

Abstract
Bacterial TrmD and eukaryotic-archaeal Trm5 form a pair of analogous tRNA methyltransferase that catalyze methyl transfer from S-adenosyl methionine (AdoMet) to N1 of G37, using catalytic motifs that share no sequence or structural homology. Here we show that natural and synthetic analogs of AdoMet are unable to distinguish TrmD from Trm5. Instead, fragments of AdoMet, adenosine and methionine, are selectively inhibitory of TrmD rather than Trm5. Detailed structural information of the two enzymes in complex with adenosine reveals how Trm5 escapes targeting by adopting an altered structure, whereas TrmD is trapped by targeting due to its rigid structure that stably accommodates the fragment. Free energy analysis exposes energetic disparities between the two enzymes in how they approach the binding of AdoMet versus fragments and provides insights into the design of inhibitors selective for TrmD.
Keywords: S-adenosyl methionine (AdoMet), adenosine, methionine, m1G37-tRNA
INTRODUCTION
Analogous enzymes are thought to be independent inventions of nature (Galperin et al. 1998), arising from different phylogenetic lineages with unrelated structures to catalyze the same reaction. These enzymes are unlike the commonly found homologous enzymes, which share similar sequence and structural folds. The existence of analogous enzymes, present in different biological domains, suggests the attractive possibility of species-specific targeting. For example, the identification of a bacterial enzyme that is structurally distinct from its human counterpart would provide a promising target for the development of new antibacterial drugs. Such a strategy would circumvent the difficulty in targeting subtle differences between bacterial and human enzymes of homologous origins. However, despite the appeal of this idea, obstacles remain high with targeting analogous enzymes. The key reason is that while these enzymes possess different catalytic structures from each other, they recognize the same substrates and cofactors using tight networks of enzyme-substrate interactions that have been well developed and adapted over time with comparable substrate binding affinities. This presents severe challenges to the conventional approach of structure-based design of substrate analogs with sufficient selectivity between analogous enzymes. To date, no successful substrate analogs have been reported that target specific members of a pair of analogous enzymes.
Here we address this challenge by evaluating the determinants of the methyl donor AdoMet for selective inhibition of the analogous tRNA methyltransferases TrmD and Trm5, which catalyze the synthesis of the m1G37 base 3′ adjacent to the anticodon. TrmD is broadly conserved in all bacterial species, whereas Trm5 is found in eukaryotes and archaea but is absent from bacteria (Bjork et al. 1989, 2001; O'Dwyer et al. 2004). While both enzymes recognize AdoMet as the methyl donor and G37-tRNA as the acceptor, they share no structural homology. TrmD functions as a homodimer and binds AdoMet using a rare trefoil-knot fold (Ahn et al. 2003; Elkins et al. 2003), whereas Trm5 functions as a monomer and binds AdoMet using the popular protein fold known as the Rossmann fold (Goto-Ito et al. 2008, 2009). Importantly, crystal structures of these enzymes show that the AdoMet structure when bound to TrmD exists in an L-shaped bent conformation (Ahn et al. 2003), where the methionine moiety bends over the adenosine moiety, whereas the AdoMet structure when bound to Trm5 exists in an extended open conformation (Fig. 1; Goto-Ito et al. 2009). The two enzymes also differ in the recognition of G37 in tRNA (Christian and Hou 2007): TrmD requires only an extended anticodon stem–loop structure for activity, whereas Trm5 requires the intact tRNA structure. Furthermore, the rate-limiting step in the TrmD-catalyzed reaction occurs during the synthesis of m1G37, whereas that for Trm5 occurs after synthesis (Christian et al. 2010b). The fundamental structural and kinetic differences between the two enzymes and their split along the evolutionary separation of bacteria from archaea and eukaryotes make this pair a paradigm to explore conceptual strategies for targeting analogous enzymes.
FIGURE 1.

Two structures of AdoMet. (A) AdoMet when bound to TrmD (Ahn et al. 2003). (B) AdoMet when bound to Trm5 (Goto-Ito et al. 2009). Drawn based on the coordinates in the structures (PDBID: 1UAK and 2ZZN) by PyMol.
tRNA modification enzymes are attractive for targeting, because of their role in the adjustment of cell fitness and viability. Extensive cellular resources are dedicated to synthesize these enzymes, which are responsible for the over 100 modified nucleotides that have been identified in tRNA. While many nucleotide modifications in tRNA can be achieved by a specific enzyme, others are achieved by multiple enzymes in successive reactions (e.g., wybutosine; Noma et al. 2006). In general, inactivation of one modification enzyme for a tRNA often has no clear growth defect, although inactivation of additional enzymes can cause synthetic lethality (e.g., Persson et al. 1997; Urbonavicius et al. 2002). The synergistic effects on growth between tRNA modification enzymes, together with the observation that hypo-modifications accelerate tRNA degradation (Alexandrov et al. 2006; Chernyakov et al. 2008), provide an underpinning for the notion that tRNA modification enzymes function collectively as a device to regulate cellular processes and as a sensor of the cellular metabolic status in both bacteria and eukaryotes (Persson 1993).
While there are other analogous pairs of tRNA modification enzymes, none are as well characterized as TrmD and Trm5. For example, the synthesis of Cm32 and Um32 (Cm and Um = ribose 2′-O-methylation of C and U) in the anticodon loop is catalyzed by TrmJ in bacteria with a trefoil-knot structure but is catalyzed by Trm7 in eukaryotes with a Rossmann-fold structure (Purta et al. 2006). The synthesis of Cm56 in the T loop (Clouet-d'Orval et al. 2005; Renalier et al. 2005) is catalyzed by a single Trm56 enzyme in most organisms but by an RNA–protein (RNP) complex in some archaea, where the proteins in the complex perform this reaction in conjunction with an RNA guide made up of C/D boxes. The synthesis of Ψ35 (Ψ, pseudouridine) in the anticodon of tRNATyr also involves analogous enzymes, with Pus7 acting as the single enzyme in eukaryotes and with an H/ACA-guided RNP in some archaea (Muller et al. 2009). However, detailed structural and kinetic information is lacking for these alternative pairs. Importantly, the reaction catalyzed by TrmD and Trm5 is rare in that it alone is required for optimal growth in bacteria (Bjork et al. 1989; Hagervall et al. 1993; O'Dwyer et al. 2004; Baba et al. 2006) and in eukaryotes (Bjork et al. 2001). This phenomenon is due to the essential role of m1G37 in the prevention of translational errors on the ribosome (Hagervall et al. 1993). Although m1A58 in the T loop of tRNA has also been shown to be essential for the viability of yeast (Anderson et al. 2000), due to its role in maintaining steady-state levels of eukaryotic initiator tRNA (Anderson et al. 1998), it is not uniformly present in human cells (Saikia et al. 2010) and is rare in bacteria, indicating that it is not a broad-based antimicrobial target. Also, while m1A58 is synthesized by the TrmI enzyme in bacteria (Roovers et al. 2004; Varshney et al. 2004) but by a Trm6–Trm61 complex in eukaryotes (Anderson et al. 1998), the bacterial TrmI shares homology with the eukaryotic Trm61, indicating an orthologous relationship between the two enzymes.
