

Abstract
4-Hydroxyphenylpyruvate dioxygenase (HPPD) catalyzes the conversion of 4-hydroxyphenylpyruvate (HPP) into homogentisate. HPPD is the molecular target of very effective synthetic herbicides. HPPD inhibitors may also be useful in treating life-threatening tyrosinemia type I and are currently in trials for treatment of Parkinson disease. The reaction mechanism of this key enzyme in both plants and animals has not yet been fully elucidated. In this study, using site-directed mutagenesis supported by quantum mechanical/molecular mechanical theoretical calculations, we investigated the role of catalytic residues potentially interacting with the substrate/intermediates. These results highlight the following: (i) the central role of Gln-272, Gln-286, and Gln-358 in HPP binding and the first nucleophilic attack; (ii) the important movement of the aromatic ring of HPP during the reaction, and (iii) the key role played by Asn-261 and Ser-246 in C1 hydroxylation and the final ortho-rearrangement steps (numbering according to the Arabidopsis HPPD crystal structure 1SQD). Furthermore, this study reveals that the last step of the catalytic reaction, the 1,2 shift of the acetate side chain, which was believed to be unique to the HPPD activity, is also catalyzed by a structurally unrelated enzyme.
Keywords: Enzyme Catalysis, Enzyme Mutation, Enzyme Structure, Metabolism, Metalloenzymes, Dioxygenase, QM/MM
Introduction
4-Hydroxyphenylpyruvate dioxygenase (HPPD)3 is an FeII-dependent non-heme oxygenase catalyzing the conversion of 4-hydroxyphenylpyruvate (HPP) into homogentisate (HGA). In most aerobic life forms, HPPD catalyzes the second step in tyrosine catabolism. In all photosynthetic organisms, HPPD also plays an anabolic role, as HGA is essential for the formation of isoprenoid redox molecules such as plastoquinone and tocochromanols (1, 2). Plant HPPD is the molecular target of several natural compounds (3, 4) and of a range of very effective synthetic herbicides that are currently used commercially (5–9). In mammals, inborn defects in this pathway give rise to metabolic disorders of different degrees of severity (5, 10). Among these, two involve HPPD. Type III tyrosinemia arises from low HPPD activity (11) caused by an alanine to valine mutation at position 268 in the human enzyme (12). Hawkinsinuria, linked with an active enzyme with almost uncoupled turnover, is a result of an alanine to threonine mutation at position 33 in the human enzyme (12). This mutant enzyme releases an arene oxide-derived intermediate excreted in large quantities in the urine (13).
Interestingly, HPPD inhibitor/herbicide molecules also act as therapeutic agents for the debilitating and lethal inborn defects associated with type I tyrosinemia. Inhibition of HPPD prevents the accumulation of toxic metabolites in this disease (5). HPPD inhibitors are also currently being used in trials for treatment of Parkinson disease, based on the premise that inhibition of tyrosine catabolism will increase tyrosine availability for conversion to 3,4-dihydroxyphenylalanine in the brain.
The reaction mechanism of HPPD is complex; it first involves the nucleophilic attack of the α-keto acid side chain by activated dioxygen leading to its decarboxylation by heterolytic cleavage. C1 hydroxylation of the aromatic ring then occurs via an electrophilic attack by an FeIV-oxene. A 1,2 shift of the acetate side chain then leads to the formation of HGA (Fig. 1). It has been shown that the substrate-binding mechanism of HPPD involves an ordered addition of substrates and an ordered release of products. HPP is the first substrate to bind and CO2 the first product to dissociate (14). The 1,2 shift of the decarboxylated side chain is consistent with that observed with P450 hydroxylases and pterin-dependent oxygenases (15). Most previous biochemical studies involving artificial substrate analogues stressed the importance of the 4-hydroxyl group. Particularly, this group supplies electrons required for hydroxylation of position C1 on the aromatic ring (16–18). Some of these biochemical studies also support the hypothesis that an arene oxide intermediate forms during the aromatic C1 hydroxylation prior to ortho-migration. The observation that some patients suffering from Hawkinsinuria excrete the unusual amino acid hawkinsin and 4-hydroxycyclohexylacetic acid (13) has been considered as further support for the involvement of an arene oxide intermediate. Finally, Gunsior et al. (17) observed a C1-hydroxylated intermediate derived from an arene oxide when using a P214T Streptomyces avermitilis HPPD mutant. However, the direct involvement of the arene oxide intermediate to the reaction mechanism remains controversial. Indeed Borowski et al. (19) proposed that it was not directly involved in the main reaction mechanism but could be a side product released by mutated forms of the enzyme.
FIGURE 1.

A, proposed mechanism of the reaction catalyzed by HPPD. When the last 1,2 rearrangement is blocked (S246A HPPD mutant), an arene oxide-derived intermediate is released as an alternative product. This compound presents very similar chemical properties than the intermediate observed by Gunsior et al. (17) with their S. avermitilis P214T HPPD mutant, which they identified by NMR after HPLC purification as an oxepinone. B, comparison of the reactions catalyzed by HPPD and HMAS. 1, enzyme-HPA-FeIV-oxene intermediate; 2, enzyme-C1-hydroxylated intermediate with a radical σ complex (black dot).
Various crystal structures of bacterial, plant, and mammalian HPPD and related enzymes in apo-form or in complex with inhibitors have been published (Pseudomonas fluorescens, 1CJX (20); Arabidopsis thaliana 1SQD (21), 1TFZ (21), and 1SP9 (22); Zea mays 1SP8 (22); S. avermitilis, 1T47 (23); and Rattus norvegicus 1SQI (21)). However, because no crystal structures of enzyme-substrate complexes are currently available, binding interactions remain hypothetical, and various models have been proposed (17, 20, 23, 30). All of these seem to agree on the α-keto acid moiety binding through bidentate coordination with the metal ion and hydrogen bonding with a conserved glutamine residue, but multiple hypotheses have been developed to explain binding of the 4-hydroxyl group of the substrate (Fig. 2).
FIGURE 2.

Three different models of HPP binding within the Arabidopsis HPPD active site (1SQD). A, binding model proposed by Serre et al. (20) consistent with that observed for other α-keto acid-dependent enzymes. This binding mode was reliant upon the following: (a) coordination of the iron metal ion by the HPP α-keto acid moiety (bidentate) and side chains of amino acids His-205, His-287, and Glu-373 within the active site (Glu-373 not shown for clarity), and (b) hydrogen bonding of HPP to conserved glutamine residues (Gln-272, Gln-286, and Gln-358). B, binding model proposed by Brownlee et al. (23), consistent with the crystallographically observed binding position of a structurally related inhibitor 2-[2-nitro-4-(trifluoromethyl)benzoyl]-1,3 cyclohexanedione in complex with HPPD (23). In this proposed binding model, the HPP α-keto acid moiety still makes bidentate contact with the metal ion and hydrogen bond with a conserved glutamine residue (Gln-358). However, its 4-hydroxyl group is no longer engaged in hydrogen bonding with amide side chains of conserved residues but is involved in π-stacking with the rings of two conserved phenylalanine residues (Phe-360 and Phe-403). C, HPP-binding position based on the structure of HMAS Co(II)-HMA complex (2R5V) (38). In this position, the HPP α-keto acid moiety again makes bidentate contact with the metal ion and hydrogen bond with a conserved glutamine residue (Gln-358). However, the 4-hydroxyl group is engaged in hydrogen bonding with Ser-246 and Asn-261. HPP is colored blue, and the active site iron is represented as a black sphere.
