

Abstract
Dehaloperoxidase (DHP) from the terebellid polychaete Amphitrite ornata is a bifunctional enzyme that possesses both hemoglobin and peroxidase activities. The bifunctional nature of DHP as a globin-peroxidase appears to be at odds with the traditional starting oxidation state for each individual activity. Namely, reversible oxygen-binding is only mediated via a ferrous heme in globins, and peroxidase activity is initiated from ferric centers and to the exclusion of the oxyferrous oxidation state from the peroxidase cycle. Thus, to address what appears to be a paradox, herein we report the details of our investigations into the DHP catalytic cycle when initiated from the deoxy- and oxyferrous states using biochemical assays, stopped-flow UV-visible and rapid-freeze-quench electron paramagnetic resonance spectroscopies, and anaerobic methods. We demonstrate the formation of Compound II directly from deoxyferrous DHP B upon its reaction with hydrogen peroxide, and show that this occurs both in the presence and absence of trihalophenol. Prior to Compound II formation, we have identified a new species which we have preliminarily attributed to a ferrous-hydroperoxide precursor that undergoes heterolysis to generate the aforementioned ferryl intermediate. Taken together, the results demonstrate that the oxyferrous state in DHP is a peroxidase competent starting species, and an updated catalytic cycle for DHP is proposed in which the ferric oxidation state is not an obligatory starting point for the peroxidase catalytic cycle of dehaloperoxidase. The data presented herein provide a link between the peroxidase and oxygen transport activities which furthers our understanding of how this bifunctional enzyme is able to unite its two inherent functions in one system.
Keywords: Dehaloperoxidase, Peroxidase, Hemoglobin, Ferrous-hydroperoxo, Ferryl, Oxyferrous
A number of sediment-dwelling marine polychaetes and hemichordates employ haloperoxidases to produce high levels of volatile brominated secondary metabolites as defense mechanisms (1, 2). Notomastus lobatus (polychaeta) (3–5) and Saccoglossus kowalevskii (hemichordata) (6, 7) are but two examples of marine worms which contaminate benthic ecosystems by secreting a diverse array of toxic haloaromatic compounds such as mono-, di-, and tribromophenols, mono- and dibromovinylphenols, and bromopyrroles. Thus, either by exposure to these toxins through contact upon burrowing or via ingestion by deposit feeders, environmental sediments that are contaminated with these biogenically-produced halometabolites represent a significant challenge to other infaunal organisms that co-inhabit coastal mudflats. One such organism, the terebellid polychaete Amphitrite ornata, has been shown to be tolerant to bromometabolite toxicity due to the production of dehalogenating enzymes, and in particular dehaloperoxidase (DHP). Evidence suggests that this bifunctional protein serves as both an intracellular, monomeric coelomic hemoglobin, and as a peroxidase that confers haloaromatic toxin resistance via oxidation of mono-, di-, and trisubstituted halophenols that possess bromine, chlorine, or fluorine substituents (8, 9). Thus, as the first hemoglobin identified to possess a biologically relevant peroxidase activity (8), and as an example of globins found in marine organisms, structural and mechanistic investigations of dehaloperoxidase may reveal new insights into the structure-function paradigms of the globin superfamily specific to bi/multi-functional proteins.
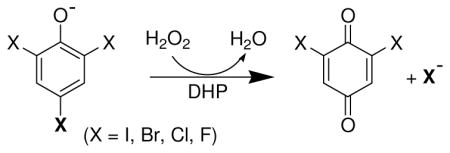
Several recent studies have focused on elucidating the structural, spectroscopic, and mechanistic details of ferric DHP (9–24). A. ornata possesses two separate genes (dhpA and dhpB) that encode for a pair of DHP isoenzymes, termed DHP A and DHP B (25), and both have been shown to catalyze the oxidative degradation of 2,4,6-trihalogenated phenols to the corresponding 2,6-dihalo-1,4-benzoquinones in the presence of hydrogen peroxide (see insert). A combination of stopped-flow UV-visible and rapid freeze-quench EPR spectroscopic methods have provided strong evidence that ferric DHP reacts with hydrogen peroxide to yield Compound ES, an iron(IV)-oxo heme center with an amino acid radical (10). The catalytic competency of that intermediate in oxidizing trihalophenol co-substrates 2,4,6-trichlorophenol (TCP) to dichloroquinone (DCQ) via a peroxidase-like catalytic cycle for DHP has been demonstrated, and evidence suggests that the overall two-electron oxidation of TCP by DHP proceeds through discrete one-electron steps (21). Interestingly, DCQ itself is not an innocent species, having been shown to react separately with both Compound ES and ferric DHP to yield oxyferrous DHP in either case (26). As deduced by spectroelectrochemistry, the unusually high reduction potential for a peroxidase of +206 mV reported for DHP B likely facilitates reactions with DCQ that ultimately favor the reduction of the heme prosthetic group and formation of oxyferrous DHP B (26). Thus, dichloroquinone chemistry may represent one possible link between the two activities of the bifunctional dehaloperoxidase by permitting the ferric state to either initiate a peroxidase pathway in the presence of TCP and H2O2, or form the oxyferrous complex in the presence of DCQ (itself generated from the aforementioned peroxidase pathway) and thus enabling O2-transport function.
While progress has been made toward understanding the DCQ-driven functional switch of how the peroxidase-active ferric DHP state can be converted to the oxygen-binding ferrous form, our understanding of the reverse, wherein oxyferrous DHP is activated toward peroxidase function, is still lacking. Given that reversible oxygen-binding is only mediated via a ferrous heme in globins, and that peroxidase activity is initiated from ferric centers and to the exclusion of the oxyferrous oxidation state from the peroxidase cycle (27), the bifunctional nature of DHP as a globin-peroxidase paradoxically appears to be at odds with the traditional starting oxidation state for each individual activity. Recently, however, in a peroxidase reaction that is unique to DHP, both we (26) and Dawson and co-workers (28) have reported that dehaloperoxidase activity has been observed when the catalytic cycle is initiated from the oxyferrous state, but that the catalytically active species is only formed in the presence of the TCP co-substrate. This observation suggests that substrate binding is the key to the functional switch of DHP from that of an oxygen-binding protein to a peroxidase. Thus, understanding how substrate interacts with DHP to bring about this functional switch is a critical question that can significantly advance our comprehension of the dehaloperoxidase mechanism.
Using data derived primarily from stopped-flow UV-visible spectroscopic methods, one possibility that has been posited by Dawson and co-workers is that oxyferrous DHP is activated by TCP• radicals, themselves formed from trace amounts of the ferric enzyme reacting with TCP and hydrogen peroxide. The TCP• radicals thus generated oxidize the oxyferrous enzyme, generating bulk ferric DHP which is then responsible for the observed dehaloperoxidase activity. This supposition has precedent with the monofunctional lignin peroxidase (LiP), where veratryl alcohol oxidation has been shown to yield radical species that oxidize oxy-LiP to the peroxidase-active form. It has also been postulated by Franzen and co-workers that both external and internal small-molecule binding sites may exist with regulatory implications for the distal histidine, His55, that may govern how DHP switches between its hemoglobin and peroxidase activities (11–13). It has been suggested that His55, which has been observed in distinct ‘open’ and ‘closed’ conformations, mediates O2 displacement from the heme in the presence of (tri)halophenol substrate, possibly serving as a trigger for peroxidase function (11). Typically, the Fe-His Nε2 distances in globins range between 4.1–4.6 Å, and 5.5–6.0 Å for peroxidases (29–32). By comparison, in dehaloperoxidase the Fe-His Nε2 distance in ferric DHP B is 5.5 Å (33) and between 4.8 and 5.5 Å in DHP A (17, 34, 35) for when the distal histidine is in the closed position, and is 5.1 Å in oxyferrous DHP A. With His55-O(1) (iron-bound) and His55-O(2) (terminal) distances of 3.2 and 2.8 Å (17), respectively, oxyferrous DHP exhibits similarities to wild-type Mb in that the distal histidine interacts with both of the oxygen atoms of the end-on bound dioxygen ligand, while at the same time ferric DHP has a longer Fe-His Nε2 distance that more resembles those of peroxidases. Thus, the distal histidine-to-heme distance in dehaloperoxidase, being intermediate between those found in globins and peroxidases, is well positioned to function as both a stabilizer of the bound dioxygen ligand in the former, and as the general acid/base that facilitates both the deprotonation and heterolytic O-O bond cleavage of hydrogen peroxide in the latter.
Although counter to conventional peroxidase thinking, one possibility, based upon the premise that a common starting point is the most efficient pathway for a bifunctional enzyme to perform its dual roles, may be that peroxidase active intermediates are formed directly from the oxyferrous/ferrous state(s) in DHP, thereby permitting a peroxidase catalytic cycle to be initiated from the oxyferrous, and not ferric, state. Thus, the focus of this report is to provide insight into how trihalophenol binding activates both deoxyferrous and oxy-DHP towards peroxidase activity. As such, we present anaerobic UV-visible spectroscopic and kinetic studies of the reaction of deoxyferrous DHP B with hydrogen peroxide, and provide evidence for the direct formation of the peroxidase-active Compound II intermediate that is formed in the absence of trihalophenol co-substrate. Evidence for a transiently formed ferrous-hydroperoxide species as a precursor to Compound II will be provided. Additionally, stopped-flow UV-visible and rapid-freeze-quench electron paramagnetic resonance spectroscopic studies for the reaction of oxyferrous DHP with H2O2 and trihalophenol, under conditions that have yet to be reported, will also be presented, thereby providing the basis for an alternative mechanism for the activation of oxyferrous DHP. Taken together, our experimental design reveals new mechanistic insights and kinetic descriptions of the dehaloperoxidase mechanism, which when interpreted in light of a mechanistic role for the conformational flexibility of the distal histidine (His55) (33), advances our understanding of how DHP performs its dual functions and resolves the dehaloperoxidase paradox.
