

Abstract
The host response to a biomaterial is characterized by both acute recruitment and attachment of cells as well as chronic encapsulating tissue reaction. The implantation procedure induces production of damage-associated molecular patterns (DAMPs) which may contribute to host recognition of the material. Toll-like receptors (TLRs) are pattern recognition receptors (PRRs) that bind not only pathogen-associated molecular patterns (PAMPs) but also DAMPs. We sought to investigate whether TLR4/DAMP interactions were involved in the acute and chronic inflammatory response to an implanted biomaterial. When PET discs were implanted intraperitoneally for 16 h, no differences were found in the number of leukocytes recruited between TLR4+ (C57BL/10J) and TLR4− (C57BL/10ScNJ) mice. However, a significant shift in the leukocyte profile on the biomaterial surface was observed for TLR4− mice. While the total number of adherent cells was the same in both strains, TLR4+ mice had a profile with equivalent neutrophil and monocyte/macrophage presence on the material surface, and TLR4− mice had a profile of predominantly neutrophils with fewer monocyte/macrophages. When implants were placed subcutaneously for 2 weeks, the fibrous capsule thicknesses were not different between TLR4+ and TLR4− mouse strains. These findings illustrate that TLR4 may play a role in the initial recognition of a biomaterial by directing the adhesive cellular profile.
Keywords: Biomaterials, Toll-like receptors, Host response, Acute inflammation
1. Introduction
Biomaterials used for medical devices elicit a response from the body that is often detrimental to the efficacy of their intended function. Simply, the host response to a biomaterial implantation includes injury, blood/material interactions, coagulation and provisional matrix deposition, acute inflammatory response, granulation tissue onset, foreign body reaction and fibrous encapsulation [1]. The implantation procedure inherently produces tissue and vasculature damage causing blood components to infiltrate the implant region. The inflammatory response toward a device is directed by these interactions between blood/tissue components and material, which results in immediate protein adsorption to the surface.
A variety of proteins mediate the localization of leukocytes to biomaterials through adhesion receptors [2]. Particularly, complement proteins [3–5] as well as a major component of the clotting cascade, fibrinogen [6–8], have been shown to mediate phagocyte adhesion to materials through β2 (CD18) integrins [3,8–10]. This adhesion is a critical step in the recognition of a biomaterial. However, there are potentially additional initial events in the inflammatory response to an implant that may prime phagocytes such as neutrophils and monocyte/macrophages for adhesion. Recent literature has brought to light the role of `danger signals' in the inflammatory response following injury, which elicit leukocyte activation primarily through binding to pattern recognition receptors (PRRs). These ligand-receptor interactions would precede (or occur simultaneously with) leukocyte adhesion, but the role of these factors in the host response to a biomaterial is unknown.
`Danger signals' or damage-associated molecular patterns (DAMPs) [11,12] are endogenous intracellular or extracellular components that are normally hidden from the environment or are present at low concentrations. Upon cellular necrosis and/or tissue damage, these occult motifs can become strong activators of inflammation, aid in the body's recognition of dying cells and can act as adjuvants through direct activation of immune cells such as dendritic cells (DCs) and macrophages [13]. Numerous endogenous DAMPs have been linked to leukocyte activation through the PRR Toll-like receptor 4 (TLR4) (for references see Rock and Kono [14]) as well as the PAMP lipopolysaccharide (LPS).
The TLR family is composed of at least ten TLRs (TLR1–TLR10) in humans [15] and are integral membrane proteins containing leucine-rich repeats in the extracellular domain which are believed to be involved in the ligand-recognition process. Immune cells (along with other cell types) possess TLRs which canonically bind conserved PAMPs to aid in the initiation of an innate immune response toward a pathogen [16]. TLR activation stimulates innate effector functions such as increases in pro-inflammatory cytokine/chemokine production [e.g. tumor necrosis factor (TNF)- α], increases in co-stimulatory molecules for antigen-presenting cells, and up-regulation of integrin expression [17]. TLR4/DAMP interactions have been investigated in the wound healing response to sterile injury in various applications ranging for hemorrhage and drug-induced lung injury to sterile skin incisions and systemic inflammation following femur fracture [18–21]. Following injury, it is believed that TLR4 initiates inflammatory signaling via cytokine communication [22] to provide necessary cues during wound repair. Neutrophil and monocyte recruitment to the peritoneal cavity has been found to be induced by DAMP presence [23]. These findings are relevant to biomaterials as many aspects of wound healing are connected to the host response to materials [1,24].
Biomaterial particulates delivered with PAMP ligands for TLR4 have been investigated for directing cancer vaccines [25,26]; however, we and others have found that biomaterials themselves are capable of acting as adjuvants inducing a strong humoral immune response toward a co-delivered antigen [27–30] in the absence of PAMP TLR ligands. Therefore, in an endotoxin-free environment, TLR4 may interact with biomaterial-associated DAMPs to propagate an innate inflammatory response and offers an explanation of the biomaterial-induced adjuvant effect previously seen.
Several groups have studied the role of TLR4 in the cellular response to biomaterials in vitro. Macrophage cytokine secretion in response to oligosaccharides of alginate is believed to be TLR4/2 dependent [31]. Pro-inflammatory cytokine production (TNF-α) but not NF-κB activation in macrophages treated with hydroxyapatite particles was found to be TLR4-dependent [32]. However, as to date no studies have examined the role of TLRs (specifically TLR4) in the in vivo host response to biomaterials. Therefore, this study sought to examine the role of TLR4 in the acute and chronic inflammatory response to a biomaterial implant.
