

Abstract
The cloning of GFP 15 years ago revolutionized cell biology by permitting visualization of a wide range of molecular mechanisms within living cells. Though initially used to make largely qualitative assessments of protein levels and localizations, fluorescence microscopy has since evolved to become highly quantitative and high-throughput. Computational image analysis has catalyzed this evolution, enabling rapid and automated processing of large datasets. Here we review studies that combine time-lapse fluorescence microscopy and automated image analysis to investigate dynamic events at the single-cell level. We highlight examples where single-cell analysis provides unique mechanistic insights into cellular processes that cannot be otherwise resolved in bulk assays. Additionally, we discuss studies where quantitative microscopy facilitates the assembly of detailed 4D lineages in developing organisms. Finally, we describe recent advances in imaging technology, focusing especially on platforms that allow the simultaneous perturbation and quantitative monitoring of biological systems.
Keywords: cell-to-cell variability, automated image analysis, lineage construction, microfluidic devices, fluorescent proteins
Introduction
In many standard biological assays there is a tradeoff among molecular specificity, spatial resolution, and temporal sampling. For instance, electron micrographs have extremely high spatial resolution, but they generally cannot image specific molecules, and the temporal dimension is lost since sample preparation requires fixation. Southern, northern, and western blots are similarly compromised: they have high molecular specificity but lack spatial resolution since they measure molecular abundance at the population level, not the single-cell level. Even PCR, which is highly sequence-specific and can be performed in single cells (Bengtsson et al 2005, Peixoto et al 2004), requires that cells be lysed, preventing sequential sampling from the same cell over time.
Genetically encoded fluorescent proteins overcome this tradeoff. They enable high molecular specificity via direct fusion to proteins of interest using molecular cloning techniques. They afford high spatial resolution: via techniques such as flow cytometry and microscopy allow detection of fluorescent proteins at the single-cell level and even permit spatial localization at effectively arbitrary resolution when sparsely distributed throughout the cell (Kural et al 2005). Finally, fluorescent proteins permit temporal sampling over a wide range of timescales in live cells, since fluorescence is generally nontoxic.
Since imaging experiments can involve several distinct molecular species (e.g., using differently colored fluorophores), thousands of individual cells, and hundreds to thousands of time points, manual attempts at quantitative analysis of time-lapse microscopy data would be tedious, error-prone, and frankly impossible in many cases. Fortunately, automated image analysis can quickly and quantitatively detect dynamic features of interest in an unbiased manner. In general, image analysis algorithms segment an image into regions based on the intensities of adjacent groups of pixels. These regions can then be rapidly classified based on many criteria, including their intensity, shape, size, velocity, pixel-to-pixel variability, colocalization with regions in other color channels, and countless more. Regions of interest can then be tracked over time, and any number of quantitative phenotypes can be extracted.
Though countless research efforts in cell and developmental biology have exploited fluorescent proteins, we restrict our focus here to papers that specifically couple time-lapse fluorescence microscopy with automated image analysis. We will first review papers in which the use of quantitative, time-lapse, single-cell imaging revealed cell-to-cell variability in the timing of cellular events. Second, we will survey papers that focus less on inter-cell comparisons and more on intra-cell dynamics, like the coordination of multiple events that overlap in space and time, the behavior of macromolecular structures in the cytoskeleton, and the various regulatory roles of feedback loops. Third, we will discuss novel insights gained from genealogical data that involves dynamically tracking single cells within a growing population. Fourth, we will highlight research that couples microfluidic devices with microscopy, allowing for exquisite control over extracellular conditions while simultaneously monitoring dynamic intracellular responses. Finally, we will feature new developments in single-cell quantitative imaging that increase resolution, throughput, and control over extracellular and intracellular conditions.
1. Inter-cell comparisons of spatiotemporal dynamics
Intro
It is now well established that a population of genetically identical cells can exhibit extensive cell-to-cell variability in the expression levels of many genes (Elowitz et al 2002, Newman et al 2006, Ozbudak et al 2002). This variability, termed “noise”, is reflected in the width of a population distribution, or histogram, representing the whole-cell fluorescence values for all single cells; mathematically, it is often defined as the ratio between the standard deviation and the mean expression level of the population. In principle, a histogram can summarize how any quantitative trait is distributed across a population of cells. Thus, in addition to noise in gene expression, noise in many other phenotypes can also be measured and studied. Since noise in gene expression is nicely reviewed elsewhere (Kaern et al 2005, Maheshri & O'Shea 2007, Raj & van Oudenaarden 2008), below we focus on papers that investigate cell-to-cell variability in other quantitative metrics that rely specifically on the spatial and/or temporal information that time-lapse microscopy provides.
1A. Temporal noise
To quantify noise in the timing of a series sequential events, histograms reflecting the time elapsed between pairwise events in a population of single cells must be compiled. The Ramanathan lab recently used quantitative microscopy to measure noise in the timing of meiotic stages in budding yeast (Nachman et al 2007). Triggered by nutrient deprivation, meiosis in diploid yeast cells is comprised of a series of sequential events: expression of meiotic regulators, a round of DNA replication, and two successive nuclear division events (called MI and MII), ultimately yielding four haploid spores. The researchers could temporally distinguish several of these meiotic stages by tracking the level and localization of the early meiotic regulator Dmc1, which they fused to YFP (“Dmc1-YFP”). Since Dmc1 binds meiotic chromosomes (Bishop 1994), automated image analysis of Dmc1-YFP clusters in single cells could determine whether a cell had undergone zero, one, or two nuclear-division events (leading to 1, 2, or 4 clusters of Dmc1-YFP, respectively) (Figure 1a). There was considerable cell-to-cell variability in the timing between nutrient deprivation and MII, effectively the first and last meiotic landmarks, respectively (“stress-to-end time” in Figure 1b). But, since the researchers had time measurements of multiple intermediate steps, they could determine to what extent variability in the entire process was influenced by variability in the intermediate processes. They found that total meiotic timing noise was dominated specifically by variability in the time between nutrient deprivation and MI; furthermore, they determined that this variability was largely accounted for by variability in the time between nutrient deprivation and the onset of expression of meiotic regulators (Figure 1b). They also used their rich single-cell dataset to demonstrate that the duration of successive stages was largely uncorrelated. This observation is noteworthy since correlation (anticorrelation) among the durations of intermediate events would increase (decrease) noise of the whole event (Figure 1c). In sum, this work by Nachman et al. highlights and critically relies upon several key advantages of quantitative time-lapse imaging and automated analysis: their phenotype of interest was defined by the subcellular localization of a fluorescent marker and required that single cells be tracked over long time periods.
Figure 1.
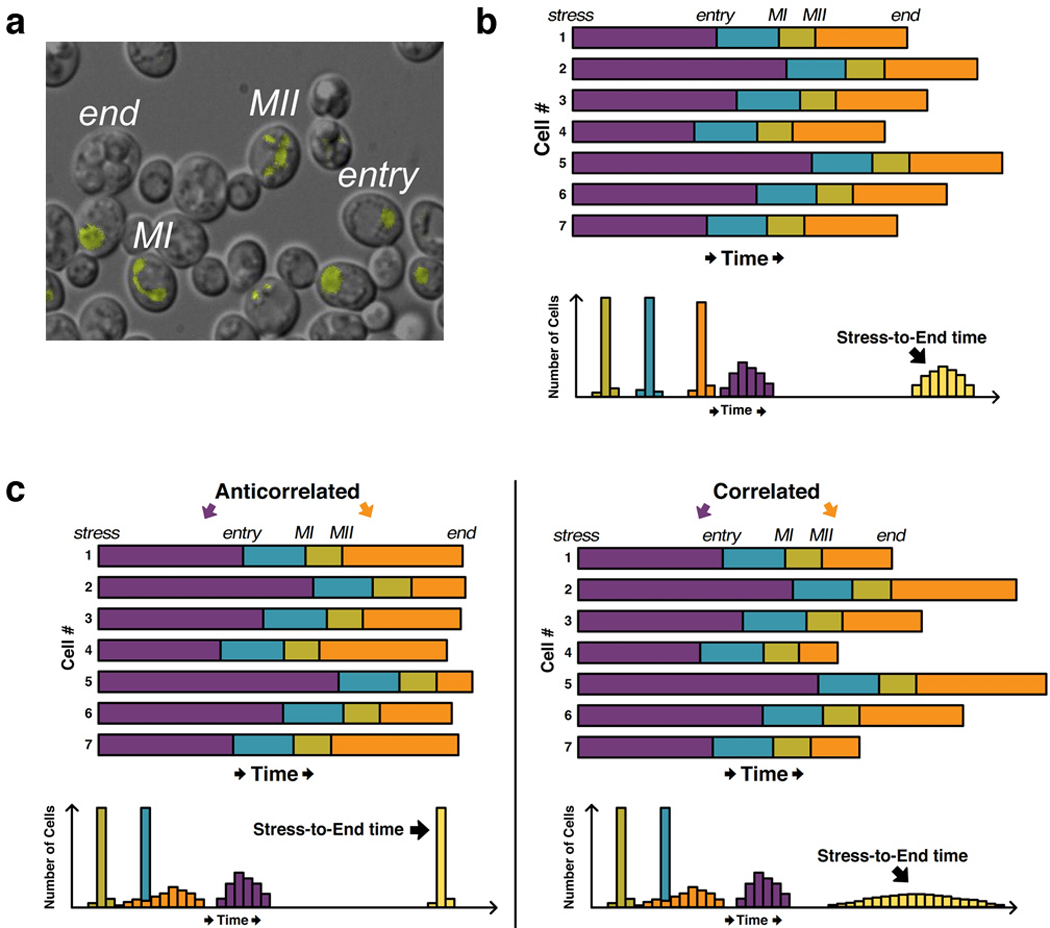
Cell-to-cell variability in the timing of meiotic events. (a) Meiotic regulator Dmc1 was fused to YFP and visualized in live budding yeast cells. Since Dmc1 associates with chromosomes, the stage of meiotic progression (e.g., entry, MI, MII, end) could be tracked using image-analysis software that effectively detects the number of Dmc1-YFP foci (e.g., entry has one focus, MI has two, and MII has four). (b) Top: Hypothetical data for seven single cells based on the results in Nachman et al., where the length of the shaded bars corresponds to the amount of time spent in each stage. Nutritional stress was applied to all cells at the same time, hence their alignment at the “stress” marker on the left. Bottom: Hypothetical histograms reflecting the time spent in each stage across many cells. Note that variability in the total time (i.e., “stress-to-end time”) is dominated by the stress-to-entry time and not by variability in the duration of other stages, as observed by Nachman et al. (c) Left: Hypothetical single-cell profiles (top) and histograms (bottom) illustrating how anticorrelation between the stress-to-entry time and the MII-to-end time lead to a reduction in stress-to-end variability as compared to (b). Right: Correlation between stress-to-entry and MII-to-end times leads to more variability in the stress-to-end time than in (b). Panel (a) reproduced from Reference (Nachman et al 2007) with permission of Cell.
The Siggia and Cross labs also elegantly investigated temporal noise in the budding yeast cell cycle, focusing on the “Start” checkpoint, which marks the beginning of the G1/S transition and commitment to cell division. Start involves the coordinated activation of many events, including transcriptional activation of a battery of genes, bud emergence, and the duplication of microtubule organizing centers (ref). To measure temporal coordination between two of these events (gene activation and bud emergence) in single cells, the researchers expressed GFP under the control of the promoter for the cyclin CLN2 gene, which is one of the genes upregulated at Start. Time-lapse fluorescence microscopy coupled with automated image analysis quantified cell-to-cell variability in Start “coherence”, defined as the time between bud emergence and peak CLN2-GFP expression (“bud-to-peak time” in Figure 2a). Interestingly, by comparing histograms compiled in wildtype and mutant cells, the authors subsequently identified Swi4 as a protein required for low cell-to-cell variability in Start coherence. This decrease in Start coherence in the absence of Swi4 is noteworthy, as it coincided with considerably higher temporal variability in cell-division times, which they tracked via a fluorescent fusion protein that marks the septin ring and fades rapidly upon cytokinesis (Figure 2a). This study is significant in that it identifies a common molecular determinant of two distinct temporal noise phenotypes (in Start coherence and cell-cycle duration) that can only be measured with quantitative microscopy. In a follow-up investigation (Di Talia et al 2007), this group further partitioned the G1 stage of the cell-cycle into two phases distinguished by the determinants of their temporal noise properties. Differences in cell volume and Cln3 levels largely accounted for variability in the first phase, whereas temporal variability in the second phase was volume-independent and arose mainly from differential expression of CLN2.
Figure 2.
