, TLR4 has a critical role in regulating myocardial NF-κB activity and cytokine peptide levels.
Previous studies showed that TNF-α depresses cardiac function in vitro [2] and in vivo [27]. In the present study, we found that TLR4 mutation improves postischemic cardiac function and reduces TNF-α and IL-1β peptide production. It is likely that regulating the production of TNF-α and IL-1β is one mechanism by which TLR4 signaling influences postischemic cardiac function. We also found that cardiac functional recovery after global ischemia/reperfusion was significantly improved in TNF-α and IL-1β knockouts, to a level comparable or close to TLR4-mutant hearts. Because TLR4 levels in TNF-α and IL-1β knockouts are similar to wild-type controls, these effects on postischemic cardiac recovery are likely the result of cytokine deficiency.
The parallel experiments, in which we treated wild-type hearts with TNF-α binding peptide (TNF-BP) or IL-1 receptor antagonist (IL-1RA), corroborated these results. These findings are consistent with the idea that TNF-α and IL-1β contribute to the multifactorial mechanism of postischemic cardiac dysfunction. To further evaluate the role of TNF-α and IL-1β in the modulation of postischemic cardiac function by TLR4 signaling, we added these two cytokines to TLR4-defective hearts during reperfusion. The addition of these two cytokines abrogated the improvement in post-ischemic cardiac functional recovery in TLR4-defective hearts. These results provide new evidence that TNF-α and IL-1β link TLR4 signaling to postischemic cardiac dysfunction.
Note that knocking out the gene for TNF-α provides superb myocardial functional protection. Although the percentage recovery of LVDP was 62.0% in the IL-1β knockout compared with 44.4% in the control, the percentage recovery of LVDP in the TNF-α knockout was 71.9%, comparable with that in TLR4-mutant hearts. TNF-α is known to induce other cytokines, including IL-18, and it has been shown that endotoxin-induced myocardial IL-18 production is reduced in TNF-α knockouts [28] and that IL-18 is involved in myocardial injury after ischemia/reperfusion [19]. Therefore the improved cardiac functional recovery observed in TNF-α knockouts is likely due to suppression of multiple cardiodepressant factors.
The mechanism by which ischemia/reperfusion activates the TLR4 pathway remains unknown. In patients undergoing cardiac operations, endotoxin is released into the circulation during the perioperative period [29]; thus, endotoxin likely contributes to the systemic and myocardial inflammatory response in these patients. Several endogenous agents, including fibronectin [30], high mobility group box 1 protein [31], heat shock protein (HSP) 60 [32], and HSP70 [33] have also been found to activate the TLR4 signaling pathway in vitro. Because HSP70 is released into the circulation during cardiac surgery [34], HSP70 or other endogenous agents released from injured cells during ischemia/reperfusion might activate myocardial TLR4 and mediate the inflammatory response.
In this study we used the isolated heart model to determine the influence of TLR4 signaling on cardiac function. Although the isolated heart model has limitations because it is an artificial experimental protocol that cannot evaluate long-term cardiac dysfunction, one strength of this model is that it eliminates the contribution of bacterial translocation and proinflammatory factors from blood cells and other tissues. It thus permits us to assess the effect of TLR4 signaling on myocardial contractility in the absence of confounding factors. Because no endotoxin was detected in the perfusion buffer by Limulus assay (data not shown), it is likely that the agents that activated TLR4 in our model are myocardial endogenous factors. Further studies are warranted to identify those factors.
In summary, this study demonstrates (1) that hearts with mutant TLR4 (defect or deletion) have improved functional recovery after global ischemia/reperfusion, (2) that TLR4 signaling regulates myocardial production of TNF-α and IL-1β peptides during global ischemia/reperfusion, and (3) that TNF-α and IL-1β mediate the effect of TLR4 signaling on cardiac function after global ischemia/reperfusion. These findings indicate that TLR4 signaling is involved in the myocardial inflammatory response after global ischemia/reperfusion and that TLR4 signaling contributes to cardiac dysfunction after global ischemia/reperfusion through its influence on myocardial production of TNF-α and IL-1β peptides.
Acknowledgments
This work was supported in part by National Institutes of Heart, Lung and Blood Grant HL079051 and National Institute of General Medical Sciences Grant GM-49222. The authors are grateful to Dr Helen Kim for critically reading this manuscript.