Despite economically significant applications in both the pharmaceutical and agricultural sectors, little has been done to identify the most catalytically relevant substrate-binding model in HPPD. The role of the conserved glutamine, asparagine, and serine residues (supplemental Fig. S1) potentially interacting with the 4-hydroxyl group, in particular, has never been addressed. In this study, we approach this question by site-directed mutagenesis of conserved residues of the active site of carrot HPPD (supplemental Fig. S1) based on the available Arabidopsis HPPD crystal structure 1SQD (21). Our results highlight important binding displacement of the phenyl ring and unravel the function of the conserved residues of the active site. The roles of these conserved residues were also confirmed by quantum mechanical/molecular mechanical (QM/MM) calculations of the enzyme-substrate complex and key reaction intermediates. Furthermore, this study reveals that the last step of the catalytic reaction, the 1,2 shift of the acetate side chain, which was believed to be unique to the 4-HPPD activity, is also catalyzed by a structurally unrelated enzyme.
EXPERIMENTAL PROCEDURES
Cloning of the Carrot HPPD Sequence into the Expression Vector pTrc99A
The pTrc99A-HPPD Car 13 plasmid coding for the carrot HPPD protein was described previously (24, 25).
Cloning of the Pseudomonas acidovorans 1,4-Dihydroxyphenylacetate Mutase (HPAM) Gene into the Expression Vector pET-28
The open reading frame of HPAM gene was amplified by PCR using the following oligonucleotides: HPAM-5′ (gcaggatgcatatgaccaccaagacc), which introduces an NdeI restriction site (underlined), and HPAM-3′ (ggacgccgccggagctcagccttcg), which introduces a SacI restriction site (underlined) downstream from the stop codon. The DNA insert was sequenced on both strands to ensure that no mutation had been introduced during the course of PCR amplification and introduced into the expression vector pET-28. The resulting plasmid was termed pET-28-HPAM-P. acidovorans.
Site-directed Mutagenesis
Site-directed mutagenesis was carried out with the pTrc99A-HPPD Car 13 vector and using the QuickChangeTM site-directed mutagenesis kit (Stratagene). Point mutation oligonucleotides were designed to replace serine 260 by alanine, glutamines 286, 300, and 372 by glutamates, and asparagine 275 by aspartate and to modify the restriction enzyme profile for the identification of mutants. Sequencing of the mutants was performed and showed no mutations other than those desired. The conserved residues Ser-260, Asn-275, Gln-286, Gln-300, and Gln-372 of the carrot HPPD mutated in this study correspond to Ser-246, Asn-261, Gln-272, Gln-286, and Gln-358 of the Arabidopsis HPPD (1SQD) (21), respectively (Table 1 and supplemental Fig. S1).
TABLE 1.
Numbering correspondence between A. thaliana HPPD (1SQD), Daucus carota HPPD, P. fluorescens HPPD (1CJX), and S. avermitilis HPPD (1T47)
Note that the first 21 amino acids of the recombinant Arabidopsis HPPD used for three-dimensional x-ray structure determination (1SQD) were deleted (21).
| D. carota | A. thaliana 1SQD | P. fluorescens 1CJX | S. avermitilis IT47 |
|---|---|---|---|
| Ser-260 | Ser-246 | Ser-201 | Ser-201 |
| Asn-275 | Asn-261 | Asn-216 | Asn-216 |
| Gln-286 | Gln-272 | Gln-225 | Gln-226 |
| Gln-300 | Gln-286 | Gln-239 | Gln-230 |
| Gln-372 | Gln-358 | Gln-309 | Gln-305 |
Overproduction and Purification of the Recombinant Proteins
Escherichia coli JM 105 cells transformed by the pTrc99A-HPPDcar or pTrc99A-HPPDS260A, pTrc99A-HPPDN275D, pTrc99A-HPPDQ286E, pTrc99A-HPPDQ300E, pTrc99A-HPPDQ372E plasmids, or E. coli BL21 cells transformed by pET-28-HPAM-P. acidovorans were grown at 37 °C in 1 liter of Luria-Bertani medium supplemented with 100 μg/ml appropriate antibiotics. When the cell growth was equivalent to an A600 of 0.6, 0.4 mm of isopropyl β-d-thiogalactoside was added in the culture medium to initiate the synthesis of the recombinant protein. The cells were further grown for 16 h at 28 °C. After harvest, the cells were centrifuged 20 min at 40,000 × g. The pellet was resuspended in buffer A containing 50 mm Na-Hepes, pH 7.5, 1 mm EDTA, pH 8.0, 1 mm dithiothreitol, 1 mm benzamidine HCl, 5 mm amino caproic acid and sonicated with a Vibra-cell disruptor (Sonics and Materials, Danbury, CT). The crude extract obtained was centrifuged for 20 min at 40,000 × g. The cell-free supernatant was submitted to a streptomycin sulfate precipitation (0.1% (w/v)) and centrifuged for 20 min at 40,000 × g. The supernatant was used for the purification of recombinant proteins. WT and mutant carrot HPPD were purified according to Ref. 24 except that an S200 gel filtration column was added at the end of the purification process. Recombinant HPAM was purified by Ni-NTA affinity chromatography.
Assay for HPPD Activity O2 Consumption Method
The activity of the purified recombinant proteins was monitored by measuring the consumption of O2 during the formation of homogentisate from HPP (25). The reaction was initiated by the addition of purified carrot HPPD, and O2 consumption was recorded versus time at 30 °C. For the determination of the apparent KM for HPP, a range of substrate concentrations from 2 μm to 2.5 mm was used.
HPLC Determination of Homogentisate
The determination of the product formed during the reaction was performed on an ODS2 C18 with an isocratic mobile phase containing H2O/acetonitrile/acetic acid (90–10-0.1) at a flow rate of 1 ml/min. The absorbance of HGA and the hydroxylated intermediate was recorded at 230 nm.
Assay for 1,4-Dihydroxyphenylacetate Mutase Activity
After 15 min of reaction, the reaction medium of S260A carrot HPPD activity was filtered on Nanosep with a cutoff of 3 kDa. 5 μg of pure recombinant HPAM protein was then added to the filtrate, and reaction was performed at room temperature for 10 min. Reaction products were then analyzed by HPLC as described previously.
Kinetic Data Analysis
Kinetic data were fitted to the appropriate theoretical equations by using the Kaleidagraph program (Abelbeck software).