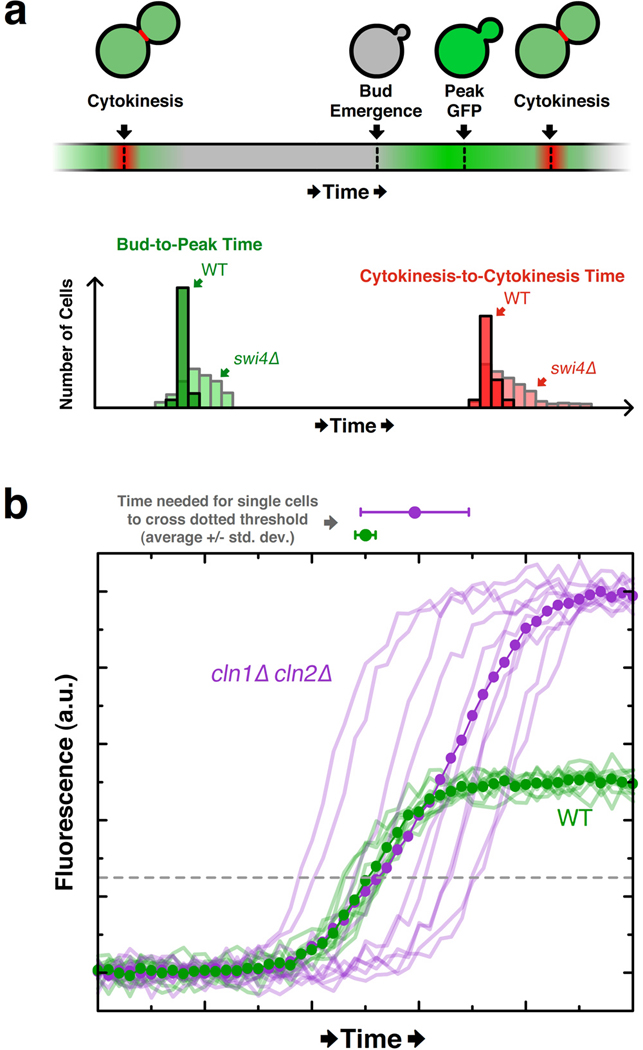
Variability in the timing of mitotic cell-cycle events and positive feedback revealed by single-cell traces. (a) Top: Schematic of single-cell events tracked via image analysis software in investigation of budding-yeast Start checkpoint. The timing of cytokinesis was determined by the rapid disappearance of the Cdc10-GFP fusion protein (here shown in red) from the bud neck. The green signal corresponds to expression of GFP under the control of the CLN2 promoter (“CLN2-GFP”). Bottom: Hypothetical histograms based on findings of Bean et al. in which swi4Δ cells exhibit considerably higher variability in the timing both between bud emergence and peak CLN2-GFP signal, and between successive cytokinesis events. (b) Hypothetical single-cell data (faded lines) illustrating how bulk measurements (dotted opaque line) mask the observation that CLN2-GFP is expressed earlier in wildtype cells than in cln1Δcln2Δ cells, as shown in Skotheim et al. The average traces overlap for much of the trajectory and give no indication that wildtype cells express CLN2-GFP earlier than mutant cells. However, the average time at which single-cell CLN2-GFP levels cross an arbitrary threshold (dashed line) is notably different between the cell lines (see dots and error boundaries above the plot).
1B. Revealed pitfalls of bulk assays
A recent paper from the Cross and Siggia labs nicely illustrated the perils of bulk measurements that single-cell analysis avoids (Skotheim et al 2008). Previous bulk measurements showed that activation of CLN2 expression occurred with similar kinetics in wildtype cells and in cln1Δ cln2Δ cells (Stuart & Wittenberg 1995), leading to the conclusion that Cln1 and Cln2 do not positively autoregulate their own expression. However, single-cell analysis of CLN2-GFP with time-lapse microscopy revealed several critical expression differences between wildtype and cln1Δ cln2Δ cells (Skotheim et al 2008). In particular, CLN2-GFP expression occurred more rapidly in wildtype cells than in mutant cells, indicative of positive feedback by Cln1 and Cln2. At the same time, though, CLN2-GFP expression was higher and had larger temporal variability in mutant cells than in wildtype cells. Together, these three effects (i.e., changes in magnitude, timing, and timing noise) mask the evidence for positive feedback when averaged over many cells (Figure 2b). In a population average, the few early and highly expressing mutant cells compensate for the late and lowly expressing cells to yield a measurement nearly identical to that of the wildtype population. Thus, Skotheim et al. used quantitative single-cell microscopy to reveal a critical positive feedback loop that they later show is required for robust bud emergence and coherence (i.e., low temporal noise) in transcriptional activation of G1/S genes.
Single-cell measurements have also demonstrated inaccuracies in our understanding of the behavior of the tumor suppressor p53 in response to DNA damage (Lahav et al 2004). Bulk western blot analysis of p53 showed that its levels undergo damped oscillations in response to DNA damage (Lev Bar-Or et al 2000). However, damped oscillations in bulk can result from several different types of single-cell behavior (Figure 3). When Lahav and colleagues fused CFP to p53 and assayed the level of p53-CFP in single human cells in response to DNA damage, they found that the damping observed in bulk was not due to a uniform decrease in the magnitude of successive pulses (Figure 3a) or decaying synchrony (Figure 3b). Instead, each cell had a discrete number of pulses of roughly equivalent magnitude (Figure 3c), and the number of cells with n pulses deceases as n increases. Increased DNA damage was found to increase the average number of pulses in each cell, rather than modulating the frequency or amplitude of each pulse. The difference between damped oscillations (Figure 3a) and a variable-length series of roughly identical pulses has major consequences on the modeling of single-cell behavior. Thus, when possible, the basis of models meant to describe single-cell dynamics should be the results of single-cell assays.
Figure 3.

Damped oscillations observed in bulk measurements can result from fundamentally different behaviors at the single-cell level. In (a), (b), and (c), all data is hypothetical; the mean is shown in purple, and single-cell traces are light green. (a) Single cells behave like the mean and undergo synchronized, damped oscillations. (b) Single cells are initially synchronized, but their synchrony decays with time. (c) Cells exhibit a discrete number of undamped and synchronized signal pulses, but the number of cells pulsing diminishes with time, consistent with the observations in Lahav et al. of p35 expression. A baseline increase to the mock signal is added in (c) such that the mean trace resembles those in (a) and (b).
1C. Subcellular localization as a proxy for posttranslational modifications
Another powerful application of single-cell microscopy and analysis is the study of a protein’s post-translational modifications. Although a fluorescent tag allows easy assessment of total protein levels (Wu & Pollard 2005), a protein’s post-translational modifications are much harder to detect, since total fluorescence remains unchanged upon modification. Protein modifications that cause major conformational changes can potentially be measured using intramolecular resonance-energy transfer (Charest et al 2005, Lohse et al 2007), but this technique requires fusion of two fluorescent molecules and serendipity in their conformation-dependent orientations. Alternatively, modifications for which specific antibodies have been developed can be measured via immunofluorescence, though cells must be fixed, thereby eliminating the possibility of detecting correlated features within individual cells across multiple time points. However, in special cases, a protein’s subcellular localization changes upon being post-translationally modified. Fluorescent fusions to such proteins, coupled with time-lapse microscopy and automated image analysis, provide a powerful tool to study dynamics in protein activity in single cells.
In elegant work from the Elowitz lab, the activation status of the yeast calcium-response regulator Crz1 was monitored in single cells via the subcellular localization of Crz1-GFP (Cai et al 2008). Normally sequestered in the cytoplasm in a phosphorylated state, Crz1 is dephosphorylated upon the introduction of extracellular calcium and rapidly translocates to the nucleus, where it directly regulates the expression of more than 100 genes (Yoshimoto et al 2002). The authors used the ratio between the cell’s brightest pixels and its mean intensity as a proxy for the activation status of Crz1. Remarkably, they found that calcium stress elicited one synchronized burst of Crz1 activity followed by an asynchronous series of bursts among individual cells (Figure 4). Averaging this response over many cells yielded an activation profile consistent with microarray results of the Crz1-regulated genes (Yoshimoto et al 2002). But, it is noteworthy that attempts to deduce single-cell behavior from the microarray results would have been highly misrepresentative (Figure 4). The authors showed that more severe calcium stress changed the frequency of bursts but not their amplitude or duration; these data suggest that burst frequency is a particularly important parameter in coordinating the downstream response. Indeed, using a simplified model and subsequent experiments, the authors argue that the frequency-modulated activation of Crz1 is optimal for the coordinate regulation of genes with diverse promoter architectures over a range of stress magnitudes. Since they also found that pulsatile activation of transcription factors is not unique to Crz1 (e.g., the activity of general stress-response factor Msn2 also pulses), it will be fascinating to see how this behavior affects other cellular processes.
Figure 4.

One synchronized pulse followed by a series of unsynchronized pulses yields an average trajectory (dotted-blue line) that fails to represent single-cell activity (faded colored lines). Mock data is shown only for four hypothetical single cells, but the average trace was calculated from a simulation of 100 single cells. This schematic resembles the data of Cai et al., who quantified activity of Crz1-GFP by using image-analysis software to measure its nuclear enrichment in single cells.
Other key studies have exploited the subcellular localization of fusion proteins to detect transcription-factor activation (Hersen et al 2008, Mettetal et al 2006); these works will be reviewed in the microfluidic section below.
2. Intra-cell comparisons of spatiotemporal dynamics
Intro
Although quantitative single-cell microscopy has provided extensive evidence for the prevalence and importance of cell-to-cell variability, the high spatiotemporal resolution of these same assays also provides information about the dynamics of processes within a single cell, irrespective of cell-to-cell variability. In the first section below, we review studies that probe the dynamics of multiple and often-overlapping events within single cells. Next, we describe quantitative fluorescent speckle microscopy, a technique that monitors the dynamics of macromolecular structures, such as the cytoskeleton, in living cells. Finally, we discuss recent works that have identified and/or characterized feedback loops that are illuminated by sensitive dynamic measurements, thereby highlighting the fact that quantitative, time-lapse single-cell analysis can reveal key insights about network structure.
2A. Timing of multiple events
The development of spectrally unique, genetically encoded GFP-variants (Ai et al 2007, Shaner et al 2004, Shaner et al 2005) has helped to elucidate mechanisms that occur in the same subcellular location and/or at the same time. For instance, two noteworthy independent investigations used GFP and RFP fusion proteins to resolve the long-standing question regarding the dynamic composition of proteins in the cisternae of the Golgi apparatus (Losev et al 2006, Matsuura-Tokita et al 2006). Prior to this research, it was unclear whether the cis, medial, and trans Golgi stacks maintained fixed protein composition over long time periods, or whether a single cisterna progressively transitioned from cis to medial to trans by dynamically adjusting its constituent proteins. In these two studies in yeast, cis- and trans-specific proteins were labeled with differently colored fluorophores in the same cell. It was shown that a cisterna first fluoresces due to the cis-specific protein for ~150 seconds (Figure 5). As the cis-specific signal falls, the trans-specific signal rises and persists for another ~200 seconds. Thus, rather than retain exclusively cis- or trans-specific proteins for a long time, cisternae rapidly mature, with secretory-processing factors progressively shuttling through them. Since these measurements were gathered in living cells over time, Losev et al. went further to demonstrate that the timescale of protein secretion (~6 minutes) was roughly equivalent to that of the observed Golgi maturation process (Losev et al 2006). These data suggest that much of the trafficking among Golgi stacks consists of Golgi-specific processing factors, and not the secreted proteins. Together, these studies elegantly demonstrate the power of quantitative time-lapse microscopy to distinguish among possible mechanisms, extract important information about their kinetic properties, and generate new hypotheses.
Figure 5.
