

Abstract
The genetic cause of late-onset focal and segmental dystonia remains unknown in most individuals. Recently, mutations in Thanatos-associated protein domain containing, apoptosis associated protein 1 (THAP1) have been described in DYT6 dystonia and associated with some cases of familial and sporadic late-onset dystonia in Caucasians. We are not aware of any previous descriptions of familial dystonia in African Americans or reports of THAP1 mutations in African Americans. Herein, we characterize an African-American (AA) kindred with late-onset primary dystonia, clinically and genetically. The clinical phenotype included cervical, laryngeal and hand-forearm dystonia. Symptoms were severe and disabling for several family members, whereas others only displayed mild signs. There were no accompanying motor or cognitive signs. In this kindred, age of onset ranged from 45 to 50 years and onset was frequently sudden, with symptoms developing within weeks or months. DYT1 was excluded as the cause of dystonia in this kindred. The entire genomic region of THAP1, including non-coding regions, was sequenced. We identified 13 sequence variants in THAP1, although none co-segregated with dystonia. A novel THAP1 variant (c.-237-3G>T/A) was found in 3/84 AA dystonia patient alleles and 3/212 AA control alleles, but not in 5,870 Caucasian alleles. In summary, although previously unreported, familial primary dystonia does occur in African Americans. Genetic analysis of the entire genomic region of THAP1 revealed a novel variant that was specific for African Americans. Therefore, genetic testing for dystonia and future studies of candidate genes must take genetic background into consideration.
Keywords: Dystonia; Genetics; African American; DYT6; THAP1; Adult-Onset Dystonias; Dystonia, Hereditary; Focal Dystonia
INTRODUCTION
Dystonia is a clinical diagnosis based on the presence of sustained involuntary muscle contractions causing twisting, repetitive movements, or abnormal postures. Focal dystonia affects a single body region and may manifest as blepharospasm, masticatory and/or lingual dystonia, spasmodic dysphonia, cervical dystonia, and hand-forearm dystonia (e.g., writer’s cramp). Dystonia may spread to contiguous anatomical segments. Primary focal dystonia is typically late-onset (>26 years) and sporadic [1]. However, approximately 10% of subjects report a positive family history of dystonia [2]. Familial late-onset dystonia may be associated with sequence variants in the Thanatos-associated protein domain containing, apoptosis associated protein 1 (THAP1, DYT6) [2, 3]. Clinical expression is variable and penetrance reduced. In many families with THAP1 mutations identified to date, at least one member had early onset dystonia [2, 3]. Familial dystonia with exclusively late-onset has been associated with the DYT7 locus [4]. DYT13 dystonia may have a late onset, but the majority of cases described had early onset [5]. The genes implicated in DYT7 and DYT13 remain unknown. Additional families without the DYT designation have also been described [6]. The overwhelming majority of studies on dystonia have been conducted with Caucasian subjects. Scarce data suggest that dystonia may be less prevalent in populations of African descent than in Caucasians [7].
We describe an African-American (AA) family in which six members were affected by dystonia with late onset (mean, 47 years). Two additional individuals with milder clinical signs were classified as probably affected. To our knowledge, this is the first report of an AA family with hereditary dystonia.
METHODS
Affected and unaffected family members were examined by A.P. or Z.K.W. at up to three time points. Blood samples were obtained and DNA was extracted from peripheral blood leucocytes as previously described [2, 8]. In addition to freely structured notes, relevant information about the participants’ history, symptoms, and signs was collected on structured questionnaires. The physical examination included tasks intended to visualize possible subclinical signs of dystonia, such as examining the head in various positions, varying phonatory tasks to enhance abductor or adductor dysphonias, and writing. Family members who were considered affected or possibly affected were videotaped. Written informed consent was obtained from all participants. Additionally, written authorization (consent-to-disclose) has been obtained from the family members on the video clip (Video Supplement), who had been given the opportunity to view the video and the manuscript. This study was approved by the institutional review board of Mayo Clinic.