High-resolution crystal structures of TrmD and Trm5 reveal that both enzymes have an extensive network of hydrogen-bonding (H-bonding) and hydrophobic interactions to recognize AdoMet. While different in spatial configuration, each network is elaborate and comprehensive in the stabilization of the N1, N6, and N7 of the adenine ring, the 2′- and 3′-OH of the ribose, and the carboxyl and amino groups of methionine in the methyl donor. These stabilizing forces are well visualized in a binary structure of TrmD with AdoMet and are preserved in a product complex with S-adenosyl homocysteine (AdoHcy) (Ahn et al. 2003; Elkins et al. 2003). They are also clearly identified in a binary structure of Trm5 with sinefungin and in a ternary structure with AdoMet and tRNA (Goto-Ito et al. 2008, 2009). The extensive stabilizing networks established for AdoMet are consistent with the comparably high affinity of both enzymes for the methyl donor. Indeed, fluorescence binding analysis of a model pair consisting of Escherichia coli TrmD and archaeal Methanococcus jannaschii Trm5 reveals their Kd for AdoMet in the range of 1–3 μM (Christian et al. 2006, 2010b), which explains their similar catalytic efficiencies in steady state and in single turnover assays (Christian et al. 2004, 2010b).
AdoMet is the major biological methyl donor in diverse methyl transfer reactions, due to the attachment of an electrophilic methionine methyl group to a positively charged sulfur atom to provide a thermodynamically favorable driving force for methyl transfer (Cantoni 1975). The two fragments of AdoMet, adenosine and methionine, are small and soluble and both are amenable to structural modifications through synthetic chemistry, thus emphasizing their potential for structure-guided drug development programs. Here we explore analogs of AdoMet in search of the determinants that are able to discriminate TrmD from Trm5, using kinetic analysis as a tool to monitor the synthesis of m1G37. A clear advantage of kinetic analysis over fluorescence or other types of binding assays is the ability to examine the contribution of binding free energy and reactivity of all of the substrates, cofactors, and discrete states of the enzyme along the reaction coordinate. Also, by focusing on the m1G37 reaction, kinetic analysis can identify AdoMet analogs that specifically inhibit the reaction, rather than those that inhibit other AdoMet-dependent reactions. For example, analogs that target TrmD can be separated by cross-reactivity analysis from those that target TrmH (Nureki et al. 2004) and TrmL (Benítez-Páez et al. 2010), which adopt the same trefoil-knot structure as in TrmD but synthesize Gm18 and Um34 on tRNA, respectively.
Using E. coli TrmD and M. jannaschii Trm5 as a pair of model enzymes, we find that neither naturally existing nor synthetic analogs of AdoMet are able to distinguish between the two enzymes; rather, we find that fragments of the methyl donor and their derivatives selectively inhibit TrmD more than Trm5. Crystal structural analysis of each enzyme in complex with the adenosine fragment reveals new insight into the molecular details of how the relatively inflexible structure of TrmD subjects the bacterial enzyme to targeting and inactivation, whereas the more fluid structure of Trm5 enables the eukaryotic-archaeal enzyme to escape targeting. Free energy analysis of binding provides evidence for an energetic penalty in the TrmD binding to AdoMet versus fragment that is not observed in the Trm5 binding. The disparities in binding energies between the two enzymes provide insight into their distinct AdoMet binding modes and suggest novel approaches toward the development of selective antibiotics targeting the bacterial enzyme. Together, this work demonstrates that the fragment-based drug discovery approach (Congreve et al. 2008; Hesterkamp and Whittaker 2008), initially developed to identify small molecules that target individual proteins or RNAs, can be successfully exploited to differentiate the disparate substrate binding determinants of analogous enzymes.
RESULTS
Kinetic analysis of AdoMet analogs
To distinguish TrmD from Trm5, we sought to identify structural analogs of AdoMet that would more effectively compete for the binding site of the methyl donor in TrmD than in Trm5, thus exhibiting selectivity against TrmD. In a competitive inhibition model, the maximum velocity (Vmax) of the reaction and the catalytic turnover kcat (= Vmax/[E], where [E] = enzyme concentration) are unchanged, whereas the apparent affinity of the substrate to the binding site is decreased, leading to an increase in the apparent Km of the substrate. We determined the inhibition constant Ki of an analog by measuring the apparent Km (AdoMet) as a function of analog concentration and fitting the data to the competitive inhibition equation 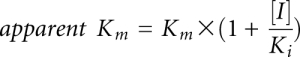 , where Km is the Michaelis constant of AdoMet and [I] is the concentration of the analog inhibitor. Because Km (AdoMet) differed substantially between TrmD and Trm5 (Fig. 2), we normalized Ki (analog) by Km (AdoMet). Mathematically, Ki/Km is equivalent to IC50/[S + Km] (Hamilton-Miller 1966) and it is an approximation of IC50/[S] at saturating substrate concentration relative to Km (AdoMet). Analog differentiation of TrmD from Trm5 was evaluated by the ratio of [Ki/Km (Trm5)]/[Ki/Km (TrmD)], which was interpreted as the fold sensitivity of TrmD to the analog relative to Trm5 (Fig. 2).
, where Km is the Michaelis constant of AdoMet and [I] is the concentration of the analog inhibitor. Because Km (AdoMet) differed substantially between TrmD and Trm5 (Fig. 2), we normalized Ki (analog) by Km (AdoMet). Mathematically, Ki/Km is equivalent to IC50/[S + Km] (Hamilton-Miller 1966) and it is an approximation of IC50/[S] at saturating substrate concentration relative to Km (AdoMet). Analog differentiation of TrmD from Trm5 was evaluated by the ratio of [Ki/Km (Trm5)]/[Ki/Km (TrmD)], which was interpreted as the fold sensitivity of TrmD to the analog relative to Trm5 (Fig. 2).
FIGURE 2.

Kinetic parameters for E. coli TrmD and M. jannaschii Trm5. Michaelis constants Km (AdoMet) and inhibition constants (Ki) for all analogs were measured in steady state and are reported as mean values of at least two independent determinations (± standard deviation values). Ki/Km is calculated from respective values for each enzyme and the ratio [Ki/Km (Trm5)]/[Ki/Km (TrmD)] is reported. Also see Supplemental Figure S1.