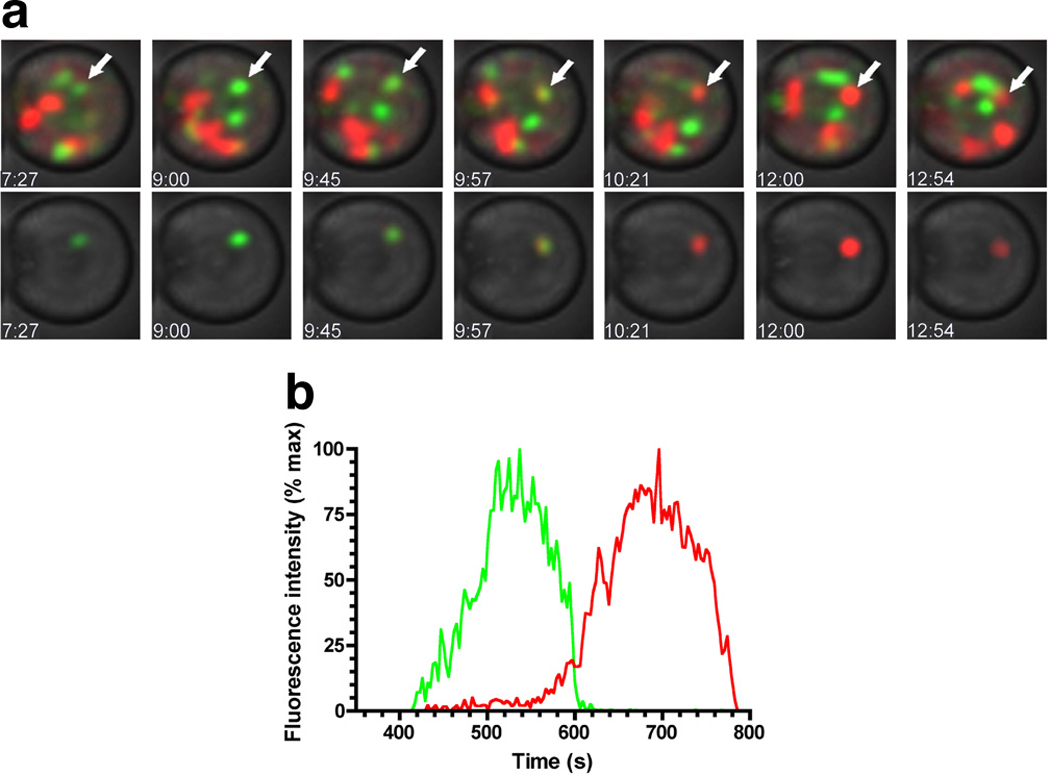
The composition of processing factors in single cisternae of the Golgi apparatus changes rapidly. (a) The early processing factor Vrg4 was tagged with GFP, and the late processing factor Sec7 was tagged with DsRed; the localization of each signal was measured dynamically (Losev et al). Image-analysis software was used to track individual cisternae. Top: One cisterna, marked by the white arrow, is followed over time as it transitions from green to red. Times shown at bottom-left of each image are in mm:ss. Bottom: Same as top, except fluorescence channels are only shown for the cisterna marked with an arrow in top. (b) Quantification of the signal in (a), indicating that the early processing factor vacates the cisterna around the time the late processing factor enters. Note that the time between entry of early factors and exit of late factors is ~7 minutes, close to the time needed for secreted proteins to undergo processing in the Golgi. Figure reproduced from Reference (Losev et al 2006) with permission of Nature.
Deducing the order of molecular events in complicated pathways is frequently challenging. Where distinct phenotypes exist, epistasis experiments can sometimes resolve whether one factor acts before another. Alternatively, biochemical time-series experiments can be performed on synchronized samples to identify which events precede others; for instance, chromatin immunoprecipitation was used to identify the order of transcription-factor recruitment to the yeast HO promoter (Cosma et al 1999). For events that cannot be resolved with epistasis or that occur on timescales faster than bulk biochemistry experiments can probe, time-lapse fluorescence microscopy can shed light on the ordering of events. For instance, Dultz et al. monitored the dynamic assembly and disassembly of the rat nuclear pore complex (NPC) (Dultz et al 2008), which is comprised of approximately 30 nucleoporin proteins (Nups) and is disassembled during mitosis. The authors created multiple cell lines, each bearing a GFP-fused Nup and DiHcRed fused to a protein (here simply called “RFP”) that leaves the nucleus upon breakdown of the nuclear envelope and reenters via the NPC upon restoration of the nuclear envelope at the end of mitosis. The position of chromosomes was also measured simultaneously via DNA staining. For each GFP-Nup fusion, the time between GFP-Nup/DNA colocalization (a proxy for incorporation into the NPC) and RFP nuclear accumulation (a proxy for NPC competence) was measured. Since the RFP construct and its nuclear entry and exit dynamics were common to all cell lines, the relative GFP-Nup/DNA colocalization times could be determined by reference to the RFP signal. Thus, for 11 Nups representing eight major subcomplexes of the NPC, nuclear incorporation could be detected with high precision at sub-minute timescales using clever quantitative analysis of three signals in parallel cell lines (Figure 6). The study showed that the order of disassembly is not simply the reverse of the assembly ordering; furthermore, the nuclear pore is partially competent for protein transport even when some Nups are not yet incorporated. These conclusions highlight the promise of time-lapse microscopy and automated image analysis for deconstructing cellular pathways, even those with fast dynamics.
Figure 6.
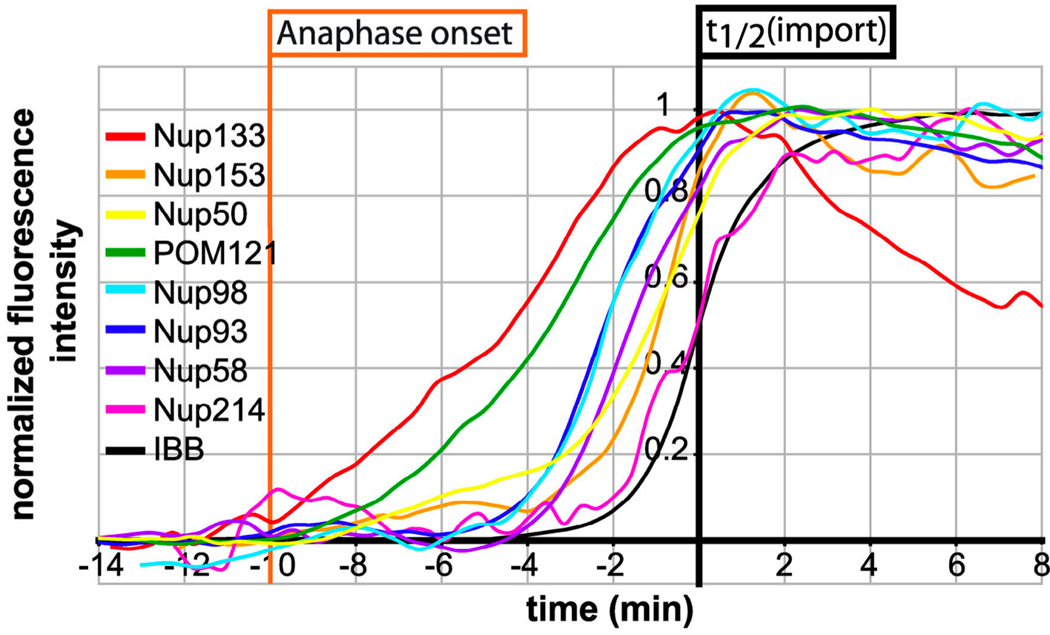
Sequential assembly of components into the nuclear-pore complex (NPC) tracked with high-temporal resolution. The thin black curve represents the nuclear localization of IBB-DiHcRed, a fusion protein imported following cell division via the NPC, and the time 0 min (i.e., “t1/2(import)”) is set when the black curve reaches half its maximal level. The relative rates of nuclear localization of GFP-tagged nucleoporins (“Nups”) and IBB-DiHcRed in strains bearing only one GFP-tagged Nup were used to determine the relative incorporation times of 11 different Nups. Figure reproduced from Reference (Dultz et al 2008) with permission of Journal of Cell Biology.
An important feature of dynamic cellular measurements is that knowledge of an event’s timescale can suggest which factors may regulate the event. This virtue is nicely illustrated by another paper from the Ellenberg lab that uses multiple fluorophores to elucidate the ordering of a pathway (Schuh & Ellenberg 2007). The authors used quantitative time-lapse microscopy to investigate the dynamics of spindle assembly in mouse oocytes. Spindle assembly in these cells is remarkable because it proceeds without the centrosomes that typically coordinate spindle assembly in other cells. By tracking the subcellular localizations of both chromosomes and microtubles, Schuh and Ellenberg find that more than 80 microtubule organizing centers (MTOCs) self-assemble to form a bipolar spindle-like structure that subsequently aligns the chromosomes. In the process of formulating this interesting event-ordering result, they use automated image-analysis to track the 3D positions of individual cytoplasmic MTOCs over time. They note that the calculated MTOC velocities, as well as their ballistic (as opposed to random) trajectory toward the nucleus, is consistent with MTOC transport by microtubule motors. Though this hypothesis of motor-protein-assisted transport may not be entirely surprising, it underscores how knowing the quantitative dynamics of a particular process via single-cell microscopic measurements can distinguish between alternate mechanisms.
2B. Quantitative fluorescence speckle microscopy
Quantitative fluorescence speckle microscopy (qFSM) combines live-cell imaging and sophisticated analysis algorithms to probe the dynamics of macromolecular structures (reviewed in(Danuser & Waterman-Storer 2006)). qFSM relies on fluorescently tagging only a small percentage (typically 0.5% to 2%) of a structure’s subunits. For instance, microtubules contain roughly 1625 tubulin dimers per 1µm (Danuser & Waterman-Storer 2006); in a qFSM experiment, GFP-tagged tubulin would be carefully expressed at a level such that only ~8–32 molecules are fluorescent per 1µm. This low level expression of fusion proteins leads not to uniform fluorescence across the microtuble, but rather to the presence of distinct and randomly distributed fluorescent foci (i.e., “speckles”). Each speckle contains roughly three to eight fluorophores {Danuser, 2003 #15} that were incorporated in close proximity during microtubule assembly. By monitoring the movement of speckles, as well as their appearance and disappearance, researchers can evaluate the motion of structures in the cytoskeleton and the kinetics of their assembly and disassembly. However, such evaluation can require the tracking of tens of thousands of speckles over time, underscoring the need for automated imaging to extract quantitative and mechanistic features from the data.
Several groups have used qFSM to investigate actin dynamics in migrating cells. Cell migration involves a series of steps: cell membrane protrusion, adhesion of the protruded membrane to the extracellular matrix, cytoskeleton-mediated pulling of the cell body against the adhesion molecules, and the severing of adhesion molecules from the rear of the cell. Monitoring nearly a thousand speckles of fluorescently tagged actin monomers in membrane protrusions (“lamellipodia”) revealed that actin polymerization is enhanced within 1µm of the lamellipodium edge (Watanabe & Mitchison 2002). A subsequent study tracked more than 10,000 actin speckles over time, determining their trajectories and lifetimes (Ponti et al 2004). Interestingly, this analysis identified partially overlapping regions of the lamellipodium in which quantitatively different actin dynamics occurred. Specifically, short-lived speckles with high velocity were enriched only near the edge; however, long-lived speckles with low-velocity were found throughout the entire lamellipodium and adjacent lamella. Exploiting these quantitative phenotypes and a range of small-molecule inhibitors, the authors showed that specific actin-associated molecules regulate the different speckle behaviors. This research is particularly noteworthy because of the temporal basis for distinguishing phenotypes: all speckles were the same color, they occupied the same subcellular localization, and their intensities were roughly identical. Thus, their lifetimes and velocities—two quantitative features extracted via automated image analysis—were the distinguishing features.
2C. Evidence for feedback loops in dynamic measurements
Analysis of large-scale gene-expression datasets and protein-protein interaction networks indicates that positive and negative feedback loops are highly over-represented motifs (Yeger-Lotem et al 2004). They serve a range of functions in controlling the dynamics of cellular processes (Brandman & Meyer 2008). For instance, negative feedback can give rise to oscillations and stability of a particular molecular state, while positive feedback frequently leads to all-or-none, switch-like behavior. The converse is also noteworthy: specifically, observation of oscillations often implies negative feedback, and switch-like behavior frequently suggests the existence of positive feedback. Since bulk measurements can disguise switch-like responses (Ferrell & Machleder 1998), time-lapse single-cell measurements are uniquely poised to reveal or verify the existence of important positive-feedback mechanisms, as a recent report from Holt and colleagues beautifully illustrates (Holt et al 2008). In light of the fact that segregation among many discrete chromosomes during anaphase is both rapid and nearly simultaneous (i.e., switch-like), the authors hypothesized that the anaphase regulatory mechanism may contain a critical positive-feedback loop. They performed biochemical analysis of the yeast protein securin, which inhibits anaphase by repressing separase, itself the factor responsible for cleaving the cohesin molecules holding sister chromatids together before separation at anaphase. They found that the phosphatase Cdc14 promotes degradation of securin. Since separase was earlier shown to activate Cdc14, the positive feedback loop consisting of one positive and two negative connections became clear: securin inhibits separase, which activates Cdc14, which effectively inhibits securin. To explore the role of this putative feedback loop in living cells, the authors labeled two individual chromosomes using GFP-fused Tet and Lac repressors, which bound to arrays of genomically incorporated operator sites on chromosomes IV and V. Chromosomes were visualized as spots in the cell, and spot tracking via automated image analysis software could identify the time at which sister chromatids segregated (i.e., one spot became two distinct spots). In wildtype cells, 90 seconds on average elapsed between the chromatid segregation of the respective chromosomes. In a cell line where positive feedback was impaired, however, the difference in segregation times nearly doubled to 170 seconds on average. In sum, the single-cell analysis performed by Holt et al. demonstrates the existence of a positive feedback loop that has a dramatic effect on the synchrony of events in anaphase.