Sequence variants in Exon 5 of TOR1A (DYT1) were excluded with high resolution melting as described by Xiao et al.[8]. Four samples (2 affected subjects, 2 unaffected subjects) from this family were used to comprehensively analyze the entire genomic region of THAP1 (8,757bp, which includes the promoter region, 5’untranslated region [UTR], Exons 1–3, Introns 1–2, and the 3’ UTR). This was done by performing two long-range polymerase chain reactions (PCR) with products of 4,920bp and 5,092bp, respectively, followed by Sanger sequencing. Long-range PCR was carried out using the SequalPrep™ Long PCR Kit from Invitrogen (Carlsbad, CA). Q-Solution from Qiagen (Valencia, CA) was added to the PCR reactions for improved specificity. Touch-down PCR was performed as follows: first, samples were denatured at 95°C for 5 minutes; then, they were subjected to 35 cycles at 94°C for 30 s, 62°C for 30 s and 72°C for 5 min; followed by a 0.2°C decrease of the annealing temperature each cycle, and ending with a 10 min extension at 72°C. PCR products were then examined on a 1% agarose gel. Specific bands of the appropriate size were cut from the gel and the DNA fragments were purified with QIAquick Gel Extraction Kit (QIAGEN). Purified DNA fragments were then sequenced using multiple primer pairs spaced approximately 600bp apart (esupp Table 1) with an Applied Biosystems 3130XL Genetic Analyzer. The sequencing data were reviewed independently by two scientists (J.X. and M.S.L.).
A variant located 5’ to Exon 1 of THAP1 (c.-237-3G>T) was detected in the proband which prompted screening of all available family members along with an additional 41 AA dystonia cases, 106 AA controls, 1569 Caucasian dystonia cases, and 1366 Caucasian unaffected and unrelated controls. To screen for this variant, high resolution melting of Exon 1 and its contiguous 5’ region was performed as previously described (2) using primers E1F and E1R (esupp Table 1). All variants were confirmed with Sanger sequencing in the forward and reverse directions. Fisher’s exact test was used for statistical analyses.
RESULTS
Eight family members were examined and blood samples were obtained from these individuals (Figure 1). Three of the examined family members were definitely affected and two were probably affected. Several family members independently reported signs of dystonia in three deceased family members. These signs were substantiated on family photographs of two members (II:3 and III:4), both showing marked cervical dystonia.
Figure 1. Family Pedigree.
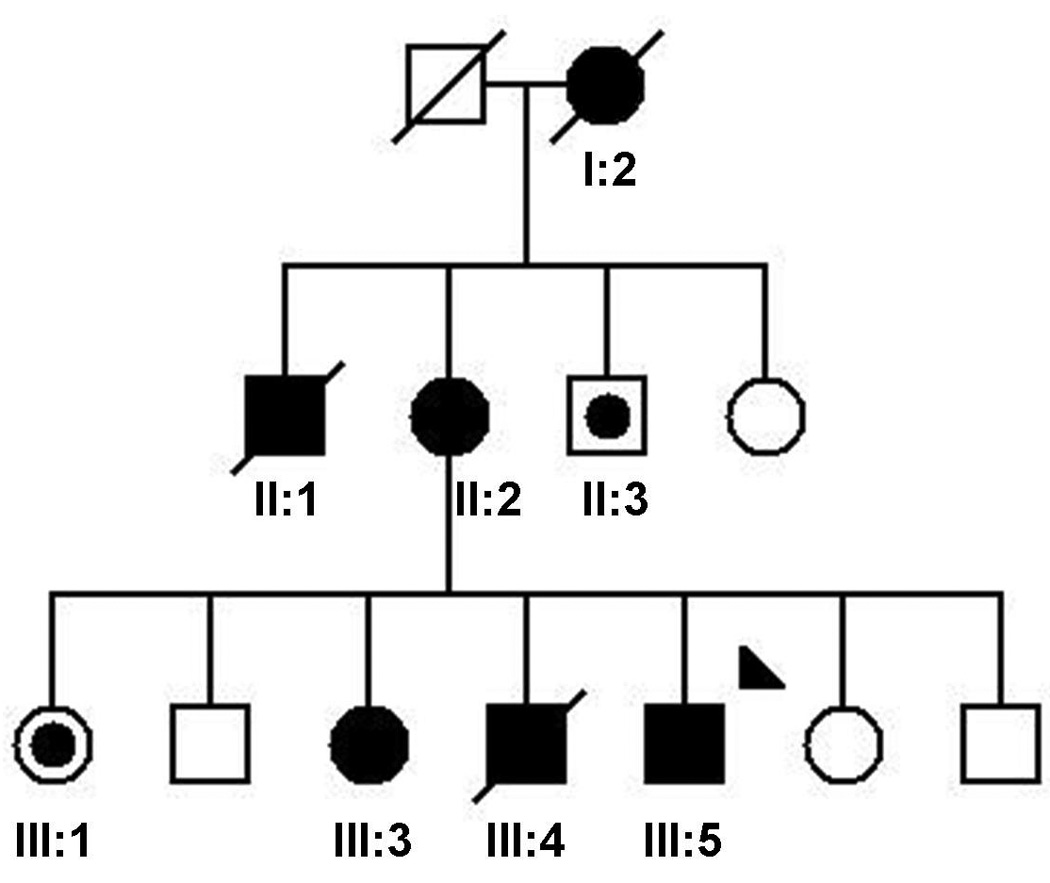
Standard symbols are used. Round symbols indicate females, squares males, diagonal lines indicate the individual is deceased. Solid black symbols denote definitive dystonia; symbols with black points denote probable dystonia. The arrowhead indicates the proband.