2. Materials and methods
2.1. Biomaterial preparation
PET implants were prepared by punching 10 mm discs from a 0.5 mm thick PET sheet (AIN Plastics, Kennesaw, GA). Discs were sterilized by rinsing with 70% ethanol for 24 h followed by three 30 min rinses in endotoxin-free water (Lonza, Basel, Switzerland). After drying in a tissue culture hood, discs were autoclaved prior to implantation. Endotoxin content of equivalently prepared PET discs (5 mm) was assessed using the Limulus Ameobocyte Lysate (LAL) assay (QCL-1000 Chromogenic LAL Endpoint assay, Lonza) in endotoxin-free test tubes (Lonza). PET discs were tested in the presence of both LAL and the chromogenic substrate and compared to a simultaneously developed endotoxin standard curve (between 0.1 and 1.0 EU/mL). Implanted PET discs tested below 0.1 EU/ml, the lower limit for this assay.
2.2. IP implantation and analysis
All animal procedures were approved by IACUC committee prior to use (Protocol 044-2008, Emory University). Mice were 6–7 weeks old and age-matched for each experiment performed across TLR4+ (C57BL/10J, The Jackson Laboratory, Bar Harbor, ME) and TLR4− (C57BL/10ScNJ, The Jackson Laboratory) strains. For intraperitoneal (IP) implantation model, survival surgeries were completed in a laminar flow hood using sterilized surgical instruments in combination with a peri-operative dose of nalbuphine (6 mg/kg) under isofluorane (Baxter, Deerfield, IL) anesthetic. Two PET discs were implanted IP per mouse for 16 h through a midline incision followed by suturing of muscle and skin (n = 7–9 mice, see Table 1). Sham surgeries with no implants (but with incision and suturing) were also performed simultaneously (see Table 1). At 16 h, mice were anesthetized and 1 ml Dulbecco's Phosphate Buffered Saline (Invitrogen, Carlsbad, CA) [D-PBS]/heparin (Abraxis Bioscience, Los Angeles, CA) (50 U/ml) was injected IP for collection of lavage by sterile transfer pipette. Discs were collected and transferred to 1 ml D-PBS on ice until analysis. Lastly, cardiac puncture (using 100 U heparin preloaded in a 1 mL syringe with a 21 g needle) was performed to subsequently determine circulating leukocyte population.
Table 1.
Number of animals (n) analyzed per strain using the indicated endpoint analyses for the three treatment groups in the IP implantation study. Lavage total cell counts and cytospin analysis were performed on all mice including those with PET implants which were also analyzed for total adherent cells and cell types (see method for detailed description). Cardiac punctures were performed on all mice to assess circulating leukocyte populations.
| Treatment | Strain | Analysis | n |
|---|---|---|---|
| Naïve | TLR4+ | Lavage | 7 |
| TLR4− | Lavage | 8 | |
| Sham | TLR4+ | Lavage | 9 |
| TLR4− | Lavage | 7 | |
| Implant | TLR4+ | Lavage | 9 |
| TLR4− | Lavage | 9 | |
| TLR4+ | Implant | 8 | |
| TLR4− | Implant | 9 |
Lavage supernatants were collected, volume estimated for concentration calculation and stored at −20 °C following centrifugation of lavages at 400 g for 5 min. Lavage cells were resuspended in 1 ml of D-PBS and a small aliquot (10 μl) was analyzed for cell counts (6–20 μm) using Multisizer 3 Coulter Counter (Beckman Coulter, Fullerton, CA). The remaining cells were resuspended at a concentration of 1 × 106 per milliliter and 200 μl used for one cytospin analysis per mouse/lavage using Shandon Cytospin 3 (Thermo Scientific, Waltham, MA). Cytospins were stained with Hema 3 (Fisher Scientific, Pittsburgh, PA) and differential cell counts (300 cells counted per sample) performed by microscopy.
The first explanted disc was stained immediately after recovery using Hema 3. From the second recovered disc, adherent cells were collected by placing the disc in trypsin/EDTA (0.25%) (Sigma–Aldrich, St. Louis, MO) at 37 °C. To assure complete removal of adherent cells, a cell scraper was lightly passed over both sides of the disc after it had been placed in RPMI 1640 media (Invitrogen) supplemented with 10% heat-inactivated FBS (Mediatech, Manassas, VA) to inactivate trypsin. Collected adherent cells were counted and stained using the identical procedure described previously for lavage cells (one cytospin analysis per disc, n = 8–9 samples per group, see Table 1). This implant was stained directly to assure complete removal of cells had been achieved. The trypsinized disc possessed no adherent cells on either side compared to the implant stained immediately after removal which was extensively covered with cells as assessed by microscopy.
Cardiac punctures were collected and immediately a blood smear prepared using HemaPrep (J.P. Gilbert Company, Boyertown, PA) and stained using Hema 3. Red blood cells were lysed with warmed ammonium chloride solution to assess total circulating leukocyte concentrations using a Multisizer 3 Coulter Counter (Beckman Coulter).
Naïve lavages and cardiac punctures were also performed in age-matched TLR4+ and TLR4− mice that had not undergone any surgical procedure (n = 7–8 mice, see Table 1).
2.3. SC implantation and analysis
For subcutaneous (SC) implantation model, survival surgeries were completed in a laminar flow hood using sterilized surgical instruments in combination with peri-operative dose of nalbuphine (6 mg/kg) and isofluorane (Baxter) anesthetic. Two PET discs were implanted SC on the dorsum (one to the left and one to the right of the spine) of TLR4+ (n = 6) or TLR4− (n = 7) mice. At 2 weeks, mice were anesthetized using isofluorane and cardiac punctures performed/analyzed as described above. Discs with surrounding tissue were explanted and immediately fixed in 10% (v/v) buffered formalin (Fisher Scientific) until processing. Tissue explants were embedded in paraffin and 5 μm sections were prepared for staining. For analysis of fibrous capsule thickness, sections were deparaffinized and stained with Van Gieson stain (American Master Tech Scientific, Lodi, CA); otherwise, sections were stained with hematoxylin and eosin (H&E). Fibrous capsule thickness (as seen by dark red collagen staining) was determined on both skin or muscle side of implants from five sections per mouse using Image Pro Plus (Media Cybernetics, Bethesda, MD) calibrated software using sections taken vertically from the center of the explanted disc.