E. coli TrmD was assayed at 37°C with an in vitro transcript of E. coli tRNALeu, while M. jannaschii Trm5 was assayed at 55°C with a transcript of M. jannaschii tRNACys, using previously established conditions (Elkins et al. 2003; Christian et al. 2004). These transcripts were chosen for their optimal sequence contexts for TrmD and Trm5, respectively, and were each refolded by annealing and verified as tRNA substrates by demonstrating a capacity for methylation to ∼70% in extended time courses (Christian et al. 2010b). Measurements of inhibition were exhaustively controlled: Enzymes were assayed at concentrations corrected by active-site titration and at levels that allowed linear synthesis of m1G37 over time, while saturation of [3H]-AdoMet and tRNA was established at concentrations 10-fold higher than the previously determined Kd of each substrate (Christian et al. 2010b).
No differentiation by natural and synthetic AdoMet analogs
The naturally existing AdoMet analogs sinefungin and AdoHcy were evaluated as inhibitors of methyl transfer (Fig. 2). Sinefungin is an antifungal and antiparasitic antibiotic produced by Streptomyces incarnates that shares with AdoMet the adenosine fragment but differs by possessing an ornithine instead of a methionine fragment, whereas AdoHcy is the product of methyl transfer that lacks the methyl group of AdoMet. Kinetic analysis showed that, while both analogs were competitive inhibitors of AdoMet, they exerted similar effects on TrmD and Trm5 without discrimination. For example, analysis of sinefungin inhibition of m1G37 synthesis as a function of AdoMet concentration revealed the same pattern for both enzymes: While increasing concentration of the inhibitor had no effect on the Vmax or kcat of the reaction, it progressively elevated the apparent Km values of AdoMet (Fig. 3A,B), indicating competition of sinefungin for the AdoMet-binding site on both enzymes. Fitting the apparent Km (AdoMet) as a function of sinefungin concentration to a linear equation derived from the competitive inhibition model returned the Km (AdoMet) for TrmD (5.0 ± 0.8 μM) and for Trm5 (0.42 ± 0.08 μM) in the absence of the analog (the y-intercepts in Fig. 3C,D), both of which are closely similar to the values reported previously (8 μM for E. coli TrmD and 1.0 ± 0.1 μM for M. jannaschii Trm5) (Elkins et al. 2003; Christian et al. 2006). Extrapolation of the linear data to the x-intercepts returned the Ki (sinefungin) for TrmD (0.62 ± 0.06 μM) and for Trm5 (33 ± 2 nM) (Fig. 3C,D). The Ki/Km value calculated for TrmD (0.12 ± 0.07) and for Trm5 (0.08 ± 0.02) was closely similar, with the value for Trm5 slightly lower than the value for TrmD by 0.7-fold. Similar inhibition of the two enzymes was also observed for AdoHcy; analysis of Ki (AdoHcy) and Km (AdoMet) showed that the Ki/Km value of TrmD (0.8 ± 0.5) is smaller than that of Trm5 (1.17 ± 0.37) by 1.5-fold.
FIGURE 3.

Sinefungin inhibition of m1G37-tRNA synthesis. (A) Analysis of initial rate of methyl transfer by TrmD (36 nM) as a function of AdoMet concentration (0–25 μM) in the presence of 20 μM E. coli tRNALeu and increasing concentration of sinefungin (0–4 μM). At each concentration of sinefungin, the value kcat (in sec−1) was calculated from Vmax/[E], where Vmax was derived from fitting the data of V0 as a function of AdoMet concentration to the Michaelis-Menten equation, while the value of apparent Km (in μM) was derived from the same fit. Values of kcat and apparent Km at each sinefungin concentration are listed. The kcat values remained constant with increasing concentration of sinefungin, while the apparent Km values increased; this indicates that sinefungin was a competitive inhibitor of AdoMet. (B) Analysis of initial rate of methyl transfer by Trm5 (25 nM) as a function of AdoMet concentration (0–5 μM) in the presence of 6 μM M. jannaschii tRNACys and increasing concentration of sinefungin (0–60 nM). (C) The concentration-dependent apparent Km (AdoMet) for TrmD was fit to a linear equation to derive Km (AdoMet) from the y-intercept and Ki (sinefungin) from the x-intercept. (D) The concentration-dependent apparent Km (AdoMet) for Trm5 was fit to a linear equation to derive Km (AdoMet) from the y-intercept and Ki (sinefungin) from the x-intercept.
Synthetic analogs with an allyl (AdoPropen) or propargyl (AdoButyn) group in place of the reactive methyl group of AdoMet were made by regiospecific chemical S-alkylation of AdoHcy (Dalhoff et al. 2006b). Both analogs have been shown as substrates for several DNA methyltransferases (Dalhoff et al. 2006a), including the N6A methyltransferase M.TaqI, the C5C methyltransferase M.HhaI, and the N4C methyltransferase M.BcnIB, for transfer of the side chains to diverse DNA substrates. While these analogs were not accommodated by E. coli TrmD or M. jannaschii Trm5 for transfer of their side chains to tRNA (data not shown), they were competitive inhibitors of AdoMet, permitting the determination of whether steric bulk in the vicinity of the sulfonium ion might engender inhibition specificity. Both analogs inhibited each enzyme more poorly than sinefungin or AdoHcy with an increased Ki, indicating that neither active site is particularly tolerant of steric bulk in the vicinity of the sulfonium ion. However, as with sinefungin and AdoHcy, the Ki/Km values for TrmD and Trm5 were closely similar, although a noticeable difference was observed with AdoButyn with a Trm5/TrmD ratio of 4.5-fold. Presumably, the rigid propargyl substituent in AdoButyn is more effective for inhibiting TrmD than Trm5.
Differentiation by fragments of AdoMet
To elucidate the determinants in AdoMet that interact with TrmD and Trm5, we tested adenosine and methionine fragments and their derivatives for competitive inhibition of methyl transfer. As expected, all of these fragments showed substantially higher Ki values for TrmD and Trm5 relative to natural or synthetic analogs of the parental methyl donor (Fig. 2), consistent with their smaller and less complex molecular structures. Importantly, all of the fragments tested, including adenosine, methionine, and most of the nucleoside and amino acid derivatives, showed selectivity against TrmD by exhibiting substantially lower Ki/Km values to TrmD relative to Trm5. For example, adenosine inhibited TrmD with a Ki/Km of 7 ± 1 and inhibited Trm5 with a markedly larger Ki/Km of 153 ± 5, a difference of 22-fold. Substitution of the 5′-OH of adenosine with 5′-[2-aminoethyl thio] as in 5′-[(2-aminoethyl)thio]-5′-deoxy-adenosine (AETA) (Goedecke et al. 2001) selectively inhibited TrmD by 17-fold. Substitution of 6-NH2 of adenosine with 6-chloro as in 6-chloropurine selectively inhibited TrmD by 14-fold. Substitution of 6-NH2 with 6-carbonyl as in inosine selectivity inhibited TrmD by 33-fold. In these nucleoside derivatives, the selectivity against TrmD is due to preferential increases in the Ki to Trm5 relative to TrmD. For example, the Ki (inosine) for Trm5 is higher than the Km (AdoMet) by 3200-fold, as compared to the 98-fold difference between the Ki (inosine) and Km (AdoMet) for TrmD. Such preferential increases in the Ki for Trm5 were also observed for the amino acid derivatives, methionine and methylthio-cysteine (MeS-cysteine). The only exception among the fragment analogs without selectivity against TrmD was MeS-adenosine, due to a smaller increase in Ki for Trm5 relative to TrmD, presumably because the methylthio-substitution in the adenosine fragment confers an important determinant of interaction for Trm5.
