

Abstract
Phospholipase C-zeta (PLCζ) is a strong candidate for the mammalian sperm-derived factor that triggers the Ca2+ oscillations required for egg activation at fertilization. PLCζ lacks a PH domain, which targets PLCδ1 to the phosphatidylinositol 4,5-bisphosphate (PtdIns(4,5)P2) substrate in the plasma membrane. Previous studies failed to detect PLCζ in the plasma membrane, hence the means of PLCζ binding to PtdIns(4,5)P2 is unclear. We find that the PLCζ XY linker, but not the C2 domain, exhibits robust binding to PtdIns(4,5)P2 or to liposomes containing near-physiological levels of PtdIns(4,5)P2. The role of positively charged residues within the XY linker was addressed by sequentially substituting alanines for three lysine residues, K374, K375 and K377. Microinjection of these mutants into mouse eggs enabled their Ca2+ oscillation-inducing activities to be compared with wild-type PLCζ. The XY-linker mutant proteins were purified and the in vitro PtdIns(4,5)P2 hydrolysis and binding properties were monitored. Successive reduction of net positive charge within the PLCζ XY linker significantly affects both in vivo Ca2+-oscillation-inducing activity and in vitro PtdIns(4,5)P2 interaction of mouse PLCζ. Our data suggest that positively charged residues within the XY linker play an important role in the PLCζ interaction with PtdIns(4,5)P2, a crucial step in generating the Ca2+ activation signal that is essential for fertilization in mammals.
Key words: Phospholipase C, Phosphoinositide signalling, Calcium oscillations, Egg activation, Fertilization
Introduction
In mammalian oocytes, the fertilizing sperm evokes a striking series of intracellular Ca2+ oscillations that are essential for the initiation of egg activation and embryonic development (Swann, 1994; Stricker, 1999; Malcuit et al., 2006). Although the detailed mechanism remains unclear, accumulating evidence suggests that sperm-specific phospholipase C-zeta (PLCζ) is delivered from the fertilizing sperm into the ooplasm, triggering cytoplasmic Ca2+ oscillations via the inositol 1,4,5-trisphosphate [Ins(1,4,5)P3] pathway (Saunders et al., 2002; Cox et al., 2002; Kouchi et al., 2004; Knott et al., 2005). Further evidence for the importance of PLCζ in mammalian fertilization has been provided by two recent clinical reports that have linked reduced expression levels and abnormal forms of PLCζ with male infertility (Yoon et al., 2008; Heytens et al., 2009). PLCζ is the smallest known mammalian PLC isozyme, possessing a similar domain organization to PLCδ1 with the notable exception that it lacks an N-terminal pleckstrin homology (PH) domain. Thus, PLCζ consists of four EF hands, the catalytic X and Y domains and a C2 domain, all of which are common to the other PLC isoforms (β, γ, δ, ε and η) (Saunders et al., 2002; Swann et al., 2006; Suh et al., 2008).
PLCs are cytosolic enzymes that require membrane association to access their phospholipid substrate. PLCδ1 binds strongly to membranes when its phosphatidylinositol 4,5-bisphosphate [PtdIns(4,5)P2] substrate is present (Pawelczyk and Lowenstein, 1993; Lomasney et al., 1996). PLCβ2 binds strongly and non-specifically to lipid membranes (Singh and Murray, 2003), whereas PLCγ targets membranes by its specific, high-affinity binding to PtdIns(3,4,5)P3 (Bae et al., 1998; Falasca et al., 1998). Membrane binding of these isoforms appears to be mediated by the PH domain. PH domains are well-defined structural modules of ~120 amino acid residues identified in >100 different proteins (Rebecchi and Pentyala, 2000). Notably, PLCζ lacks a PH domain, leaving unresolved the exact mechanism of how PLCζ targets its membrane substrate. Studies using ‘Venus’ GFP–PLCζ indicate that it does not specifically localize to the plasma membrane, but is apparently distributed throughout the cytoplasm (Yoda et al., 2004). This implies that only a small fraction of PLCζ can bind to plasma-membrane-located PtdIns(4,5)P2 and/or that PLCζ might interact with cytoplasmic vesicles. In either case, it is anticipated that a specific domain(s) within PLCζ binds to PtdIns(4,5)P2. Given the presumed roles of the catalytic X and Y and the N-terminal EF hand domains, there are two putative candidates that might be involved in targeting of PLCζ to PtdIns(4,5)P2: the C-terminal C2 domain and the intervening region separating the catalytic X and Y domains (the ‘XY linker’).