2.4. TNF-α ELISA analysis
Cleared lavage supernatants from implant, sham or naïve treatment groups were assessed for TNF-α concentration using an ELISA (R&D Systems, Minneapolis, MN). Samples were run undiluted and analyzed following manufacturer's instructions. Values that were below blank of the assay were taken as zero.
2.5. Statistical analysis
All statistical analysis was performed using one-way ANOVA (Prism v5.0a, GraphPad Software Inc, La Jolla, CA) with animals nested within treatment. Comparison across groups was accomplished using a Tukey post-test with a 95% confidence interval (p < 0.05 was considered significant).
3. Results
Using an established TLR4-deficient mouse strain, C57BL/10ScN [33–35], which possesses a complete deletion of the tlr4 gene, along with an appropriate wild-type control strain (C57BL/10) as recipients of biomaterial implants, the role of TLR4 in the host response to a biomaterial implant was examined. Discs of PET, as a model synthetic implant, have been used extensively [6,8,36–39] and yield robust phagocyte accumulation (neutrophils and monocyte/macrophages) in response to biomaterial in the peritoneal cavity at 16 h [7]. To study the role of TLR4/DAMP interactions in the acute inflammatory response to biomaterials, PET discs were implanted IP for 16 h in TLR4− or TLR4+ mice and total leukocyte concentrations and differential leukocyte profiles were analyzed in IP lavages and adherent cells harvested from the discs. Sham surgeries and naïve mice were used as controls to isolate the role TLR4/DAMP interaction plays in the recruitment of leukocytes due to the biomaterial itself. The TLR4-dependence of the fibrous capsule formation was assessed following 2 weeks of PET disc implantation subcutaneously in TLR4− or TLR4+ mice.
Total leukocyte concentrations were determined in IP lavages collected from TLR4+ or TLR4− mice receiving a PET disc implant, sham surgery or in the naïve control group (Fig. 1). Implantation of a PET disc induced recruitment of leukocytes into the peritoneal cavity of TLR4− mice with higher total leukocyte concentrations as compared to that of the respective naïve mice. This trend was also present upon PET disc implantation into TLR4+ mice but was not found to be statistically significant. Sham surgery induced leukocyte recruitment into the peritoneal cavity of either mouse strain to levels which were between those found for naïve or implant groups of either mouse strain, though not-statistically different from either group (Fig. 1).
Fig. 1.
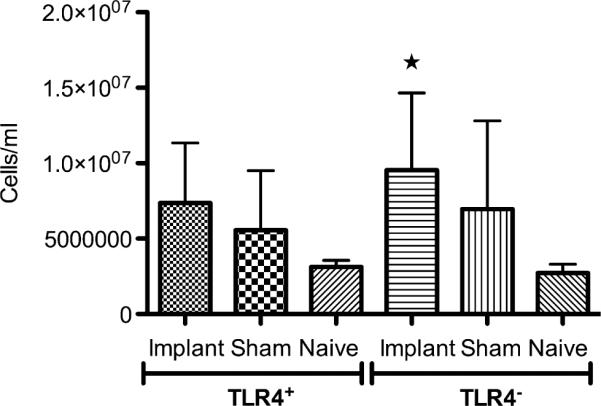
Total leukocyte concentrations in IP lavages for TLR4+ or TLR4− mice receiving a PET disc implant, sham surgery or in the naïve control group.★; p < 0.05 in comparison to either naïve group, n = 7–9 mice per group. Bars represent mean + SD.
The differential leukocyte profiles in IP lavages for TLR4+ or TLR4− mice were determined and found to be similar for both mouse strains for the various treatment groups (Fig. 2). The majority of leukocytes in the peritoneal cavity at 16 h were neutrophils for both mouse strains regardless of treatment; however, there was a significantly lower fraction of neutrophils in naïve TLR4− than all TLR4+ treatments (Fig. 2A). TLR4− mice tended to have a slightly higher fraction of monocyte/macrophage than TLR4+ mice (Fig. 2B); however, no significant differences were found between implant groups of the two strains. Eosinophil and lymphocyte fractions from both strains were found to be similar (Fig. 2C and D, respectively). To examine if a TLR4 deficiency affected circulating leukocyte concentrations, which would certainly have affected recruitment into the IP space, total circulating leukocyte concentrations were determined in lysed whole blood obtained from cardiac punctures. No differences were found across all treatments (data not shown) for both TLR4+ and TLR4− mice.
Fig. 2.

Differential leukocyte profiles showing fractions of (A) Neutrophils, (B) Monocyte/Macrophages, (C) Eosinophils, (D) Lymphocytes in IP lavages for TLR4+ or TLR4− mice receiving a PET disc implant, sham surgery or naïve control. Brackets indicate significant differences between groups (p < 0.05), n = 7–9 mice per group. Bars represent mean + SD.
As a potent pro-inflammatory cytokine often produced upon TLR4 signaling, TNF-α controls numerous cellular actions in the acute inflammatory response. The concentration of TNF-α in peritoneal cavity was determined to assess if it was induced in a TLR4-dependent manner following biomaterial implantation for 16 h. TNF-α was only significantly detected in the peritoneal cavity upon implantation of a PET disc since little to no TNF-α was detected in IP lavages of sham or naïve groups from both TLR4+ and TLR4− mice (Fig. 3). However, this response was found to be TLR4-independent as both strains elicited similar production of the cytokine upon PET disc implantation.
Fig. 3.