Mass Spectrum Analysis
Mass spectrum analyses were performed using a VG QUATTRO II high resolution instrument (Waters) connected to a LC-Packings-Dionex Waters X-Terra-MS column (3 μm 1 × 10 mm), flow rate 40 μl/min, mobile phase isocratic H2O/ACN/HCOOH (87:13:0.1), injection volume 5 μl, UV detector 230 and 290 nm. The tuning parameters were as follows: source, capillary 2.45 kV; cone, 22 V; source block temperature, 90 °C; desolvation temperature, 120 °C; MS, low mass resolution 15; high mass resolution, 15; multiplier, 650 V.
Protein Determination
Protein content was measured either by the method of Bradford (26) (for crude extracts only) with bovine-γ-globulin as standard or by measuring A205 as described by Scopes (27).
Theoretical Computations
We have performed a series of theoretical calculations on models of key structures to illustrate the possible interactions of substrate/intermediates with the residues surrounding the active site. Four structures were studied as follows: (i) the binary complex enzyme-HPP; (ii) the ternary enzyme-HPP-dioxygen complex; (iii) the reaction intermediate complex before the FeIV-oxene electrophilic attack at the aromatic C1 position; and (iv) the reaction intermediate complex after the FeIV-oxene attack. Calculations were performed within the framework of the quantum mechanics/molecular mechanics (QM/MM) approach (28, 29). In this approach, the active site and potentially interacting residues are modeled with quantum-based methods. The remaining atoms are described with a molecular mechanics force field.
In our calculation Programs used were the Gaussian03 package (30) for the quantal parts (QM parts hereafter) and Tinker 4.2 (31) for the part described by molecular mechanics (MM part). Dangling bonds between the QM and MM subsystems were treated using the Link-Atom scheme (29) as implemented in Ref. 32. For the QM part, the B3LYP density functional method (33–35) with a double-ζ basis set was used. This basis set was built so as to reproduce the basis set used by Borowski et al. (19) in QM-only calculations. The MM part was treated with the Charmm27 force field (36, 37). Initial structure for QM/MM calculations was directly taken from the x-ray structure of Arabidopsis HPPD 1SQD (21). This structure was selected because the one for carrot is not available. Missing residues and hydrogens were added to complete the structure. All histidines were protonated on their Nδ atoms, whereas aspartate and glutamate residues were kept unprotonated (negative charge). All arginine and lysine residues were positively charged. In addition to iron or iron-oxygen and substrate or intermediates, the QM part consisted of side chains of His-205, His-287, Glu-373 (equivalent in size to the QM-only model used by Borowski et al. (19)) together with side chains of Ser-246, Asn-261, Gln-272, Gln-286, and Gln-358, which could interact with the substrate. All MM atoms belonging to residues within a distance of 3 Å of the QM part were allowed to relax during geometry optimizations, and the remaining structure was kept frozen.
Following the results of Ref. 19, all the calculations were carried out with a quintet spin state and zero charge for the QM region. Further details for the model, methods, programs used and associated references can be found in the supplemental material.
RESULTS
Identification of Key Residues Involved in Substrate Binding and Catalytic Activities
In the three enzyme-substrate models proposed in the literature (Fig. 2), HPP carboxylate and keto oxygen atoms show bidentate interaction with the active site metal ion. The carboxylate moiety of the substrate is further stabilized through hydrogen bonding with the amino group of Gln-358 (1SQD) and a water molecule (20, 23, 38). We chose not to consider the substrate-binding model based on crystallography of HPPD with 2-[2-nitro-4-(trifluoromethyl)benzoyl]-1,3 cyclohexanedione, an HPP structurally related inhibitor (1T47) (23), as this type of inhibitor does not have a corresponding hydroxyl group, and in this position the 4-hydroxyl group of the substrate does not therefore interact with active site residues (Fig. 2B). In the two other models, the 4-hydroxyl group forms two hydrogen bonds either with Gln-272 and Gln-286 (1SQD) (Fig. 2A) or with Ser-246 and Asn-261 (1SQD) (Fig. 2C). Furthermore, Asn-209, Ser-246, Asn-261, and Gln-286 (1SQD) are involved in a hydrogen-bonding network stabilizing these residues in catalytic positions. To see how these highly conserved residues (supplemental Fig. S1) affect substrate binding and reaction mechanism, we made the following mutations to carrot HPPD: S260A, N275D, Q286E, Q300E, and Q372E. For the clarity of this study, these mutated residues were numbered according to their corresponding position in Arabidopsis HPPD crystal structure 1SQD (21) (S246A, N261D, Q272E, Q286E, and Q358E) (Table 1) in all the following sections.
Purification and Characterization of Wild-type and Mutant Carrot HPPD
Recombinant WT and mutant HPPDs were purified to apparent homogeneity by a set of three chromatography steps, as reported previously (24). All mutant enzymes behaved similarly to the WT enzyme throughout the purification procedure. Furthermore, S200 gel filtration chromatography confirmed that, like the WT enzyme, mutant enzymes all behaved as homodimers. Altogether, these results suggested that the point mutations in these HPPD mutants did not alter protein folding.
Kinetic Properties of Mutant HPPD
HPPD enzymatic activity was assessed by two different methods as follows: (i) measuring O2 consumption during HGA formation from HPP and (ii) monitoring HGA formation by HPLC (24, 25). For the five carrot HPPD mutants a plot of v (steady-state O2 consumption rate) versus HPP concentration fits the Michaelis-Menten hyperbolic equation (supplemental Fig. S2). The kinetic parameters are displayed in Table 2.
TABLE 2.
Kinetic parameters of WT and mutant carrot HPPD
For clarity, mutated residues are numbered according to the Arabidopsis crystal structure 1SQD (21). The conserved residues Ser-260, Asn-275, Gln-286, Gln-300, and Gln-372 of the carrot HPPD mutated in this study correspond thus to Ser-246, Asn-261, Gln-272, Gln-286, and Gln-358 of the Arabidopsis HPPD (1SQD), respectively (see Table 1).
| Enzyme | kcat | KM-HPP |
|---|---|---|
| s−1 | μm | |
| WT | 1.8 | 7.5 ± 2.5 |
| Q358E | 2.1 | 286 ± 22 |
| Q286E | 0.1 | 551 ± 76 |
| Q272E | 0.2 | 452 ± 63 |
| N261D | 0.3 | 153 ± 32 |
| S246A | 0.2 | 9 ± 3 |
The apparent KM-HPP for the WT enzyme was 7.5 ± 2.5 μm, and the apparent Vm was 2.0 ± 0.3 μmol of O2·min−1·mg−1 of purified HPPD, giving a kcat of 1.8 s−1 (based on a molecular mass of 48 kDa for the monomer (24, 25)). Saturation curves revealed that the mutant enzyme Q358E retained similar kcat compared with the WT enzyme (2.1 s−1). However, its KM-HPP (286 ± 22 μm) was more than 30 times higher than the KM-HPP of the WT enzyme. Activity of the mutant enzyme Q286E was highly affected in both its KM-HPP (551 ± 76 μm) and kcat (0.1s−1) values. Q272E activity was also highly affected (KM-HPP of 452 ± 63 μm and kcat of 0.2 s−1). N261D retained some activity (kcat of 0.35 s−1) but was affected in its KM-HPP (153 ± 32 μm). In contrast, the mutant enzyme S246A presented nearly the same affinity for HPP as the WT enzyme (KM-HPP of 9 ± 3 μm), but it retained only 10% of the HPPD activity (kcat of 0.22 s−1).