The C2 domain, comprising ~120 amino acid residues, has been identified in numerous proteins, including all isoforms of protein kinase C, phospholipase A, synaptotagmin and PLC. First identified in protein kinase C, the C2 domain was functionally implicated in Ca2+-dependent phospholipid interactions (Nalefski and Falke, 1996). The C2 domain has since been characterized as a membrane-associating and intermolecular interaction domain in a variety of proteins (Medkova and Cho, 1999). Most C2 domains bind to Ca2+, a crucial determinant for the associated enzyme activity (Zheng et al., 2000). The PLCδ1 C2 domain interacts with phosphatidylserine (PtdSer) to form a C2-Ca2+-PtdSer ternary complex, which enhances enzyme activity (Lomasney et al., 1999). The PLCζ C2 domain appears to have an essential role in cellular function because deletion of this domain leads to inability of the truncated PLCζ to cause Ca2+ oscillations in intact eggs, although enzyme activity is retained (Nomikos et al., 2005; Kouchi et al., 2005). There is currently no evidence for PLCζ C2 domain binding to membrane phospholipids through Ca2+-dependent or -independent mechanisms, although screening phosphoinositides for interaction with the C2 domain of PLCζ revealed that it can bind to both phosphatidylinositol 3-phosphate (PtdIns3P) and phosphatidylinositol 5-phosphate (PtdIns5P) (Kouchi et al., 2005).
An alternative PLCζ region that could be involved in association with biological membranes is the XY-linker segment, separating the X and Y catalytic domains. In contrast to PLCδ1, PLCζ contains a more extended XY linker that is notably rich in positively charged residues (Saunders et al., 2002; Cox et al., 2002). The study reported here investigated the potential importance of the C2 domain and XY linker in PLCζ association with PtdIns(4,5)P2. A protein–lipid overlay and a liposome-binding assay were employed to examine the interaction of the PLCζ XY linker and C2 domain with PtdIns(4,5)P2. In addition, a series of full-length PLCζ mutants was prepared that sequentially neutralized several positively charged lysines that are clustered within the XY linker. The Ca2+ oscillation-inducing properties of these lysine mutants and wild-type PLCζ were compared by microinjecting them into unfertilized mouse eggs and analysing their enzymatic activity using an in vitro PtdIns(4,5)P2 hydrolysis assay. Furthermore, the binding properties of wild-type and mutant PLCζ to PtdIns(4,5)P2 was examined using a liposome binding assay, in parallel with a number of control PLC constructs. Our studies suggest that the PLCζ XY linker, but not the C2 domain, possesses significant affinity for PtdIns(4,5)P2 and that sequential reduction of the XY-linker net positive charge significantly affects both in vivo Ca2+-oscillation-inducing activity and also the in vitro interaction of PLCζ with PtdIns(4,5)P2. Thus, we propose that the XY-linker region plays a major role in the binding of PLCζ to PtdIns(4,5)P2-enriched membranes.
Results
Binding of the XY linker and C2 domain of PLCζ to PtdIns(4,5)P2
To examine the ability of the PLCζ XY linker and C2 domain to bind PtdIns(4,5)P2, we employed a protein–lipid overlay and a liposome-binding assay to assess distinct GST fusion proteins of both the XY linker or C2 domain of PLCζ, with the PH domain of PLCδ1 as a positive control. Fig. 1A schematically illustrates the three GST fusion proteins: the PLCδ1 PH domain, the PLCζ C2 domain and XY linker, and their corresponding sequence coordinates. These fusion proteins were expressed in Escherichia coli Rosetta (DE3) cells and purified by glutathione affinity chromatography. Optimal recombinant protein expression conditions required maintaining cultures at 37°C until absorbance at 600 nm (A600) reached 0.5, followed by induction of expression with 0.1 mM isopropyl β-D-thiogalactopyranoside (IPTG) for 18 hours at 16°C. Fig. 1B shows the glutathione affinity-purified GST fusion proteins analyzed by SDS-PAGE. The predicted molecular mass, including the GST protein (26 kDa), for GST–XYlinkζ, GST–C2ζ and GST–PHδ1, was 34, 39 and 40 kDa, respectively. For each expression plasmid the corresponding protein with expected molecular mass was observed as the major band. In addition, a minor, lower band of 26 kDa was also present, consistent with the mass of GST alone, suggesting that some protein degradation had occurring during the protein purification process.
Fig. 1.

Expression of the XY linker and C2 domains of PLCζ and the PH domain of PLCδ1. (A) Schematic representation of the GST fusion protein constructs with the PLCδ1 PH domain, PLCζ C2 domain and PLCζ XY linker, with numbers denoting their amino acid coordinates. (B) SDS-PAGE of glutathione affinity-purified recombinant PLCζ and PLCδ1 domains (2 μg) analyzed by 12% SDS-PAGE and Coomassie Brilliant Blue staining.