The kinetic analysis is relevant for inhibition in vivo. The intracellular concentration of AdoMet in bacteria (∼400 μM) (Javor 1993) is saturating relative to Km (AdoMet) of E. coli TrmD (5.0 ± 0.8 μM, Fig. 2). Similarly, the intracellular concentration of AdoMet in archaea and eukaryotes is likely saturating relative to Km (AdoMet) of M. jannaschii Trm5 (0.42 ± 0.08 μM, Fig. 2), based on the known intracellular concentration of AdoMet in humans (∼60 μM; de Ferra and Baglioni 1983). In both cases, the parameter Ki/Km can be simplified as IC50/[S] to allow calculation of the IC50 value from (Ki/Km) × [S] (the intracellular concentration of AdoMet). For example, in the case of adenosine, the IC50 (adenosine) calculated for TrmD (7 × 400 μM) is smaller than IC50 (adenosine) calculated for Trm5 (153 × 60 μM) by more than threefold in the respective cellular conditions, suggesting that the fragment has the capacity to selectively inhibit TrmD in vivo. This analysis reveals that, due to the ∼6-fold difference in the intracellular AdoMet concentration in bacteria and in humans, analogs that are selective against TrmD must exhibit at least a sixfold difference in Ki/Km of TrmD relative to Trm5 to offset the difference in the intracellular concentrations of AdoMet in vivo. Given that a 10-fold difference in binding affinity is desirable for a selective inhibitor, this requires a 60-fold difference in Ki/Km when considering the cellular concentrations of AdoMet in humans and bacteria.
Kinetic Kd of adenosine to TrmD and Trm5
To independently evaluate the selective inhibition by fragments of AdoMet, we focused on the adenosine fragment and determined its affinity to E. coli TrmD and M. jannaschii Trm5 by measuring the kinetic Kd as a different probe than the steady-state Ki/Km. The measurements of kinetic Kd were performed by rapid mixing of an enzyme-inhibitor complex with saturating AdoMet and tRNA to allow the formation of enzyme-substrate and enzyme-inhibitor complexes in rapid equilibria, so that the extent of methyl transfer in one turnover would provide a direct readout of the amount of the uninhibited enzyme capable of synthesis of m1G37 (Fig. 4A,B). The time periods permitted for one turnover of E. coli TrmD and M. jannaschii Trm5 were respectively 11 and 8 sec, based on the single-turnover rate constant of each enzyme (Christian et al. 2010b). As the inhibitor concentration increased, the amount of the active enzyme available for methyl transfer progressively decreased. The data of m1G37 synthesis as a function of adenosine concentration were well fit to a quadratic equation, revealing the kinetic Kd for TrmD (372 ± 37 μM) and for Trm5 (910 ± 58 μM) (Fig. 4A,B). As a control, the kinetic Kd (AdoMet) for E. coli TrmD was determined (5.3 ± 0.4 μM) by a similar assay but without the inhibitor (Supplemental Fig. S2), while the kinetic Kd (AdoMet) for M. jannaschii Trm5 was previously determined (0.44 ± 0.09 μM) (Christian et al. 2010b). For each enzyme, normalization of Kd (adenosine) by Kd (AdoMet) was used to evaluate the competence of the inhibitor to compete for the AdoMet-binding site during the rapid binding equilibria of one methyl transfer. Analysis of this normalized ratio revealed a preferential binding of adenosine to TrmD than to Trm5 by 29-fold (Fig. 4C), similar to the 22-fold preference in the steady-state analysis of [Ki (adenosine)]/[Km (AdoMet)] (Fig. 2).
FIGURE 4.

Determination of kinetic Kd (adenosine). (A) Determination of kinetic Kd (adenosine) for E. coli TrmD. Kinetic analysis was performed in single turnover conditions by rapid mixing of TrmD (0.4 μM) and adenosine (0.08–8 mM) in one syringe with tRNA (10 μM) and AdoMet (60 μM) in the second syringe (shown in the scheme). The reaction was quenched after one turnover (11 sec) by acid and the amount of product synthesis was determined. Fitting the data of concentration-dependent product synthesis to a quadratic binding equation revealed the Kd (adenosine) for TrmD. (B) Determination of kinetic Kd (adenosine) for M. jannaschii Trm5. Kinetic analysis was performed in single turnover conditions by rapid mixing of Trm5 (0.4 μM) and adenosine (0.2–17 mM) in one syringe with tRNA (16 μM) and AdoMet (60 μM) in the second syringe (shown in the scheme). The reaction was quenched after one turnover (8 sec) and the amount of product synthesis was determined. Fitting the data of the concentration-dependent product synthesis to a quadratic binding equation revealed the Kd (adenosine) for Trm5. Error bars correspond to standard deviation from three independent measurements. The inset in each graph shows the order of mixing of reagents and the reaction time. (C) A summary of Kd (AdoMet) and Kd (adenosine) for E. coli TrmD and M. jannaschii Trm5. The Kd (AdoMet) for Trm5 was previously determined (Christian et al. 2010b). The ratio of Kd (adenosine)/Kd (AdoMet) was calculated for both enzymes to reflect the competitive binding of adenosine for the AdoMet binding site. Values in parentheses correspond to the normalized ratios of Trm5 relative to TrmD. Also see Supplemental Figure S2.