Accumulation of an Arene Oxide-derived C1-hydroxylated Intermediate with Mutants S246A and N261D
HPLC and LC/MS analyses were carried out to check whether intermediates accumulated during reactions catalyzed by the mutant enzymes. HGA was the only product detected in the reaction medium of enzymatic reactions catalyzed by Q272E, Q286E, or Q358E mutants. In contrast, only a small amount of HGA (15% based on peak area) could be detected as a reaction product with the S246A mutant enzyme (Fig. 3). The major product (85% based on peak area) formed by mutant S246A is not HGA but a molecule with a shorter retention time, 5.3 min as opposed to 7.2 min for HGA (HPLC analyses, Fig. 3) or 6.3 min as opposed to 8.9 min for HGA (LC/MS analyses, Fig. 4A). This new product also has a different absorption spectrum, with a maximum at 230 nm and no maximum at 292 nm, as observed for HGA (Fig. 4A). LC/MS analyses revealed that this alternative product presents the same m/z (169) as HGA (Fig. 4A). It therefore derives from the C1-hydroxylated reaction intermediate. Its HPLC and LC/MS characteristics are very similar to those of the C1-hydroxylated arene oxide-derived intermediate observed by Gunsior et al. (17) with a P214T S. avermitilis HPPD mutant. This alternative product was also detected at lower levels (<20%) when using N261D mutant enzyme (Fig. 3).
FIGURE 3.
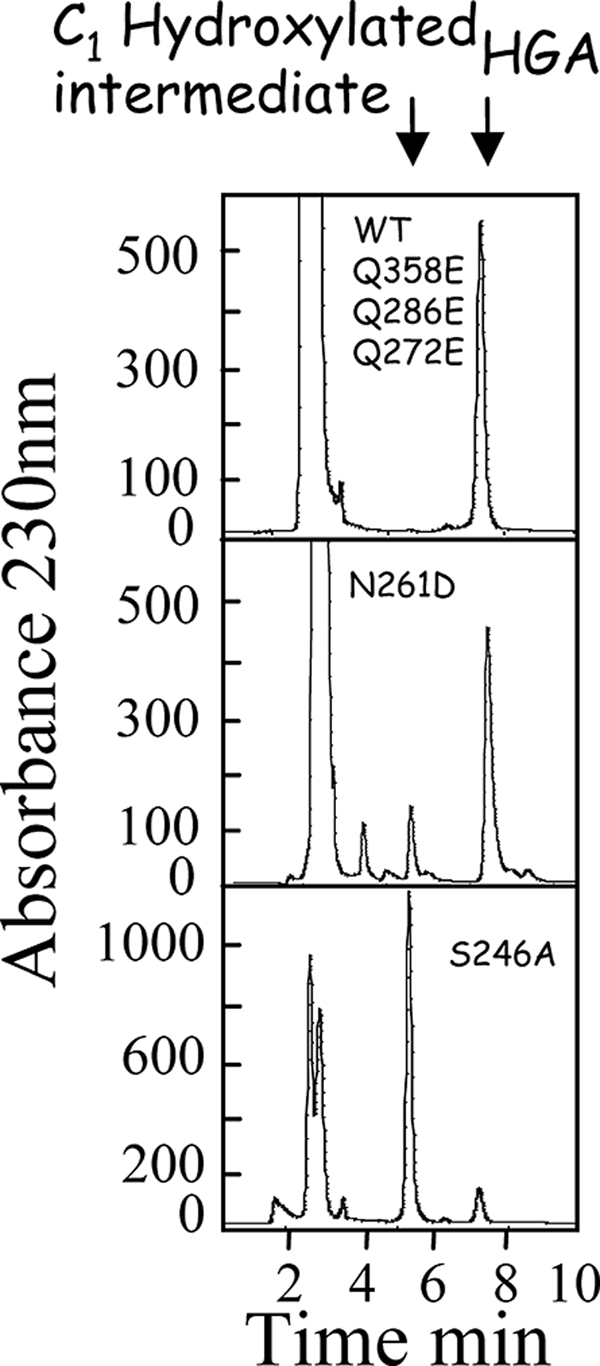
Analysis of the product formed by WT and HPPD mutants. Representative HPLCs illustrating the product formed by WT and HPPD mutants. HPLC was run on an ODS2 C18 with an isocratic mobile phase containing H2O/acetonitrile/acetic acid (90–10-0.1) at a flow rate of 1 ml/min. Absorbance was recorded at 230 nm. C1-hydroxylated intermediate corresponds to the alternative product released by S246A and N261D HPPD mutants.
FIGURE 4.

Analyses of the intermediate released by the S246A mutant. A, LC/MS analysis. Panel I, HPLC recorded at 230 and 290 nm of 50 μl of S246A reaction medium. The absorption spectrum of the C1-hydroxylated intermediate and HGA are presented in the left and right insets, respectively. Panel II, MS chromatogram. Panel III, LC/MS spectrum of the elution pick at 6.03 min (C1-hydroxylated intermediate). Panel IV, LC/MS spectrum of the elution pick at 8.5 min (HGA). Note that although the two products have different retention times and absorption spectra, they have the same m/z of 169. B, C1-hydroxylated intermediate released by the S246A mutant is transformed into HGA by HPAM. Panel I, HPLC recorded at 230 and 290 nm after incubation of the S246A mutant reaction medium with HPAM (injected volume 25 μl). Absorption spectrum of HGA is presented in the inset. Note that all the C1-hydroxylated intermediate was transformed into HGA. Panel II, MS chromatogram. Panel III, LC/MS spectrum of the elution pick at 6.13 min (C1-hydroxylated intermediate). Panel IV, LC/MS spectrum of the elution pick at 6.13 min (HGA). Note that no product could be detected at 6.3 min. C, schematic representation of the reaction catalyzed by the P. acidovorans 4-HPA-1-hydroxylase two-enzyme system constituted by an HPAH and a new HPAM. D, purification of recombinant P. acidovorans HPAM by Ni-NTA affinity column. Lane 1, 50 μg of total protein extract; lane 2, 50 μg of soluble protein extract; lane 3, run off; lane 4, wash with 50 mm imidazole; lane 5, elution with 250 mm imidazole. MM, molecular mass. Arene oxide, proposed C1-hydroxylated product released by HPAH.