Materials and Methods
Materials
Buffer salts were purchased from Fisher Scientific. All other reagents and biochemicals, unless otherwise specified, were of the highest grade available from Sigma-Aldrich. Solutions of trihalogenated phenols were freshly prepared prior to each experiment in 100 mM potassium phosphate (KPi) buffer, pH 7 and kept at 4 °C and protected against light. UV-visible spectra were recorded periodically to ensure that the co-substrate had not degraded by monitoring its absorbance: trifluorophenol, 270 nm (1027 M−1cm−1) (26); trichlorophenol, 312 nm (3752 M−1cm−1) (10); tribromophenol, 316 nm (5055 M−1cm−1) (26). Hydrogen peroxide solutions were also freshly made prior to each experiment: initially, a 10 mM stock solution of H2O2 was prepared and maintained on ice (typically less than 15 minutes), during which all other protein/substrate solutions were loaded into the stopped-flow apparatus or utilized for kinetic assays. When prepared in this manner, the stock H2O2 solution did not exhibit any degradation over this time period as determined by UV-visible spectroscopic analysis of the hydrogen peroxide absorbance at 240 nm (ε240 = 43.6 M−1 cm−1) (36). For stopped-flow/freeze-quench experiments, the stock H2O2 solution was then diluted to the appropriate pre-mixing concentration and immediately loaded into the stopped-flow apparatus. WT DHP isoenzymes A and B (6xHis-tagged protein) expression and purification procedures were performed as previously described (10, 12), and only protein samples that exhibited Reinheitzahl values (Rz) greater than 4.0 were utilized in this study. Optical spectra were recorded on either an Agilent 8453 diode array spectrophotometer or a Cary 50 UV-visible spectrophotometer, both equipped with thermostated cell holders at 25 °C. 31P NMR spectra were acquired on a Bruker-300 MHz NMR spectrometer, and chemical shifts are reported as δ values referenced to an external standard of trimethylphosphate (δ = 0 ppm). Ferrous DHP B samples for spectroscopic characterization were prepared in an MBraun LabMaster 130 inert atmosphere (<1 ppm O2, <1 ppm H2O) glovebox under nitrogen atmosphere. The spin trap DIPPMO was synthesized and characterized according to published procedures (37).
Preparation of Oxyferrous DHP B
Oxyferrous DHP B was generated through incubation of the ferric enzyme with 5 equivalents of ascorbic acid in 100 mM KPi (variable pH). Excess reducing agent was removed by using a PD-10 desalting column prepacked with Sephadex G-25 medium. The protein was concentrated using an Amicon Ultra centrifugal device equipped with a 10 kDa cutoff molecular weight membrane. Protein concentration was determined using a heme molar absorption coefficient of 117.6 mM−1cm−1 (λmax = 407 nm) for both the ferric (26) and oxyferrous states of the enzyme. The formation of the oxyferrous species was monitored by its UV-visible spectrum [418 (Soret), 542, 578 nm] (10, 26).
Preparation of Ferrous DHP B
Ferrous DHP B was prepared by purging a ferric DHP B solution (100 mM KPi, variable pH) for at least 20 minutes with argon. Then, the degassed enzyme solution was placed inside the glove box and titrated with sub-stoichiometric aliquots of sodium dithionite, itself dissolved in anaerobic buffer. Formation of ferrous DHP B was followed by UV-visible spectroscopy using an all quartz immersion probe (Hellma, Germany) that was connected to the spectrometer located outside of the glovebox via fiber optic cable.
Stopped-flow UV-Visible Spectrophotometric Studies
Experiments were performed on a Bio-Logic SFM-400 Triple Mixing Stopped-Flow instrument equipped with a diode array UV-visible spectrophotometer, and were carried out at 20 °C in 100 mM KPi buffer (variable pH). Constant temperature of 20 °C was maintained using a circulating water bath. Data were collected (900 scans total) over a three time-domain regime (2.5 ms, 25 ms, 250 ms; 300 scans each) using the Bio Kinet32 software package (Bio-Logic). Experiments were performed in single-mixing mode as indicated: (i) oxyferrous DHP B at a final concentration of 10 μM was mixed with 2.5–25 equivalents of H2O2, and (ii) a solution containing 10 μM final concentration of oxyferrous DHP B and 1–30 equivalents of TCP was reacted with 2.5–25 equivalents of H2O2. Experiments were performed in double-mixing mode using an aging line prior to the second mixing step. The design of the experiments allowed for the mixing of oxyferrous DHP B containing 1 equiv TCP with 2.5 equiv H2O2 for various aging times, followed by the second mix with an additional 2.5–25 equiv TCP: i) oxyferrous DHP B + 1 equiv TCP + 2.5 equiv H2O2 → Delay → + 2.5–25 equiv TCP. All data were evaluated using the Specfit Global Analysis System software package (Spectrum Software Associates) and fit to exponential functions as either onestep, two species or two-step, three species irreversible mechanisms, where applicable. Kinetics data were baseline corrected using the Specfit autozero function.
Benchtop Optical Measurements
UV-visible absorption spectra were collected at room temperature. Oxyferrous DHP B (10 μM) was incubated with 1, 15 or 30 equivalents TCP in 100 mM KPi buffer solution at pH 8. Reaction was initiated by addition of H2O2 (2.5–25 equiv). Spectra were collected every 0.1 minutes for a period of 5 minutes.
Preparation of Reaction Intermediates by Freeze-Quench Methods
Rapid freeze-quench experiments were performed with a BioLogic SFM 400 Freeze-Quench apparatus by mixing a 50 μM oxyferrous DHP B solution (final concentration) containing 2,4,6-trichlorophenol (1 and 15 equiv) with a 10-fold excess of H2O2 in 100 mM KPi (pH 8) at 25 °C. A standard 4 mm O.D. quartz EPR tube was connected to a Teflon funnel, and both the tube and the funnel were completely immersed in an isopentane bath at −110 °C. The reaction mixtures were quenched by spraying them into the cold isopentane, and the frozen material so obtained was packed at the bottom of the quartz tube using a packing rod equipped with a Teflon plunger. In this manner, a packing factor of 60 ± 2% was consistently achieved. Reaction times were as follows: 100 ms, 1, and 30 s (1 equiv TCP) or 100 ms and 10 s (15 equiv TCP). Samples were then transferred to a liquid nitrogen storage dewar until analyzed. Up to 200 individual scans were averaged to achieve a sufficient signal-to-noise ratio.
X-band EPR Spectroscopy
EPR spectra were recorded with an X-band (9 GHz) Varian E-9 EPR spectrometer (Varian, El Palo, CA). A standard 4 mm o.d. quartz EPR tube containing the sample was placed into a quartz finger dewar insert filled with liquid nitrogen. The temperature of the samples was maintained at 77 K for the duration of the data acquisition, which required periodic refilling of the dewar due to the evaporation of the liquid nitrogen during longer acquisition runs. The typical spectrometer settings were as follows: field sweep 200 G, scan rate 3.33 Gauss/s, modulation frequency 100 KHz, modulation amplitude 4.0 G, and microwave power 2 mW. The exact resonant frequency of each EPR experiment was measured by an EIP-578 (PhaseMatrix, San Jose, CA) in-line microwave frequency counter.
TCP Radical Spin Trapping Experiments
500 μL of solution of 5.0 mM TCP in DMF/100 mM KPi pH 7 (1:1) with 10 mM H2O2 was added over a period of 3 h using a microsyringe pump to 500 μL of a solution of DIPPMO (1.5 mM) and ferric or oxyferrous DHP B (60 μM), or horseradish peroxidase. For the 31P NMR samples, 300 μL of the reaction were added to 100 μL of D2O with chromium chloride as a relaxation agent and trimethylphosphate as the internal standard (37).
RESULTS
Formation and UV-visible Characterization of Oxyferrous DHP B
Oxyferrous DHP was generated using different reducing agents (see Supporting Information). Addition of sodium dithionite (Na2S2O4; 25 equiv) [Figure SD1A, E°′ ~ −660 mV (38)] or tris(2-carboxyethyl)phosphine (TCEP; 1 mM) [Figure SD1B, E°′ = ~ −0.05 V (39)] to ferric DHP [UV-visible: 407 (Soret), 504, 538, 635 nm] both resulted in the clean formation of oxyferrous DHP [UV-visible: 417–8 (Soret), 543, 578 nm], which was found to be relatively stable under the conditions examined. Interestingly, while TCEP is able to reduce DHP B [E°′(FeIII/FeII) = +0.206 V vs. NHE (26)], it is not able to reduce HhMb [E°′(FeIII/FeII) = +0.046 V vs. NHE (40)], highlighting an electrochemical difference between these two globins with potential functional implications that have been discussed previously (26).
Recombinant oxyferrous DHP B was also obtained by treatment of ferric DHP B with ascorbic acid [E°′ = 0.058 V vs SHE, pH 7 (41)], followed by the use of size exclusion chromatography to remove excess of ascorbic acid. The electronic absorption spectra of oxyferrous DHP B prepared in this manner at pH 8 in the presence and absence of TCP is presented in Figure SD2. Other than the absorption maxima at 245 and 312 nm being observed for TCP, its addition did not alter the spectral features of the oxyferrous species. Oxyferrous DHP exhibited a typical spectrum of 6cLS ferrous heme with a Soret absorption band at 417 nm and the corresponding visible bands at 542 and 578 nm as noted previously (10, 17, 26, 28). The ferric DHP B spectrum at this pH value showed the spectrum of a hydroxide-bound heme protein as previously reported (16).
Enzymatic Activity of Oxyferrous DHP B
The hydrogen peroxide-dependent oxidative dehalogenation of 2,4,6-tribromophenol and 2,4,6-trifluorophenol as catalyzed by oxyferrous DHP B at pH 7 was monitored by UV-visible spectroscopy, and the kinetic parameters (kcat, Km, and kcat/Km) determined as a function of TXP co-substrate are presented in Table 1 after the experimental data was analyzed using Michaelis-Menten kinetics with the method of initial rates. The reaction mixture yielded the corresponding 2,6-dihalo-1,4-benzoquinone (DXQ) products, as expected, when reactions were initiated upon the addition of H2O2. Both enzyme and TXP co-substrate concentrations were held constant while H2O2 concentrations were varied. The known molar absorptivity of TBP at pH 7 was utilized to compute d[S]/dt in order to obtain the resulting kcat value for the reaction of this co-substrate. As we have noted before for TFP (26), neither -d[S]/dt or d[P]/dt were determined as the absorption maximum for the TFP co-substrate (λmax = 330 nm) underwent a shift during catalytic turnover, and the molar absorptivity of the DFQ product is unknown. Consequently, the corresponding kcat value for TFP oxidation could not be determined by these means.
Table 1.