Similar analysis of spatiotemporal data performed by Ozbudak et al. supports the existence of a negative feedback loop that regulates bud formation in yeast (Ozbudak et al 2005). The authors tracked the membrane-specific localization of a fusion protein that binds activated Cdc42, which in yeast marks the site of bud formation by becoming localized at a particular patch of the cell periphery. This aggregation of the Cdc42 signal (the “Cdc24 polar cap”) had been previously shown to result from a positive feedback loop (Irazoqui et al 2003, Wedlich-Soldner et al 2003). A positive feedback loop alone, however, would predict that as soon as a Cdc42 polar cap nucleates, its intensity could increase as a function of time but its location should not change. Remarkably, in cells where the mechanisms that typically program the yeast budding pattern were disabled, the researchers found that the Cdc42 polar cap freely traveled around the periphery of the cell (Figure 7), and these spatial deviations continued up to ~15 minutes before bud emergence. A concise quantitative model was developed to analyze the cap dynamics, and it was shown that positive feedback alone was insufficient to explain the system behavior, but the addition of a negative feedback loop could capture the observed cap migration. Thus, a phenomenon only detectable via time-lapse imaging at the single-cell level revealed the likely existence of a novel network feature, whose molecular basis can be subsequently investigated.
Figure 7.
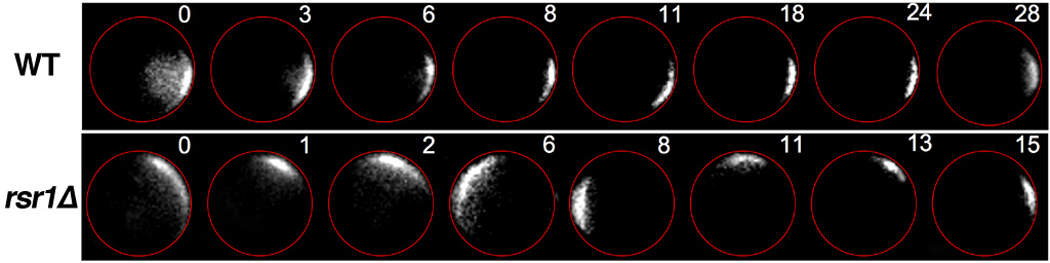
In the absence of bud-site landmarks, the polar cap of active Cdc42 wanders around the cell periphery. The CRIB domain of Gic2, which binds specifically to active Cdc42, was tagged with GFP and monitored in wildtype cells and in mutant cells lacking Rsr1 (a.k.a. “Bud1”). Rsr1 marks the site of budding, and active Cdc42 recruits factors that mediate the budding process. In rsr1Δ cells, bud formation still occurs but at a random location. These time-lapse images (where the number at upper-left is the number of minutes) indicate that the polar cap of active Cdc42 rapidly traverses the cell periphery instead of randomly picking one location and remaining fixed there. Figure reproduced from Reference (Ozbudak et al 2005) with permission of Developmental Cell.
3. Construction of 4D developmental lineages using quantitative microscopy
The decade-long and Nobel-Prize-winning effort of John Sulston and his colleagues determined the entire embryonic lineage of Caenorhabditis elegans (Sulston et al 1983). Since this tremendous achievement, C. elegans has become a key model organism for the study of development. In recent years, it has become possible to determine the lineage of a range of organisms with considerably less effort by exploiting the power of fluorescence time-lapse microscopy coupled with automated image-analysis techniques. Below we discuss studies that span a wide range of organismal complexity, each of which offers exciting insights into developmental processes.
3A. Yeast
Though yeast are unicellular organisms and thus not generally considered a model organism for development, Kaufmann, Yang and colleagues show that tracing the pedigree of a growing colony can reveal important insight into the molecular “memory” passed down through generations (Kaufmann et al 2007). Budding yeast cells expressing a fluorescent reporter of Gal-network activation were tracked via microscopy for upwards of 12 hours, leading to expansion of a colony from one cell to ~20 cells. The authors used a strain in which the switch-like activity of the Gal network was mutated such that activation (“switching on”) occurred stochastically with a probability of ~10% per generation; this allowed for switching events to occur within the 12-hour observation period (Figure 8a). By simultaneously tracking reporter gene expression and a cell’s ancestry, it was shown that mothers and daughters maintain a level of synchrony in their switching times over multiple generations (Figure 8b). As expected, this memory was shown to decay as a function of time, but, remarkably, the correlation persisted for four doublings. This long-term memory was consistent with a model in which Gal-regulatory factors are expressed in large but infrequent bursts. The frequency and amplitude of gene-expression bursts are well-characterized parameters of gene expression noise (Maheshri & O'Shea 2007, Raj & van Oudenaarden 2008). It will be interesting to see the extent to which noise regulation in the development of multicellular organisms affects synchronized gene expression programs across the cells in a pedigree.
Figure 8.
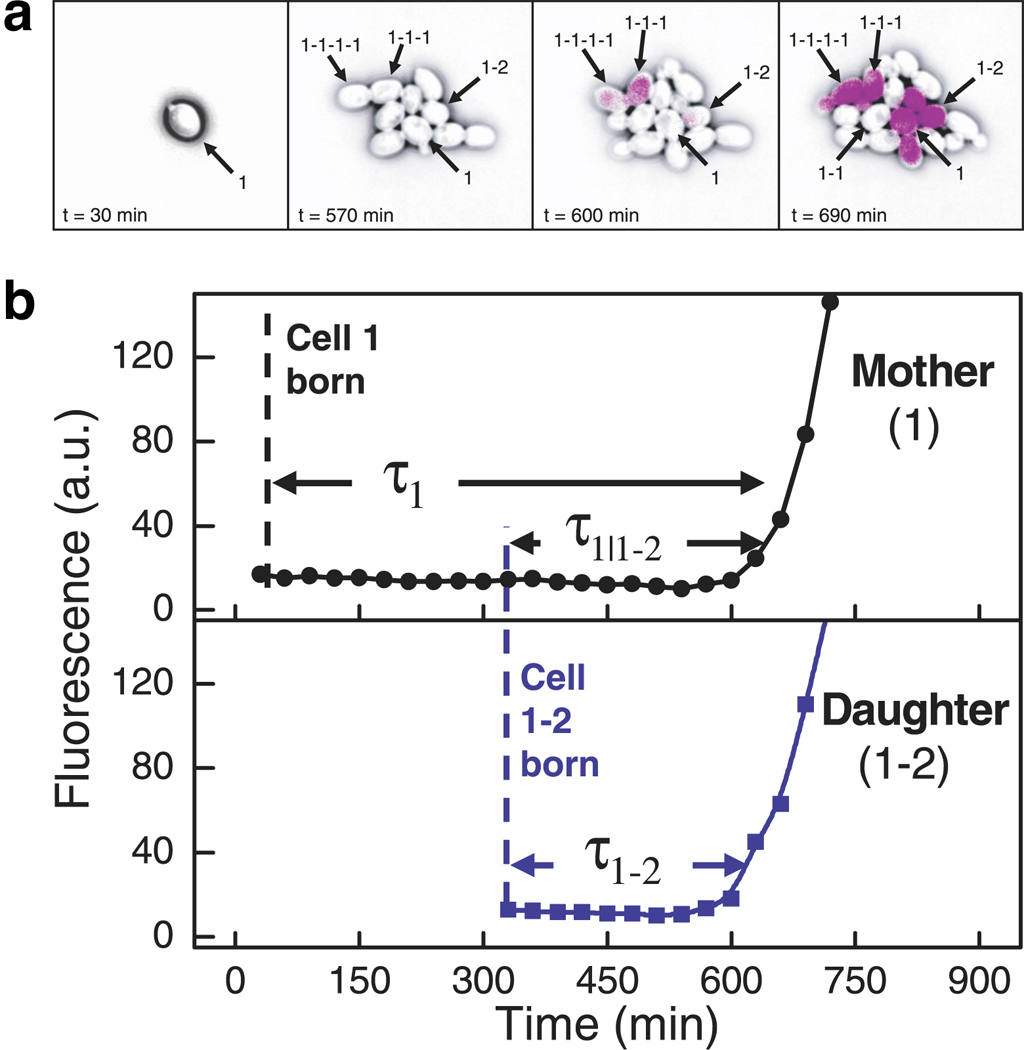
Correlated switching times among closely related single cells revealed via simultaneous monitoring of lineage and gene expression. (a) YFP expressed under control of the GAL1 promoter (GAL1-YFP) is shown in purple. Genealogy is indicated by the numbering scheme in which hyphens separate generations (e.g., 1-1-1 is the daughter of 1-1 and the granddaughter of 1), and the number indicates siblings (e.g., 1–2 is the sister of 1-1 and the second daughter of 1). Although all cells in the rightmost panel are close relatives of cell 1, only a subset expresses GAL1-YFP. (b) Quantification of GAL1-YFP expression in a mother and daughter shows that both switch to an expressing state nearly simultaneously. It was shown that related cells separated by up to four generations tended to activate GAL1-YFP expression at a similar time. Figure reproduced from Reference (Kaufmann et al 2007) with permission of PLoS Biology.
3B. Worms
In their automated-lineage analyses of developing C. elegans embryos (Bao et al 2006, Zhao et al 2008), the Waterston lab provides an illustrative example of the comparisons that can be made using rich phenotypes. Their general approach was to label histone proteins with fluorescent proteins (Murray et al 2006). Since histones are ubiquitously associated with DNA, the nucleus of every cell in the organism up to the ~350-cell stage is resolvable via microscopy. Furthermore, since nuclei are approximately spherical, and the fluorescent-histone signal fills the nucleus, resolving individual cells via image-analysis software is relatively straightforward, as compared to the difficulty involved in segmentation using cell boundaries rather than nuclei for cells with a range of shapes. Due to the relatively large size of the embryo, a z-stack of 2D images must be captured in rapid succession in order for sophisticated image-analysis routines (Murray et al 2006) to assemble a 3D depiction of nuclei positions. The results of this method are quite striking: an entire lineage can be constructed in just six hours of image acquisition and two-to-four hours of image analysis. Of course, the lineage has amazingly high temporal precision, with full-embryo sampling every minute. But, it also contains tremendous spatial information—the dynamic position of every cell is known throughout development. Like the work in yeast performed by Kauffman et al., Waterston and his colleagues mapped gene expression data onto the lineage by driving histone-RFP expression with developmentally relevant promoters (a histone-GFP fusion under control of the endogenous histone promoter was still present to trace the entire lineage) (Murray et al 2008). Among other findings, they showed that neural-development regulator cnd-1 was expressed earlier and in a much broader range of cells than previously characterized. A separate study from the Waterston group highlighted the possibility of quantitatively comparing lineage characteristics across species (Zhao et al 2008). The authors constructed lineages for embryos of C. elegans and the related worm Caenorhabditis briggsae. Remarkably, despite extensive genomic differences between the species, their development appeared to be quite similar (Figure 9). There was a one-to-one correspondence in the lineages between the species up to at least the 350-cell stage. The timing of divisions was also highly conserved among most branches of the lineage, but the assay was precise enough to identify a particular subset of cells with a different cell-division rate. Just as inter-species comparisons of genome sequence have enabled extensive evolutionary analyses, large-scale comparative lineage analyses have the potential to broadly impact the study of development and evolution.
Figure 9.

Lineage comparison reveals developmental similarities between C. elegans (a) and C. briggsae (b) embryos. Spots represent nuclei, which were visualized via GFP-labeled histones and tracked computationally in 3D over time. Color-coding indicates descendants of a common precursor cell. Figure reproduced from Reference (Zhao et al 2008) with permission of Developmental Biology.