Computational Results
To confirm the potential implications of the conserved amino acids Ser-246, Asn-261, Gln-272, Gln-286, and Gln-358, four key enzyme-substrate or enzyme-intermediate structures have been modeled by QM/MM using the crystal structure of the Arabidopsis enzyme 1SQD (Fig. 5).
FIGURE 5.

Structures resulting from QM/MM calculations of substrate and key intermediate enzyme complexes. Fig. 5 shows the extract of the full structure corresponding to the active region of the QM/MM calculation. The iron atom is represented by a light cyan sphere and oxygen by a red sphere. The amino acids involved in the coordination of iron (His-205, Glu-287, and Glu-373) together with the amino acids involved in the catalytic cycle of HPP (Ser-246, Asn-261, Gln-272, Gln-286, and Gln-358) are shown as sticks. The side chains of these amino acids, the iron or iron-oxygen and HPP or intermediates, constitute the QM part of the system. The remaining amino acids are described with a molecular mechanics force field and are depicted as lines. Amino acids engaged in hydrogen bonds with substrate or intermediates are colored in light blue, and distances are indicated in Å. Substrate or intermediates are colored in blue. A, binary complex enzyme-HPP. HPP carboxylate and keto oxygen atoms show bidentate interaction with the active site metal ion. The carboxylate moiety of the substrate is further stabilized through hydrogen bonding with the amino group of Gln-358. HPP phenyl ring is stabilized in the active site by hydrogen bonding with two glutamine residues Gln-272 and Gln-286. Hydrogen bonds between HPP and Gln-286-Hϵ, Gln-272-Oϵ, and Gln-358-Hϵ are indicated (distances in Å). B, ternary complex enzyme-HPP-dioxygen. Computational calculation shows that HPP adopts a very similar position to that in the binary complex enzyme-HPP. C, enzyme-FeIV-oxene-HPA intermediate complex. At this step of the reaction HPA carboxylate moiety is now in a monodentate interaction with the catalytic iron but is still stabilized by hydrogen bonding with Gln-358 by a weak interaction (2.18 Å). The HPA 4-hydroxyl group is now in strong interaction with Ser-246 and Asn-261. The phenyl C1 is still sp2 and is at 3.85 Å from the oxygen. D, enzyme-C1-hydroxylated intermediate (labeled INT on the figure). The electrophilic attack at C1 results in a strong conformational change, and the planar sp2 C1 carbon distorts to tetrahedral sp3 geometry to form the new C–O bond (1.49 Å). This distortion is accompanied by a shift of the carboxylate, which is no longer stabilized by Gln-358. The four images were generated with the PyMOL program (Molecular Simulations Inc.).
The modeled structure of the enzyme-HPP binary complex (Fig. 5A) shows a strong interaction of the substrate with both the iron active site and the three residues Gln-272, Gln-286, and Gln-358. The pyruvate moiety of the HPP substrate interacts with iron via the α-keto oxygen atom and the carboxylic oxygen on the same side at distances of 2.15 and 2.23 Å, respectively. The second carboxylic oxygen forms a hydrogen bond with Gln-358 with a distance of 1.93 Å. The 4-hydroxyl group is engaged in hydrogen bonds with two glutamine residues, Gln-272 and Gln-286 at a distance of 1.74 and 1.88 Å, respectively.
The modeled structure of the enzyme-HPP-dioxygen bound to iron ternary complex before the nucleophilic attack (Fig. 5B) shows very similar geometric properties; the second carboxylic oxygen forms a hydrogen bond with Gln-358 with a distance of 1.94 Å. The 4-hydroxyl group is still engaged in hydrogen bonds with residues Gln-272 and Gln-286 at a distance of 1.74 and 1.93 Å, respectively. These two model structures thus confirm the key role played by Gln-272 and Gln-286 in the binding of the 4-hydroxyl group of HPP and the first nucleophilic attack.
Fig. 5C shows the structure of the ternary complex enzyme-HPA-FeIV-oxene, which corresponds to the immediate intermediate preceding the hydroxylation on the aromatic C1 atom by the oxygen atom of the FeIV-oxene moiety. This structure is closer to the transition state of the hydroxylation reaction than that proposed by Borowski et al. (see Fig. 4 of Ref. 19) because the C1-oxene distance is already reduced to 3.86 Å. Our structure is comparable with the model of Neidig et al. (39) in which HPA is bound to iron in a monodentate interaction and with a C1-oxene distance of 3.69 Å before electrophilic attack (Fig. 2 of Ref. 39). The carboxylate moiety is bound to both iron (Fe-O distance of 1.91 Å) and Gln-358 via a weak hydrogen bond (O···H distance of 2.18 Å). On the opposite side of the substrate, the 4-hydroxyl group is involved in a hydrogen bonding network with residues Ser-246 as acceptor at a distance of 1.69 Å and Asn-261 as donor at a distance of 1.65 Å. This structure thus highlights the implication of Asn-261 and Ser-246 in the interaction with the 4-hydroxyl group of the intermediate. These interactions position the aromatic C1 atom for the electrophilic attack by the oxygen atom of the FeIV-oxene moiety.
In the last structure, shown in Fig. 5D, hydroxylation has occurred. C1 is now hybridized sp3 and bound to oxygen at a distance of 1.49 Å. This strong conformational evolution implies a displacement of the carboxylate moiety, which now interacts with iron only. This conformational change is very similar to the one observed in Fig. 2 of Ref. 39. The 4-hydroxyl group is still involved in H-bonds with Ser-246 and Asn-261 at distances of 1.69 and 1.56 Å, respectively. This structure thus highlights the implication of Ser-246 and Asn-261 in the interaction with the 4-hydroxyl group of the intermediate. These interactions facilitate the 1,2 shift of the acetate side chain. Finally, in the four optimized structures detailed in Fig. 5, the distances between iron and the atoms of its three ligands (N of His-205 and His-287 and O of Glu-373) remain quite constant, varying by 0.06 Å at most.
1,4-Dihydroxyphenylacetate Mutase (HPAM) from P. acidovorans Converts the Arene Oxide-derived C1-hydroxylated Intermediate Released by S246A HPPD into HGA
Microorganisms like P. acidovorans or Rhodococcus erythropolis can transform HPA into HGA using an FAD-dependent HPAH (40, 41). Sailland and co-workers (42) identified a two-enzyme system required for the hydroxylation of HPA into HGA in P. acidovorans. Their results suggested that a first polypeptide corresponding to a mono-component FAD hydroxylase (HPAH) (EC 1.14.13.-.) catalyzes hydroxylation of the aromatic C1 of HPA. A second polypeptide is required for the production of HGA. It was supposed that this second polypeptide (HPAM) catalyzes the 1,2 shift of the acetate substituent, giving rise to HGA (Fig. 4C). The similarity between this dual enzyme system and HPPD in terms of reaction mechanism (see under “Discussion”) led us to hypothesize that they might share the same C1-hydroxylated intermediate. To test this, we cloned and expressed the second polypeptide (HPAM) in Escherichia coli (Fig. 4D). The corresponding recombinant protein was purified by Ni-NTA affinity chromatography. It presents an apparent molecular mass of 42 kDa (Fig. 4D). After 15 min of reaction with the S246A HPPD mutant, the reaction medium was filtered, and 5 μg of the purified recombinant protein was added. LC/MS analyses of the reaction medium 10 min after the addition of the HPAM recombinant protein (Fig. 4B) revealed that the C1-hydroxylated intermediate released by the S246A mutant enzyme was totally converted into HGA. Indeed, this new reaction product now presents the same 292- nm absorption maximum, the same retention time of 8.9 min, and still has the same m/z of 169 than HGA (Fig. 4B).