Patient descriptions
With the exception of one subject who possibly began to manifest cervical dystonia in his twenties, affected family members remained unaffected until they developed symptoms at age 45 to 63 years of age. Then, signs of dystonia became manifest within weeks to months, leading to marked impairment, and, in the proband’s case, to an inability to continue working in his profession. Symptoms were varied and different forms of focal, segmental or generalized dystonia were found in the affected family members. The clinical data are summarized in Table 2. Representative videos of three affected family members can be viewed in the Video Supplement.
Table 2. Summary of clinical characteristics.
See Methods section for detailed descriptions, and Video Supplement for videos of family members as indicated. FHx, family history.
| ID | Gender | Section in Video Supplement | DNA | Examined | Age at Examination |
Age at Initial Symptom |
Blepharospasm | Oromandbular | Torticollis, Head Tremor |
Dysphonia | Hand-Forearm | Lower Limb | Diagnosis |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| I:2 | F | Deceased | 50 | X | Dystonia (acc to FHx) | ||||||||
| II:1 | M | Deceased | 50 | X | Dystonia (acc to FHx) | ||||||||
| II:2 | F | 2 | Y | Y | 78 | 50 | X | X | X | X | Segmental Dystonia | ||
| II:3 | M | Y | Y | 75 | ? | X | X | Probable Segmental Dystonia | |||||
| III:1 | F | Y | Y | 64 | 63 | X | X | X | Probable Generalized Dystonia | ||||
| III:2 | M | Y | Y | 61 | unaffected | ||||||||
| III:3 | F | 3 | Y | Y | 53 | 50 | X | X | X | X | Segmental Dystonia | ||
| III:4 | M | Deceased | 20's | X | Dystonia (acc to FHx) | ||||||||
| III:5 | M | 1 | Y | Y | 46 | 45 | X | Focal Dystonia | |||||
| III:6 | F | Y | Y | 48 | unaffected | ||||||||
| III:7 | M | Y | Y | 43 | unaffected |
The proband (III:5) is an AA man whose head started to turn towards the right side at the age of 45 years. He also developed a head tremor. There were no precipitating events, and, in particular, no trauma. Significant worsening of his symptoms occurred within weeks, causing severe neck pain. Touching his cheek with his hand helped to ameliorate the involuntary movements. Similarly, warmth (from a heating pad), and lying down improved the situation. On examination, almost constant head turning towards the right side and an irregular “no-no” phasic head tremor were noted. There was palpable hypertrophy of the left sternocleidomastoid muscle. While walking, his head was turning almost 90 degrees towards the right side, which made his gait slow and cautious.