3C. Plants
Automated lineage analysis has also been performed in developing Arabidopsis thaliana meristems (Reddy et al 2007, Reddy et al 2004). In a recent study, a technique similar to the one used for lineage analysis was applied to explore the relationship between microtubule alignment and morphological stress across cell walls in developing plant meristems (Hamant et al 2008). In A. thaliana, cortical microtubules form a striated bundle in each single cell with a clear orientation (Figure 10a). Others had proposed that anisotropic stress across the cell wall exerted by neighboring cells played a role in microtubule orientation, so the authors investigated this possible mechanism via time-lapse microscopy in a developing shoot apical meristem. They fused GFP to a microtubule-associated protein and tracked the microtubule orientation over time in the developing plant via analysis software. They found that microtubule-orientation fluctuations changed over time. Specifically, at the tip of the meristem, microtubule orientations shifted frequently in the first ~12 hours of a cell-cycle, but stabilized (i.e., stopped rotating) in the ~4 hours preceding cell division. The stabilized orientation was consistent with the predictions of a model the authors proposed to deduce cell-wall stress from the 3D shape of the developing meristem. Importantly, to confirm this suggested link between orientation and stress, the authors performed two key experiments, one using laser ablation and the other involving application of direct compression force. In the laser-ablation experiment, an individual cell was rapidly killed such that cell-wall stress on the adjacent cells was diminished; within six hours, the authors observed reorientation of microtubules in nearby cells, indicative of adjustment to a new cell-wall stress landscape. In the direct-compression experiment, the plant was pinched between two teflon blades, dramatically altering the direction of stress across cell walls. Shortly after the stress began, microtubule orientation destabilized, and within six hours, most cells in proximity to the blades had reoriented their microtubules such that they were in alignment with the imposed cell-wall stress. Together, these experiments demonstrate how quantitative lineage analysis, coupled with the ability to make perturbations and sensitively measure their effects, can significantly advance our understanding of development.
Figure 10.
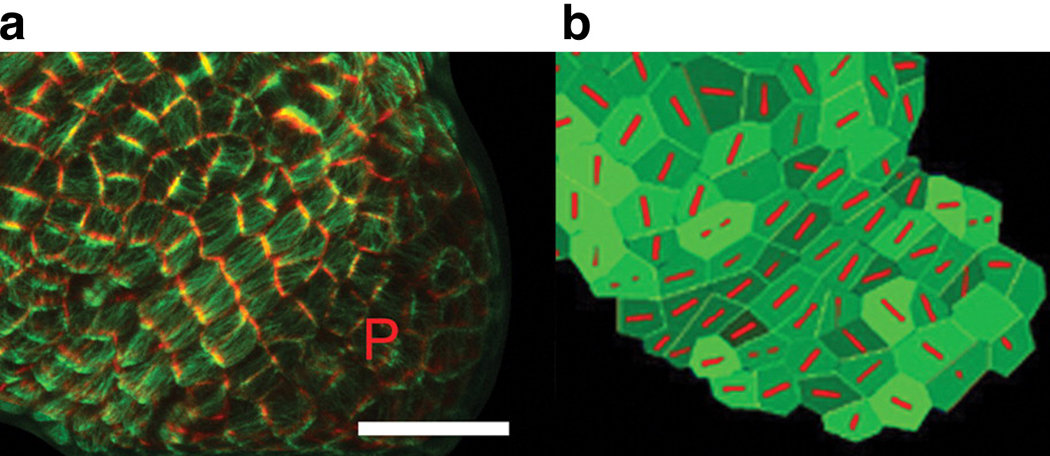
Orientation of microtubule bundles in A. thaliana corresponds highly with predictions from model of cell-wall stress. (a) The microtubule-binding domain of a microtuble-associated protein was tagged with GFP. GFP signal above a high threshold signified cell boundaries (pseudocolored red), and signal below the threshold indicated cell-traversing microtubules (pseudocolored green). The letter P indicates the position of a developing primordium, a position characterized by rapid cell growth and a lack of alignment in microtubule orientation. To the upper-left of the primordium is a region where microtubules are tightly aligned in the southwest-to-northeast direction. Scale bar = 20 µm. (b) A model that predicts stress across the cell wall can also accurately predict microtubule orientations (red). Inputs to the model include the 3D tissue shape and cell boundaries from (a). Note the consistency between the model’s predictions and actual microtubule orientations from (a) in the region to the upper-left of P. Figure reproduced from Reference (Hamant et al 2008) with permission of Science.
3D. Flies
Tracking the dynamics of development in living organisms via fluorescently labeled histone proteins was recently performed in Drosophila embryos. Stathopoulos and colleagues focused on the creation of the mesoderm during gastrulation (McMahon et al 2008). The mesoderm is formed from a tube-shaped invagination of the ectoderm that subsequently flattens and spreads along the ectoderm to form two distinct but adjacent cell layers. Though this sequence of events is evident from antibody staining of fixed embryos, dynamic data in a living embryo provided much higher temporal and spatial resolution, thereby elucidating novel features of the development pathway. Individual nuclei were tracked for more than two hours and sampled every ~50 seconds; approximately 100 mesoderm and 1500 ectoderm nuclei were followed in each embryo. Interestingly, since all cells bore the same fluorescent histone marker, mesoderm and ectoderm cells could only be distinguished at later time points by tracing their trajectories back to their initial positions in the embryo (i.e., before the flattening of the mesoderm); this underscores the need for sophisticated image analysis and a long sampling duration. The authors quantitatively verified a previous qualitative observation suggesting that movement of ectoderm and mesoderm cells was coupled. To reach this conclusion, they examined the correlation between the velocity of a mesoderm cell and its six nearest ectoderm neighbors. Remarkably, of the three velocity components (i.e., anterior-to-posterior, left-to-right, and center-to-ectoderm), only the anterior-to-posterior velocity displayed a coupling between mesoderm and ectoderm movement. This highly specific finding suggests a common mechanism for this directed motion but also the existence of independent mechanisms regulating the other motion components. The single-cell traces along the left-to-right axis suggest that some controlling mechanism is at work—as opposed to simple diffusion—since the relative left-to-right positions of cells were largely maintained through two cell divisions and the formation of a flattened mesoderm. Finally, in another beautiful quantitative analysis, the authors showed via lineage tracing that anomalous mesoderm development in flies with defective FGF signaling is due to a very specific cell-migration problem. Cells initially in the lower furrow (i.e., the ectoderm-proximal lower half of the tube-shaped, pre-flattened mesoderm structure), and thus close to the ectoderm, underwent normal migration; however, those in the upper furrow far from the ectoderm had trajectories dissimilar to wildtype cells, leading to a very different flattened mesoderm pattern. In all, the work of McMahon et al. very nicely illustrates how the rich phenotypes measured via quantitative lineage analysis can refine our understanding of developmental mechanisms and also suggest the presence of novel forms of regulation.
3E. Fish
An exciting recent paper about zebrafish development (Keller et al 2008) sheds light on how far lineage analysis can take us in unraveling developmental mechanisms of higher organisms. Keller et al. developed a novel optical technique that permitted high-resolution 3D scanning of the entire zebrafish embryo every 90 seconds. Again exploiting the fluorescent labeling of histones to find nuclei, the authors tracked every nucleus position from shortly after fertilization until the embryo contained ~16,000 cells (Figure 11)! The enormity of data generated and analyzed cannot be understated, as four high-resolution images were captured every second for 24 hours. After image analysis and computational 3D reconstruction of the embryo, a breathtaking movie of the developing embryo was constructed (Online movie M1). Aesthetics aside, it also contains an inordinate amount of information that can be analyzed for years to come. The authors perform a few analyses in their initial report, such as identifying the time of symmetry breaking and exploring the specific events in a mutant zebrafish that lead to defective mesendoderm formation. In the future, it will be fascinating to use these data and techniques to assess the robustness of development, discover the effects of a range of mutations, and perform inter-species comparisons among higher vertebrates.
Figure 11.
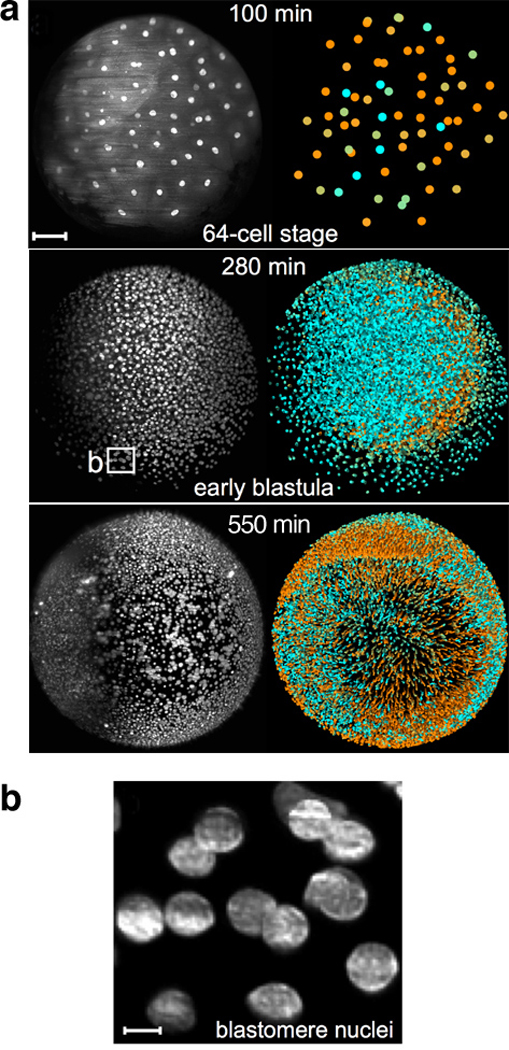
Development of the zebrafish embryo imaged using GFP-labeled histones. (a) Positions and velocities of nuclei represented at three time points. At each time point, a 3D image is generated by acquiring a stack of 2D images (x and y dimensions) across a range of z positions. In the black-and-white images at left, the maximum intensity across the whole z-stack for every pixel in x,y space is plotted. Shading in the colored images at right indicates the velocities (cyan = slow; orange = fast) of single nuclei as determined by their relative position in adjacent frames. Scale bar = 100µm. (b) Individual nuclei from 280-minute sample in (a) can be imaged at very high resolution. Scale bar = 10µm. Figure reproduced from Reference (Keller et al 2008) with permission from Science.
4. Microfluidic devices
The ability to perturb cell’s surroundings dynamically can be critical for unraveling regulatory mechanisms. Additionally, while monitoring cells on short timescales is vitally important, so too is the capability to measure cellular properties over very long timescales, potentially days or weeks using microscopy. Described below are advances in both extracellular manipulation and long-term microscopy made possible by utilizing microfluidic devices.
4A. Specialized stimuli
Several recent studies have powerfully exploited microfluidic devices to measure living cells’ dynamic responses to an exquisitely controlled extracellular environment. Though two works we will mention utilize the device to alter the surroundings temporally, we will also review a study that uses a device to control the spatial distribution of a signaling input.
Using microfluidics for temporal control of a stimulatory signal, Mettetal et al. and Hersen et al. both investigated dynamics of the Hog1 MAP kinase cascade that controls the hyperosmotic shock response in budding yeast (Hersen et al 2008, Mettetal et al 2006). In its inactive state, Hog1 is primarily cytoplasmic, but it translocates to the nucleus rapidly upon hyperosmotic shock. In both studies, the authors fused a fluorescent protein to Hog1 and used custom image-analysis software to measure the nuclear accumulation of Hog1 as a proxy for its activation state in living cells. Additionally, in both studies yeast cells were affixed to a coverslip and placed in a microfluidic device that could rapidly switch between media of different salt concentrations. Hersen et al. focused their analysis on the system’s bandwidth, which corresponds to the upper limit of input frequencies (i.e., oscillating pulses of salt) below which the cells can accurately track the input. For example, for low frequency signals below the bandwidth, Hog1 activity oscillates with a similar frequency as the input (Figure 12a). However, for high frequency signals above the bandwidth, the Hog1 osmotic-stress response is not fast enough to reflect the input fluctuations and instead responds to the average of the quickly fluctuating signal (Figure 12b). Such analysis is important because it reflects the responsiveness constraints of the system and contains important mechanistic information as well. For instance, all steps in the response must be faster than the bandwidth. To identify the rate-limiting step, the authors adjusted the duration of high-salt and low-salt periods. They found that the system’s slowest reaction, which is primarily responsible for setting the bandwidth frequency, regulates deactivation of the Hog1 response. Importantly, the approach of Hersen et al. is very general: it can be applied to any system where the input is well known and the output can be measured at a rate faster than the frequency of fluctuations in the input signal.
Figure 12.

Illustration of bandwidth as a measure of pathway responsiveness. (a) In both Hersen et al. and Mettetal et al., a microfluidic device delivered pulses of media with differing osmolyte concentrations (“Input” panel) to yeast cells, causing the activation and nuclear enrichment of the fluorescently tagged MAP kinase Hog1 (“Output” panel). Below the bandwidth frequency, the output tracks the input signal. (b) For input frequencies above the bandwidth, however, the cells cannot reliably process the input, leading to an “Effective Input” shown schematically in blue, which is approximately the integral of the input. The output very poorly represents the fluctuations in the input signal.