X-ray structural analysis of adenosine-bound TrmD and Trm5 complexes
To gain structural insight into the selective binding affinity of adenosine to TrmD rather than to Trm5, crystal structures of the two enzymes were solved in complex with the fragment (detailed information summarized in Supplemental Table S1). The TrmD structure was solved at 2.25 Å resolution with the Haemophilus influenzae enzyme, which shares 83% sequence identity and 91% similarity with E. coli TrmD. As seen in the previous structures of TrmD (Ahn et al. 2003; Elkins et al. 2003), the two active sites in the dimeric H. influenzae enzyme were symmetrically assembled in the dimer interface, with each consisting of residues from the AdoMet-binding motifs in the N-terminal domain of one monomer and residues from the tRNA-binding motifs in the C-terminal domain of the second monomer. A comparison with the previous AdoMet-bound structure of TrmD (Ahn et al. 2003) revealed that the two adenosine molecules bound in the new structure were in virtually overlapping positions as the adenosine moiety of the previous structure (Fig. 5A). The overlapping occupancy of adenosine in the adenosine-pocket of the AdoMet-binding site was also preserved in the AdoHcy-bound structures of TrmD (Ahn et al. 2003; Elkins et al. 2003). The network of H-bonds that stabilizes AdoMet was maintained in its entirety to stabilize adenosine in the new structure, including those linking adenosine N1, N6, and N7 with, respectively, the amide group of I133, the carbonyl groups of G134 and Y136, and the amide of L138 in the main chain. As in the previous structures, the ribose oxygen of adenosine in the new structure was stabilized by the main-chain amide of G140 and G141, while the 2′- and 3′-OH groups were stabilized respectively by the main-chain of L87 and side-chain of Y86. However, minor changes were observed in the new structure relative to the previous structures, which were primarily associated with the conformation of the residues involved in the stabilization of the methionine moiety of AdoMet, including S170, D177, and H180, all of which made adjustments to occupy the space left open by the absence of the amino acid. Overall, a pairwise superposition of the Cα-atoms of the AdoMet- and adenosine-bound complexes revealed identical structures (Fig. 5C) with a root-mean-square deviation (RMSD) of 0.18 Å for 217 of the 236 atoms aligned. The conformation of the adenosine-bound N-terminal domain was essentially unchanged in the two structures (RMSD of 0.13 Å for the 147 atoms aligned), while the movement of the C-terminal domain was slightly more pronounced (RMSD of 0.28 Å for the 66 atoms aligned). The latter movement may be due to the disordering of residues 161–171 in both structures. The superposition also showed that adenosine binding to TrmD created few changes to the extensive dimer interface that forms the active site. Thus, the structure of TrmD appears rigid enough that it can use the same active site and overall conformation to accommodate adenosine in the adenosine pocket of the AdoMet- or AdoHcy-binding site.
FIGURE 5.

Structural analysis of adenosine-bound TrmD and Trm5 complexes. (A) Superposition of the Cα atoms of adenosine in the adenosine-bound (magenta) complex onto the structure of the AdoMet-bound complex of H. influenzae TrmD (orange, PDB ID: 1UAK). Adenosine and AdoMet are colored in blue and cyan, respectively. Enzyme residues that form the interacting network with AdoMet are depicted, showing H-bonds (dashed lines) with the methyl donor. These residues are in overlapping positions in the two structures. (B) Superposition of the Cα atoms of adenosine in the adenosine-bound (magenta) complex onto the sinefungin-bound complex of M. jannaschii Trm5 (orange, PDB ID: 2YX1). Adenosine and sinefungin are colored in blue and cyan, respectively. Enzyme residues that form the interacting network with sinefungin are depicted, showing H-bonds (dashed lines) with the AdoMet analog. The superposition reveals a shift of adenosine toward the methionine pocket by 0.6 Å with the sugar flexed away from the active site by 1.35 Å. Residues with the most significant shifts are those that contact G37-tRNA in the D2 domain, including Y177 and R136. (C) A ribbon diagram showing the superposition of the overall adenosine-bound and AdoMet-bound structures of H. influenzae TrmD. (D) A ribbon diagram showing the superposition of the overall adenosine-bound and sinefungin-bound structures of M. jannaschii Trm5.
The adenosine-bound Trm5 structure was solved at a 3.05 Å resolution with the M. jannaschii enzyme. Trm5 consists of three domains: an N-terminal D1 domain that contacts the tertiary core (elbow) region of the tRNA L shape in a ternary complex of the enzyme with tRNA and AdoMet (Goto-Ito et al. 2009); a D2 domain that recognizes G37 and the anticodon loop; and a D3 domain that stabilizes AdoMet binding and catalyzes methyl transfer. Due to the lack of a Trm5 structure bound to AdoMet, the new structure was compared to the sinefungin-bound structure of the enzyme (Goto-Ito et al. 2008). This comparison revealed that, in contrast to TrmD, significant misalignment of adenosine with the sinefungin site of Trm5 was observed, primarily due to a 0.6 Å translation and 0.5 Å downward shift of adenosine toward the methionine pocket (Fig. 5B). The movement of adenosine disturbed the H-bond and hydrophobic network established for the adenosine fragment in the sinefungin-bound structure by weakening or abolishing the enzyme interactions with the adenine ring through D251, V252, and I224 and the interactions with the ribose through F203, D223, and L266. More substantial disruptions were observed in the conformation of the residues in the D2–D3 domain responsible for recognition of G37-tRNA, including N265, P267, and Y177 that played a role in stabilizing the purine base, R136, R181, and R186 that stabilized the conformation of the anticodon loop, and E185 that was proposed to deprotonate N1 of G37 before methyl transfer (Christian et al. 2010a). The other catalytic residue R145 that presumably stabilized O6 of G37 during deprotonation (Christian et al. 2010a) was in a disordered region. All of these residues are important for the reaction mechanism of Trm5, and their displacements from the positions in the AdoMet- or sinefungin-bound complex due to adenosine binding indicated a coordinated dismantling of the active site. Displacement in the D1 domain was also observed (Fig. 5D), most notably for residues that recognized the backbone structure of the tRNA tertiary core (e.g., K10, R16, and K27). However, because D1 is flexibly linked to the D2–D3 domain, its displacement in the new structure from the sinefungin-bound structure might be due to crystal packing effects. An overall pairwise superposition was thus performed only with the Cα atoms of the D2–D3 domains in the present structure relative to the sinefungin-bound structure, which revealed a 0.46 Å RMSD (based on alignment of 236 out of 253 residues), significantly higher than the 0.18 Å value obtained with TrmD. The sinefungin position in the sinefungin-bound structure differed slightly from the AdoMet position in the ternary complex of Trm5–AdoMet–tRNA. An overall pairwise superposition performed with the Cα atoms of the D2–D3 domains in the present structure relative to the ternary complex structure revealed an even higher RMSD value of 0.65 Å.
DISCUSSION
While analogous enzymes exist in unrelated structures, they recognize the same substrates using distinct but well-developed interacting networks to catalyze the same reaction. Identifying substrate analogs that can preferentially bind to one enzyme but not the other has proven to be a significant challenge. The challenge is clearly illustrated for the TrmD and Trm5 pair, which as we show here cannot be distinguished by several previously well-characterized natural and synthetic AdoMet analogs. Instead, fragments of AdoMet, particularly adenosine and its purine derivatives, show discerning and specific inhibition of TrmD. To our best knowledge, this work is the first and successful application of the fragment-based structure-activity analysis to differentiate the substrate binding determinants of analogous enzymes.