Kinetics data for the oxidative dehalogenation reaction of TFP, TCP, and TBP (150 μM) as catalyzed by 0.5 μM oxyferrous DHP B in the presence of varying H2O2 concentrations at pH 7.
| Co-Substrate | KMH2O2 (μM) | kcat (s−1) | kcat/KMH2O2 (μ −1 s−1) |
|---|---|---|---|
| TFP | 69 ± 12 | N/A | N/A |
| TCPa | 35 ± 6 | 1.17 ± 0.05 | 0.034 |
| TBP | 17 ± 1 | 0.70 ± 0.04 | 0.041 |
N/A = not available;
obtained from reference [(26)]
In agreement with our previous observations, the oxyferrous DHP B activity observed for TBP, TCP, and TFP was not significantly different from the enzymatic activity of the ferric enzyme (10, 26). These results were expected based on our proposed mechanism (vide infra) and that of Dawson and co-workers (28), as both mechanisms invoke the ferric form during substrate turnover.
Stopped-Flow UV-Visible Characterization of DHP B Compound II
Formation of high valent iron-oxo intermediates generated from the reaction of oxyferrous DHP B with hydrogen peroxide were investigated by single-mixing stopped-flow UV-visible spectroscopy at pH 8. Upon rapid mixing (2 ms) of oxyferrous DHP B species [UV-visible spectrum: 418 (Soret), 541, 577 nm] with either a 10- or 25-fold excess (Figure SD3) of H2O2, no intermediates were observed over the 85 second observation period, but a slight decrease in Soret band intensity was noted. Dawson and co-workers have reported that oxyferrous DHP A converts to Compound RH in the presence of a 10-fold excess of H2O2 (28), with this chemistry occurring on a reaction timescale of 2000 s, and as such may explain the minor spectral changes observed on the 85 s timescale employed in this work. We attempted other conditions to evaluate the reproducibility of these results with several reaction modifications, including varying H2O2 concentration, pH (6–8), and different ways to form the oxyferrous species. Under all conditions examined, the oxyferrous form was not reactive with H2O2 as the oxidant within the time scale under observation (data not shown).
However, when the oxyferrous DHP B solution was incubated with at least one equivalent of TCP as the co-substrate prior to being rapidly mixed with 10 equivalents of H2O2, substantial spectral changes were observed (Figure 1). The oxyferrous form converted to a species whose spectral features [UV-visible spectrum: 420 (Soret), 546, 588 nm] closely resembled that of a ferryl-containing intermediate (e.g., Compound II or ES). The assignment of the DHP B intermediate observed here to that of a ferryl-containing species is based upon our previous identification of such reaction intermediates in DHP (10, 16, 19, 26, 42), as well as the spectroscopic properties of other Fe(IV)-oxo species commonly found in typical peroxidases. Myoglobin (43, 44) and hemoglobin (45, 46) both exhibit similar red-shifted Soret features and have visible bands that appear generally at 540–545 and 580–590 nm for their respective ferrylheme intermediates. It is also important to note that Dawson and co-workers (28) observed a similar intermediate (under slightly different reaction conditions) which they attributed to either Compound ES or II, both of which are ferryl-containing species. Assignment of this species as a traditional Compound I intermediate was ruled out based on the lack of hypochromicity (large decrease in the Soret band intensity) and absence of a strong red shift of the Soret band that is typical for an iron(IV)-oxo porphyrin π-cation radical (47–49), whereas Compound 0 (ferric-hydroperoxide) was also ruled out due to time-scale considerations (50, 51) and the lack of an observed hyperporphyrin spectrum (52, 53). As Compound ES and Compound II, both ferryl-containing intermediates, are not distinguishable by UV-visible spectroscopy, our assignment for the intermediate described here as Compound II was based upon the results of our EPR spectroscopic study (vide infra).
Figure 1.

(A) Stopped-flow UV-visible spectroscopic monitoring of the reaction (900 scans, 85 sec) between oxyferrous DHP B (10 μM) containing one equivalent TCP (10 uM) and a 10-fold excess of H2O2 at pH 8. (B) Calculated UV-visible spectra for both oxyferrous (black) and Compound II (red) are shown; the rapid-scanning data from panel A were fit to a one-step, two species sequential irreversible model using Specfit global analysis software. (C) Relative concentration profile determined from the three component fit used in (B).
The experimental values of kobs for Compound II formation (Scheme 1, step i) varied linearly with TCP in the range of 1 – 30 molar equivalents. From this dependence, the bimolecular rate constant was determined to be (1.60 ± 0.14) × 103 M−1s−1 (Figure SD4). When 1 equivalent of TCP was employed, the formation of Compound II was observed over the 85 second observation period without conversion or decay to either the ferric enzyme or Compound RH (the decay product of Compound ES in the absence of TXP). When the concentration of TCP was increased to 15 equivalents versus that of the enzyme (10-fold excess of H2O2 maintained), a partial conversion of the Compound II intermediate to ferric DHP B was noted (data not shown), which was more clearly observed at 30 equivalents of TCP (Figure SD5), suggesting that TCP is a competent reducing agent for Compound II.
Scheme 1.

Proposed catalytic cycle for (de)oxyferrous dehaloperoxidase B.a
a Adapted from reference [(26)]. The pathways forming Compounds RH and P426, as well as monohalophenol inhibition, have been omitted for clarity.
Several differences between the reactivity of Compound II and that of Compound ES are notable. First, the bimolecular rate constant for the formation of Compound II [Scheme 1, step i; (1.60 ± 0.14) × 103 M−1s−1] was significantly slower than Compound ES formation (Scheme 1, step iii; ~5.1 × 104 M−1s−1) when that latter species was generated from the ferric enzyme in the absence of TCP at pH 8 (Figure SD6). Second, the reaction of ferric DHP B incubated with 30 equivalents TCP and reacted with a 10-fold excess of H2O2 did not generate an observable Compound ES intermediate (26), a result that was attributed to the rapid in situ reduction of the Compound ES species in the presence of TCP. However, our results here demonstrate that Compound II is significantly less reactive than Compound ES, as the reaction of oxyferrous DHP B incubated with 30 equivalents TCP with a 10-fold excess of H2O2 did yield a meta-stable (~15 s; Figure SD6) Compound II species prior to its reduction to the ferric enzyme.
To further investigate the reactivity of DHP B Compound II, sequential double-mixing stopped-flow studies were employed to monitor the reaction of pre-formed Compound II with TCP substrate by UV-visible spectroscopy (Figure SD7). Oxyferrous DHP B containing 1 equivalent TCP was reacted with 2.5 molar equivalents of H2O2 at pH 8, incubated for 85 sec to allow for the maximum accumulation of Compound II, and subsequently mixed with an additional 2.5 equiv (Figure SD7A), 10 equiv (Figure SD7B) and 25 molar equivalents of TCP (Figure SD7C), which resulted in the regeneration of ferric DHP B. The disappearance of Compound II (Scheme 1, step ii) and the formation of the ferric enzyme were linearly dependent on the concentration of TCP co-substrate, yielding a bimolecular rate constant of (4.1 ± 0.3) × 102 M−1s−1 (Figure SD7D). At the highest concentration of TCP investigated (25 equiv; Figure SD7C), the ferric form of the enzyme was further found to partially convert to oxyferrous DHP due to the presence of DCQ generated from the reaction of Compound II with TCP, in accordance with the known reactivity of ferric DHP B with DCQ (26).
Characterization of Compound II by EPR
Rapid-freeze-quench methods were employed to stabilize the ferryl intermediate observed in the above reaction for subsequent characterization by continuous wave (CW) EPR spectroscopy. Specifically, X-band EPR spectra at various quench times (100 ms, 1 and 30 sec) were obtained for the reaction of oxyferrous DHP B with a 10-fold excess of H2O2 in the presence of one equivalent of TCP, thereby generating the putative Compound II intermediate. No detectable EPR signal was observed (Figure 2), indicating that no protein radical was present. For comparison, and as a control for verifying the experimental and spectroscopic apparatus, the EPR signal of Compound ES that resulted from the rapid mixing (500 ms) of ferric DHP B with 10 equivalents of H2O2 at pH 8 (generated side-by-side with the oxyferrous reaction samples) is also shown. As previously reported, the free radical concentration in Compound ES is nearly quantitative with respect to the enzyme concentration (~0.93 spin/heme) in DHP (54). As the EPR spectroscopic method presented here provides an unambiguous means of differentiating between the two ferryl-containing intermediates, Compound ES and II, our spectroscopic data unequivocally rule out the intermediate observed above in the stopped-flow studies as Compound ES. The lack of a spectral signal obtained here strongly suggests either a diamagnetic or even-spin species, the latter being consistent with the assignment of Compound II as the intermediate generated between the reaction of oxyferrous DHP B and hydrogen peroxide in the presence of TCP.
Figure 2.

EPR spectrum of DHP B Compound II at a freeze-quench time of 100 ms (pH 8). Data collected at 1 and 30 second quench times for DHP B Compound II similarly showed no signal. For comparative purposes, the EPR spectrum of Compound ES is also shown. Rapid freeze-quench samples were prepared from the reaction of oxyferrous DHP B (50 μM final) containing 1 equivalent of TCP with a 10-fold excess of H2O2 at 25 °C, and rapidly frozen in an isopentane slurry.
To eliminate the possible interpretation that the lack of an observed radical signal was due to a Compound ES intermediate that was rapidly quenched by TCP, RFQ-EPR spectroscopy was employed to investigate the reaction of a 10-fold excess of H2O2 with ferric DHP B pre-incubated with 1 or 1.2 equivalents of TCP at a freeze-quench time of 100 ms (pH 8). Under these conditions, a radical signal was observed (Figure SD8). Although the radical intensity in the presence of TCP was ~10% (0.09 spin/heme) that of Compound ES itself in the absence of TCP, and the shape of the signal differed as well, nonetheless the result of an observable radical formed under these conditions contrasts markedly with the lack of a radical observed from the above reaction of oxyferrous DHP B with a 10-fold excess of H2O2 in the presence of one equivalent of TCP. Thus, we exclude the possibility that the mechanism of DHP proceeds through a ferric state that yields a Compound ES species whose amino acid radical is quickly quenched by the TCP co-substrate.