Recent work by Mettetal et al. used engineering principles to gain insight into the network topology of the Hog1 system (Mettetal et al 2006). They exposed cells to a range of different frequencies of salt stress and measured the amplitude and phase shift of the corresponding oscillations in Hog1 activity. Similar to the bandwidth analysis of Hersen et al, the frequencies at which dramatic changes in the output response occur correspond to important reaction rates that dominate the entire system’s dynamics. The authors found two such dramatic changes, implying that the network dynamics could be modeled with only two differential equations (as opposed to tens of differential equations, one for every reaction in the network). The equations deduced from their frequency analysis suggested a network structure highly similar to the known architecture of this well-characterized system; thus, frequency analysis could prove very enlightening for systems with unknown architecture but that otherwise have well known and measurable inputs and outputs. Together, the experiments of Hersen et al. and Mettetal et al. demonstrate that microfluidic devices coupled with quantitative time-lapse microscopy can yield important insights into the dynamics and topology of cellular networks.
Aside from temporal-signal manipulation, microfluidic devices can also create a very precise spatial gradient. Hao et al. used a microfluidic device to create a linear concentration gradient of yeast mating pheromone (Hao et al 2008). Yeast growing in the device exhibited three distinct states of growth: vegetative growth occurred in the region with low pheromone, chemotropic growth at intermediate pheromone, and shmoo formation at high pheromone. In wildtype cells undergoing chemotropic growth, cell division occurs in the direction of higher pheromone. The authors measured the angle of budding relative to the gradient, which was made possible because the gradient orientation was very precisely controlled and cells could be imaged as they grow over time. Using this quantitative phenotype, they found that only one of the two pheromone-responsive MAP kinases could yield directed growth toward high pheromone. The work of Hao et al. is particularly interesting because they observe several distinct cellular phenotypes (e.g., vegetative growth, chemotropic growth, and shmoo formation) as a function of concentration in just one experiment. This approach could be extended to perform a range of titration experiments using a microscope: researchers could scan the device for the phenotype of interest and then determine the precise extracellular conditions causing the phenotype based on the position within the device.
4B. Highly parallel, long-term imaging
Microfluidics are also powerful because they prevent stress and permit long-term imaging (Melin & Quake 2007). Quake and colleagues demonstrated that Escherichia coli could be cultured for up to 200 hours in a device that permitted simultaneous imaging by microscopy (Balagaddé et al 2005). Their device is particularly noteworthy since E. coli typically form biofilms when cultured to high density, thereby complicating more traditional long-term microscopy methods. The authors engineered a microfluidic device where cells grow in a looped chamber with 16 segments. At specified times, one segment can be cordoned off, washed with lysis buffer to remove biofilm-nucleating cells, rinsed of lysis buffer, and then reincorporated into the loop of flowing media. In a subsequent study the authors showed that 96 independently controllable chambers on one microscope slide could be used to culture mammalian cells for more than a week while imaging simultaneously (Gómez-Sjöberg et al 2007). We expect the use of microfluidics to grow rapidly as an experimental platform that allows highly parallel, long-term imaging of cells in an environment that can be very tightly controlled.
5. The Road Ahead
In this review, we have discussed pioneering studies that specifically couple quantitative, time-lapse fluorescence microscopy in living single cells with automated image-analysis techniques to explore the temporal and spatial properties of biological systems. In the near future, we anticipate tremendous progress in this field on multiple fronts: enhanced ability to perturb the cells’ interior and exterior, a wider range of fluorophores and imaging technologies, higher spatial resolution, greater parallelization, and more refined image-analysis software. Below we highlight examples of such progress.
The ability to perturb a system is critical for distinguishing correlation from causation. As described earlier, microfluidic devices and flow chambers permit precise control of the extracellular environment, but other new technologies are needed to affect intracellular processes on short timescales. One such example is chromophore-assisted laser inactivation (CALI) (Bulina et al 2006a, Bulina et al 2006b, Liao et al 1994, Tanabe et al 2005, Wang et al 2008). The basic principle of CALI is that excitation of specific fluorophores (e.g., KillerRed, a variant RFP molecule) fused to a protein of interest creates free radicals that inactivate the protein of interest within minutes (refs). Though CALI can obviously be used to assess the rapid knockdown of a protein, it could also inactivate a repressor, thereby leading to sharp accumulation of a protein of interest. Since the microscope itself perturbs the system, data can be gathered at all time points before and after the stress; thus, even the short-timescale function of a protein can be detected.
The refinement and development of new fluorophore technologies has been rampant in recent years (Wang et al 2008), and we expect progress to persist. Quantum dots (Dahan et al 2003, Giepmans et al 2006) and the resonance-energy transfer technologies (e.g., FRET, BRET, BiFC)—largely excluded from this review because they have been nicely reviewed elsewhere (Joo et al 2008)—are increasingly being used in live-cell assays, especially those classifying protein-protein interactions in vivo (Kerppola 2008). Development of spectrally distinct, genetically encoded fluorophores (Shaner et al 2005) has also significantly advanced the field, since more factors can be monitored in the same cell concurrently. Particularly interesting from the standpoint of tracking dynamic events in live cells are photoswitchable fluorophores (Patterson 2008) and so-called fluorescence timers (FTs) (Subach et al 2009). The former can be converted among two colors (or light and dark states) via UV light exposure either reversibly (Ando et al 2004) or irreversibly (Chudakov et al 2004, Patterson & Lippincott-Schwartz 2002); the latter convert from one color to another color, without intervention, over a range of timescales. These technologies will allow researchers to effectively timestamp particular molecules in either a switch-like or graded manner.
Though imaging with light microscopy reveals a wealth of dynamic information within single cells, its spatial resolution has traditionally been capped by the diffraction limit, which is roughly half the fluorophores’s excitation wavelength (typically 200nm at best). Several revolutionary technologies have shattered the diffraction limit, yet they impressively still use far-field light microscopy (Hell 2007, Hell 2009). Stimulated emission depletion (STED) (Hein et al 2008, Klar et al 2000, Nägerl et al 2008), stochastic optical reconstruction microscopy (STORM) (Bates et al 2007, Huang et al 2008, Rust et al 2006) and the related photoactivated localization microscopy (PALM) (Betzig et al 2006, Shroff et al 2008) effectively allow imaging at an arbitrary resolution, and already-published reports have resolved features separated by <20nm (Betzig et al 2006, Eggeling et al 2008, Rust et al 2006). The comparison between STED- and STORM-acquired images and those from confocal microscopy are striking (Figure 13). In addition to high resolution in the x,y-plane, sub-diffraction-limit imaging in the z-direction permits 3D imaging using both STED (Nägerl et al 2008) and STORM (Huang et al 2008). Multiple colors can be detected with both techniques, but thus far only STED can use genetically encoded fluorophores (Willig et al 2006); STORM and PALM both require special fluorophores that can be cycled between light and dark states repeatedly. Also, though STORM and PALM can capture only a few high-resolution images per minute at best, the temporal sampling of STED is remarkable (e.g., 28 frames per second in (Westphal et al 2008)). The major drawback of all of these technologies is that they require very high-power illumination sources that are potentially damaging to the cells over long imaging periods. Critically, however, they can all image events in live cells, setting them apart from other impressive sub-diffraction-limit approaches, such as structured-illumination microscopy (Schermelleh et al 2008), that currently require cell fixation.
Figure 13.
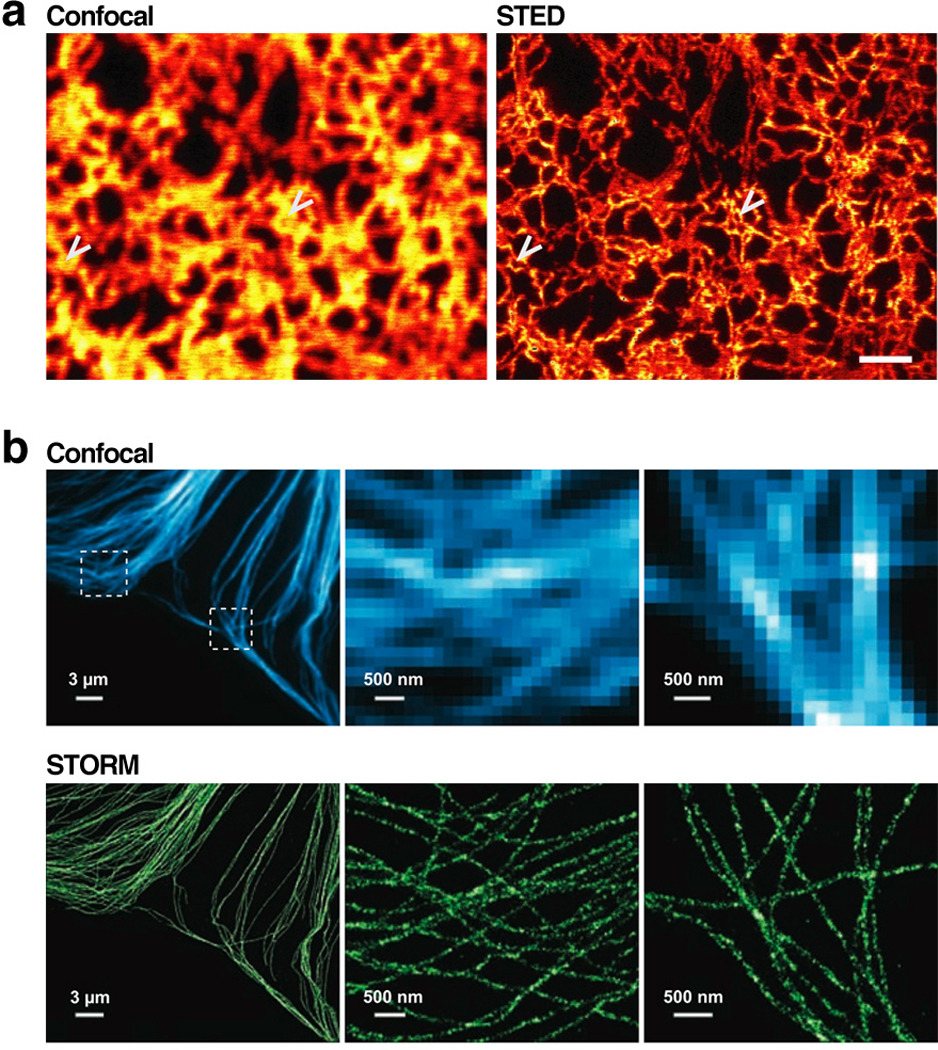
Fluorescence imaging below the diffraction limit highlights features occluded even using confocal microscopy. (a) A yellow fluorescent protein fused to a sequence that targeted it to the endoplasmic reticulum was imaged using confocal microscopy (left) and stimulated emission depletion (STED, right). Arrows indicate positions where STED detects a ring in the ER structure not visible by confocal imaging. Scale bar = 1µm. (b) Comparison of immunofluorescence staining of microtubules using confocal microscopy (top) and stochastic optical reconstruction microscopy (STORM, bottom). The middle and rightmost panels are zoomed portrayals of the dotted boxes in the upper-left panel. The pixelation of these zoomed panels is apparent in the confocal images but not STORM, since STORM imaging can identify fluorophore positioning with considerably higher spatial resolution as compared to confocal imaging. Part (a) of figure reproduced from Reference (Hein et al 2008) with permission of PNAS, and part (b) reproduced from Reference (Bates et al 2007) with permission of Science.
We expect the high temporal and spatial resolution of these techniques to lead to significant advances in quantitative systems modeling. For instance, even with standard far-field imaging, parameters measured from live-cell, microscopy-based assays have been instrumental in quantitative modeling (Marco et al 2007). Higher temporal and spatial resolution will only enable better parameter estimation, thereby facilitating dynamic modeling efforts. STED, STORM, PALM, and their variants should allow very sensitive measurement of a range of reaction rates in vivo, effectively permitting in vivo biochemistry.
Inter-cell differences are easily detected with current microscopic methods, but high-throughput comparison of multiple samples (e.g., different mutants, species, drug treatments) is less straightforward. As discussed earlier, microfluidic devices are one tool that can make automated microscopy more parallel, and their use will inevitably become more common in the future. Tissue arrays are another platform that shows promise (Sauter et al 2003). Imaging adherent cells within 384-well plates has been used to measure phenotypic profiles of a range of drugs (Perlman et al 2004), and microarrays with thousands of tissue samples spotted on a microscope slide allow for massively parallel microscopy-based phenotyping (Sauter et al 2003). Improvements will be needed, however, before tissue microarrays can be used for live-cell imaging.