We found that after preparative HPLC purification, this alternative product was no longer a substrate of recombinant HPAM. This reveals a high instability, which most certainly results in its transformation into the oxepinone compound identified previously by Gunsior et al. (17) (Fig. 1) with their P214T S. avermitilis HPPD mutant.
DISCUSSION
Gln-272, Gln-286, and Gln-358 Play a Central Role in Substrate Binding and the First Step of the Reaction
Among the three binding models of enzyme-HPP complex reported in the literature, only two were considered in this study. The first one proposed by Serre et al. (20) (Fig. 2A) involved the participation of Gln-272, Gln-286, and Gln-358. The other one proposed by Brownlee et al. (38) (Fig. 2C) was based on the crystal structure of the structurally related enzyme HMAS in complex with either its substrate HPP or product HMA (2R5V) (38). HMAS catalyzes the committed step in the formation of 4-hydroxyphenylglycine, a precursor of antibiotics such as vancomycin. HMAS uses the same substrates as HPPD (HPP and O2) and also catalyzes a dioxygenation reaction. The difference between the two reactions lies in the insertion of the second oxygen atom. HMAS allows the insertion of this atom onto the benzylic carbon of the substrate side chain, whereas HPPD hydroxylates the aromatic C1 (Fig. 1B). The crystal structure of HMAS was solved from crystals grown in the presence of HPP (2R5V) (38). This crystal structure of HMAS revealed that hydrogen bonds form between the 4-hydroxyl group and a serine residue, which is conserved in HPPD (Ser-246 in 1SQD). According to these data, and previous spectroscopic comparisons of HMAS and HPPD enzyme-substrate complexes (39), it was proposed that the phenol ring of HPP adopts a similar position in the HPPD active site than in the HMAS-binding site (38, 39). Its 4-hydroxyl group interacts with the conserved serine together with an asparagine residue (Asn-261 in 1SQD) (Fig. 2C) (38).
Our QM/MM modeling of HPP within the active site of Arabidopsis HPPD (1SQD) is in agreement with the model proposed by Serre et al. (20) (Fig. 5A) involving interaction of Gln-358 with the carboxylate moiety of HPP and interactions of Gln-272 and Gln-286 with the 4-hydroxyl group of HPP. By contrast, QM/MM theoretical calculations of HPP within the active site diverge from the model proposed by Brownlee et al. (38) (Fig. 2C). Indeed, QM/MM modeling failed to find a binding position of HPP in interaction with Gln-358, Ser-246, and Asn-261 (data not shown) as proposed by Brownlee et al. (38) (Fig. 2C).
In addition, site-directed mutagenesis experiments conducted in this study provide a biochemical confirmation to our QM/MM theoretical calculations. Indeed, kinetic studies of Q272E, Q286E, and Q358E mutant enzymes clearly confirm the key role of these conserved glutamine residues in the formation of the enzyme-substrate complex. The amino group of Gln-358 stabilizes the carboxylate of the HPP α-keto acid moiety by hydrogen bonding. Replacing it with a glutamate modifies this interaction and results in a strong decrease in apparent affinity of the Q358E mutant for the substrate, without affecting the kcat of the mutant enzyme (Table 2 and supplemental Fig. S2). Gln-358 thus participates in substrate stabilization but does not play a catalytic role. Replacing Gln-272 or Gln-286 by a glutamate residue modifies their capacity to create hydrogen bonds with the 4-hydroxyl group. The strong effect of these two Gln/Glu mutations on both the KM-HPP and the kcat of the Q272E and Q286E mutant HPPD (Table 2, supplemental Fig. S2) confirmed that their interactions through hydrogen bond with the substrate 4-hydroxyl group play an important role both in the formation of the enzyme-HPP complex and the first nucleophilic attack.
The eventual participation of Ser-246 and Asn-261 in forming the binary enzyme-HPP complex was also evaluated by site-directed mutagenesis. Replacing Ser-246 with an alanine residue, unable to form hydrogen bonds, does not alter enzyme-substrate apparent affinity, although it does have a strong impact on the kcat (Table 2 and supplemental Fig. S2). Thus, Ser-246 does not play any significant role in substrate binding. Replacement of Asn-261 by an aspartate residue affects both the KM and kcat of the corresponding HPPD mutant. This indicates that Asn-261 also participates in the catalytic reaction. However, QM/MM modeling of HPP within the active site of Arabidopsis HPPD (1SQD) shows that the 4-hydroxyl group cannot interact with Asn-261 and Gln-272 at the same time (Fig. 5A). This excludes any participation of Asn-261 in the formation of the binary enzyme-HPP complex and the first nucleophilic attack. Our site-directed mutagenesis supported by QM/MM calculations revealed that HPP binding and the first nucleophilic attack require interactions of the substrate 4-hydroxyl group with Gln-272 and Gln-286 (Fig. 5, A and B).
Ser-246 and Asn-261 Are Involved in Electrophilic Attack at the Aromatic C1 of the 4-Hydroxyphenylacetate Intermediate
HPP decarboxylation gives rise to HPA. A study carried out on Fe2+/α-ketoglutarate-dependent dioxygenases revealed that α-ketoglutarate decarboxylation results in monodentate binding of succinate to the catalytic iron cation (39, 43). Similarly, our QM/MM modeling of the HPA intermediate in the active site of Arabidopsis HPPD (Fig. 5C) and the kinetic data for the N261D and S246A mutants, presented here, suggest that the carboxylate group of HPA interacts in a monodentate manner with iron and is stabilized by Gln-358. In this position, the 4-hydroxyl group interacts with Asn-261 and Ser-246 (Fig. 5C). These interactions of the 4-hydroxyl group, in particular with Asn-261, which is conserved in all HPPD and replaced by isoleucine in S. avermitilis HMAS (17), properly orient the HPA intermediate for the FeIV-oxene electrophilic attack at the aromatic C1 position in HPPD, whereas a hydroxylation at the Cβ position is catalyzed by HMAS. This electrophilic attack on the C1 atom results in strong conformational changes of the intermediate because it is no longer aromatic (C1 changes from a planar sp2 to a tetrahedral sp3 hybridization). Furthermore, the carboxylate moiety rotates and is no longer H-bonded to Gln-358 (Fig. 5D).