H2O2-Reactivity of Ferrous DHP B
The reactivity of ferrous DHP B with H2O2 was investigated using UV-visible spectroscopy under anaerobic conditions to avoid the formation of the oxyferrous species upon oxygen binding to the reduced enzyme. The UV-visible spectra were monitored inside a glove box using a fiber optic dip probe. Due to the poor signal-to-noise ratio of the fiber optic setup, two enzyme concentrations, 10 and 50 μM, were employed for the data in Figure 3 to provide resolution of both the Soret and the visible bands (as shown in the inset), respectively. The reduced enzyme was obtained from the titration of ferric DHP B with sub-stoichiometric additions of sodium dithionite until fully formed [UV-vis features: 432 (Soret), 558 nm (17, 26)] (Figure SD9). Ferrous DHP B (10 or 50 μM) thus generated was mixed with excess H2O2 (2.5 – 10 equiv), and the resulting changes in the electronic absorption spectrum were recorded immediately after mixing (~2–3 seconds). Overall, two intermediates were observed prior to the formation of the final product (either as ferric DHP B or Compound RH, depending upon the reaction conditions employed). Ferrous DHP B first converted to a new species [UV-vis features: ~421–3 (Soret), 488, 505 (sh) nm; kobs > 0.6 s−1] (Figures 3 and SD10) which was found to be unstable, and interconverted (kobs = ~0.036 s−1) to a second intermediate whose spectral features [UV-vis features: 420 (Soret), 546, 586 nm] we attribute to the ferryl-containing intermediate Compound II based upon i) spectral similarity to other ferrylhemes (vide supra), ii) the results of our EPR spectroscopic study (vide supra), and iii) the known reactivity of ferrous heme proteins with H2O2 (see Discussion). The amount observed of this unknown intermediate appeared to be proportional to the amount of hydrogen peroxide that was initially reacted with ferrous DHP B (Figure 3), although the temporal resolution of the anaerobic dip probe/UV-visible benchtop spectrometer did not allow for a quantitative assessment to be made. The reaction was repeated at reduced temperature (~ 4 °C) to stabilize the intermediate and obtain the spectrum shown in Figure 4. The visible bands [488, 505 (sh) nm] clearly are distinct from ferric DHP B, Compound I or II/ES, and ferrous DHP B in terms of both absorption maxima and molar absorptivity. The Soret band (~421–3 nm) is also distinct when compared to the ferrous (432 nm) or ferric (410 nm) forms of the enzyme at this pH. Overall, the UV-visible spectroscopic data suggest the presence of an intermediate prior to the formation of Compound II, with the latter peroxidase-active species being formed from the ferrous enzyme in the absence of TCP. Addition of TCP (10 equiv) to the Compound II intermediate formed here in the absence of co-substrate yielded the ferric state of the enzyme as expected (data not shown).
Figure 3.

UV-visible spectroscopic monitoring (10 scans, 0.1 min interval/scan, 60 sec total) of the reaction between ferrous DHP B (10 μM) with (A) 2.5 equiv (B) 10 equiv and (C) 25 equiv H2O2 yielding Compound II at pH 8. Inset: Same experimental conditions as described above except that the ferrous DHP B concentration was 50 μM.
Figure 4.

Comparison of the visible spectroscopic features of ferrous DHP B (10 μM, black), ferric DHP B (green), Compound II (red), and the putative ferrous-hydroperoxide intermediate (blue) that was formed prior to Compound II in 100 mM KPi (pH 8) at 4 °C.
In the presence of one-equivalent TCP, the reaction of ferrous DHP B with hydrogen peroxide was qualitatively similar to that described above for the reaction in the absence of trihalophenol in that the formation of an intermediate was observed prior to the identification of Compound II (Figure SD11). Again, this intermediate interconverted to form Compound II [UV-vis features: 420 (Soret), 546, 586 nm], but the process was ~5-fold faster than when TCP was absent. Under the conditions examined, Compound II was stable for duration of the 60 s observation window, a marked contrast to the instability of Compound ES under similar conditions (10, 26). However, when the TCP concentration in the reaction was increased to 30 equivalents, the Compound II intermediate converted to ferric DHP B (Figure SD12), with a concomitant decrease in the TCP absorbance (312 nm) that was proportional to the amount of hydrogen peroxide initially reacted (Figure SD12A – 2.5 equiv H2O2; SD12B – 10 equiv H2O2). The TCP-dependent reactivity of Compound II observed here was consistent with that noted for the reaction of oxyferrous DHP B with hydrogen peroxide from our stopped-flow study (vide supra).
Reactivity Studies of Oxyferrous DHP B
The reaction of oxyferrous DHP B (10 μM) with hydrogen peroxide (2.5, 10, and 25 equiv) in the presence of TCP co-substrate yielding DCQ was further explored by UV-visible spectroscopy. No reaction was observed upon the addition of 100 μM H2O2 to a solution of oxyferrous DHP B containing a sub-stoichiometric amount of TCP (0.5 equiv, data not shown). As such, the TCP concentration was varied over a range of 10, 150 and 300 μM (1 – 30 equiv, Figures SD13-SD15) to evaluate the effect of co-substrate on Compound II formation and reactivity. With one equivalent TCP, the DHP B Compound II intermediate was found to be stable over the reaction time of 60 seconds (Figure SD13). Neither TCP loss (312 nm) nor DCQ formation (274 nm) were able to be observed when only one equivalent of TCP was initially reacted. As was observed in the stopped-flow study (vide supra), when the amount of TCP (15 – 30 equiv) greatly exceeded that of H2O2, the ferryl-containing intermediate converted to a species whose spectral features matched those of ferric DHP B (Figures SD14 and SD15), concomitant with TCP loss and DCQ formation, the extent for both being well-correlated with the amount of H2O2 initially employed in the reaction. Reaction conditions that generated both ferric DHP B and a relatively large amount of DCQ (i.e., [TCP] = 300 μM and [H2O2] = 100 μM, Figure SD15B) exhibited re-formation of the oxyferrous state at longer reaction times (5 min, data not shown). This observation was consistent with our previous report that incubation of the ferric form of the enzyme with DCQ generates oxyferrous DHP B (26). At higher concentrations of H2O2, bleaching of the heme cofactor was noted. The apparent increase in the baseline between 450 – 550 nm when high concentrations of DCQ are formed may be attributable to the formation of 2,6-dichloro-3-hydroxy-1,4-benzoquinone (DCHB) as a secondary product (λmax = 524 nm; ε = 2530 ± 60 M−1 cm−1) (55, 56).
Trapping of TCP Radicals During Enzymatic Turnover
TCP radicals generated from the reaction of ferric or oxyferrous DHP B (30 μM) and H2O2 (5 mM) in the presence of 1.25 mM TCP at pH 7 were trapped using the radical trap 5-diisopropoxy-phosphoryl-5-methyl-1-pyrroline-N-oxide (DIPPMPO). As an alternative to traditional EPR spectroscopic methods, the results were analyzed by 31P-NMR spectroscopy as previously described (37). DIPPMO alone exhibited a resonance at δ 22.2 ppm (Figure SD16A). The 31P-NMR spectra acquired after the reaction employing either ferric (Figure SD16B) or oxyferrous DHP B (Figure SD16C) both exhibited two peaks at δ 22.2 and 25.1 ppm, which correspond to the native spin trap signal and the covalent adduct of TCP-DIPPMO, respectively. These NMR spectroscopic results here for DHP B were indistinguishable from the previous identification (and re-confirmed here as a control, Figure SD16D) of TCP radicals generated from the reaction of horseradish peroxidase with H2O2 in the presence of TCP (37), and provide unequivocal evidence for the formation of TCP radicals that have been postulated to form during dehaloperoxidase turnover (21, 26).
DISCUSSION
As both an oxygen-transport globin and a peroxidase, dehaloperoxidase from the terebellid polychaete Amphitrite ornata possesses two inherent functions that occur at a single heme active site. Given that reversible oxygen-binding is only mediated via a ferrous heme in globins, and that peroxidase activity is initiated from ferric centers and to the exclusion of the oxyferrous oxidation state from the peroxidase cycle, the bifunctional nature of DHP as a globin-peroxidase appears to be at odds with the traditional starting oxidation state for each individual activity. Our understanding of how the enzyme rectifies this apparent paradox, wherein the peroxidase-active ferric DHP state can be converted to the oxygen-binding ferrous form, or the reverse, wherein oxyferrous DHP is activated toward peroxidase function, is still lacking. Thus, as the means of resolving this paradox, the primary focus of this report is to provide the details of dehaloperoxidase chemistry which have yet to be reported for the deoxyferrous state of the enzyme, and to augment those with additional studies of oxyferrous DHP.
The activity studies presented herein further demonstrate that dehaloperoxidase B is able to catalyze the oxidative dehalogenation of 2,4,6-trihalogenated phenols to their corresponding 2,6-dihalo-1,4-benzoquinones in the presence of hydrogen peroxide when the catalytic cycle was initiated from the oxyferrous state. This is of critical importance given that the oxyferrous state is normally a catalytically incompetent species for monofunctional peroxidases (57), and represents a partial solution to the aforementioned paradox. As it is possible that dehaloperoxidase may have evolved its peroxidase function to begin from the oxyferrous state, which is the normal oxidation state for this hemoglobin in A. ornata, the reaction of oxyferrous DHP with hydrogen peroxide was further investigated using stopped-flow UV-visible spectroscopy. In the presence of as little as one equivalent of TCP co-substrate, the first intermediate observed exhibited spectral features which matched those for a ferryl-containing species that lacked a porphyrin π-cation radical (10), suggestive of the formation of either Compound ES or Compound II. However, Compound ES was ruled out on the basis of electron counting, as it is three electrons oxidized with respect to the starting ferrous oxidation state, and hydrogen peroxide is only a two-electron oxidant when cleaved heterolytically. Furthermore, RFQ-CW-EPR spectroscopic studies presented here also ruled out this intermediate as Compound ES, as the presence of a protein radical was not observed, consistent with the formulation of the ferryl-containing intermediate as DHP B Compound II.
When oxyferrous DHP was reacted under conditions where [TCP] > [H2O2], it was observed that Compound II converted to the ferric form of the enzyme, concomitant with TCP loss and DCQ formation. Sequential double-mixing stopped-flow studies demonstrated that DHP B Compound II was directly capable of oxidizing TCP. These results were consistent with TCP serving as a one-electron reducing agent. Dawson and co-workers (21) recently reported evidence supporting a peroxidase cycle for DHP A involving two sequential one-electron oxidations of TCP to yield DCQ when preformed Compound ES was employed (presumably via a transiently formed Compound II species), and it appears that TCP here is serving a similar function in terms of reducing Compound II to the ferric state. Ferric DHP B was not the final ‘resting’ state of the enzyme, however, as experimental conditions that favored the formation of DCQ also led to the regeneration of the starting oxyferrous DHP B species. We attribute this to the previously characterized reaction between ferric DHP B with DCQ (26), which was shown to form oxyferrous DHP B. The unusually high reduction potential of +206 mV for a peroxidase likely favors reduction of the ferric heme and formation of oxyferrous DHP B in the presence of DCQ.