Finally, we expect the maturation of image-analysis software to continue. Many software packages are already available that range in both price and complexity. Matlab and the freeware ImageJ are two particularly popular software suites for image analysis, but there are many other commercial packages available as well. Since no program could possibly perform every quantitative analysis one would want, it is important for image-analysis software to have scripting capabilities so that the user can define custom metrics. We are eager for speed increases in automated image processing as well as better integration between analysis and acquisition software. Progress on these two fronts could potentially allow on-the-fly analysis of images that then informs the acquisition process. For instance, since fluorophore bleaching after many exposures is a common problem with time-lapse microscopy, image-processing software could partially alleviate this problem by dynamically adjusting the image-acquisition rate based on how quickly the parameter of interest is changing. A similar process is used to minimize processing time when numerically integrating differential equations. Image-processing software could similarly be programmed to intervene (e.g., switching the extracellular media, deactivating proteins via CALI, switching the color of photoswitchable dyes, etc.) only when a particular event is detected. Thus, in silico feedback on the in vivo system will allow the profiling of a large variety of phenotypes in response to complex system inputs.
Summary Points.
Automated image-processing software is an invaluable and flexible tool for extracting quantitative phenotypes from microscopy datasets.
Gene expression noise is just one example of widespread variability among genetically identical cells in a population. Any quantitative trait—such as event timing or transcription-factor enrichment in the nucleus—can have noise properties that reflect underlying regulatory mechanisms and potentially refute the results of bulk assays.
Measuring dynamic processes at high temporal and spatial resolution in a single living cell can resolve the ordering of events in complicated pathways and identify important network properties, such as feedback loops.
Quantitative microscopy can track spatiotemporal and ancestral trajectories at the single-cell level in a broad range of developing organisms.
Microfluidic devices allow concurrent imaging of cells and manipulation of the extracellular environment. Microscopy experiments using microfluidics can be highly parallel an long-lasting (e.g., more than one week).
Future Issues.
Fluorophore development will continue its rapid growth, providing molecules that range in color, brightness, maturation time, photostability, switching ability, and other qualities we cannot even anticipate.
Sub-diffracion-limit imaging techniques will become more accessible.
Repositories for raw microscopy data will be established, providing a resource for cell biologists similar to the protein data bank for structural biologists.
Cell and developmental biologists, physicists, engineers, chemists, computer scientists, statisticians, and applied mathematicians will increasingly work together to extend the frontiers of quantitative imaging in biology.
Supplementary Material
Acknowledgements
We apologize to all researchers whose relevant studies could not be included here due to space constraints. We are grateful to Suzanne Komili, Arjun Raj, Jeff Gore, Hyun Youk, and John Tsang for their helpful comments on the manuscript. This work was supported by an NSF Graduate Research Fellowships to D.M. and NIH grants R01-GM068957 and 5 R90 DK071511-01 to A.v.O.
References
- Ai HW, Shaner NC, Cheng Z, Tsien RY, Campbell RE. Exploration of new chromophore structures leads to the identification of improved blue fluorescent proteins. Biochemistry. 2007;46:5904–5910. doi: 10.1021/bi700199g. [DOI] [PubMed] [Google Scholar]
- Ando R, Mizuno H, Miyawaki A. Regulated fast nucleocytoplasmic shuttling observed by reversible protein highlighting. Science. 2004;306:1370–1373. doi: 10.1126/science.1102506. [DOI] [PubMed] [Google Scholar]
- Balagaddé FK, You L, Hansen CL, Arnold FH, Quake SR. Long-term monitoring of bacteria undergoing programmed population control in a microchemostat. Science. 2005;309:137–140. doi: 10.1126/science.1109173. [DOI] [PubMed] [Google Scholar]
- Bao Z, Murray JI, Boyle T, Ooi SL, Sandel MJ, Waterston RH. Automated cell lineage tracing in Caenorhabditis elegans. Proc Natl Acad Sci USA. 2006;103:2707–2712. doi: 10.1073/pnas.0511111103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bates M, Huang B, Dempsey GT, Zhuang X. Multicolor super-resolution imaging with photo-switchable fluorescent probes. Science. 2007;317:1749–1753. doi: 10.1126/science.1146598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bengtsson M, Stahlberg A, Rorsman P, Kubista M. Gene expression profiling in single cells from the pancreatic islets of Langerhans reveals lognormal distribution of mRNA levels. Genome Res. 2005;15:1388–1392. doi: 10.1101/gr.3820805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betzig E, Patterson GH, Sougrat R, Lindwasser OW, Olenych S, et al. Imaging intracellular fluorescent proteins at nanometer resolution. Science. 2006;313:1642–1645. doi: 10.1126/science.1127344. [DOI] [PubMed] [Google Scholar]
- Bishop DK. RecA homologs Dmc1 and Rad51 interact to form multiple nuclear complexes prior to meiotic chromosome synapsis. Cell. 1994;79:1081–1092. doi: 10.1016/0092-8674(94)90038-8. [DOI] [PubMed] [Google Scholar]
- Brandman O, Meyer T. Feedback loops shape cellular signals in space and time. Science. 2008;322:390–395. doi: 10.1126/science.1160617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulina ME, Chudakov DM, Britanova OV, Yanushevich YG, Staroverov DB, et al. A genetically encoded photosensitizer. Nat Biotechnol. 2006a;24:95–99. doi: 10.1038/nbt1175. [DOI] [PubMed] [Google Scholar]
- Bulina ME, Lukyanov KA, Britanova OV, Onichtchouk D, Lukyanov S, Chudakov DM. Chromophore-assisted light inactivation (CALI) using the phototoxic fluorescent protein KillerRed. Nat Protoc. 2006b;1:947–953. doi: 10.1038/nprot.2006.89. [DOI] [PubMed] [Google Scholar]
- Cai L, Dalal CK, Elowitz MB. Frequency-modulated nuclear localization bursts coordinate gene regulation. Nature. 2008;455:485–490. doi: 10.1038/nature07292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charest PG, Terrillon S, Bouvier M. Monitoring agonist-promoted conformational changes of beta-arrestin in living cells by intramolecular BRET. EMBO Rep. 2005;6:334–340. doi: 10.1038/sj.embor.7400373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chudakov DM, Verkhusha VV, Staroverov DB, Souslova EA, Lukyanov S, Lukyanov KA. Photoswitchable cyan fluorescent protein for protein tracking. Nat Biotechnol. 2004;22:1435–1439. doi: 10.1038/nbt1025. [DOI] [PubMed] [Google Scholar]
- Cosma MP, Tanaka T, Nasmyth K. Ordered recruitment of transcription and chromatin remodeling factors to a cell cycle- and developmentally regulated promoter. Cell. 1999;97:299–311. doi: 10.1016/s0092-8674(00)80740-0. [DOI] [PubMed] [Google Scholar]
- Dahan M, Lévi S, Luccardini C, Rostaing P, Riveau B, Triller A. Diffusion dynamics of glycine receptors revealed by single-quantum dot tracking. Science. 2003;302:442–445. doi: 10.1126/science.1088525. [DOI] [PubMed] [Google Scholar]
- Danuser G, Waterman-Storer CM. Quantitative fluorescent speckle microscopy of cytoskeleton dynamics. Annual review of biophysics and biomolecular structure. 2006;35:361–387. doi: 10.1146/annurev.biophys.35.040405.102114. [DOI] [PubMed] [Google Scholar]
- Di Talia S, Skotheim JM, Bean JM, Siggia ED, Cross FR. The effects of molecular noise and size control on variability in the budding yeast cell cycle. Nature. 2007;448:947–951. doi: 10.1038/nature06072. [DOI] [PubMed] [Google Scholar]
- Dultz E, Zanin E, Wurzenberger C, Braun M, Rabut G, et al. Systematic kinetic analysis of mitotic dis- and reassembly of the nuclear pore in living cells. J Cell Biol. 2008;180:857–865. doi: 10.1083/jcb.200707026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eggeling C, Ringemann C, Medda R, Schwarzmann G, Sandhoff K, et al. Direct observation of the nanoscale dynamics of membrane lipids in a living cell. Nature. 2008 doi: 10.1038/nature07596. [DOI] [PubMed] [Google Scholar]
- Elowitz MB, Levine AJ, Siggia ED, Swain PS. Stochastic gene expression in a single cell. Science. 2002;297:1183–1186. doi: 10.1126/science.1070919. [DOI] [PubMed] [Google Scholar]
- Ferrell JE, Machleder EM. The biochemical basis of an all-or-none cell fate switch in Xenopus oocytes. Science. 1998;280:895–898. doi: 10.1126/science.280.5365.895. [DOI] [PubMed] [Google Scholar]
- Giepmans BN, Adams SR, Ellisman MH, Tsien RY. The fluorescent toolbox for assessing protein location and function. Science. 2006;312:217–224. doi: 10.1126/science.1124618. [DOI] [PubMed] [Google Scholar]
- Gómez-Sjöberg R, Leyrat AA, Pirone DM, Chen CS, Quake SR. Versatile, fully automated, microfluidic cell culture system. Anal Chem. 2007;79:8557–8563. doi: 10.1021/ac071311w. [DOI] [PubMed] [Google Scholar]
- Hamant O, Heisler MG, Jönsson H, Krupinski P, Uyttewaal M, et al. Developmental patterning by mechanical signals in Arabidopsis. Science. 2008;322:1650–1655. doi: 10.1126/science.1165594. [DOI] [PubMed] [Google Scholar]
- Hao N, Nayak S, Behar M, Shanks RH, Nagiec MJ, et al. Regulation of cell signaling dynamics by the protein kinase-scaffold Ste5. Mol Cell. 2008;30:649–656. doi: 10.1016/j.molcel.2008.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hein B, Willig KI, Hell SW. Stimulated emission depletion (STED) nanoscopy of a fluorescent protein-labeled organelle inside a living cell. Proc Natl Acad Sci USA. 2008;105:14271–14276. doi: 10.1073/pnas.0807705105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hell SW. Far-field optical nanoscopy. Science. 2007;316:1153–1158. doi: 10.1126/science.1137395. [DOI] [PubMed] [Google Scholar]
- Hell SW. Microscopy and its focal switch. Nat Methods. 2009;6:24–32. doi: 10.1038/nmeth.1291. [DOI] [PubMed] [Google Scholar]
- Hersen P, McClean MN, Mahadevan L, Ramanathan S. Signal processing by the HOG MAP kinase pathway. Proc Natl Acad Sci USA. 2008;105:7165–7170. doi: 10.1073/pnas.0710770105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holt LJ, Krutchinsky AN, Morgan DO. Positive feedback sharpens the anaphase switch. Nature. 2008;454:353–357. doi: 10.1038/nature07050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang B, Wang W, Bates M, Zhuang X. Three-dimensional super-resolution imaging by stochastic optical reconstruction microscopy. Science. 2008;319:810–813. doi: 10.1126/science.1153529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irazoqui JE, Gladfelter AS, Lew DJ. Scaffold-mediated symmetry breaking by Cdc42p. Nat Cell Biol. 2003;5:1062–1070. doi: 10.1038/ncb1068. [DOI] [PubMed] [Google Scholar]
- Joo C, Balci H, Ishitsuka Y, Buranachai C, Ha T. Advances in single-molecule fluorescence methods for molecular biology. Annu Rev Biochem. 2008;77:51–76. doi: 10.1146/annurev.biochem.77.070606.101543. [DOI] [PubMed] [Google Scholar]
- Kaern M, Elston TC, Blake WJ, Collins JJ. Stochasticity in gene expression: from theories to phenotypes. Nat Rev Genet. 2005;6:451–464. doi: 10.1038/nrg1615. [DOI] [PubMed] [Google Scholar]
- Kaufmann BB, Yang Q, Mettetal JT, van Oudenaarden A. Heritable stochastic switching revealed by single-cell genealogy. PLoS Biol. 2007;5:e239. doi: 10.1371/journal.pbio.0050239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller PJ, Schmidt AD, Wittbrodt J, Stelzer EH. Reconstruction of zebrafish early embryonic development by scanned light sheet microscopy. Science. 2008;322:1065–1069. doi: 10.1126/science.1162493. [DOI] [PubMed] [Google Scholar]