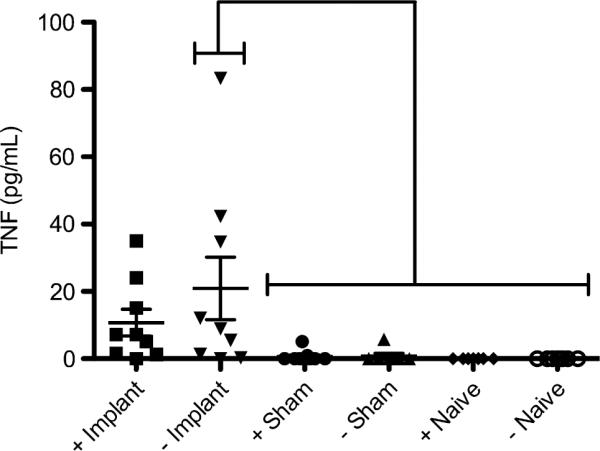
TNF-a concentrations in IP lavages for TLR4+ (+) or TLR4− (−) mice receiving a PET disc implant (Implant), sham surgery (Sham) or naïve control (naïve). Individual mouse concentrations are plotted as points with horizontal lines representing mean with standard deviations. Brackets indicate statistical difference (p < 0.05) between group, n = 7–9 mice per group.
The role of TLR4 in controlling the adherent leukocyte profile on the IP-implanted PET disc was assessed by determining the total number and differential profiles of leukocytes harvested by trypsinization from discs. The total numbers of leukocytes recovered as adherent to the biomaterial were similar in TLR4+ and TLR4− mice (Fig. 4A). However, the differential leukocyte profiles recovered from the surface of the material were distinct between strains (Fig. 4B). Implants in TLR4+ mice possessed equivalent fractions of harvested adherent neutrophils and monocyte/macrophages while implants in TLR4− mice had significantly higher fractions of adherent neutrophils than monocyte/macrophages (Fig. 4B and Fig. 5). Furthermore, both of these fractions were significantly different between the two strains (Fig. 4B).
Fig. 4.

Adherent leukocyte profiles on PET discs following 16 h of IP implantation into TLR4+ or TLR4− mice. (A) Total number of adherent leukocytes collected from implants in either strain. No statistical difference was found between strains (p = 0.27); n = 7–9 mice per group. (B) Adherent leukocyte profiles showing fractions of neutrophils, monocyte/macrophages, eosinophils, and lymphocytes. Brackets indicate significant differences between groups (p < 0.05); n = 7–9 mice per group. All treatments significantly different from each other except for lymphocyte and eosinophil fractions from either strain as well as neutrophil and monocyte/macrophage fractions from TLR4+ strain.
Fig. 5.

Representative cytospins of adherent leukocytes collected from PET discs implanted for 16 h in TLR4+ (A) or TLR4− (B) mice. Bar indicates 10 μm.
To examine whether TLR4 presence affected the fibrous encapsulation of an implanted biomaterial, PET discs were implanted SC in TLR4+ or TLR4− for 2 weeks and the resultant thicknesses assessed in histological sections. Tissue surrounding the implants was stained with Van Gieson to highlight the collagen content of the capsule. TLR4+ and TLR4− mice displayed no noticeable differences in collagen content at the implant interfaces. Both strains elicited a strong tissue reaction (Fig. 6A–D) as well as a thick fibrous capsule on the dermal side as opposed to the muscle, which was thin (Fig. 6E, F & Fig. 7). The capsule on the dermal side was determined to be 16 ± 4 μm for TLR4+ mice and 18 ± 2 μm for TLR4− mice while the capsule adjacent to muscle was 8 ± 2 μm for TLR4+ mice and 9 ± 2 μm for TLR4− mice. There were no differences in the thicknesses of the fibrous capsules on either side between the two strains (Fig. 7).
Fig. 6.

Representative H&E and Van Gieson stained tissue sections of PET discs implanted SC for 2 weeks. Shown are 10× images of representative H&E stained sections from a TLR4+ (A) or a TLR4− (B) mouse (bar indicates 100 μm) as well as 40μ magnifications TLR4+ (C) or a TLR4− (D) (bar indicates 10 μm). Also shown are 10μ images of Van Gieson (collagen) stained sections from a TLR4+ (E) or a TLR4− (F) mouse (bar indicates 100 μm). No noticeable differences in fibrous capsule formation were found between the two strains.
Fig. 7.
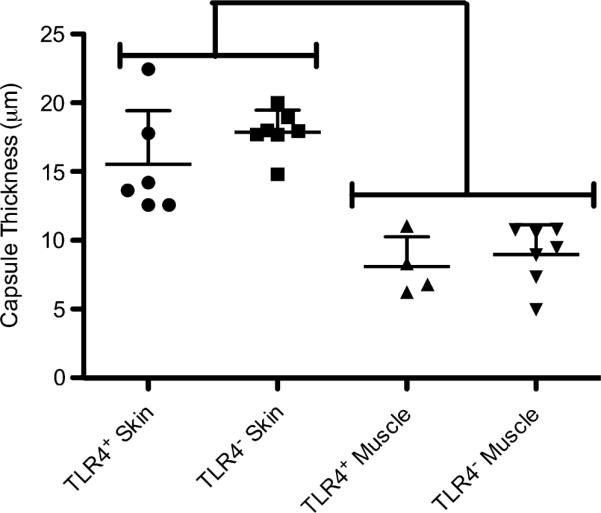
Fibrous capsule thickness surrounding PET discs implanted for 2 weeks SC in TLR4+ or TLR4− mice assessed on the skin and muscle sides. Thicknesses were averaged for the skin side or the muscle side of the implant for each animal and individual determinations are plotted as points with horizontal lines representing mean with standard deviations; n = 4–7 mice per group. Brackets indicate statistical difference between skin and muscle thicknesses only (p < 0.05).