The protein–lipid overlay assay, using membrane-spotted arrays of inositol phospholipids, showed that GST–XYlinkζ was able to bind to all the phosphoinositides though not to PtdIns (Fig. 2A). The strongest interaction occurred with the polyvalent phosphoinositides, PtdIns(4,5)P2 and PtdIns(3,4,5)P3 even at the lower concentrations (e.g. at 3.13 pmol). By contrast, GST–C2ζ bound very weakly only to PtdIns3P and PtdIns5P at the highest concentrations (50–100 pmol), whereas GST–PHδ1 exhibited strong binding specifically to the three monophosphates, and preferential binding to the bisphosphate, PtdIns(4,5)P2, as anticipated (Lomasney et al., 1996). The inositol phospholipid array results suggest that the PLCζ XY-linker region has the ability to interact with a wide range of phosphoinositides spanning mono, bis and trisphosphates, whereas the PLCζ C2 domain displays negligible binding to inositol lipids.
Fig. 2.

In vitro binding of PLCζ XY linker and C2 domain to PtdIns(4,5)P2. (A) PLC domain protein–lipid overlay assays. Recombinant protein binding to spotted phosphoinositides on the PIP Arrays was detected using a polyclonal anti-GST antibody. PI, phosphoinositide (e.g. PI(4,5)P2 is PtdIns(4,5)P2.(B) Liposome ‘pull-down’ assay of PLC domains. Unilamellar liposomes comprising PtdCho:CHOL:PtdEtn (4:2:1) with or without 1% PtdIns(4,5)P2 were incubated with recombinant protein. Following centrifugation, both the supernatant (s) and liposome pellet (p) were subjected to SDS-PAGE and Coomassie Brilliant Blue staining. Previous control experiments showed that GST alone does not bind to phosphoinositides (Nomikos et al., 2007).
The ability of GST–XYlinkζ and GST–C2ζ to bind PtdIns(4,5)P2 was further monitored using unilamellar liposomes composed of phosphatidylcholine:cholesterol:phosphatidyethanolamine (PtdCho:CHOL:PtdEtn; 4:2:1) with incorporation of either 0 or 1% PtdIns(4,5)P2 (Fig. 2B). For this binding assay, GST–PHδ1 provided the positive control whereas the GST moiety was a negative control. Thus, GST–PHδ1 bound robustly to liposomes containing 1% PtdIns(4,5)P2 (Fig. 2B, top panel) and remained in the supernatant in the absence of PtdIns(4,5)P2, whereas the GST negative control did not exhibit any specific liposome binding with or without PtdIns(4,5)P2 (Fig. 2B, bottom panel). The majority (~90%) of GST–XYlinkζ, was detected in the supernatant of liposomes that did not contain PtdIns(4,5)P2. By contrast, the presence of a near-physiological concentration of 1% PtdIns(4,5)P2 in the liposomes resulted in GST–XYlinkζ binding strongly, with all of this protein being detected in the liposome pellet (Fig. 2B, third panel). However, the binding of GST–C2ζ to liposomes either with or without 1% PtdIns(4,5)P2 was not detected (Fig. 2B, second panel). Thus, the results obtained using two different phosphoinositide binding interaction assays has provided congruent evidence that only the PLCζ XY linker and not the C2 domain has the ability to bind to PtdIns(4,5)P2.