- Kerppola TK. Bimolecular fluorescence complementation (BiFC) analysis as a probe of protein interactions in living cells. Annu Rev Biophys. 2008;37:465–487. doi: 10.1146/annurev.biophys.37.032807.125842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klar TA, Jakobs S, Dyba M, Egner A, Hell SW. Fluorescence microscopy with diffraction resolution barrier broken by stimulated emission. Proc Natl Acad Sci USA. 2000;97:8206–8210. doi: 10.1073/pnas.97.15.8206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kural C, Kim H, Syed S, Goshima G, Gelfand VI, Selvin PR. Kinesin and dynein move a peroxisome in vivo: a tug-of-war or coordinated movement? Science. 2005;308:1469–1472. doi: 10.1126/science.1108408. [DOI] [PubMed] [Google Scholar]
- Lahav G, Rosenfeld N, Sigal A, Geva-Zatorsky N, Levine AJ, et al. Dynamics of the p53-Mdm2 feedback loop in individual cells. Nat Genet. 2004;36:147–150. doi: 10.1038/ng1293. [DOI] [PubMed] [Google Scholar]
- Lev Bar-Or R, Maya R, Segel LA, Alon U, Levine AJ, Oren M. Generation of oscillations by the p53-Mdm2 feedback loop: a theoretical and experimental study. Proc Natl Acad Sci USA. 2000;97:11250–11255. doi: 10.1073/pnas.210171597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao JC, Roider J, Jay DG. Chromophore-assisted laser inactivation of proteins is mediated by the photogeneration of free radicals. Proc Natl Acad Sci USA. 1994;91:2659–2663. doi: 10.1073/pnas.91.7.2659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohse MJ, Bunemann M, Hoffmann C, Vilardaga JP, Nikolaev VO. Monitoring receptor signaling by intramolecular FRET. Curr Opin Pharmacol. 2007;7:547–553. doi: 10.1016/j.coph.2007.08.007. [DOI] [PubMed] [Google Scholar]
- Losev E, Reinke CA, Jellen J, Strongin DE, Bevis BJ, Glick BS. Golgi maturation visualized in living yeast. Nature. 2006;441:1002–1006. doi: 10.1038/nature04717. [DOI] [PubMed] [Google Scholar]
- Maheshri N, O'Shea EK. Living with noisy genes: how cells function reliably with inherent variability in gene expression. Annu Rev Biophys Biomol Struct. 2007;36:413–434. doi: 10.1146/annurev.biophys.36.040306.132705. [DOI] [PubMed] [Google Scholar]
- Marco E, Wedlich-Soldner R, Li R, Altschuler SJ, Wu LF. Endocytosis optimizes the dynamic localization of membrane proteins that regulate cortical polarity. Cell. 2007;129:411–422. doi: 10.1016/j.cell.2007.02.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuura-Tokita K, Takeuchi M, Ichihara A, Mikuriya K, Nakano A. Live imaging of yeast Golgi cisternal maturation. Nature. 2006;441:1007–1010. doi: 10.1038/nature04737. [DOI] [PubMed] [Google Scholar]
- McMahon A, Supatto W, Fraser SE, Stathopoulos A. Dynamic analyses of Drosophila gastrulation provide insights into collective cell migration. Science. 2008;322:1546–1550. doi: 10.1126/science.1167094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melin J, Quake SR. Microfluidic large-scale integration: the evolution of design rules for biological automation. Annu Rev Biophys Biomol Struct. 2007;36:213–231. doi: 10.1146/annurev.biophys.36.040306.132646. [DOI] [PubMed] [Google Scholar]
- Mettetal JT, Muzzey D, Pedraza JM, Ozbudak EM, van Oudenaarden A. Predicting stochastic gene expression dynamics in single cells. Proc Natl Acad Sci USA. 2006;103:7304–7309. doi: 10.1073/pnas.0509874103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray JI, Bao Z, Boyle TJ, Boeck ME, Mericle BL, et al. Automated analysis of embryonic gene expression with cellular resolution in C. elegans. Nat Methods. 2008;5:703–709. doi: 10.1038/nmeth.1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray JI, Bao Z, Boyle TJ, Waterston RH. The lineaging of fluorescently-labeled Caenorhabditis elegans embryos with StarryNite and AceTree. Nat Protoc. 2006;1:1468–1476. doi: 10.1038/nprot.2006.222. [DOI] [PubMed] [Google Scholar]
- Nachman I, Regev A, Ramanathan S. Dissecting timing variability in yeast meiosis. Cell. 2007;131:544–556. doi: 10.1016/j.cell.2007.09.044. [DOI] [PubMed] [Google Scholar]
- Newman JR, Ghaemmaghami S, Ihmels J, Breslow DK, Noble M, et al. Single-cell proteomic analysis of S. cerevisiae reveals the architecture of biological noise. Nature. 2006;441:840–846. doi: 10.1038/nature04785. [DOI] [PubMed] [Google Scholar]
- Nägerl UV, Willig KI, Hein B, Hell SW, Bonhoeffer T. Live-cell imaging of dendritic spines by STED microscopy. Proc Natl Acad Sci USA. 2008;105:18982–18987. doi: 10.1073/pnas.0810028105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozbudak EM, Becskei A, van Oudenaarden A. A system of counteracting feedback loops regulates Cdc42p activity during spontaneous cell polarization. Dev Cell. 2005;9:565–571. doi: 10.1016/j.devcel.2005.08.014. [DOI] [PubMed] [Google Scholar]
- Ozbudak EM, Thattai M, Kurtser I, Grossman AD, van Oudenaarden A. Regulation of noise in the expression of a single gene. Nat Genet. 2002;31:69–73. doi: 10.1038/ng869. [DOI] [PubMed] [Google Scholar]
- Patterson GH. Photoactivation and imaging of photoactivatable fluorescent proteins. Curr Protoc Cell Biol. 2008;21 doi: 10.1002/0471143030.cb2106s38. Chapter Unit 21 6. [DOI] [PubMed] [Google Scholar]
- Patterson GH, Lippincott-Schwartz J. A photoactivatable GFP for selective photolabeling of proteins and cells. Science. 2002;297:1873–1877. doi: 10.1126/science.1074952. [DOI] [PubMed] [Google Scholar]
- Peixoto A, Monteiro M, Rocha B, Veiga-Fernandes H. Quantification of multiple gene expression in individual cells. Genome Res. 2004;14:1938–1947. doi: 10.1101/gr.2890204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perlman ZE, Slack MD, Feng Y, Mitchison TJ, Wu LF, Altschuler SJ. Multidimensional drug profiling by automated microscopy. Science. 2004;306:1194–1198. doi: 10.1126/science.1100709. [DOI] [PubMed] [Google Scholar]
- Ponti A, Machacek M, Gupton SL, Waterman-Storer CM, Danuser G. Two distinct actin networks drive the protrusion of migrating cells. Science. 2004;305:1782–1786. doi: 10.1126/science.1100533. [DOI] [PubMed] [Google Scholar]
- Raj A, van Oudenaarden A. Nature, nurture, or chance: stochastic gene expression and its consequences. Cell. 2008;135:216–226. doi: 10.1016/j.cell.2008.09.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy GV, Gordon SP, Meyerowitz EM. Unravelling developmental dynamics: transient intervention and live imaging in plants. Nat Rev Mol Cell Biol. 2007;8:491–501. doi: 10.1038/nrm2188. [DOI] [PubMed] [Google Scholar]
- Reddy GV, Heisler MG, Ehrhardt DW, Meyerowitz EM. Real-time lineage analysis reveals oriented cell divisions associated with morphogenesis at the shoot apex of Arabidopsis thaliana. Development. 2004;131:4225–4237. doi: 10.1242/dev.01261. [DOI] [PubMed] [Google Scholar]
- Rust MJ, Bates M, Zhuang X. Sub-diffraction-limit imaging by stochastic optical reconstruction microscopy (STORM) Nat Methods. 2006;3:793–795. doi: 10.1038/nmeth929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauter G, Simon R, Hillan K. Tissue microarrays in drug discovery. Nature reviews Drug discovery. 2003;2:962–972. doi: 10.1038/nrd1254. [DOI] [PubMed] [Google Scholar]
- Schermelleh L, Carlton PM, Haase S, Shao L, Winoto L, et al. Subdiffraction multicolor imaging of the nuclear periphery with 3D structured illumination microscopy. Science. 2008;320:1332–1336. doi: 10.1126/science.1156947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuh M, Ellenberg J. Self-organization of MTOCs replaces centrosome function during acentrosomal spindle assembly in live mouse oocytes. Cell. 2007;130:484–498. doi: 10.1016/j.cell.2007.06.025. [DOI] [PubMed] [Google Scholar]
- Shaner NC, Campbell RE, Steinbach PA, Giepmans BN, Palmer AE, Tsien RY. Improved monomeric red, orange and yellow fluorescent proteins derived from Discosoma sp. red fluorescent protein. Nat Biotechnol. 2004;22:1567–1572. doi: 10.1038/nbt1037. [DOI] [PubMed] [Google Scholar]
- Shaner NC, Steinbach PA, Tsien RY. A guide to choosing fluorescent proteins. Nat Methods. 2005;2:905–909. doi: 10.1038/nmeth819. [DOI] [PubMed] [Google Scholar]
- Shroff H, Galbraith CG, Galbraith JA, Betzig E. Live-cell photoactivated localization microscopy of nanoscale adhesion dynamics. Nat Methods. 2008;5:417–423. doi: 10.1038/nmeth.1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skotheim JM, Di Talia S, Siggia ED, Cross FR. Positive feedback of G1 cyclins ensures coherent cell cycle entry. Nature. 2008;454:291–296. doi: 10.1038/nature07118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart D, Wittenberg C. CLN3, not positive feedback, determines the timing of CLN2 transcription in cycling cells. Genes Dev. 1995;9:2780–2794. doi: 10.1101/gad.9.22.2780. [DOI] [PubMed] [Google Scholar]
- Subach FV, Subach OM, Gundorov IS, Morozova KS, Piatkevich KD, et al. Monomeric fluorescent timers that change color from blue to red report on cellular trafficking. Nat Chem Biol. 2009;5:118–126. doi: 10.1038/nchembio.138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sulston JE, Schierenberg E, White JG, Thomson JN. The embryonic cell lineage of the nematode Caenorhabditis elegans. Dev Biol. 1983;100:64–119. doi: 10.1016/0012-1606(83)90201-4. [DOI] [PubMed] [Google Scholar]
- Tanabe T, Oyamada M, Fujita K, Dai P, Tanaka H, Takamatsu T. Multiphoton excitation-evoked chromophore-assisted laser inactivation using green fluorescent protein. Nat Methods. 2005;2:503–505. doi: 10.1038/nmeth770. [DOI] [PubMed] [Google Scholar]
- Wang Y, Shyy JY, Chien S. Fluorescence proteins, live-cell imaging, and mechanobiology: seeing is believing. Annual review of biomedical engineering. 2008;10:1–38. doi: 10.1146/annurev.bioeng.010308.161731. [DOI] [PubMed] [Google Scholar]
- Watanabe N, Mitchison TJ. Single-molecule speckle analysis of actin filament turnover in lamellipodia. Science. 2002;295:1083–1086. doi: 10.1126/science.1067470. [DOI] [PubMed] [Google Scholar]
- Wedlich-Soldner R, Altschuler S, Wu L, Li R. Spontaneous cell polarization through actomyosin-based delivery of the Cdc42 GTPase. Science. 2003;299:1231–1235. doi: 10.1126/science.1080944. [DOI] [PubMed] [Google Scholar]
- Westphal V, Rizzoli SO, Lauterbach MA, Kamin D, Jahn R, Hell SW. Video-rate far-field optical nanoscopy dissects synaptic vesicle movement. Science. 2008;320:246–249. doi: 10.1126/science.1154228. [DOI] [PubMed] [Google Scholar]
- Willig KI, Kellner RR, Medda R, Hein B, Jakobs S, Hell SW. Nanoscale resolution in GFP-based microscopy. Nat Methods. 2006;3:721–723. doi: 10.1038/nmeth922. [DOI] [PubMed] [Google Scholar]
- Wu JQ, Pollard TD. Counting cytokinesis proteins globally and locally in fission yeast. Science. 2005;310:310–314. doi: 10.1126/science.1113230. [DOI] [PubMed] [Google Scholar]
- Yeger-Lotem E, Sattath S, Kashtan N, Itzkovitz S, Milo R, et al. Network motifs in integrated cellular networks of transcription-regulation and protein-protein interaction. Proc Natl Acad Sci U S A. 2004;101:5934–5939. doi: 10.1073/pnas.0306752101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshimoto H, Saltsman K, Gasch AP, Li HX, Ogawa N, et al. Genome-wide analysis of gene expression regulated by the calcineurin/Crz1p signaling pathway in Saccharomyces cerevisiae. J Biol Chem. 2002;277:31079–31088. doi: 10.1074/jbc.M202718200. [DOI] [PubMed] [Google Scholar]
- Zhao Z, Boyle TJ, Bao Z, Murray JI, Mericle B, Waterston RH. Comparative analysis of embryonic cell lineage between Caenorhabditis briggsae and Caenorhabditis elegans. Dev Biol. 2008;314:93–99. doi: 10.1016/j.ydbio.2007.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.