4. Discussion
This research demonstrates that TLR4 plays a role in the host response to a biomaterial in vivo. Specifically, mice lacking TLR4 had an altered differential leukocyte profile recovered from PET surfaces following a 16 h intraperitoneal implantation as compared to mice possessing wild-type TLR4 (Fig. 4B). Hence, TLR4 appears to affect the differential profile of biomaterial-adherent leukocytes while not having an effect on the differential profile recruited into the peritoneal cavity (Figs. 1 and 2). While there were differences in the profile of adherent leukocytes following an IP implantation for 16 h, in the SC site, after 2 weeks of implantation of a PET disc, there was no dependence on TLR4 of the resultant fibrous capsule (Fig. 6 and Fig. 7). Therefore, TLR4 may be necessary for optimal activation and adhesion of early responders such as neutrophils, but the chronic inflammatory response is unaffected by the absence of TLR4.
It was hypothesized that a lack of TLR4 in the context of a biomaterial implant would lead to lower recruitment of leukocytes following an implantation procedure as neutrophil accumulation and TNF-α production following hemorrhage-induced lung injury was lessened in TLR4-defective mice [20]. Also, macrophage-mediated inflammation following acute lung injury occurs via TLR4/TLR2 interactions with hyaluronic acid fragments [21]. However, mice possessing wild-type TLR4 or lacking TLR4 elicited similar leukocyte recruitment to the peritoneal cavity following biomaterial implantation (Fig. 1). Sham and implant treatment groups for both strains were also similar (Fig. 1) implying that surgery alone may have accounted for a significant aspect of the inflammatory response. However, no statistical difference was found for leukocyte concentrations for lavages from sham or naïve groups. Others have noted that the total number of macrophages and neutrophils following a sham surgery procedure at 18 h was found to be similar to naive mice [37] and did not account for the majority of inflammation to PET implanted IP. However, in this study lavages (which exhibited similar total leukocyte concentrations) from sham surgeries or biomaterial implantation contained similar profiles of neutrophils (Fig. 2A) and monocyte/macrophages (Fig. 2B) for both TLR4+ and TLR4− mice.
It was unanticipated that only in the presence of a biomaterial implant would TNF-α be detected IP (Fig. 3), furthermore in a TLR4-independent manner, wherein sham and naïve mice had essentially no detectable levels of the pro-inflammatory cytokine. Others have found that peritoneal macrophages from TLR4-defective mice produce significantly less TNF-α mRNA in vitro in response to hydroxyapatite particles than TLR4+ mice [32]. In vivo a biomaterial likely presents many other DAMPs than presented by a biomaterial in vitro, and these may activate cytokine production through other PRRs such as TLR2 [40–42]. In this study, however, using an IP biomaterial implantation model such a strong response to injury alone was not observed. These findings are supported by others who have shown that TLR4 does not account for inflammation (recruitment of neutrophils) IP following injection of dead cells [43]. Overall, these findings imply that TLR4 was not responsible for the recruitment of leukocytes to the IP cavity or TNF-α production.
Conversely, a differential profile of adherent phagocytes on IP-implanted PET discs was observed that was TLR4-dependent. It was surprising that an altered differential leukocyte profile was observed for TLR4− mice as compared to TLR4+ (Fig. 4B). Implants in TLR4− mice had a significantly higher fraction of recovered adherent neutrophils (~0.58) and a lower fraction of recovered adherent monocyte/macrophage (~0.36) as compared to that of TLR4+ mice (Fig. 4B). In contrast, TLR4+ mice had equivalent fractions of recovered adherent neutrophils and monocyte/macrophages (about 0.47 for both) (Fig. 4B) from discs. This was observed even though the total number of leukocytes attaching to PET disc was equivalent between TLR4+ and TLR4− mice (Fig. 4A), and the IP milieu surrounding the implants wherein neutrophil and monocyte/macrophage fractions were equivalent in TLR4+ and TLR4− mice (Fig. 2A and B). In support of a role for TLR4 in controlling the adhesive leukocyte profile, another common wild-type TLR4 strain, C57BL/6, exhibited the same neutrophil and monocyte/macrophage profile at 16 h on PET to that of the TLR4+ strain used here, C57BL/10 (unpublished data).
There are two possible explanations for the observed TLR4-dependence of the adherent phagocyte profile on PET discs implanted IP. First, as neutrophils account for the majority of inflammatory cells recruited during acute inflammation in response to an implanted biomaterial within the time frame studied here, endogenous ligands present on the material surface may trigger TLR4-induced activation of neutrophils to subsequently progress the inflammatory cascade (including stimulating monocyte/macrophages). Without TLR4, neutrophils would not be fully activated by proteins on the PET surface. In fact, TLR signaling also induces up-regulation of integrin (CD11b/CD18) expression in neutrophils [17,44] which may even control adhesion, along with optimal activation, to a material [3,9] and explain why an apparent “delayed” inflammatory response is seen in TLR4− mice. Numerous proteins adsorb to biomaterials from physiological fluids [2]. Most of these are considered adhesive substrates, but plasmin [45], fibrinogen [22] and fibronectin [46] are also ligands for TLR4. Hence, such biomaterial-adsorbed proteins may concurrently induce the inflammatory cascade. Interestingly, firm adhesions via integrins also leads to optimal TLR4 signaling [47] in macrophages further supporting their activity. Therefore, upon implantation of a biomaterial, a foreign surface may offer the perfect environment for TLR4/DAMP interactions to lead to leukocyte activation: by acting as a depot for DAMPs to elicit a `danger' response from the host as well as offering an adhesive substrate.
Lastly, TLR4-mediated phagocyte accumulation on the surface may not be a product of direct interaction with DAMPs on biomaterials but as a result from the absence of TLR4 on many other cells in TLR4− mice. TLR4 is expressed on various cell types including most immune cells, some epithelial cells and even endothelial cells [34]. The adhesion to the material surface may be indirectly controlled, for example, by endothelial cells which may act to prime neutrophils and monocyte/macrophages for adhesion to a material surface. It is well documented that endothelial cells stimulate neutrophil activation and integrin-mediated adhesion during an inflammatory response through secretion of chemokines. It is also believed that endothelial cells sense DAMPs (such as hyaluronic acid fragments) through TLR4 which induce chemokine (IL-8) secretion [48]. Therefore, endogenously activated endothelial cells may ready neutrophils for adhesion through paracrine chemokine interactions.