Free energy analysis
To gain insight into the AdoMet binding determinants of TrmD and Trm5, the free energy of binding the methyl donor by these enzymes relative to the analogs studied here was calculated and summarized (Table 1), based on the experimentally determined Km and Ki values (Fig. 2). Although TrmD and Trm5 differ in the rate-limiting step in the control of their catalytic turnover, they display the same catalytic turnover rate (Christian et al. 2010b), suggesting that the use of Km and Ki values for free energy calculation was reasonable for both enzymes. In addition, the Km and Kd values of AdoMet are closely similar for both enzymes, and the Ki/Km ratios of adenosine are closely similar to the Kd ratios (Figs. 2, 4). Free energy calculations indicate that TrmD interacts with AdoMet with lower affinity than does Trm5 (−7.5 vs. −9.6 kcal/mol, respectively), and that the difference in the apparent binding free energy between the two enzymes remains more or less the same in all of the natural and synthetic analogs as well as in the MeS-adenosine analog, which is the only fragment that lacks selectivity. Interestingly, free energy calculations indicate that TrmD interacts with similar affinity as does Trm5 with fragments or fragment derivatives that show selectivity. However, additivity analysis of TrmD reveals that the sum of the free energies of binding the adenosine and methionine fragments ([−6.3] + [−2.5] = −8.8 kcal/mol) is smaller than the free energy of binding AdoMet by 1.3 kcal/mol, suggesting the existence of an energetic penalty in the binding of AdoMet relative to fragments. In contrast, additivity analysis of Trm5 reveals that the sum of the free energies of binding adenosine and methionine ([−6.3] + [−3.3] = −9.6 kcal/mol) is the same as the free energy of binding AdoMet, suggesting that the fragment free energies of interaction for Trm5 are additive. The disparity in free energy additivity of TrmD and Trm5 is likely attributable to the unusual bent conformation of AdoMet when bound to TrmD in contrast to the extended conformation when bound to Trm5 (Fig. 1). The bent conformation of AdoMet is also observed in the structures of sinefungin and AdoHcy when bound to TrmD (Ahn et al. 2003; Elkins et al. 2003), while the extended conformation is preserved in the structure of sinefungin when bound to Trm5 (Goto-Ito et al. 2008). The consistent bent conformation in AdoMet and in natural analogs when bound to TrmD suggests that the energetic penalty in the binding of these compounds by TrmD is involved in the entropic costs associated with stabilizing the bent conformation, preventing the enzyme from full utilization of the binding energy of its component fragments. In contrast, the additivity of binding by Trm5 suggests that the enzyme can capture the full spectrum of the binding free energies from the component fragments in a way that is facilitated by the extended conformation of the bound AdoMet.
TABLE 1.
Calculation of binding free energy

The different binding determinants of AdoMet versus fragments to TrmD and to Trm5 provide a basis to understand the fragment selectivity. Even if a fragment binds with the same binding energy to both enzymes, the overall energetic contribution of the fragment relative to AdoMet binding is different for the two enzymes. For example, while adenosine binds with the same energy to both TrmD and Trm5 (−6.3 kcal/mol), it accounts for 84% of the total binding energy of AdoMet to TrmD (−7.5 kcal/mol), but only 66% of the total binding energy of AdoMet to Trm5 (−9.6 kcal/mol). The greater contribution of adenosine binding to the TrmD–AdoMet binding interaction than to the Trm5–AdoMet interaction suggests a more stable binding of the fragment to TrmD than to Trm5. Analysis of the thermodynamic Kd as determined by kinetic measurements under rapid binding equilibrium (Fig. 4) confirms that the adenosine affinity to TrmD is 2.4-fold higher than the affinity to Trm5 (Kd = 372 ± 37 and 910 ± 58 μM, respectively). This difference, after normalization by the inherent 12-fold difference of AdoMet affinity to the two enzymes (Kd = 5.2 ± 0.4 and 0.44 ± 0.09 μM, respectively), is enlarged to 29-fold, meaning that the fragment is 29-fold more stable in the AdoMet-binding site on TrmD than on Trm5 each time it outcompetes against AdoMet for the binding site. Due to the rapid binding equilibria under which the binding constants were measured, the Kd (= koff/kon) values can be used to calculate koffs assuming that kon is diffusion controlled. This calculation suggests that the adenosine fragment on TrmD is more stable than on Trm5 with a 29-fold longer duration time. Direct measurements of fragment binding kinetics to TrmD and Trm5 will more precisely assess the difference in binding stability. Nonetheless, part of the stability of the fragment on TrmD may be provided by the inherent structural rigidity of the trefoil-knot fold, as suggested by a biophysical analysis of a model knotted protein (King et al. 2010). Conversely, part of the instability of the fragment on Trm5 may derive from the inherent structural fluidity of the enzyme, which is indicated by a recent study of Trm5 during catalysis (Christian et al. 2010a) and is supported by the higher B factors associated with Trm5 relative to TrmD in structural analysis (Supplemental Table S1).
Combining free energy analysis with structural analysis, we propose a model to explain the selectivity of adenosine targeting to TrmD. In this model (Fig. 6), the adenosine fragment binds to the adenosine pocket of TrmD in an overlapping position, trapping the enzyme in an inactive state that is likely stabilized by the rigidity of the enzyme structure. In contrast, the fragment binds to the adenosine pocket of Trm5 in a misfit position, which is corroborated with global structural perturbation in the active site and throughout the D2–D3 domains. The inherent structural flexibility of Trm5 may promote rapid release of the fragment, facilitating the return to the native structure and explaining the escape from targeting.
FIGURE 6.

A model of adenosine targeting TrmD and Trm5. In the active state, TrmD and Trm5 each possess an AdoMet-binding site, consisting of an adenosine and a methionine pocket. The AdoMet site is located in a dimer interface of TrmD comprised of the N-terminal domain of monomer 1 (N1) and the C-terminal domain of monomer 2 (C2), whereas it is located between the D2 and D3 domains of the monomeric Trm5. In the adenosine-bound inactive state, TrmD maintains the same structure and binds adenosine in the already-established adenosine pocket, whereas Trm5 shifts its adenosine pocket and binds adenosine in a misfit conformation, leading to structural alterations of the enzyme. In TrmD, the stable binding of adenosine causes delayed release of the ligand, whereas in Trm5 the unstable binding of adenosine can lead to rapid release of the fragment, allowing refolding of the enzyme to its native structure. The chemical structure of AdoMet is depicted below with a blue box showing the adenosine fragment and a red box showing the methionine fragment. The AdoMet-binding site is shown in white, whereas the adenosine ligand is shown in dark blue.
Insight into selective targeting TrmD
Fragments of enzyme substrates, due to their smaller chemical space and easier accessibility to exploration, have been suggested as an attractive starting point to build fragment libraries for drug screening that may have the potential to supersede traditional high throughput screening of larger molecules. Fragment hits usually achieve only low mM to high μM affinities with the possibility for further elaboration into more potent inhibitors. The elaboration can take place in one of four directions: evolution of initial fragments by building larger and more complex molecules that form additional interactions with the target protein; chemically linking two or more fragments that bind to separate but close enough sites on the target protein to generate a larger and higher-affinity molecule; using the target protein as a template to assemble fragments with complementary functional groups so as to react together to generate a larger molecule with a higher potency; and optimization of initial fragments based on properties other than just the binding potency. Recent work has seen successful examples in all of these approaches each with the potential to develop from initial fragments into high-affinity leads (Rees et al. 2004).