Detailed neurological examination revealed no other abnormalities; specifically, there were no eye movement abnormalities, ataxia or Parkinsonism. The dystonia was functionally debilitating and he was unable to continue in his occupation as a firefighter. Blood ceruloplasmin was slightly elevated, arguing against Wilson’s disease. Electromyographic (EMG) mapping of dystonia revealed either intermittent or constant phasic and tonic activity in the neck musculature, consistent with cervical dystonia. Magnetic resonance imaging (MRI) of the brain and MRI angiography of the cervical and intracranial blood vessels was normal. A trial of levodopa, titrated to a maximum dose of 750mg/d, was discontinued after 2 weeks due to lack of efficacy.
The patient received polypharmacotherapy with clonazepam, trihexyphenidyl, tizanidine and gabapentin, which improved the patient’s dystonia minimally, but caused unwanted side effects. The patient received several sets of injections of botulinum toxin type A (180 – 250 units) into the cervical paraspinal and sternocleidomastoid muscles and into the right splenius capitis and upper trapezius muscles. Responses were variable and incomplete, but did allow the patient to move his head to neutral position and hold it there, which had been impossible before the injections (Video Supplement).
The proband’s mother (II:2) developed head shakes, speech problems, and facial “twitches” at 50 years of age. She had never received any treatment for this problem. On examination, she had oral-facial dystonia with blepharospasm. She also had mild torticollis and marked spasmodic dysphonia of the adductor type which made her speech almost unintelligible at times (Video Supplement).
The proband’s sister (III:3) noted a strained voice at the age of 50 years, which slowly progressed in intensity. She complained about one-sided shoulder tension, and, on examination, had dysphonia (abductor type), perioral dyskinesias, and slight torticollis.
The patient reported she was able to speak with normal voice immediately after waking up in the morning, but then her voice quality deteriorating during the remainder of the day (Video Supplement).
The proband’s brother (III:4), maternal uncle (II:3) and maternal grandmother (I:2) are deceased but had very similar signs and symptoms according to family history.
Two individuals (II:3 and III:1) had mild signs of focal or segmental dystonia on examination and were considered probably affected. II:3 had difficulty writing, using unusually high muscle force to hold and guide the pen, indicative of focal hand-forearm dystonia (writer’s cramp), and had a strained voice suggestive of spasmodic dysphonia. III:1 was unable to rotate her head quickly from one side to another and had slight head tremor and torticollis. During ambulation, her left foot tended to rotate outward in comparison with her right foot.
Genetic studies
A novel variant located 5’ to Exon 1 of THAP1, (c.-237-3G>T), was initially detected in the proband and his affected mother, which prompted screening of an AA case-control series. The c.-237-3G>T variant was found in one unrelated control. At the same location, a c.-237-3G>A variant was present in one subject with laryngeal dysphonia (adductor subtype) and two unrelated controls. Taken together, in the AA cohort, a c.-237-3G>T or c.-237-3G>A substitution was present in 3/84 (3.6%) alleles from dystonia patients and 3/212 (1.4%) alleles from unaffected controls (p=0.36). None of these variants was detected in 3,138 alleles from Caucasian patients or 2,732 alleles from Caucasian controls. Subsequently, as more samples from the family described here became available, the THAP1 (c.-237-3G>T) variant was found in 3/4 additional unaffected family members but not in the proband’s affected sister. More detailed analysis with comprehensive sequencing of the entire THAP1 genomic region revealed thirteen sequence variants in four members in this family (esupp Table 3). Five of these variants had already been reported in the NCBI SNP database (http://www.ncbi.nlm.nih.gov/snp). Although eight variants were novel, none co-segregated with dystonia.