In this study, we were able to very clearly and accurately identify the inflammatory cells recruited into the peritoneal cavity and recovered from discs upon IP implantation of the biomaterial disc by using cytospins and light microscopy to identify the cells based on their morphology. A limitation of this study is that trypsin removal of biomaterial-adherent leukocytes did not allow for direct cell staining using immunofluorescence for cell surface antigens or activation markers due to anticipated cleavage of these surface receptors by the trypsin. This prevents the use of flow cytometry to better characterize the inflammatory cell population/phenotype which would be beneficial in future studies. Similarly, as discussed next, for the subcutaneously implanted discs, the purpose was to assess the host tissue reaction namely, fibrous encapsulation. This objective was well achieved using histological assessment and the staining techniques (e.g. Van Gieson to highlight the collagen content of the capsule). Future immunohistochemical analysis can examine the types of cells present or their expression of activation/phenotypic markers.
The lack of TLR4-dependence on the fibrous capsule formation at 2 weeks to PET implanted subcutaneously was anticipated (Figs. 6 and 7) as other TLRs (such as TLR2) may compensate for DAMP interactions. A significant decrease in tissue reaction to an implanted biomaterial would be anticipated in mice which lack MyD88 (a common adapter molecule required for nearly all TLR family member signaling, with the exception of TLR3). MyD88−/− mice had an almost completely abrogated inflammatory response to DAMP-mediated inflammation [43]. Interestingly, IL-1R (which shares identical signaling pathway with TLRs) was, and has been shown by others, linked to these effects. Specifically, intracellular inflammasome activation of IL-1/IL-1R signaling may be the cause for `danger'-induced effects including the adjuvanticity of alum [49] and other biomaterial particulates [30] used for vaccines. However, the potential for a flat and large surface such as the PET disc implanted herein to interact intracellularly with inflammasome components to induce IL-1 activation in the absence of internalization of the material due to size remains to be determined but seems unlikely.
5. Conclusion
TLR4 plays a role in determining the adhesive leukocyte profile on biomaterials during the acute inflammatory response. This may be mediated by a direct interaction with DAMPs adsorbed on the material surface or indirectly affected by other cell types which prime leukocytes for adhesion. TLR4 blocking may be beneficial if trying to direct an acute host response that minimizes monocyte/macrophage adhesion to a biomaterial. Fibrous encapsulation following SC implantation of a biomaterial propagates normally in the absence of TLR4 indicating other compensatory receptors for activation and support of the foreign body tissue response. Therefore, the window of activity for TLR4 in the host response to a biomaterial is only in the initial recognition of the foreign entity.
Acknowledgements
The authors would like to thank Dr. Andrés García of Georgia Institute of Technology for providing the PET material for implantation. This work was supported by funding from the Georgia Tech/Emory Center (GTEC) for the Engineering of Living Tissues, an Engineering Research Council (ERC) program of the National Science Foundation under award number EEC-9731643.
Appendix
Figures with essential colour discrimination. Certain figures in this article, in particular Figs. 5 and 6, are difficult to interpret in black and white. The full colour images can be found in the online version, at doi:10.1016/j.biomaterials.2009.09.077.
References
- [1].Anderson JM, Rodriguez A, Chang DT. Foreign body reaction to biomaterials. Semin Immunol. 2008;20:86–100. doi: 10.1016/j.smim.2007.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Wilson CJ, Clegg RE, Leavesley DI, Pearcy MJ. Mediation of biomaterial-cell interactions by adsorbed proteins: a review. Tissue Eng. 2005;11:1–18. doi: 10.1089/ten.2005.11.1. [DOI] [PubMed] [Google Scholar]
- [3].McNally AK, Anderson JM. Complement C3 participation in monocyte adhesion to different surfaces. Proc Natl Acad Sci U S A. 1994;91:10119–23. doi: 10.1073/pnas.91.21.10119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Brodbeck WG, Colton E, Anderson JM. Effects of adsorbed heat labile serum proteins and fibrinogen on adhesion and apoptosis of monocytes/macrophages on biomaterials. J Mater Sci Mater Med. 2003;14:671–5. doi: 10.1023/a:1024951330265. [DOI] [PubMed] [Google Scholar]
- [5].Gorbet MB, Sefton MV. Biomaterial-associated thrombosis: roles of coagulation factors, complement, platelets and leukocytes. Biomaterials. 2004;25:5681–703. doi: 10.1016/j.biomaterials.2004.01.023. [DOI] [PubMed] [Google Scholar]