In light of the spectroscopic and biochemical findings presented here, we propose the following updated mechanism for the in vitro peroxide-dependent oxidation of trihalophenol as catalyzed by DHP B from A. ornata (Scheme 1): In the presence of trihalophenol, (de)oxyferrous DHP B reacts with H2O2 to form the peroxidase-active species Compound II (step i), which is subsequently reduced in the presence of excess TXP to the ferric enzyme (step ii). The ferric enzyme can proceed along a traditional peroxidase reaction pathway, reacting with hydrogen peroxide to generate Compound ES via a transiently formed Compound I species (step iii), and returning to the ferric state via two consecutive one-electron reduction steps as postulated by Dawson and co-workers (21) (steps iv and ii). Dihaloquinone can be formed from either two consecutive one-electron oxidations of TXP (via a TXP radical), or from the disproportionation of TXP radicals. The DXQ product thus formed, either due to its inherent instability or as an alternative co-substrate for the enzyme, leads to the reduction of the ferric state to the ferrous, generating oxyferrous DHP B in the presence of dioxygen (step v). Alternatively, Compound ES may directly react with DCQ as a co-substrate, leading to the generation of the (de)oxyferrous species (step vi). The proposed mechanism therefore resolves the dehaloperoxidase paradox wherein the bifunctional nature of a globin-peroxidase appears to be at odds with the traditional starting oxidation state for each individual activity. Namely, (de)oxyferrous DHP is a catalytically competent state for initiating a peroxidase catalytic cycle, and is the first such example that has a relevant biological function. Moreover, as discussed previously (26) and noted here in additional detail, dihaloquinone generated from the peroxidase cycle is itself a substrate for both ferric and Compound ES DHP B, leading to formation of oxyferrous DHP B. Thus, the product of the peroxidase cycle regenerates the globin-active species, which is also a viable oxidation state for initiating peroxidase chemistry. Taken together, these two reactivities observed here highlight the unique chemistry of dehaloperoxidase which enables both globin and peroxidase discrete functions from what appears to be a limited set of resources.
Interestingly, in the absence of TCP, oxyferrous DHP B was unreactive towards hydrogen peroxide with respect to the formation of observable high-valent iron-oxo intermediates (i.e., Compound II), and therefore a role for trihalophenol needs to be postulated that is consistent with the above mechanism. Dawson and co-workers have suggested that the observed conversion of oxyferrous DHP to Compound II/ES was due to the presence of a trace amount of ferric DHP that undergoes a traditional peroxidase cycle, thereby generating TCP radicals that oxidize the bulk oxyferrous DHP to the ferric enzyme (28). Subsequent reaction of the resulting ferric DHP with hydrogen peroxide generates the observed ferryl-containing species. In that work, the typical reaction conditions employed an excess of TCP with respect to H2O2. Although precedent for the oxyferrous to ferric conversion as catalyzed by substrate radicals has been demonstrated for lignin peroxidase, the reaction conditions employed in our RFQ-EPR spectroscopic study and stoichiometric considerations argue against this mechanism. Specifically, 10 equivalents of H2O2 were reacted with oxyferrous DHP B in the presence of one equivalent of TCP. Assuming trace ferric DHP B initially reacted to generate TCP radicals, which then oxidized oxyferrous DHP B to the ferric form (thereby regenerating TCP in the process), a one-half equivalent of hydrogen peroxide would have then been consumed, leaving 9.5 equivalents H2O2 to subsequently react with the ferric enzyme and TCP in solution (one equivalent each). This reaction would generate DHP B Compound ES in situ, a two-electron oxidant which has been shown to react rapidly with TCP to yield DCQ (one equivalent) and the ferric enzyme (26). The ferric enzyme would then react with the remaining H2O2, reforming Compound ES, whose subsequent reaction with DCQ is slow enough to have allowed for identification of the amino acid radical of this intermediate by RFQ-EPR spectroscopy. As no protein radical was observed in our EPR spectroscopic study, we conclude that the observed oxyferrous reactivity yielding Compound II cannot be attributable to a mechanism invoking a transiently formed ferric state, and that the oxidation of oxyferrous DHP B via TCP radicals is unlikely under the reaction conditions examined ([H2O2] > [TCP]).
Thus, an alternative mechanism that involves trihalophenol binding, but not TXP radical formation, is needed to describe the reaction of oxyferrous DHP with hydrogen peroxide yielding Compound II. Based on our anaerobic studies, we suggest that the reaction of deoxyferrous DHP B with H2O2 is responsible for the formation of the observed Compound II intermediate, and that trihalophenol binding plays a role in the activation of oxyferrous DHP to the deoxy state when hydrogen peroxide is present. Specifically, we have observed the formation of Compound II upon the reaction of deoxyferrous DHP B with hydrogen peroxide in the absence of trihalophenol. The conversion of deoxyferrous hemoproteins to Compound II in what is generally regarded as a single-step, two-electron oxidation has been noted and/or postulated in a number of other systems, including horseradish peroxidase (58), leghemoglobin (59), lactoperoxidase (60, 61), myeloperoxidase (62), and KatG (63). Importantly, none of aforementioned systems invoked substrate radicals as mechanistically relevant to account for their observed chemistry. Furthermore, the importance of this observation is also underscored by the in vivo role of dehaloperoxidase as an oxygen-transport protein, wherein the globin exists in both the oxy- and deoxyferrous states, and it is reasonable to surmise that the initiation of a peroxidase pathway should not be limited to only one of the two major states of a globin.
The reaction of deoxyferrous DHP B with hydrogen peroxide surprisingly revealed an intermediate that formed prior to the full formation of Compound II. The spectral features of this species did not match any for the known intermediates of dehaloperoxidase (e.g., Compounds I/II/ES/III) (10, 26). One possibility is that this intermediate preliminarily represents the ferrous-hydroperoxide adduct that must form prior to heterolytic cleavage of the hydrogen peroxide that ultimately yields Compound II. The observation of the Fe(II)-OOH precursor prior to Compound II formation is analogous to the Compound 0 species, Fe(III)-OOH, that has been shown to form prior to Compound I in traditional heme chemistry (52, 53, 64–68). Typically, such ferric-hydroperoxide species are short-lived (< 200 μs) (64), however examples of peroxidase (69) and myoglobin mutants (68), as well as native cytochrome c552 (65), have been reported that exhibit more reasonable lifetimes (ms – sec), with the consensus opinion that the lack of a suitable proton donor in the hydrophobic heme cavities lowers the rate of heterolytic O-O bond cleavage, thereby stabilizing Compound 0. A similar argument can be posited to account for the relative stability of the putative ferrous-hydroperoxide intermediate of DHP B, as the distal histidine-to-heme distance in dehaloperoxidase [Fe-His Nε2 = 5.5 Å (33), ferric DHP B, His55 in the closed position] is intermediate between those found in globins (4.1–4.6 Å) and peroxidases (5.5–6.0 Å) (29–32), and as such may not be ideally positioned to facilitate H2O2 heterolysis. Further, when TCP was present prior to the reaction of deoxyferrous DHP B with H2O2, the stability of the Fe(II)-OOH intermediate was lowered, and we surmise that the position of the distal histidine (and the nature of the tautomeric bonds of the imidazole ring) thus facilitates O-O bond cleavage when trihalophenol is bound. This is consistent with the postulate that both external and internal small-molecule binding sites may exist with regulatory implications linked to the conformation flexibility of the distal histidine, His55, that may govern how DHP switches between its hemoglobin and peroxidase activities (11–13, 33). An additional consideration that may contribute to the stability of the ferrous-hydroperoxide precursor is the very positive Fe(III)/Fe(II) reduction potential (ca. ~ +200 mV) of DHP (26). Rather than the nearly ubiquitous Asp-His-Fe or Glu-His-Fe charge relays that are found in peroxidases and have been shown to impart a partially anionic character to the proximal histidine, dehaloperoxidase lacks such a catalytic triad. Instead, DHP possesses a proximal histidine that is in van der Waals contact with Met86 (35), and vibrational studies have suggested the presence of a neutral proximal histidine that is unlikely to activate bound peroxide via a push effect (70). Consequently, the lack of a push effect in DHP as well as a high reduction potential are two additional factors related to the presence of Met86 that likely contribute to the stability of the putative Fe(II)-OOH species.
The lack of reactivity observed for oxyferrous DHP B may have a physiological role when trihalophenol co-substrate is absent, preventing formation of Compound RH (with its attenuated dehaloperoxidase activity) when exposed to hydrogen peroxide for short reaction times (< 60 s), and also minimizing the deleterious effects of unwanted peroxidase activity in the absence of a reducing substrate. In the case of the different reactivity observed for ferrous and oxyferrous DHP B in the absence of trihalophenol, it is likely that the bound oxygen molecule acts as an inhibitor of the reaction by blocking H2O2 from binding to the heme iron. As described by Alayash and co-workers (71) for the reaction of oxyferrous swMb with hydrogen peroxide, the rate constant that describes the initial oxidation of the oxyferrous heme by hydrogen peroxide reflects the competition between the two ligands, O2 and H2O2, for the ferrous iron. Under conditions of high H2O2 and low O2 concentrations, the apparent rate of the oxyferrous reaction approaches that of the intrinsic rate constant for heterolytic cleavage of the bound peroxide, whereas under reaction conditions of high O2 and low H2O2 concentrations, the rate is proportional to KH2O2/KO2, the ratio of the binding constants of the two ligands. The kinetic observations by Dawson and co-workers for the reaction of oxyferrous DHP with hydrogen peroxide (TCP present) (28), when re-interpreted in light of our proposed mechanism of direct (de)oxyferrous oxidation to Compound II (Schemes 1 & 2), appear to be consistent with the kinetic scheme of Alayash and co-workers with respect to the observed lag times that were presented as evidence for the conversion of oxyferrous DHP to the ferric state via substrate radicals (generated from trace ferric DHP). However, further studies exploring both mechanistic interpretations will be necessary to fully elucidate the details of this process.
Scheme 2.