DISCUSSION
To our knowledge, this is the first description of an AA family with late-onset primary dystonia. Within this kindred, clinical manifestations ranged from very mild to severely disabling. The distribution of affected individuals within the pedigree suggests autosomal dominant inheritance, but no causal variants were identified in THAP1 (DYT6) or Exon 5 of TOR1A (DYT1). Our study sheds light on dystonia in non-Caucasians, which so far has been markedly underreported and understudied. The previous concentration of dystonia research on Caucasians populations may have compromised our search for new disease genes and associated molecular pathological mechanisms.
Very little is known about dystonia in non-Caucasians. In Oslo, Norway, dystonia prevalence was based on how many individuals of European or of non-European descent had sought medical care for their condition. This value was almost 10-fold lower for subjects of Asian or African descent than for those of European descent [7]. Dystonia in AAs, who comprise 12.9 percent (36.2 million people) of the total population of the USA [9], has been assessed in a few previous publications. For example, AA individuals with sporadic or familial AA dystonia patients have been described in case reports [10, 11]. These reported cases had severe early-onset generalized dystonia. A medical record survey was used to investigate the incidence of torticollis in a Northern California population that included 8% AAs [12]. Ethnicity was known for 57 individuals with a torticollis diagnosis in their medical records. None of these cases was AA. Direct comparison of AA, Ashkenazi and non-Jewish Caucasian dystonia patients seen in a movement disorder clinic was performed in one study. Compared to the other two groups, AA dystonia patients tended to have an intermediate age at onset, more common cranial and laryngeal onset, and less frequent leg onset. However, only 29 AA patients were studied and the differences did not reach statistical significance [13]. All of these studies were based on data from individuals who had contact with health services. However, as long as dystonia symptoms are not debilitating, many sufferers may not consult a health service provider, as was the case in several members of the family we describe herein. Thus, these studies could not resolve the question of whether there is a true difference in the age of onset, presentation or incidence of dystonia among the ethnic groups studied, or whether the findings were a consequence of ethnic differences in health-care utilization patterns, referral bias and ascertainment bias. At this time, we are not aware of a truly population-based (e.g. door-to-door) study directly comparing these dystonia variables among the different ethnic groups living in the USA.
With the exception of one family with American-Indian ancestry [14] and a clinical description of an AA family with definitive dystonia in one member and possible formes frustes in two others [15], all published individuals with familial late-onset primary dystonia are of Caucasian origin. Although DYT1 dystonia is most commonly associated with early-onset dystonia in a leg, late-onset primary dystonia can also be caused by the same GAG deletion in Exon 5 of TOR1A [8, 14]. Other loci have been associated with familial late-onset dystonia (DYT7 and DYT13). DYT7 is a predominantly focal dystonia with an age of onset from 28 to 70 years, whose locus has been mapped to chromosome 18p [4]. One family with initially focal dystonia and ages at onset of 5 to 40 years was designated as DYT13 [16], and, at follow-up, dystonia had spread to other body parts [5]. Additional published families with familial early-onset primary dystonia have been reviewed [6]. Recently, Fuchs et al. identified mutations in THAP1 as the cause of DYT6 dystonia [3]. In this report, affected subjects had age at onset ranging from 5 to 38 years, and carriers had focal, segmental, multifocal or generalized dystonia phenotypes. The original mutations described were a point substitution and an insertion/deletion [3]. In the interim, over 40 sequencing variants have been reported when screening dystonia patients [2, 17–21], but similarly, all but one [21] was Caucasian. Orofacial involvement and spasmodic dysphonia are frequently observed in patients with THAP1 mutations, and myoclonic jerks have been described as another DYT6 feature [2, 17–22].