- [6].Tang L, Wu Y, Timmons RB. Fibrinogen adsorption and host tissue responses to plasma functionalized surfaces. J Biomed Mater Res. 1998;42:156–63. doi: 10.1002/(sici)1097-4636(199810)42:1<156::aid-jbm19>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- [7].Tang L, Eaton JW. Fibrin(ogen) mediates acute inflammatory responses to biomaterials. J Exp Med. 1993;178:2147–56. doi: 10.1084/jem.178.6.2147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Hu WJ, Eaton JW, Ugarova TP, Tang L. Molecular basis of biomaterial-mediated foreign body reactions. Blood. 2001;98:1231–8. doi: 10.1182/blood.v98.4.1231. [DOI] [PubMed] [Google Scholar]
- [9].McNally AK, Anderson JM. Beta1 and beta2 integrins mediate adhesion during macrophage fusion and multinucleated foreign body giant cell formation. Am J Pathol. 2002;160:621–30. doi: 10.1016/s0002-9440(10)64882-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Tang L, Ugarova TP, Plow EF, Eaton JW. Molecular determinants of acute inflammatory responses to biomaterials. J Clin Invest. 1996;97:1329–34. doi: 10.1172/JCI118549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Matzinger P. Tolerance, danger, and the extended family. Annu Rev Immunol. 1994;12:991–1045. doi: 10.1146/annurev.iy.12.040194.005015. [DOI] [PubMed] [Google Scholar]
- [12].Matzinger P. The danger model: a renewed sense of self. Science. 2002;296:301–5. doi: 10.1126/science.1071059. [DOI] [PubMed] [Google Scholar]
- [13].Gallucci S, Lolkema M, Matzinger P. Natural adjuvants: endogenous activators of dendritic cells. Nat Med. 1999;5:1249–55. doi: 10.1038/15200. [DOI] [PubMed] [Google Scholar]
- [14].Rock KL, Kono H. The inflammatory response to cell death. Annu Rev Pathol. 2008;3:99–126. doi: 10.1146/annurev.pathmechdis.3.121806.151456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Akira S, Uematsu S, Takeuchi O. Pathogen reconition and innate immunity. Cell. 2006;124:783–801. doi: 10.1016/j.cell.2006.02.015. [DOI] [PubMed] [Google Scholar]
- [16].Janeway CA, Jr, Medzhitov R. Innate immune recognition. Annu Rev Immunol. 2002;20:197–216. doi: 10.1146/annurev.immunol.20.083001.084359. [DOI] [PubMed] [Google Scholar]
- [17].Sabroe I, Prince LR, Jones EC, Horsburgh MJ, Foster SJ, Vogel SN, et al. Selective roles for Toll-like receptor (TLR)2 and TLR4 in the regulation of neutrophil activation and life span. J Immunol. 2003;170:5268–75. doi: 10.4049/jimmunol.170.10.5268. [DOI] [PubMed] [Google Scholar]
- [18].Levy R, Prince J, Yang R, Mollen K, Liao H, Watson G, et al. Systemic inflammation and remote organ damage following bilateral femur fracture requires Toll-like receptor 4. Am J Physiol Regul Integr Comp Physiol. 2006;291:R970–6. doi: 10.1152/ajpregu.00793.2005. [DOI] [PubMed] [Google Scholar]
- [19].Bettinger D, Pellicane J, Tarry W, Yager D, Diegelmann R, Lee R, et al. The role of inflammatory cytokines in wound healing: accelerated healing in endotoxin-resistant mice. J Trauma. 1994;36:810–4. doi: 10.1097/00005373-199406000-00010. [DOI] [PubMed] [Google Scholar]
- [20].Barsness K, Arcaroli J, Harken A, Abraham E, Banerjee A, Reznikov L, et al. Hemorrhage-induced acute lung injury is TLR-4 dependent. Am J Physiol Regul Integr Comp Physiol. 2004;281:R592–9. doi: 10.1152/ajpregu.00412.2003. [DOI] [PubMed] [Google Scholar]
- [21].Jiang D, Liang J, Fan J, Yu S, Chen S, Luo Y, et al. Regulation of lung injury and repair by Toll-like receptors and hyaluronan. Nat Med. 2005;11:1173–9. doi: 10.1038/nm1315. [DOI] [PubMed] [Google Scholar]
- [22].Mollen K, Anand R, Tsung A, Prince J, Levy R, Billiar T. Emerging paradigm: toll-like receptor 4-sentinel for the detection of tissue damage. Shock. 2006;26:430–7. doi: 10.1097/01.shk.0000228797.41044.08. [DOI] [PubMed] [Google Scholar]
- [23].Chen CJ, Shi Y, Hearn A, Fitzgerald K, Golenbock D, Reed G, et al. MyD88-dependent IL-1 receptor signaling is essential for gouty inflammation stimulated by monosodium urate crystals. J Clin Invest. 2006;116:2262–71. doi: 10.1172/JCI28075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Anderson JM. Biological responses to materials. Annu Rev Mater Res. 2001;31:81–110. [Google Scholar]
- [25].Hamdy S, Molavi O, Ma Z, Haddadi A, Alshamsan A, Gobti Z, et al. Co-delivery of cancer-associated antigen and Toll-like receptor 4 ligand in PLGA nano-particles induces potent CD8 T cell-mediated anti-tumor immunity. Vaccine. 2008;26:5046–57. doi: 10.1016/j.vaccine.2008.07.035. [DOI] [PubMed] [Google Scholar]
- [26].Schlosser E, Mueller M, Fischer S, Basta S, Busch DH, Gander B, et al. TLR ligands and antigen need to be coencapsulated into the same biodegradable microsphere for the generation of potent cytotoxic T lymphocyte responses. Vaccine. 2008;26:1626–37. doi: 10.1016/j.vaccine.2008.01.030. [DOI] [PubMed] [Google Scholar]
- [27].Matzelle M, Babensee J. Humoral immune responses to model antigen co-delivered with biomaterials used in tissue engineering. Biomaterials. 2004;25:295–304. doi: 10.1016/s0142-9612(03)00531-3. [DOI] [PubMed] [Google Scholar]
- [28].Babensee JE. Interaction of dendritic cells with biomaterials. Semin Immunol. 2008;20:101–8. doi: 10.1016/j.smim.2007.10.013. [DOI] [PubMed] [Google Scholar]