References
- 1.Meldrum DR, Meng X, Dinarello CA, et al. Human myocardial tissue TNFα expression following acute global ischemia in vivo. J Mol Cell Cardiol. 1998;30:1683–9. doi: 10.1006/jmcc.1998.0776. [DOI] [PubMed] [Google Scholar]
- 2.Cain BS, Meldrum DR, Dinarello CA, et al. Tumor necrosis factor-alpha and interleukin-1beta synergistically depress human myocardial function. Crit Care Med. 1999;27:1309–18. doi: 10.1097/00003246-199907000-00018. [DOI] [PubMed] [Google Scholar]
- 3.Tomasdottir H, Hjartarson H, Ricksten A, Wasslavik C, Bengtsson A, Ricksten SE. Tumor necrosis factor gene polymorphism is associated with enhanced systemic inflammatory response and increased cardiopulmonary morbidity after cardiac surgery. Anesth Analg. 2003;97:944–9. doi: 10.1213/01.ANE.0000078574.76915.11. [DOI] [PubMed] [Google Scholar]
- 4.Meng X, Ao L, Song Y, Raeburn CD, Fullerton DA, Harken AH. Signaling for myocardial depression in hemorrhagic shock: roles of Toll-like receptor 4 and p55 TNF-alpha receptor. Am J Physiol Regul Integr Comp Physiol. 2005;288:R600–6. doi: 10.1152/ajpregu.00182.2004. [DOI] [PubMed] [Google Scholar]
- 5.Zhai Y, Shen XD, O'Connell R, et al. TLR4 activation mediates liver ischemia/reperfusion inflammatory response via IFN regulatory factor 3-dependent MyD88-independent pathway. J Immunol. 2004;173:7115–9. doi: 10.4049/jimmunol.173.12.7115. [DOI] [PubMed] [Google Scholar]
- 6.Caso JR, Pradillo JM, Hurtado O, Lorenzo P, Moro MA, Lizasoain I. Toll-like receptor 4 is involved in brain damage and inflammation after experimental stroke. Circulation. 2007;115:1599–608. doi: 10.1161/CIRCULATIONAHA.106.603431. [DOI] [PubMed] [Google Scholar]
- 7.Chong AJ, Shimamoto A, Hampton CR, et al. Toll-like receptor 4 mediates ischemia/reperfusion injury of the heart. J Thorac Cardiovasc Surg. 2004;128:170–9. doi: 10.1016/j.jtcvs.2003.11.036. [DOI] [PubMed] [Google Scholar]
- 8.Shimamoto A, Chong AJ, Yada M, et al. Inhibition of Toll-like receptor 4 with eritoran attenuates myocardial ischemia-reperfusion injury. Circulation. 2006;114:I270–4. doi: 10.1161/CIRCULATIONAHA.105.000901. [DOI] [PubMed] [Google Scholar]
- 9.Oyama J, Blais C, Jr, Liu X, et al. Reduced myocardial ischemia-reperfusion injury in toll-like receptor 4-deficient mice. Circulation. 2004;109:784–9. doi: 10.1161/01.CIR.0000112575.66565.84. [DOI] [PubMed] [Google Scholar]
- 10.Stapel H, Kim SC, Osterkamp S, et al. Toll-like receptor 4 modulates myocardial ischaemia-reperfusion injury: Role of matrix metalloproteinases. Eur J Heart Fail. 2006;8:665–72. doi: 10.1016/j.ejheart.2006.03.005. [DOI] [PubMed] [Google Scholar]
- 11.Gurevitch J, Frolkis I, Yuhas Y, et al. Anti-tumor necrosis factor-alpha improves myocardial recovery after ischemia and reperfusion. J Am Coll Cardiol. 1997;30:1554–61. doi: 10.1016/s0735-1097(97)00328-8. [DOI] [PubMed] [Google Scholar]
- 12.Meldrum DR, Dinarello CA, Shames BD, et al. Ischemic preconditioning decreases postischemic myocardial tumor necrosis factor-α production: potential ultimate effector mechanism of preconditioning. Circulation. 1998;98:II214–9. [PubMed] [Google Scholar]
- 13.Wan S, DeSmet JM, Barvais L, Goldstein M, Vincent JL, LeClerc JL. Myocardium is a major source of proinflammatory cytokines in patients undergoing cardiopulmonary bypass. J Thorac Cardiovasc Surg. 1996;112:806–11. doi: 10.1016/S0022-5223(96)70068-5. [DOI] [PubMed] [Google Scholar]
- 14.Deten A, Volz HC, Briest W, Zimmer HG. Differential cytokine expression in myocytes and non-myocytes after myocardial infaction in rats. Mol Cell Biochem. 2003;242:47–55. [PubMed] [Google Scholar]
- 15.Chandrasekar B, Colston JT, Freeman GL. Induction of proinflammatory cytokine and antioxidant enzyme gene expression following brief myocardial ischemia. Clin Exp Immunol. 1997;108:346–51. doi: 10.1046/j.1365-2249.1997.d01-1017.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Meldrum DR, Cleveland JC, Cain BS, Meng X, Harken AH. Increased myocardial tumor necrosis factor-α in a crystalloid-perfused model of cardiac ischemia-reperfusion injury. Ann Thorac Surg. 1998;65:439–43. doi: 10.1016/s0003-4975(97)01297-6. [DOI] [PubMed] [Google Scholar]