Proposed tautomerization of the distal histidine in oxyferrous DHP B upon trihalophenol binding. A direct interaction between His55 and TXP is depicted, however binding may occur elsewhere, leading to the proposed tautomerization.
It is clear that trihalophenol binding triggers the activation of oxyferrous DHP B to Compound II in the presence of hydrogen peroxide. It has been suggested that His55, which has been observed in distinct ‘open’ and ‘closed’ conformations, mediates O2 displacement from the heme in the presence of (tri)halophenol substrate, possibly serving as this trigger for peroxidase function (11). Given this, and in light of the results described herein, we have considered the role of the distal histidine as integral to the substrate-dependent activation of oxyferrous DHP B, and propose the following mechanism for the activation step of the dehaloperoxidase cycle (Scheme 2): Upon binding of trihalophenol to oxyferrous DHP, tautomerization of the distal His55 occurs, forming a hydrogen-bond between the His-Nδ and the phenolic hydrogen of the co-substrate, and disrupting the H-bond between the His-Nε2 and the bound oxygen ligand that had stabilized the oxyferrous species. Evidence for the destabilization of oxyferrous DHP upon trihalophenol binding comes from Dawson and co-workers who observed a 2.6-fold increase in the rate of autoxidation of the oxyferrous enzyme in the presence of TCP when compared to its absence (28). The destabilized oxyferrous DHP is therefore activated for reaction with hydrogen peroxide, which displaces the now weakly bound oxygen ligand, generating the Fe(II)-OOH intermediate that was observed in the deoxyferrous studies. Heterolysis of this ferrous-hydroperoxide intermediate, facilitated by the tautomer that now positions the His-Nε2 as a general base, yields Compound II, which can now initiate a peroxidase-cycle as described in Scheme 1. It is important to note that although the trihalophenol binding site is believed to be external to the heme active site (11–13), it is not known whether bound TXP can directly interact with the distal histidine. Thus, tautomerization may result from either a direct or indirect (allosteric) binding event, and this ambiguity is schematically represented in Scheme 2. There are several examples in the literature in which histidine tautomerization has played an important role during enzymatic function for both heme and non-heme proteins. These include i) human carbonic anhydrase II (hCAII), where the tautomers of His64 (tautomeric equilibrium constant of 1.0) act as both a general acid and base, mediating the transfer of both protons and water molecules at a neutral pH with high efficiency (72), and ii) myoglobin (73–79), where the distal histidine, His64, has been postulated to play a role in the discrimination of oxygen vs. CO binding. It is thought that ligand entry into the heme pocket of Mb is controlled by the distal histidine gate, and hydrogen bond donation through the neutral NδH tautomer of His64 regulates oxygen affinity. In our system, we propose that TXP co-substrate binding similarly affects the stability of the oxyferrous enzyme, enabling reaction with hydrogen peroxide, and allowing for the distal histidine to serve as the general base required for H2O2 heterolysis.
Conclusion
We have shown that the reaction of deoxyferrous DHP with hydrogen peroxide forms Compound II, a peroxidase active species, in the absence of trihalophenol. Literature precedents for other heme systems support such a direct two-electron oxidation of the deoxyferrous heme without the need for invoking a ferric oxidation state that has been the traditional starting point for initiating the peroxidase catalytic cycle. Further, although not altogether excluding a role for substrate radicals in effecting an oxyferrous to Compound II conversion, the proposed mechanism suggests that substrate radicals need not be considered for this process to occur, and that a simple displacement of the bound oxygen ligand upon simultaneous trihalophenol and H2O2 binding suffices. We have also shown preliminary evidence for a ferrous-hydroperoxide intermediate that forms prior to Compound II. Given the relative stability of this species, future studies will be directed towards its characterization and reactivity. When combined with the known DCQ-driven reduction of ferric DHP to the oxyferrous enzyme, the chemistry reported herein establishes oxyferrous DHP as both a starting and end point for peroxidase chemistry that maintains a globin-active center, resolves the oxidation state paradox of globin-peroxidases, and furthers our understanding of how this bifunctional enzyme is able to unite its two inherent functions in one system.
Supplementary Material
Abbreviations
- DHP
dehaloperoxidase
- Hb
hemoglobin
- Mb
myoglobin
- HhMb
horse heart myoglobin
- DCP
2,4-dichlorophenol
- TBP
2,4,6-tribromophenol
- TCP
2,4,6-trichlorophenol
- TFP
2,4,6-trifluorophenol
- TXP
trihalophenol
- DXQ
dihalophenol
- RFQ-CW-EPR
rapid-freeze-quench continuous wave electron paramagnetic resonance
- Compound I
two-electron oxidized heme center when compared to the ferric form, commonly as an FeIV= O porphyrin π-cation radical
- Compound II
one-electron oxidized heme center when compared to the ferric form, commonly as an FeIV=O or FeIV–OH
- Compound III
oxyferrous [FeII-O2 or FeIII-(O2−)] state of the enzyme
- Compound ES
two-electron-oxidized state containing both a ferryl center [FeIV=O] and an amino acid (tryptophanyl or tyrosyl) radical, analogous to Compound ES in cytochrome c peroxidase
- Compound RH
‘Reversible Heme’ state of dehaloperoxidase, formed from the decay of Compound ES in the absence of co-substrate
- 5cHS
five-coordinate high spin heme
- 6cHS
six-coordinate high spin heme
- 6cLS
six-coordinate low spin heme
Footnotes
This project was supported by the North Carolina State University Molecular Biotechnology Training Program through an NIH T32 Biotechnology Traineeship grant (J. D.) and the Army Research Office Grant 57861-LS (R. A. G.).
Supporting Information Available – Methods for the preparation of oxyferrous DHP B from TCEP or sodium dithionite (Fig. SD1), UV-visible spectra of oxyferrous DHP B in the presence and absence of TCP (Fig. SD2), stopped-flow UV-visible spectroscopic monitoring of the reaction between oxyferrous DHP B and a 10-fold excess of H2O2 (Fig. SD3), dependence of kobs as a function of TCP concentration for the formation of DHP B Compound II (Fig. SD4), stopped-flow UV-visible spectroscopic monitoring of the reaction between oxyferrous DHP B containing 30 equivalent TCP and a 10-fold excess of H2O2 (Fig. SD5), stopped-flow UV-visible spectroscopic monitoring of the reaction between ferric DHP B and a 10-fold excess of H2O2 (Fig. SD6), sequential stopped-flow double-mixing reaction between preformed Compound II and 2.5–25 equiv of TCP at pH 8 (Fig. SD7), EPR spectrum of the radical formed from the reaction between ferric DHP B containing 1 equiv TCP with a 10-fold excess of H2O2 (Fig. SD8), titration of ferric DHP B with sodium dithionite (Fig. SD9), UV-visible spectroscopic monitoring of the reaction between ferrous DHP B with H2O2 yielding Compound II in the absence (Fig. SD10) and presence (Figs. SD 11 & 12) of TCP, UV-visible spectroscopic monitoring of the reaction between oxyferrous DHP B containing TCP reacted with H2O2 yielding Compound II (Figs. SD13–15), and 31P-NMR spectra of DIPPMO in the presence of H2O2, TCP, and ferric DHP B, oxyferrous DHP B, and HRP (Fig. SD16). This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Woodin SA. Recruitment of Infauna - Positive or Negative Cues. Am Zool. 1991;31:797–807. [Google Scholar]
- 2.Woodin SA, Walla MD, Lincoln DE. Occurrence of Brominated Compounds in Soft-Bottom Benthic Organisms. J Exp Mar Biol Ecol. 1987;107:209–217. [Google Scholar]
- 3.Chen YP, Lincoln DE, Woodin SA, Lovell CR. Purification and Properties of a Unique Flavin-Containing Chloroperoxidase from the Capitellid Polychaete Notomastus-Lobatus. J Biol Chem. 1991;266:23909–23915. [PubMed] [Google Scholar]
- 4.Roach MP, Chen YP, Woodin SA, Lincoln DE, Lovell CR, Dawson JH. Notomastus lobatus chloroperoxidase and Amphitrite ornata dehaloperoxidase both contain histidine as their proximal heme iron ligand. Biochem. 1997;36:2197–2202. doi: 10.1021/bi9621371. [DOI] [PubMed] [Google Scholar]
- 5.Lincoln DE, Fielman KT, Marinelli RL, Woodin SA. Bromophenol accumulation and sediment contamination by the marine annelids Notomastus lobatus and Thelepus crispus. Biochem Syst Ecol. 2005;33:559–570. [Google Scholar]
- 6.King GM. Inhibition of Microbial Activity in Marine-Sediments by a Bromophenol from a Hemichordate. Nature. 1986;323:257–259. [Google Scholar]
- 7.Fielman KT, Targett NM. Variation of 2,3,4-Tribromopyrrole and Its Sodium Sulfamate Salt in the Hemichordate Saccoglossus-Kowalevskii. Mar Ecol-Prog Ser. 1995;116:125–136. [Google Scholar]