Ser-246 Is Absolutely Required in the Ortho-migration of the Acetate Chain
In contrast with the Q272E, Q286E, and Q358E HPPD mutants, which all catalyze a complete turnover resulting in synthesis of HGA as the sole reaction product, S246A mutant releases a different main compound. It has a shorter retention time and a completely different absorption spectrum than HGA (Figs. 3 and 4A). This new compound presents a very similar absorption spectrum, HPLC behavior, and MS m/z properties to the arene oxide-derived intermediate identified by NMR by Gunsior et al. (17) in their attempt to produce HMAS activity using a P214T S. avermitilis HPPD mutant. This strongly suggests that these two compounds derive from the same C1-hydroxylated reaction intermediate. The release of the 1,4-dihydroxylated intermediate and the decrease in kcat when using mutant S246A show that Ser-246 is absolutely required for the last step of the reaction, i.e. ortho-migration of the acetate side chain. Mutant N261D also releases a small amount of 1,4-dihydroxylated intermediate, indicating that Asn-261 also participates in the ortho-migration of the acetate chain thanks to hydrogen bonding with the 4-hydroxyl group and/or indirect stabilization of Ser-246. Our analysis shows that the position of the 4-hydroxyl group in the QM/MM theoretical model of the C1-hydroxylated intermediate, strongly bound to Ser-246 and Asn-261 (Fig. 5D), properly orients the hydroxylated intermediate for the final ortho-rearrangement. In particular, the geometrical constraints induced by Ser-246 interaction with the 4-hydroxyl group favors the C1–Cβ bond cleavage and explains the efficient rearrangement of this intermediate to HGA against the thermodynamic trap of arene oxide intermediate formation.
Indeed, arene oxide formation has been shown to be energetically favorable over C1–Cβ bond cleavage in the absence of geometrical constraints (17, 19). This explains why removing the interaction of Ser-246 with the 4-hydroxyl group in S246A mutant results in the release of an arene oxide-derived product.
Ortho-migration of Acetate Side Chain Is Not Unique to HPPD
In their attempt to engineer plants resistant to HPPD inhibitors by by-passing HPPD, Sailland and co-workers (42, 44) identified a two-enzyme component 4-HPA hydroxylase system from P. acidovorans that catalyzes the transformation of HPA into HGA. Although the reaction mechanism has not yet been characterized, the conversion of HPA into HGA by this two-enzyme system shares similarities with catalysis of the HPA intermediate by HPPD, involving aromatic C1 hydroxylation and acetate side chain ortho-migration. Indeed in both cases, hydroxylation at the aromatic C1 position of HPA is the result of an electrophilic attack via a hydroperoxoflavin intermediate for HPAH or an FeIV oxene for HPPD followed by a 1,2 shift of the acetate side chain. Given that HPPD mutant S246A releases a C1-hydroxylated intermediate, we investigated whether HPPD and the FAD-dependent HPAH, although structurally unrelated, could give rise to the same hydroxylated intermediate. This proved to be the case. The intermediate produced by the HPPD mutant S246A is indeed transformed into HGA by a recombinant form of the second enzymatic component of P. acidovorans HPAM (Fig. 4B). Thus, this polypeptide catalyzes the ortho-migration of the acetate side chain giving rise to HGA (Fig. 4C). The catalytic convergence between HPPD and the two component 4-HPA hydroxylase from P. acidovorans has important implications. First, the ortho-migration of the acetate side chain of the C1-hydroxylated intermediate is not unique to HPPD. Second, whatever its direct (16, 17, 45) or indirect (this study and see Ref. 19) involvement in the reaction mechanism, the arene oxide deriving C1-hydroxylated intermediate released by the HPPD mutant S246A is able to undergo further enzymatic transformation after its release into the reaction medium. Interestingly, the 1,2 shift rearrangement step of the reaction alone cannot be catalyzed by WT HPPD (data not shown). The aromatic C1-hydroxylated intermediate probably cannot bind the active site iron of the free enzyme.
Concluding Remarks
Study of the interactions between substrate or intermediates and key residues in the active site of HPPD reveals the successive involvements of amino acids Gln-358, Gln-286, Gln-272, Asn-261, and Ser-246 during catalysis. Site-directed mutagenesis supported by QM/MM calculations allowed us to decipher the catalytic role of these conserved residues in the different catalytic steps (Fig. 5 and supplemental movie Fig. S3). Our results definitively clarify the structure of the enzyme-substrate binary complex. They highlight the central role of Gln-272, Gln-286, and Gln-358 in HPP binding and the first nucleophilic attack. These results highlight the important movement of the aromatic ring during the reaction, and the key role played by Asn-261 and Ser-246 in C1 hydroxylation and the final ortho-rearrangement steps. In addition and very interestingly, this study reveals that the 1,2 shift of the acetate side chain, which was believed to be unique to the 4-HPPD activity (19), is also catalyzed by a structurally unrelated enzyme. The 4-HPA hydroxylase two-enzyme system from P. acidovorans is the first example of two distinct and independent enzymatic components catalyzing aromatic 1-hydroxylation and side chain 1,2 shift. To our knowledge, this latter reaction corresponds to a new mutase reaction. We thus propose to name the enzyme performing this catalysis as 1,4-dihydroxyphenylacetate mutase (EC 5.4.99.-).
Supplementary Material
Acknowledgments
We greatly appreciate the gift of P. acidovorans strain from Anne Roland (Bayer CropScience, Lyon, France).

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1 and S2, movie Fig. S3, and additional references.
- HPPD
- 4-hydroxyphenylpyruvate dioxygenase
- HPP
- 4-hydroxyphenylpyruvate
- HGA
- homogentisate
- HMAS
- 4-hydroxymandelate synthase
- HMA
- 4-hydroxymandelate
- HPA
- 4-hydroxyphenylacetate
- HPAH
- monocomponent FAD-dependent HPA hydroxylase
- HPAM
- 1, 4-dihydroxyphenylacetate mutase
- QM/MM
- quantum mechanics/molecular mechanics
- Ni-NTA
- nickel-nitrilotriacetic acid.