- 17.Shames BD, Barton HH, Reznikov LL, et al. Ischemia alone is sufficient to induce TNF-alpha mRNA and peptide in the myocardium. Shock. 2002;17:114–9. doi: 10.1097/00024382-200202000-00006. [DOI] [PubMed] [Google Scholar]
- 18.Kurrelmeyer KM, Michael LH, Baumgarten G, et al. Endogenous tumor necrosis factor protects the adult cardiac myocyte against ischemic-induced apoptosis in a murine model of acute myocardial infarction. Proc Natl Acad Sci U S A. 2000;97:5456–61. doi: 10.1073/pnas.070036297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pomerantz BJ, Reznikov LL, Harken AH, Dinarello CA. Inhibition of caspase 1 reduces human myocardial ischemic dysfunction via inhibition of IL-18 and IL-1beta. Proc Natl Acad Sci U S A. 2001;98:2871–6. doi: 10.1073/pnas.041611398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fantuzzi G, Zheng H, Faggioni R, et al. Effect of endotoxin in IL-1 beta-deficient mice. J Immunol. 1996;157:291–6. [PubMed] [Google Scholar]
- 21.Ao L, Song Y, Fullerton DA, Dinarello CA, Meng X. The interaction between myocardial depressant factors in endotoxemic cardiac dysfunction: Role of TNF-alpha in TLR4-mediated ICAM-1 expression. Cytokine. 2007;38:124–9. doi: 10.1016/j.cyto.2007.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Meng X, Ao L, Meldrum DR, et al. TNF-α and myocardial depression in endotoxemic rats: temporal discordance of an obligatory relationship. Am J Physiol. 1998;275:R502–8. doi: 10.1152/ajpregu.1998.275.2.R502. [DOI] [PubMed] [Google Scholar]
- 23.Shames BD, Meldrum DR, Selzman CH, et al. Increased levels of myocardial IkB-α protein promotes tolerance to endotoxin. Am J Physiol. 1998;275:H1084–91. doi: 10.1152/ajpheart.1998.275.3.H1084. [DOI] [PubMed] [Google Scholar]
- 24.Rosenau C, Emery D, Kaboord B, Qoronfleh MW. Development of a high-throughput plate-based chemiluminescent transcription factor assay. J Biomol Screen. 2004;9:334–42. doi: 10.1177/1087057103261446. [DOI] [PubMed] [Google Scholar]
- 25.Appleyard RF, Cohn LH. Myocardial stunning and reperfusion injury in cardiac surgery. J Card Surg. 1993;8(2 suppl):316–24. doi: 10.1111/j.1540-8191.1993.tb01332.x. [DOI] [PubMed] [Google Scholar]
- 26.Lüss H, Schäfers M, Neumann J, et al. Biochemical mechanisms of hibernation and stunning in the human heart. Cardiovasc Res. 2002;56:411–21. doi: 10.1016/s0008-6363(02)00596-5. [DOI] [PubMed] [Google Scholar]
- 27.Pagani FD, Baker LS, Hsi C, et al. Left ventricular systolic and diastolic dysfunction after infusion of tumor necrosis factor-α in conscious dogs. J Clin Invest. 1992;90:389–98. doi: 10.1172/JCI115873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Raeburn C, Dinarello CA, Zimmerman MA, et al. Neutralization of IL-18 attenuates lipopolysaccharide-induced myocardial dysfunction. Am J Physiol Heart Circ Physiol. 2002;283:H650–7. doi: 10.1152/ajpheart.00043.2002. [DOI] [PubMed] [Google Scholar]
- 29.Casey WF, Hauser GJ, Hannallah RS, Midgley FM, Khan WN. Circulating endotoxin and tumor necrosis factor during pediatric cardiac surgery. Crit Care Med. 1992;20:1090–6. doi: 10.1097/00003246-199208000-00004. [DOI] [PubMed] [Google Scholar]
- 30.Okamura Y, Watari M, Jerud ES, et al. The extra domain A of fibronectin activates Toll-like receptor 4. J Biol Chem. 2001;276:10229–33. doi: 10.1074/jbc.M100099200. [DOI] [PubMed] [Google Scholar]
- 31.Park JS, Gamboni-Robertson F, He Q, et al. High mobility group box 1 protein interacts with multiple Toll-like receptors. Am J Physiol Cell Physiol. 2006;290:C917–24. doi: 10.1152/ajpcell.00401.2005. [DOI] [PubMed] [Google Scholar]
- 32.Ohashi K, Burkart V, Flohe S, Kolb H. Cutting edge: heat shock protein 60 is a putative endogenous ligand of the toll-like receptor-4 complex. J Immunol. 2000;164:558–61. doi: 10.4049/jimmunol.164.2.558. [DOI] [PubMed] [Google Scholar]
- 33.Asea A, Rehli M, Kabingu E, et al. Novel signal transduction pathway utilized by extracellular HSP70: role of toll-like receptor (TLR) 2 and TLR4. J Biol Chem. 2002;277:15028–34. doi: 10.1074/jbc.M200497200. [DOI] [PubMed] [Google Scholar]
- 34.Dybdahl B, Wahba A, Lien E, et al. Inflammatory response after open heart surgery: release of heat-shock protein 70 and signaling through toll-like receptor-4. Circulation. 2002;105:685–90. doi: 10.1161/hc0602.103617. [DOI] [PubMed] [Google Scholar]