In this study, after excluding variants in Exon 5 of TOR1A, we employed long-range PCR to facilitate sequencing the entire genomic region of THAP1 and detected 13 sequence variants that did not co-segregate with the dystonia (esupp Table 3). One novel THAP1 variant (c.-237-3G>T/A) was found in the AA population but not in Caucasians, despite extensive screening. Although the rs71521601 variant has been considered as a possible dystonia risk allele in Caucasians [21], we only detected it in 1/4 alleles of dystonia patients in this study, thus its association with dystonia in other populations requires additional investigation. The c.-237_236GA>TT sequence variant was reported as a dystonia risk allele [18] in one report but not replicated in an independent study [23]; it was not found in our 4 subjects.
The clinical phenotype described in this family appears to differ from the known phenotypes discussed above. First of all, the dystonia manifest in our AA kindred may be more severe, on average, than the dystonias associated with DYT6, DYT7 and DYT13, as several family members were significantly impaired by their symptoms. The proband, III:5, was disabled such that he discontinued working, and, I:2 and II:1, of whom we only saw photographs, were also severely affected. The voice of II:2, and, sometimes also of II:3, was significantly impaired. Second, in contrast to the limb onset typical of DYT1, symptoms in this family were concentrated in the craniocervical region. Although DYT6 is a diagnostic consideration, mutations in THAP1 were largely excluded by Sanger sequencing. Moreover, unlike DYT7 dystonia, none of our affected patients showed evidence of a hand tremor. Thirdly, the mean age of onset in our AA kindred was much latter than that described for DYT13 and most DYT6 families.
Further studies of this and other AA families with dystonia may lead to the discovery of new disease-associated genes and to increased awareness of dystonia in the AA population in the United States. It appears prudent to include individuals of various ethnic backgrounds in the search for new genes associated with dystonia and other movement disorders, bearing in mind that our concept of race may be based largely on superficial traits such as skin color, which only poorly define genetically distinct populations [24]. Our data also highlight that future studies of candidate genes and genetic risk factors in dystonia must take genetic background into consideration.
Supplementary Material
Tm, melting temperature.
†, N1–N8 denote novel variants that have not been reported in the NCBI SNP database.
The video shows three affected family members. Section 1 is mute; sections 2 and 3 include sound.
Section 1: Three weeks after the last injection of botulinum toxin type A, the proband (III:5) has marked torticollis and head tremor, and shows how his head deviates to the right and starts to shake. The patient demonstrates how touching his right cheek with his hand abolishes the dystonic tremor and slightly improves the posturing (sensory trick).
Section 2: The proband’s mother (II:2) pronounces the days of the week, but is severely affected by adductor type of spasmodic dysphonia. Especially after she speaks, oromandibular dystonic movements can clearly be seen.
Section 3: The voice of the proband’s sister (III:3) and her description that it is normal after a night’s rest, is clearly indicative of spasmodic dysphonia (abductor subtype).
ACKNOWLEDGEMENTS