In the pursuit of selective targeting of TrmD, we show here that the enzyme has the ability to fully accommodate adenosine to the adenosine pocket in a conformation indistinguishable from that in AdoMet or AdoHcy, suggesting that the energetic penalty upon binding AdoMet is due to the inability to completely capture the free energy of binding the methionine portion of the molecule. Indeed, the methionine portion of AdoMet when bound to TrmD adopts an unusual conformation that places the sulfonium ion in close proximity to the bridging 4-oxo group of the ribose, resulting in the bent conformation. This raises the possibility that a fragment-linking approach that links the adenosine fragment with derivatives of the methionine fragment in structural or topological constraints that stabilize the bent conformation of AdoMet may offer an attractive route to develop selective targeting. This possibility provides a strong impetus for further studies aimed at solving additional TrmD structures in complex with methionine. The new structural information should provide a basis for a fragment-linking approach to generate analogs of AdoMet that exhibit enhanced affinity to TrmD, or reduced affinity to Trm5, or a combination of the two.
MATERIALS AND METHODS
Synthesis of m1G37-tRNA was monitored by the incorporation of the [3H-methyl] group of AdoMet into tRNA and acid precipitation on filter pads as described (Christian et al. 2010a). Trm5 was assayed with the transcript of M. jannaschii tRNACys, while TrmD was assayed with transcript of E. coli tRNALeu. Km (AdoMet) was determined from steady-state analysis of initial rate (V0) of methyl transfer as a function of AdoMet concentration and fitting the data to the Michaelis-Menten equation. Ki (analog) was determined from analysis of the apparent Km (AdoMet) as a function of the analog concentration and fitting the data to the competitive inhibition equation. Determination of Kd (adenosine) was performed in single turnover conditions upon rapid mixing of a solution of saturating tRNA and AdoMet with a solution of enzyme and adenosine on a Kintek RQF-3 apparatus. The reaction was quenched exactly after one single turnover cycle and the amount of m1G37-tRNA was plotted against adenosine concentration. The data were fit to a quadratic equation to derive Kd (adenosine). Determination of Kd (AdoMet) for TrmD was performed similarly in single turnover conditions but in the absence of adenosine. Details of structural analysis of the adenosine-bound TrmD and Trm5 structures are described in Supplemental Table S1.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
ACKNOWLEDGMENTS
This work was supported by NIH grant GM081601 to Y.M.H. We thank Dr. Saulius Klimasauskas for AdoPropen and AdoButyn, Dr. Elmar Weinhold for AETA, Dr. Se Won Suh for the H. influenzae TrmD expression plasmid, and Drs. Charles P. Scott, Saulius Klimasauskas, and Howard Gamper for productive discussion.
Authors' contributions: G.L., S.Y., and Y.M.H. designed the research; G.L., S.G.I., K.Y., and T.I. performed the research; all authors analyzed data and wrote the paper.
Footnotes
Article published online ahead of print. Article and publication date are at http://www.rnajournal.org/cgi/doi/10.1261/rna.2706011.
REFERENCES
- Ahn HJ, Kim HW, Yoon HJ, Lee BI, Suh SW, Yang JK 2003. Crystal structure of tRNA(m1G37)methyltransferase: Insights into tRNA recognition. EMBO J 22: 2593–2603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexandrov A, Chernyakov I, Gu W, Hiley SL, Hughes TR, Grayhack EJ, Phizicky EM 2006. Rapid tRNA decay can result from lack of nonessential modifications. Mol Cell 21: 87–96 [DOI] [PubMed] [Google Scholar]
- Anderson J, Phan L, Cuesta R, Carlson BA, Pak M, Asano K, Bjork GR, Tamame M, Hinnebusch AG 1998. The essential Gcd10p–Gcd14p nuclear complex is required for 1-methyladenosine modification and maturation of initiator methionyl-tRNA. Genes Dev 12: 3650–3662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson J, Phan L, Hinnebusch AG 2000. The Gcd10p/Gcd14p complex is the essential two-subunit tRNA(1-methyladenosine) methyltransferase of Saccharomyces cerevisiae. Proc Natl Acad Sci 97: 5173–5178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baba T, Ara T, Hasegawa M, Takai Y, Okumura Y, Baba M, Datsenko KA, Tomita M, Wanner BL, Mori H 2006. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: The Keio collection. Mol Syst Biol 2: 2006.0008 doi: 10.1038/msb4100050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benítez-Páez A, Villarroya M, Douthwaite S, Gabaldon T, Armengod ME 2010. YibK is the 2′-O-methyltransferase TrmL that modifies the wobble nucleotide in Escherichia coli tRNALeu isoacceptors. RNA 16: 2131–2143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjork GR, Wikstrom PM, Bystrom AS 1989. Prevention of translational frameshifting by the modified nucleoside 1-methylguanosine. Science 244: 986–989 [DOI] [PubMed] [Google Scholar]
- Bjork GR, Jacobsson K, Nilsson K, Johansson MJ, Bystrom AS, Persson OP 2001. A primordial tRNA modification required for the evolution of life? EMBO J 20: 231–239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantoni GL 1975. Biological methylation: Selected aspects. Annu Rev Biochem 44: 435–451 [DOI] [PubMed] [Google Scholar]
- Chernyakov I, Baker MA, Grayhack EJ, Phizicky EM 2008. Chapter 11. Identification and analysis of tRNAs that are degraded in Saccharomyces cerevisiae due to lack of modifications. Methods Enzymol 449: 221–237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christian T, Hou YM 2007. Distinct determinants of tRNA recognition by the TrmD and Trm5 methyl transferases. J Mol Biol 373: 623–632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christian T, Evilia C, Williams S, Hou YM 2004. Distinct origins of tRNA(m1G37) methyltransferase. J Mol Biol 339: 707–719 [DOI] [PubMed] [Google Scholar]