- 8.Chen YP, Woodin SA, Lincoln DE, Lovell CR. An unusual dehalogenating peroxidase from the marine terebellid polychaete Amphitrite ornata. J Biol Chem. 1996;271:4609–4612. [PubMed] [Google Scholar]
- 9.Osborne RL, Taylor LO, Han KP, Ely B, Dawson JH. Amphitrite ornata dehaloperoxidase: enhanced activity for the catalytically active globin using MCPBA. Biochem Biophys Res Commun. 2004;324:1194–1198. doi: 10.1016/j.bbrc.2004.09.174. [DOI] [PubMed] [Google Scholar]
- 10.Feducia J, Dumarieh R, Gilvey LB, Smirnova T, Franzen S, Ghiladi RA. Characterization of dehaloperoxidase compound ES and its reactivity with trihalophenols. Biochem. 2009;48:995–1005. doi: 10.1021/bi801916j. [DOI] [PubMed] [Google Scholar]
- 11.Chen Z, de Serrano V, Betts L, Franzen S. Distal histidine conformational flexibility in dehaloperoxidase from Amphitrite ornata. Acta Crystallogr D Biol Crystallogr. 2009;65:34–40. doi: 10.1107/S0907444908036548. [DOI] [PubMed] [Google Scholar]
- 12.Davis MF, Gracz H, Vendeix FA, de Serrano V, Somasundaram A, Decatur SM, Franzen S. Different modes of binding of mono-, di-, and trihalogenated phenols to the hemoglobin dehaloperoxidase from Amphitrite ornata. Biochem. 2009;48:2164–2172. doi: 10.1021/bi801568s. [DOI] [PubMed] [Google Scholar]
- 13.Smirnova TI, Weber RT, Davis MF, Franzen S. Substrate binding triggers a switch in the iron coordination in dehaloperoxidase from Amphitrite ornata: HYSCORE experiments. J Am Chem Soc. 2008;130:2128–2129. doi: 10.1021/ja0772952. [DOI] [PubMed] [Google Scholar]
- 14.Miksovska J, Horsa S, Davis MF, Franzen S. Conformational dynamics associated with photodissociation of CO from dehaloperoxidase studied using photoacoustic calorimetry. Biochem. 2008;47:11510–11517. doi: 10.1021/bi8012033. [DOI] [PubMed] [Google Scholar]
- 15.Nienhaus K, Nickel E, Davis MF, Franzen S, Nienhaus GU. Determinants of substrate internalization in the distal pocket of dehaloperoxidase hemoglobin of Amphitrite ornata. Biochem. 2008;47:12985–12994. doi: 10.1021/bi801564r. [DOI] [PubMed] [Google Scholar]
- 16.Franzen S, Gilvey LB, Belyea JL. The pH dependence of the activity of dehaloperoxidase from Amphitrite ornata. Biochim Biophys Acta. 2007;1774:121–130. doi: 10.1016/j.bbapap.2006.09.019. [DOI] [PubMed] [Google Scholar]
- 17.de Serrano V, Chen Z, Davis MF, Franzen S. X-ray crystal structural analysis of the binding site in the ferric and oxyferrous forms of the recombinant heme dehaloperoxidase cloned from Amphitrite ornata. Acta Crystallogr D Biol Crystallogr. 2007;63:1094–1101. doi: 10.1107/S0907444907043417. [DOI] [PubMed] [Google Scholar]
- 18.Franzen S, Jasaitis A, Belyea J, Brewer SH, Casey R, MacFarlane AWt, Stanley RJ, Vos MH, Martin JL. Hydrophobic distal pocket affects NO-heme geminate recombination dynamics in dehaloperoxidase and H64V myoglobin. J Phys Chem B. 2006;110:14483–14493. doi: 10.1021/jp056790m. [DOI] [PubMed] [Google Scholar]
- 19.Franzen S, Belyea J, Gilvey LB, Davis MF, Chaudhary CE, Sit TL, Lommel SA. Proximal cavity, distal histidine, and substrate hydrogen-bonding mutations modulate the activity of Amphitrite ornata dehaloperoxidase. Biochem. 2006;45:9085–9094. doi: 10.1021/bi060020z. [DOI] [PubMed] [Google Scholar]
- 20.Belyea J, Belyea CM, Lappi S, Franzen S. Resonance Raman study of ferric heme adducts of dehaloperoxidase from Amphitrite ornata. Biochem. 2006;45:14275–14284. doi: 10.1021/bi0609218. [DOI] [PubMed] [Google Scholar]
- 21.Osborne RL, Coggins MK, Raner GM, Walla M, Dawson JH. The mechanism of oxidative halophenol dehalogenation by Amphitrite ornata dehaloperoxidase is initiated by H2O2 binding and involves two consecutive one-electron steps: role of ferryl intermediates. Biochem. 2009;48:4231–4238. doi: 10.1021/bi900367e. [DOI] [PubMed] [Google Scholar]
- 22.Osborne RL, Coggins MK, Walla M, Dawson JH. Horse heart myoglobin catalyzes the H2O2-dependent oxidative dehalogenation of chlorophenols to DNA-binding radicals and quinones. Biochem. 2007;46:9823–9829. doi: 10.1021/bi700684u. [DOI] [PubMed] [Google Scholar]
- 23.Osborne RL, Raner GM, Hager LP, Dawson JH. C. fumago chloroperoxidase is also a dehaloperoxidase: oxidative dehalogenation of halophenols. J Am Chem Soc. 2006;128:1036–1037. doi: 10.1021/ja056213b. [DOI] [PubMed] [Google Scholar]
- 24.Osborne RL, Sumithran S, Coggins MK, Chen YP, Lincoln DE, Dawson JH. Spectroscopic characterization of the ferric states of Amphitrite ornata dehaloperoxidase and Notomastus lobatus chloroperoxidase: His-ligated peroxidases with globin-like proximal and distal properties. J Inorg Biochem. 2006;100:1100–1108. doi: 10.1016/j.jinorgbio.2006.02.008. [DOI] [PubMed] [Google Scholar]
- 25.Han K, Woodin SA, Lincoln DE, Fielman KT, Ely B. Amphitrite ornata, a marine worm, contains two dehaloperoxidase genes. Mar Biotechnol (NY) 2001;3:287–292. doi: 10.1007/s10126-001-0003-8. [DOI] [PubMed] [Google Scholar]
- 26.D’Antonio J, D’Antonio EL, Thompson MK, Bowden EF, Franzen S, Smirnova T, Ghiladi RA. Spectroscopic and mechanistic investigations of dehaloperoxidase B from Amphitrite ornata. Biochem. 2010;49:6600–6616. doi: 10.1021/bi100407v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dunford HB, Stillman JS. Function and Mechanism of Action of Peroxidases. Coord Chem Rev. 1976;19:187–251. [Google Scholar]
- 28.Du J, Sono M, Dawson JH. Functional Switching of Amphitrite ornata Dehaloperoxidase from O2-Binding Globin to Peroxidase Enzyme Facilitated by Halophenol Substrate and H2O2. Biochem. 2010;49:6064–6069. doi: 10.1021/bi100741z. [DOI] [PubMed] [Google Scholar]
- 29.Matsui T, Ozaki S, Liong E, Phillips GN, Jr, Watanabe Y. Effects of the location of distal histidine in the reaction of myoglobin with hydrogen peroxide. J Biol Chem. 1999;274:2838–2844. doi: 10.1074/jbc.274.5.2838. [DOI] [PubMed] [Google Scholar]
- 30.Finzel BC, Poulos TL, Kraut J. Crystal structure of yeast cytochrome c peroxidase refined at 1.7-A resolution. J Biol Chem. 1984;259:13027–13036. [PubMed] [Google Scholar]
- 31.Phillips GN, Jr, Arduini RM, Springer BA, Sligar SG. Crystal structure of myoglobin from a synthetic gene. Proteins. 1990;7:358–365. doi: 10.1002/prot.340070407. [DOI] [PubMed] [Google Scholar]
- 32.Gajhede M, Schuller DJ, Henriksen A, Smith AT, Poulos TL. Crystal structure of horseradish peroxidase C at 2.15 A resolution. Nat Struct Biol. 1997;4:1032–1038. doi: 10.1038/nsb1297-1032. [DOI] [PubMed] [Google Scholar]
- 33.de Serrano V, D’Antonio J, Franzen S, Ghiladi RA. Structure of dehaloperoxidase B at 1.58 A resolution and structural characterization of the AB dimer from Amphitrite ornata. Acta Crystallogr D Biol Crystallogr. 2010;66:529–538. doi: 10.1107/S0907444910004580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.LaCount MW, Zhang E, Chen YP, Han K, Whitton MM, Lincoln DE, Woodin SA, Lebioda L. The crystal structure and amino acid sequence of dehaloperoxidase from Amphitrite ornata indicate common ancestry with globins. J Biol Chem. 2000;275:18712–18716. doi: 10.1074/jbc.M001194200. [DOI] [PubMed] [Google Scholar]
- 35.Lebioda L, LaCount MW, Zhang E, Chen YP, Han K, Whitton MM, Lincoln DE, Woodin SA. An enzymatic globin from a marine worm. Nature. 1999;401:445. doi: 10.1038/46728. [DOI] [PubMed] [Google Scholar]
- 36.Beers RF, Jr, Sizer IW. A spectrophotometric method for measuring the breakdown of hydrogen peroxide by catalase. J Biol Chem. 1952;195:133–140. [PubMed] [Google Scholar]
- 37.Zoia L, Argyropoulos DS. Phenoxy radical detection using P-31 NMR spin trapping. J Phys Org Chem. 2009;22:1070–1077. [Google Scholar]
- 38.Mayhew SG. The redox potential of dithionite and SO-2 from equilibrium reactions with flavodoxins, methyl viologen and hydrogen plus hydrogenase. Eur J Biochem. 1978;85:535–547. doi: 10.1111/j.1432-1033.1978.tb12269.x. [DOI] [PubMed] [Google Scholar]
- 39.Kizek R, Vacek J, Trnkova L, Jelen F. Cyclic voltammetric study of the redox system of glutathione using the disulfide bond reductant tris(2-carboxyethyl)phosphine. Bioelectrochem. 2004;63:19–24. doi: 10.1016/j.bioelechem.2003.12.001. [DOI] [PubMed] [Google Scholar]
- 40.Heineman WR, Meckstroth ML, Norris BJ, Su CH. Optically transparent thin layer electrode techniquesfor the study of biological redox systems. J Electroanal Chem. 1979;104:577–585. [Google Scholar]
- 41.Ball EG. Studies on Oxidation-Reduction XXIII: Ascorbic Acid. J Biol Chem. 1937;118:219–239. [Google Scholar]
- 42.Belyea J, Gilvey LB, Davis MF, Godek M, Sit TL, Lommel SA, Franzen S. Enzyme function of the globin dehaloperoxidase from Amphitrite ornata is activated by substrate binding. Biochem. 2005;44:15637–15644. doi: 10.1021/bi051731k. [DOI] [PubMed] [Google Scholar]
- 43.Lardinois OM, Ortiz de Montellano PR. Autoreduction of ferryl myoglobin: discrimination among the three tyrosine and two tryptophan residues as electron donors. Biochem. 2004;43:4601–4610. doi: 10.1021/bi036241b. [DOI] [PubMed] [Google Scholar]