- [29].Bennewitz N, Babensee J. The effect of the physical form of poly(lactic-coglycolic acid) carriers on the humoral immune response to co-delivered antigens. Biomaterials. 2005;26:2991–9. doi: 10.1016/j.biomaterials.2004.08.023. [DOI] [PubMed] [Google Scholar]
- [30].Sharp FA, Ruane D, Claass B, Creagh E, Harris J, Malyala P, et al. Uptake of particulate vaccine adjuvants by dendritic cells activates the NALP3 inflammasome. Proc Natl Acad Sci U S A. 2009;106:870–5. doi: 10.1073/pnas.0804897106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Iwamoto M, Kurachi M, Nakashima T, Kim D, Yamaguchi K, Oda T, et al. Structure-activity relationship of alginate oligosaccharides in the induction of cytokine production from RAW264.7 cells. FEBS Lett. 2005;579:4423–9. doi: 10.1016/j.febslet.2005.07.007. [DOI] [PubMed] [Google Scholar]
- [32].Grandjean-Laquerriere A, Tabary O, Jacquot J, Richard D, Frayssinet P, Guenounou M, et al. Involvement of toll-like receptor 4 in the inflammatory reaction induced by hydroxyapatite particles. Biomaterials. 2007;28:400–4. doi: 10.1016/j.biomaterials.2006.09.015. [DOI] [PubMed] [Google Scholar]
- [33].Poltorak A, He X, Smirnova I, Liu M, Huffel C, Du X, et al. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science. 1998;282:2085–8. doi: 10.1126/science.282.5396.2085. [DOI] [PubMed] [Google Scholar]
- [34].Takeda K, Kaisho T, Akira S. Toll-like receptors. Annu Rev Immunol. 2003;21:335–76. doi: 10.1146/annurev.immunol.21.120601.141126. [DOI] [PubMed] [Google Scholar]
- [35].Paterson HM, Murphy TJ, Purcell EJ, Shelley O, Kriynovich SJ, Lien E, et al. Injury primes the innate immune system for enhanced Toll-like receptor reactivity. J Immunol. 2003;171:1473–83. doi: 10.4049/jimmunol.171.3.1473. [DOI] [PubMed] [Google Scholar]
- [36].Tang L, Jiang W, Welty SE. The participation of P- and E-selectins on biomaterial-mediated tissue responses. J Biomed Mater Res. 2002;62:471–7. doi: 10.1002/jbm.10271. [DOI] [PubMed] [Google Scholar]
- [37].Busuttil SJ, Ploplis VA, Castellino FJ, Tang L, Eaton JW, Plow EF. A central role for plasminogen in the inflammatory response to biomaterials. J Thromb Haemost. 2004;2:1798–805. doi: 10.1111/j.1538-7836.2004.00916.x. [DOI] [PubMed] [Google Scholar]
- [38].Keselowsky B, Bridges A, Burns K, Tate C, Babensee J, LaPlaca M, et al. Role of plasma fibronectin in the foreign body response to biomaterials. Biomaterials. 2007;28:3626–31. doi: 10.1016/j.biomaterials.2007.04.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Zdolsek J, Eaton J, Tang L. Histamine release and fibrinogen adsorption mediate acute inflammatory responses to biomaterial implants in humans. J Transl Med. 2007;5:31. doi: 10.1186/1479-5876-5-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Yu M, Wang H, Ding A, Golenbock D, Latz E, Czura C, et al. HMGB1 signals through Toll-like receptor (TLR) 4 and TLR2. Shock. 2006;26:174–9. doi: 10.1097/01.shk.0000225404.51320.82. [DOI] [PubMed] [Google Scholar]
- [41].Tsan MF, Gao B. Endogenous ligands of Toll-like receptors. J Leukoc Biol. 2004;76:514–9. doi: 10.1189/jlb.0304127. [DOI] [PubMed] [Google Scholar]
- [42].Maitra R, Clement CC, Crisi GM, Cobelli N, Santambrogio L. Immunogenecity of modified alkane polymers is mediated through TLR1/2 activation. PLoS ONE. 2008;3:e2438. doi: 10.1371/journal.pone.0002438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Chen C, Kono H, Golenbock D, Reed G, Akira S, Rock K. Identifcation of a key pathway required for the sterile inflammatory response triggered by dying cells. Nat Med. 2007;13:851–6. doi: 10.1038/nm1603. [DOI] [PubMed] [Google Scholar]
- [44].Ding ZM, Babensee JE, Simon SI, Lu H, Perrard JL, Bullard DC, et al. Relative contribution of LFA-1 and Mac-1 to neutrophil adhesion and migration. J Immunol. 1999;163:5029–38. [PubMed] [Google Scholar]
- [45].Ward J, Dower S, Whyte M, Buttle D, Sabroe I. Potentiation of TLR4 signaling by plasmin activity. Biochem Biophys Res Commun. 2006;341:299–303. doi: 10.1016/j.bbrc.2005.12.188. [DOI] [PubMed] [Google Scholar]
- [46].Okamura Y, Watari M, Jerud ES, Young DW, Ishizaka ST, Rose J, et al. The extra domain A of fibronectin activates Toll-like receptor 4. J Biol Chem. 2001;276:10229–33. doi: 10.1074/jbc.M100099200. [DOI] [PubMed] [Google Scholar]
- [47].Monick MM, Powers L, Butler N, Yarovinsky T, Hunninghake GW. Interaction of matrix with integrin receptors is required for optimal LPS-induced MAP kinase activation. Am J Physiol Lung Cell Mol Physiol. 2002;283:L390–402. doi: 10.1152/ajplung.00437.2001. [DOI] [PubMed] [Google Scholar]
- [48].Taylor KR, Trowbridge JM, Rudisill JA, Termeer CC, Simon JC, Gallo RL. Hyaluronan fragments stimulate endothelial recognition of injury through TLR4. J Biol Chem. 2004;279:17079–84. doi: 10.1074/jbc.M310859200. [DOI] [PubMed] [Google Scholar]
- [49].Li H, Nookala S, Re F. Aluminum hydroxide adjuvants activate caspase-1 and induce IL-1beta and IL-18 release. J Immunol. 2007;178:5271–6. doi: 10.4049/jimmunol.178.8.5271. [DOI] [PubMed] [Google Scholar]