REFERENCES
- 1. Thomas G., Threlfall D. R. (1974) Biochem. J. 142, 437–440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Soll J., Kemmerling M., Schultz G. (1980) Arch. Biochem. Biophys. 204, 544–550 [DOI] [PubMed] [Google Scholar]
- 3. Prisbylla M. P., Onisko B. C., Shribbs J. M., Adams D. O., Liu Y., Ellis M. K., Hawkes T. R., Mutter L. C. (1993) Weeds: British Crop Protection Council, pp. 731–738, Surrey, UK [Google Scholar]
- 4. Romagni J. G., Meazza G., Nanayakkara N. P., Dayan F. E. (2000) FEBS Lett. 480, 301–305 [DOI] [PubMed] [Google Scholar]
- 5. Lindstedt S., Holme E., Lock E. A., Hjalmarson O., Strandvik B. (1992) Lancet 340, 813–817 [DOI] [PubMed] [Google Scholar]
- 6. Schulz A., Ort O., Beyer P., Kleinig H. (1993) FEBS Lett. 318, 162–166 [DOI] [PubMed] [Google Scholar]
- 7. Secor J. (1994) Plant Physiol. 106, 1429–1433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ellis M. K., Whitfield A. C., Gowans L. A., Auton T. R., Provan W. M., Lock E. A., Smith L. L. (1995) Toxicol. Appl. Pharmacol. 133, 12–19 [DOI] [PubMed] [Google Scholar]
- 9. Luscombe B. M., Pallett K. E., Loubiere P., Millet J. C., Melgarejo J., Vrabel T. E. (1995) Proc. Brighton Crop Prot. Conf. Weeds 2, 35–42 [Google Scholar]
- 10. Lindblad B., Lindstedt S., Steen G. (1977) Proc. Natl. Acad. Sci. U.S.A. 74, 4641–4645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Endo F., Kitano A., Uehara I., Nagata N., Matsuda I., Shinka T., Kuhara T., Matsumoto I. (1983) Pediatr. Res. 17, 92–96 [DOI] [PubMed] [Google Scholar]
- 12. Tomoeda K., Awata H., Matsuura T., Matsuda I., Ploechl E., Milovac T., Boneh A., Scott C. R., Danks D. M., Endo F. (2000) Mol. Genet. Metab. 71, 506–510 [DOI] [PubMed] [Google Scholar]
- 13. Niederwieser A., Wadman S. K., Danks D. M. (1978) Clin. Chim. Acta 90, 195–200 [DOI] [PubMed] [Google Scholar]
- 14. Rundgren M. (1977) J. Biol. Chem. 252, 5094–5099 [PubMed] [Google Scholar]
- 15. Guroff G., Daly J. W., Jerina D. M., Renson J., Witkop B., Udenfriend S. (1967) Science 157, 1524–1530 [DOI] [PubMed] [Google Scholar]
- 16. Crouch N. P., Adlington R. M., Baldwin J. E., Lee M. H., MacKinnon C. H. (1997) Tetrahedron 53, 6993–7010 [Google Scholar]
- 17. Gunsior M., Ravel J., Challis G. L., Townsend C. A. (2004) Biochemistry 43, 663–674 [DOI] [PubMed] [Google Scholar]
- 18. Johnson-Winters K., Purpero V. M., Kavana M., Nelson T., Moran G. R. (2003) Biochemistry 42, 2072–2080 [DOI] [PubMed] [Google Scholar]
- 19. Borowski T., Bassan A., Siegbahn P. E. (2004) Biochemistry 43, 12331–12342 [DOI] [PubMed] [Google Scholar]
- 20. Serre L., Sailland A., Sy D., Boudec P., Rolland A., Pebay-Peyroula E., Cohen-Addad C. (1999) Structure 7, 977–988 [DOI] [PubMed] [Google Scholar]
- 21. Yang C., Pflugrath J. W., Camper D. L., Foster M. L., Pernich D. J., Walsh T. A. (2004) Biochemistry 43, 10414–10423 [DOI] [PubMed] [Google Scholar]
- 22. Fritze I. M., Linden L., Freigang J., Auerbach G., Huber R., Steinbacher S. (2004) Plant Physiol. 134, 1388–1400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Brownlee J. M., Johnson-Winters K., Harrison D. H., Moran G. R. (2004) Biochemistry 43, 6370–6377 [DOI] [PubMed] [Google Scholar]
- 24. Garcia I., Rodgers M., Pepin R., Hsieh T. F., Matringe M. (1999) Plant Physiol. 119, 1507–1516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Garcia I., Job D., Matringe M. (2000) Biochemistry 39, 7501–7507 [DOI] [PubMed] [Google Scholar]
- 26. Bradford M. M. (1976) Anal. Biochem. 72, 248–254 [DOI] [PubMed] [Google Scholar]
- 27. Scopes R. K. (1974) Anal. Biochem. 59, 277–282 [DOI] [PubMed] [Google Scholar]
- 28. Warshel A., Levitt M. (1976) J. Mol. Biol. 103, 227–249 [DOI] [PubMed] [Google Scholar]
- 29. Field M. J., Bash P. A., Karplus M. (1990) J. Comput. Chem. 11, 700–733 [Google Scholar]
- 30. Frisch M. J., Trucks G. W., Schlegel H. B., Scuseria G. E., Robb, et al. (2004), Gaussian 03, revision B. 05, Gaussian, Inc., Wallingford, CT [Google Scholar]
- 31. Ponder J. W. (2001) TINKER: Software Tools for Molecular Design, Version 4.2, Washington University School of Medicine, St Louis, MO [Google Scholar]
- 32. Ferré N., Olivucci M. (2003) J. Am. Chem. Soc. 125, 6868–6869 [DOI] [PubMed] [Google Scholar]
- 33. Becke A. D. (1988) Phys. Rev. A 38, 3098–3100 [DOI] [PubMed] [Google Scholar]
- 34. Lee C., Yang W., Parr R. G. (1988) Phys. Rev. B Condens Matter 37, 785–789 [DOI] [PubMed] [Google Scholar]
- 35. Becke A. D. (1993) Chem. Phys. 98, 5648–5652 [Google Scholar]
- 36. Foloppe N., MacKerell A. D. (2000) J. Comput. Chem. 21, 86–104 [Google Scholar]
- 37. Brooks B. R., Bruccoleri R. E., Olafson B. D., States D. J., Swaminathan S., Karplus M. (1983) J. Comput. Chem. 4, 187–217 [Google Scholar]
- 38. Brownlee J., He P., Moran G. R., Harrison D. H. (2008) Biochemistry 47, 2002–2013 [DOI] [PubMed] [Google Scholar]
- 39. Neidig M. L., Decker A., Choroba O. W., Huang F., Kavana M., Moran G. R., Spencer J. B., Solomon E. I. (2006) Proc. Natl. Acad. Sci. U.S.A. 103, 12966–12973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hareland W. A., Crawford R. L., Chapman P. J., Dagley S. (1975) J. Bacteriol. 121, 272–285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Suemori A., Nakajima K., Kurane R., Nakamura Y. (1996) J. Ferment. Bioeng. 81, 133–137 [Google Scholar]
- 42.Zink O., Paget E., Rolland A., Sailland A., Freyssinet G. Patent application FR 00/13942 (2000)
- 43. Solomon E. I., Brunold T. C., Davis M. I., Kemsley J. N., Lee S. K., Lehnert N., Neese F., Skulan A. J., Yang Y. S., Zhou J. (2000) Chem. Rev. 100, 235–350 [DOI] [PubMed] [Google Scholar]
- 44. Matringe M., Sailland A., Pelissier B., Rolland A., Zink O. (2005) Pest Manag. Sci. 61, 269–276 [DOI] [PubMed] [Google Scholar]
- 45. Pascal R. A., Jr., Oliver M. A., Chen Y. C. (1985) Biochemistry 24, 3158–3165 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.