The authors wish to express their gratitude to all members of this family who participated in the study. We thank Audrey Strongosky, Mayo Clinic in Florida, for preparing the graphics of the pedigree (Figure 1). Andreas Puschmann received funding from the Swedish Parkinson Academy, AFA Insurance, The Swedish Parkinson Foundation, Apotekare Hedbergs Foundation, Elsa Schmitz Foundation, and Lund University Research Foundation. The Swedish Parkinson Academy, The Research Foundation of the Swedish Parkinson’s Disease Association, Lund University Research Fund, and The Royal Physiographic Society in Lund, Sweden. Mark S. LeDoux was supported by the Neuroscience Institute at the University of Tennessee Health Science Center, Dystonia Medical Research Foundation, NIH grants R01NS048458 and R01NS069936, NIH U54 Dystonia Coalition (1U54NS065701) Pilot Projects Program, and the Parkinson’s & Movement Disorder Foundation. Zbigniew K. Wszolek is partially supported by the NIH/NINDS 1RC2NS070276, NS057567, P50NS072187, Mayo Clinic Florida (MCF) Research Committee CR programs (MCF #90052030 and MCF #90052030), and the gift from Carl Edward Bolch, Jr., and Susan Bass Bolch (MCF #90052031/PAU #90052).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.de Carvalho Aguiar PM, Ozelius LJ. Classification and genetics of dystonia. Lancet Neurol. 2002 Sep;1(5):316–325. doi: 10.1016/s1474-4422(02)00137-0. [DOI] [PubMed] [Google Scholar]
- 2.Xiao J, Zhao Y, Bastian RW, Perlmutter JS, Racette BA, Tabbal SD, et al. Novel THAP1 sequence variants in primary dystonia. Neurology. 2010 Jan 19;74(3):229–238. doi: 10.1212/WNL.0b013e3181ca00ca. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fuchs T, Gavarini S, Saunders-Pullman R, Raymond D, Ehrlich ME, Bressman SB, et al. Mutations in the THAP1 gene are responsible for DYT6 primary torsion dystonia. Nat Genet. 2009 Mar;41(3):286–288. doi: 10.1038/ng.304. [DOI] [PubMed] [Google Scholar]
- 4.Leube B, Hendgen T, Kessler KR, Knapp M, Benecke R, Auburger G. Evidence for DYT7 being a common cause of cervical dystonia (torticollis) in Central Europe. Am J Med Genet. 1997 Sep 19;74(5):529–532. doi: 10.1002/(sici)1096-8628(19970919)74:5<529::aid-ajmg15>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- 5.Bentivoglio AR, Ialongo T, Contarino MF, Valente EM, Albanese A. Phenotypic characterization of DYT13 primary torsion dystonia. Mov Disord. 2004 Feb;19(2):200–206. doi: 10.1002/mds.10634. [DOI] [PubMed] [Google Scholar]
- 6.Defazio G, Berardelli A, Hallett M. Do primary adult-onset focal dystonias share aetiological factors? Brain. 2007 May;130(Pt 5):1183–1193. doi: 10.1093/brain/awl355. [DOI] [PubMed] [Google Scholar]
- 7.Le KD, Nilsen B, Dietrichs E. Prevalence of primary focal and segmental dystonia in Oslo. Neurology. 2003 Nov 11;61(9):1294–1296. doi: 10.1212/01.wnl.0000090463.05980.59. [DOI] [PubMed] [Google Scholar]
- 8.Xiao J, Bastian RW, Perlmutter JS, Racette BA, Tabbal SD, Karimi M, et al. High-throughput mutational analysis of TOR1A in primary dystonia. BMC Med Genet. 2009;10:24. doi: 10.1186/1471-2350-10-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McKinnon J, Bennett C. In: We the People: Blacks in the United States. Bureau USC, U.S. Department of Commerce EaSA, editor. 2005. [Google Scholar]
- 10.Zeman W, Dyken P. Dystonia musculorum deformans. Clinical, genetic and pathoanatomical studies. Psychiatr Neurol Neurochir. 1967 Mar–Apr;70(2):77–121. [PubMed] [Google Scholar]
- 11.Golden GS. Dystonia in the black and Puerto Rican population. Adv Neurol. 1976;14:121–124. [PubMed] [Google Scholar]
- 12.Marras C, Van den Eeden SK, Fross RD, Benedict-Albers KS, Klingman J, Leimpeter AD, et al. Minimum incidence of primary cervical dystonia in a multiethnic health care population. Neurology. 2007 Aug 14;69(7):676–680. doi: 10.1212/01.wnl.0000267425.51598.c9. [DOI] [PubMed] [Google Scholar]
- 13.Almasy L, Bressman S, de Leon D, Risch N. Ethnic variation in the clinical expression of idiopathic torsion dystonia. Mov Disord. 1997 Sep;12(5):715–721. doi: 10.1002/mds.870120515. [DOI] [PubMed] [Google Scholar]