- Christian T, Evilia C, Hou YM 2006. Catalysis by the second class of tRNA(m1G37) methyl transferase requires a conserved proline. Biochemistry 45: 7463–7473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christian T, Lahoud G, Liu C, Hoffmann K, Perona JJ, Hou YM 2010a. Mechanism of N-methylation by the tRNA m1G37 methyltransferase Trm5. RNA 16: 2484–2492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christian T, Lahoud G, Liu C, Hou YM 2010b. Control of catalytic cycle by a pair of analogous tRNA modification enzymes. J Mol Biol 400: 204–217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clouet-d'Orval B, Gaspin C, Mougin A 2005. Two different mechanisms for tRNA ribose methylation in Archaea: A short survey. Biochimie 87: 889–895 [DOI] [PubMed] [Google Scholar]
- Congreve M, Chessari G, Tisi D, Woodhead AJ 2008. Recent developments in fragment-based drug discovery. J Med Chem 51: 3661–3680 [DOI] [PubMed] [Google Scholar]
- Dalhoff C, Lukinavicius G, Klimasauskas S, Weinhold E 2006a. Direct transfer of extended groups from synthetic cofactors by DNA methyltransferases. Nat Chem Biol 2: 31–32 [DOI] [PubMed] [Google Scholar]
- Dalhoff C, Lukinavicius G, Klimasauskas S, Weinhold E 2006b. Synthesis of S-adenosyl-L-methionine analogs and their use for sequence-specific transalkylation of DNA by methyltransferases. Nat Protoc 1: 1879–1886 [DOI] [PubMed] [Google Scholar]
- de Ferra F, Baglioni C 1983. Increase in S-adenosylhomocysteine concentration in interferon-treated HeLa cells and inhibition of methylation of vesicular stomatitis virus mRNA. J Biol Chem 258: 2118–2121 [PubMed] [Google Scholar]
- Elkins PA, Watts JM, Zalacain M, van Thiel A, Vitazka PR, Redlak M, Andraos-Selim C, Rastinejad F, Holmes WM 2003. Insights into catalysis by a knotted TrmD tRNA methyltransferase. J Mol Biol 333: 931–949 [DOI] [PubMed] [Google Scholar]
- Galperin MY, Walker DR, Koonin EV 1998. Analogous enzymes: Independent inventions in enzyme evolution. Genome Res 8: 779–790 [DOI] [PubMed] [Google Scholar]
- Goedecke K, Pignot M, Goody RS, Scheidig AJ, Weinhold E 2001. Structure of the N6-adenine DNA methyltransferase M.TaqI in complex with DNA and a cofactor analog. Nat Struct Biol 8: 121–125 [DOI] [PubMed] [Google Scholar]
- Goto-Ito S, Ito T, Ishii R, Muto Y, Bessho Y, Yokoyama S 2008. Crystal structure of archaeal tRNA(m1G37)methyltransferase aTrm5. Proteins 72: 1274–1289 [DOI] [PubMed] [Google Scholar]
- Goto-Ito S, Ito T, Kuratani M, Bessho Y, Yokoyama S 2009. Tertiary structure checkpoint at anticodon loop modification in tRNA functional maturation. Nat Struct Mol Biol 16: 1109–1115 [DOI] [PubMed] [Google Scholar]
- Hagervall TG, Tuohy TM, Atkins JF, Bjork GR 1993. Deficiency of 1-methylguanosine in tRNA from Salmonella typhimurium induces frameshifting by quadruplet translocation. J Mol Biol 232: 756–765 [DOI] [PubMed] [Google Scholar]
- Hamilton-Miller JM 1966. A novel method for evaluating K/K and its application to the competitive inhibition of staphylococcal penicillinase by cephalosporins. Biochem J 101: 40C–42C [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hesterkamp T, Whittaker M 2008. Fragment-based activity space: Smaller is better. Curr Opin Chem Biol 12: 260–268 [DOI] [PubMed] [Google Scholar]
- Javor GT 1993. Depression of adenosylmethionine content of Escherichia coli by thioglycerol. Antimicrob Agents Chemother 24: 860–867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- King NP, Jacobitz AW, Sawaya MR, Goldschmidt L, Yeates TO 2010. Structure and folding of a designed knotted protein. Proc Natl Acad Sci 107: 20732–20737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller S, Urban A, Hecker A, Leclerc F, Branlant C, Motorin Y 2009. Deficiency of the tRNATyr:ψ 35-synthase aPus7 in Archaea of the Sulfolobales order might be rescued by the H/ACA sRNA-guided machinery. Nucleic Acids Res 37: 1308–1322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noma A, Kirino Y, Ikeuchi Y, Suzuki T 2006. Biosynthesis of wybutosine, a hyper-modified nucleoside in eukaryotic phenylalanine tRNA. EMBO J 25: 2142–2154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nureki O, Watanabe K, Fukai S, Ishii R, Endo Y, Hori H, Yokoyama S 2004. Deep knot structure for construction of active site and cofactor binding site of tRNA modification enzyme. Structure 12: 593–602 [DOI] [PubMed] [Google Scholar]
- O'Dwyer K, Watts JM, Biswas S, Ambrad J, Barber M, Brule H, Petit C, Holmes DJ, Zalacain M, Holmes WM 2004. Characterization of Streptococcus pneumoniae TrmD, a tRNA methyltransferase essential for growth. J Bacteriol 186: 2346–2354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Persson BC 1993. Modification of tRNA as a regulatory device. Mol Microbiol 8: 1011–1016 [DOI] [PubMed] [Google Scholar]
- Persson BC, Jager G, Gustafsson C 1997. The spoU gene of Escherichia coli, the fourth gene of the spoT operon, is essential for tRNA (Gm18) 2′-O-methyltransferase activity. Nucleic Acids Res 25: 4093–4097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purta E, van Vliet F, Tkaczuk KL, Dunin-Horkawicz S, Mori H, Droogmans L, Bujnicki JM 2006. The yfhQ gene of Escherichia coli encodes a tRNA:Cm32/Um32 methyltransferase. BMC Mol Biol 7: 23 doi: 10.1186/1471-2199-7-23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rees DC, Congreve M, Murray CW, Carr R 2004. Fragment-based lead discovery. Nat Rev Drug Discov 3: 660–672 [DOI] [PubMed] [Google Scholar]
- Renalier MH, Joseph N, Gaspin C, Thebault P, Mougin A 2005. The Cm56 tRNA modification in archaea is catalyzed either by a specific 2′-O-methylase, or a C/D sRNP. RNA 11: 1051–1063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roovers M, Wouters J, Bujnicki JM, Tricot C, Stalon V, Grosjean H, Droogmans L 2004. A primordial RNA modification enzyme: The case of tRNA (m1A) methyltransferase. Nucleic Acids Res 32: 465–476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saikia M, Fu Y, Pavon-Eternod M, He C, Pan T 2010. Genome-wide analysis of N1-methyl-adenosine modification in human tRNAs. RNA 16: 1317–1327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urbonavicius J, Durand JM, Bjork GR 2002. Three modifications in the D and T arms of tRNA influence translation in Escherichia coli and expression of virulence genes in Shigella flexneri. J Bacteriol 184: 5348–5357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varshney U, Ramesh V, Madabushi A, Gaur R, Subramanya HS, RajBhandary UL 2004. Mycobacterium tuberculosis Rv2118c codes for a single-component homotetrameric m1A58 tRNA methyltransferase. Nucleic Acids Res 32: 1018–1027 [DOI] [PMC free article] [PubMed] [Google Scholar]