- 44.Giulivi C, Cadenas E. Ferrylmyoglobin: formation and chemical reactivity toward electron-donating compounds. Methods Enzymol. 1994;233:189–202. doi: 10.1016/s0076-6879(94)33022-0. [DOI] [PubMed] [Google Scholar]
- 45.Herold S, Rehmann FJ. Kinetics of the reactions of nitrogen monoxide and nitrite with ferryl hemoglobin. Free Radic Biol Med. 2003;34:531–545. doi: 10.1016/s0891-5849(02)01355-2. [DOI] [PubMed] [Google Scholar]
- 46.Giulivi C, Davies KJ. Hydrogen peroxide-mediated ferrylhemoglobin generation in vitro and in red blood cells. Methods Enzymol. 1994;231:490–496. doi: 10.1016/0076-6879(94)31032-7. [DOI] [PubMed] [Google Scholar]
- 47.Yadav RK, Dolai S, Pal S, Adak S. Role of tryptophan-208 residue in cytochrome c oxidation by ascorbate peroxidase from Leishmania major-kinetic studies on Trp208Phe mutant and wild type enzyme. Biochim Biophys Acta. 2008;1784:863–871. doi: 10.1016/j.bbapap.2008.02.006. [DOI] [PubMed] [Google Scholar]
- 48.Marquez LA, Quitoriano M, Zilinskas BA, Dunford HB. Kinetic and spectral properties of pea cytosolic ascorbate peroxidase. FEBS Lett. 1996;389:153–156. doi: 10.1016/0014-5793(96)00562-5. [DOI] [PubMed] [Google Scholar]
- 49.Hewson WD, Hager LP. Oxidation of horseradish peroxidase compound II to compound I. J Biol Chem. 1979;254:3182–3186. [PubMed] [Google Scholar]
- 50.Belevich I, Borisov VB, Verkhovsky MI. Discovery of the true peroxy intermediate in the catalytic cycle of terminal oxidases by real-time measurement. J Biol Chem. 2007;282:28514–28519. doi: 10.1074/jbc.M705562200. [DOI] [PubMed] [Google Scholar]
- 51.Shintaku M, Matsuura K, Yoshioka S, Takahashi S, Ishimori K, Morishima I. Absence of a detectable intermediate in the compound I formation of horseradish peroxidase at ambient temperature. J Biol Chem. 2005;280:40934–40938. doi: 10.1074/jbc.M503472200. [DOI] [PubMed] [Google Scholar]
- 52.Baek HK, Van Wart HE. Elementary steps in the formation of horseradish peroxidase compound I: direct observation of compound 0, a new intermediate with a hyperporphyrin spectrum. Biochem. 1989;28:5714–5719. doi: 10.1021/bi00440a003. [DOI] [PubMed] [Google Scholar]
- 53.Egawa T, Yoshioka S, Takahashi S, Hori H, Nagano S, Shimada H, Ishimori K, Morishima I, Suematsu M, Ishimura Y. Kinetic and spectroscopic characterization of a hydroperoxy compound in the reaction of native myoglobin with hydrogen peroxide. J Biol Chem. 2003;278:41597–41606. doi: 10.1074/jbc.M210383200. [DOI] [PubMed] [Google Scholar]
- 54.Thompson MK, Franzen S, Ghiladi RA, Reeder BJ, Svistunenko DA. Compound ES of Dehaloperoxidase Decays via Two Alternative Pathways Depending on the Conformation of the Distal Histidine. J Am Chem Soc. 2010;132:17501–17510. doi: 10.1021/ja106620q. [DOI] [PubMed] [Google Scholar]
- 55.Lente G, Espenson JH. Photoreduction of 2,6-dichloroquinone in aqueous solution - Use of a diode array spectrophotometer concurrently to drive and detect a photochemical reaction. J Photoch Photobio A. 2004;163:249–258. [Google Scholar]
- 56.Lente G, Espenson JH. A kinetic study of the early steps in the oxidation of chlorophenols by hydrogen peroxide catalyzed by a water-soluble iron(III) porphyrin. New J Chem. 2004;28:847–852. [Google Scholar]
- 57.Dunford HB. Heme Peroxidases. Wiley-VCH; New York: 1999. [Google Scholar]
- 58.Noble RW, Gibson QH. The reaction of ferrous horseradish peroxidase with hydrogen peroxide. J Biol Chem. 1970;245:2409–2413. [PubMed] [Google Scholar]
- 59.Aviram I, Wittenberg A, Wittenberg JB. The reaction of ferrous leghemoglobin with hydrogen peroxide to form leghemoglobin(IV) J Biol Chem. 1978;253:5685–5689. [PubMed] [Google Scholar]
- 60.Kohler H, Taurog A, Dunford HB. Spectral studies with lactoperoxidase and thyroid peroxidase: interconversions between native enzyme, compound II, and compound III. Arch Biochem Biophys. 1988;264:438–449. doi: 10.1016/0003-9861(88)90309-8. [DOI] [PubMed] [Google Scholar]
- 61.Jantschko W, Furtmuller PG, Zederbauer M, Neugschwandtner K, Jakopitsch C, Obinger C. Reaction of ferrous lactoperoxidase with hydrogen peroxide and dioxygen: an anaerobic stopped-flow study. Arch Biochem Biophys. 2005;434:51–59. doi: 10.1016/j.abb.2004.10.014. [DOI] [PubMed] [Google Scholar]
- 62.Jantschko W, Georg Furtmuller P, Zederbauer M, Lanz M, Jakopitsch C, Obinger C. Direct conversion of ferrous myeloperoxidase to compound II by hydrogen peroxide: an anaerobic stopped-flow study. Biochem Biophys Res Commun. 2003;312:292–298. doi: 10.1016/j.bbrc.2003.10.117. [DOI] [PubMed] [Google Scholar]
- 63.Jakopitsch C, Wanasinghe A, Jantschko W, Furtmueller PG, Obinger C. Kinetics of interconversion of ferrous enzyme, compound II and compound III of wild-type synechocystis catalase-peroxidase and Y249F. Proposal for the catalatic mechanism. J Biol Chem. 2005;280:9037–9042. doi: 10.1074/jbc.M413317200. [DOI] [PubMed] [Google Scholar]
- 64.Tanaka M, Matsuura K, Yoshioka S, Takahashi S, Ishimori K, Hori H, Morishima I. Activation of hydrogen peroxide in horseradish peroxidase occurs within approximately 200 micro s observed by a new freeze-quench device. Biophys J. 2003;84:1998–2004. doi: 10.1016/s0006-3495(03)75008-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ichikawa Y, Nakajima H, Watanabe Y. Characterization of peroxide-bound heme species generated in the reaction of thermally tolerant cytochrome c552 with hydrogen peroxide. Chembiochem. 2006;7:1582–1589. doi: 10.1002/cbic.200600135. [DOI] [PubMed] [Google Scholar]
- 66.Denisov IG, Dawson JH, Hager LP, Sligar SG. The ferric-hydroperoxo complex of chloroperoxidase. Biochem Biophys Res Commun. 2007;363:954–958. doi: 10.1016/j.bbrc.2007.09.085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Denisov IG, Makris TM, Sligar SG. Formation and decay of hydroperoxo-ferric heme complex in horseradish peroxidase studied by cryoradiolysis. J Biol Chem. 2002;277:42706–42710. doi: 10.1074/jbc.M207949200. [DOI] [PubMed] [Google Scholar]
- 68.Brittain T, Baker AR, Butler CS, Little RH, Lowe DJ, Greenwood C, Watmough NJ. Reaction of variant sperm-whale myoglobins with hydrogen peroxide: the effects of mutating a histidine residue in the haem distal pocket. Biochem J. 1997;326(Pt 1):109–115. doi: 10.1042/bj3260109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ozaki S, Inada Y, Watanabe Y. Characterization of polyethylene glycolated horseradish peroxidase in organic solvents: Generation and stabilization of transient catalytic intermediates at low temperature. J Am Chem Soc. 1998;120:8020–8025. [Google Scholar]
- 70.Franzen S, Roach MP, Chen YP, Dyer RB, Woodruff WH, Dawson JH. The unusual reactivities of Amphirite ornata dehaloperoxidase and Notomastus lobatus chloroperoxidase do not arise from a histidine imidazolate proximal heme iron ligand. J Am Chem Soc. 1998;120:4658–4661. [Google Scholar]
- 71.Alayash AI, Ryan BA, Eich RF, Olson JS, Cashon RE. Reactions of sperm whale myoglobin with hydrogen peroxide. Effects of distal pocket mutations on the formation and stability of the ferryl intermediate. J Biol Chem. 1999;274:2029–2037. doi: 10.1074/jbc.274.4.2029. [DOI] [PubMed] [Google Scholar]
- 72.Shimahara H, Yoshida T, Shibata Y, Shimizu M, Kyogoku Y, Sakiyama F, Nakazawa T, Tate S, Ohki SY, Kato T, Moriyama H, Kishida K, Tano Y, Ohkubo T, Kobayashi Y. Tautomerism of histidine 64 associated with proton transfer in catalysis of carbonic anhydrase. J Biol Chem. 2007;282:9646–9656. doi: 10.1074/jbc.M609679200. [DOI] [PubMed] [Google Scholar]
- 73.Jewsbury P, Kitagawa T. The distal residue-CO interaction in carbonmonoxy myoglobins: a molecular dynamics study of two distal histidine tautomers. Biophys J. 1994;67:2236–2250. doi: 10.1016/S0006-3495(94)80708-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Rovira C, Schulze B, Eichinger M, Evanseck JD, Parrinello M. Influence of the heme pocket conformation on the structure and vibrations of the Fe-CO bond in myoglobin: a QM/MM density functional study. Biophys J. 2001;81:435–445. doi: 10.1016/S0006-3495(01)75711-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.De Angelis F, Jarzecki AA, Car R, Spiro TG. Quantum chemical evaluation of protein control over heme ligation: CO/O2 discrimination in myoglobin. J Phys Chem B. 2005;109:3065–3070. doi: 10.1021/jp0451851. [DOI] [PubMed] [Google Scholar]
- 76.Lukin JA, Simplaceanu V, Zou M, Ho NT, Ho C. NMR reveals hydrogen bonds between oxygen and distal histidines in oxyhemoglobin. Proc Natl Acad Sci U S A. 2000;97:10354–10358. doi: 10.1073/pnas.190254697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Phillips GN, Teodoro ML, Li TS, Smith B, Olson JS. Bound CO is a molecular probe of electrostatic potential in the distal pocket of myoglobin. J Phys Chem B. 1999;103:8817–8829. [Google Scholar]
- 78.Rovira C. Role of the His64 residue on the properties of the Fe-CO and Fe-O-2 bonds in myoglobin. A CHARMM/DFT study. J Mol Struc-TheoChem. 2003;632:309–321. [Google Scholar]
- 79.Rovira C. The structure and dynamics of the Fe-CO bond in myoglobin. J Phys: Condens Matter. 2003;15:S1809–S1822. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.