- 14.Ozelius LJ, Hewett JW, Page CE, Bressman SB, Kramer PL, Shalish C, et al. The early-onset torsion dystonia gene (DYT1) encodes an ATP-binding protein. Nat Genet. 1997 Sep;17(1):40–48. doi: 10.1038/ng0997-40. [DOI] [PubMed] [Google Scholar]
- 15.Weir R. Torsion dystonia: a case report. J Natl Med Assoc. 1977 Feb;69(2):99–101. [PMC free article] [PubMed] [Google Scholar]
- 16.Bentivoglio AR, Del Grosso N, Albanese A, Cassetta E, Tonali P, Frontali M. Non-DYT1 dystonia in a large Italian family. J Neurol Neurosurg Psychiatry. 1997 Apr;62(4):357–360. doi: 10.1136/jnnp.62.4.357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bressman SB, Raymond D, Fuchs T, Heiman GA, Ozelius LJ, Saunders-Pullman R. Mutations in THAP1 (DYT6) in early-onset dystonia: a genetic screening study. Lancet Neurol. 2009 May;8(5):441–446. doi: 10.1016/S1474-4422(09)70081-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Djarmati A, Schneider SA, Lohmann K, Winkler S, Pawlack H, Hagenah J, et al. Mutations in THAP1 (DYT6) and generalised dystonia with prominent spasmodic dysphonia: a genetic screening study. Lancet Neurol. 2009 May;8(5):447–452. doi: 10.1016/S1474-4422(09)70083-3. [DOI] [PubMed] [Google Scholar]
- 19.Bonetti M, Barzaghi C, Brancati F, Ferraris A, Bellacchio E, Giovanetti A, et al. Mutation screening of the DYT6/THAP1 gene in Italy. Mov Disord. 2009 Dec 15;24(16):2424–2427. doi: 10.1002/mds.22861. [DOI] [PubMed] [Google Scholar]
- 20.Van Gerpen JA, Ledoux MS, Wszolek ZK. Adult-onset leg dystonia due to a missense mutation in THAP1. Mov Disord. 2010 Jul 15;25(9):1306–1307. doi: 10.1002/mds.23086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Houlden H, Schneider SA, Paudel R, Melchers A, Schwingenschuh P, Edwards M, et al. THAP1 mutations (DYT6) are an additional cause of early-onset dystonia. Neurology. 2010 Mar 9;74(10):846–850. doi: 10.1212/WNL.0b013e3181d5276d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Clot F, Grabli D, Burbaud P, Aya M, Derkinderen P, Defebvre L, et al. Screening of the THAP1 gene in patients with early-onset dystonia: myoclonic jerks are part of the dystonia 6 phenotype. Neurogenetics. 2011 Feb;12(1):87–89. doi: 10.1007/s10048-010-0264-3. [DOI] [PubMed] [Google Scholar]
- 23.Xiao J, Zhao Y, Bastian RW, Perlmutter JS, Racette BA, Tabbal SD, et al. The c.-237_236GA>TT THAP1 sequence variant does not increase risk for primary dystonia. Mov Disord. 2011 Mar;:2. doi: 10.1002/mds.23551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Relethford JH. Race and global patterns of phenotypic variation. Am J Phys Anthropol. 2009 May;139(1):16–22. doi: 10.1002/ajpa.20900. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Tm, melting temperature.
†, N1–N8 denote novel variants that have not been reported in the NCBI SNP database.
The video shows three affected family members. Section 1 is mute; sections 2 and 3 include sound.
Section 1: Three weeks after the last injection of botulinum toxin type A, the proband (III:5) has marked torticollis and head tremor, and shows how his head deviates to the right and starts to shake. The patient demonstrates how touching his right cheek with his hand abolishes the dystonic tremor and slightly improves the posturing (sensory trick).
Section 2: The proband’s mother (II:2) pronounces the days of the week, but is severely affected by adductor type of spasmodic dysphonia. Especially after she speaks, oromandibular dystonic movements can clearly be seen.
Section 3: The voice of the proband’s sister (III:3) and her description that it is normal after a night’s rest, is clearly indicative of spasmodic dysphonia (abductor subtype).