Analysis of XY-linker mutations on PLCζ-mediated Ca2+ oscillations in mouse eggs
To investigate the potential importance of the cluster of basic residues within the PLCζ XY linker (Fig. 3A), we performed site-directed mutagenesis to produce a panel of three cumulative mutations within this positively charged region of mouse PLCζ. Thus, the three lysines, K374, K375 and K377, were sequentially replaced with the neutral amino acid, alanine, to create single (PLCζK374A), double (PLCζK374,5AA) and triple (PLCζK374,5,7AAA) substituted PLCζ mutants. In order to test the ability of PLCζK374A, PLCζK374,5AA and PLCζK374,5,7AAA to trigger Ca2+ oscillations and to verify their expression upon microinjection of the corresponding cRNA into eggs, we generated luciferase fusion constructs of each of these mutants. This approach enables the accurate quantification of relative protein expression by luminescence detection for each of the luciferase-tagged PLCζ proteins (Nomikos et al., 2005). A catalytically inactive PLCζ mutant (PLCζD210R) served as a control in these experiments in order to establish the degree of inhibition effected by the K-to-A mutations. We have previously reported that point mutation of Asp210 (D210R) in the XY catalytic domain of PLCζ results in loss of PLCζ Ca2+-oscillation inducing activity in the oocytes (Saunders et al., 2002; Nomikos et al., 2011). Fig. 3B and Table 1 summarize the results of the wild-type and mutant PLCζ–luciferase cRNA microinjection experiments. Prominent Ca2+ oscillations (19 spikes in the first 2 hours) were observed in the wild-type PLCζ cRNA-injected eggs, with the first Ca2+ spike occurring ~30 minutes after microinjection at a luminescence corresponding to PLCζ protein expression of ~35 fg/egg. Microinjection of cRNA encoding the PLCζK374A single mutant also caused Ca2+ oscillations in mouse eggs, exhibiting a similar potency to wild-type PLCζ (17 spikes in 2 hours) with the first Ca2+ spike detected at a protein expression level of ~33 fg/egg. By contrast, egg microinjection with cRNA of either PLCζK374,5AA or PLCζK374,5,7AAA resulted in a significant reduction in the frequency of Ca2+ oscillations compared with wild-type PLCζ, causing 8 and 2.6 spikes in 2 hours, respectively. Moreover, there was also a significant increase in time required for initiation of Ca2+ oscillations for the double and triple PLCζ mutants, with the first Ca2+ spike appearing at ~65 minutes (Fig. 3B; Table 1). Furthermore, the luminescence level required to produce the first Ca2+ spike was equivalent to protein expression of ~55 and ~68 fg/egg for the double and triple mutant, respectively. Finally, microinjection of cRNA encoding the PLCζD210R mutant failed to cause any Ca2+ oscillations even though it was expressed at much higher levels than wild-type PLCζ and PLCζ K-to-A mutants (Table 1). These data indicate that the substitution of two or more alanines for lysines within the positively charged cluster of the PLCζ XY linker dramatically alters their Ca2+-oscillation-inducing activity in mouse eggs by reducing the Ca2+ spike frequency and increasing both the time and amount of mutant PLCζ expression required to initiate the first spike.
Fig. 3.

Effect of PLCζ XY-linker mutations on Ca2+-oscillation-inducing activity in mouse eggs. (A) Schematic representation of mouse PLCζ domain structure (comprising the N-terminal EF hands, X and Y catalytic domains and C-terminal C2 domain) identifying the successive K-to-A mutations between residues 374 and 379 in the XY-linker region, as well as the position of the D210R mutation in the X catalytic domain (arrows). (B) Fluorescence and luminescence recordings reporting the Ca2+ changes (black traces; Ca2+) and luciferase expression (red traces; Lum, in counts per second; cps) in unfertilized mouse eggs following microinjection of cRNA encoding luciferase-tagged, wild-type PLCζ or the indicated single, double, triple K-to-A and D210R mutants of PLCζ (left panels). The average luminescence level (cps) achieved in mouse eggs (n is number of eggs) is indicated for each microinjected cRNA, e.g. PLCζwt Lum=7.33 (top trace). Panels on the right are the integrated luminescence images of individual mouse eggs following cRNA microinjection of either wild-type or mutant PLCζ (Table 1). Two eggs at the bottom left of the PLCζ wt panel were poorly fluorescing and not included in the analysis of Ca2+ oscillations. All eggs were microinjected with 1.5 g/l cRNA.
Table 1.
Summary of the properties of Ca2+ oscillations observed in mouse eggs following cRNA microinjection of the luciferase-tagged wild-type PLCζ and various PLCζ XY-linker K-to-A mutants (see Fig. 3B)

Enzymatic properties of PLCζ XY-linker mutations
The PLCζK374A, PLCζK374,5AA and PLCζK374,5,7AAA mutants were expressed as GST fusion proteins and purified by glutathione affinity chromatography. The optimal protein production for the full-length PLCζ constructs, PLCζ C2 and XY linker, required induction with 0.1 mM IPTG for 18 hours at 16°C. Fig. 4A shows the four glutathione affinity-purified GST–PLCζ fusion proteins analyzed by SDS-PAGE (left panel) and immunoblotting with anti-GST antibody (right panel). The predicted molecular mass for the GST–PLCζ and mutants, including the GST tag is 100 kDa. A protein of ~100 kDa was observed as the major band in both the SDS gels and the immunoblots (Fig. 4A). A protein migrating at 26 kDa in both the gels and immunoblots is consistent with cleaved GST, which, along with several fainter intermediate molecular mass proteins detected by the GST antibody, is probably the result of protease degradation occurring during protein purification.
Fig. 4.
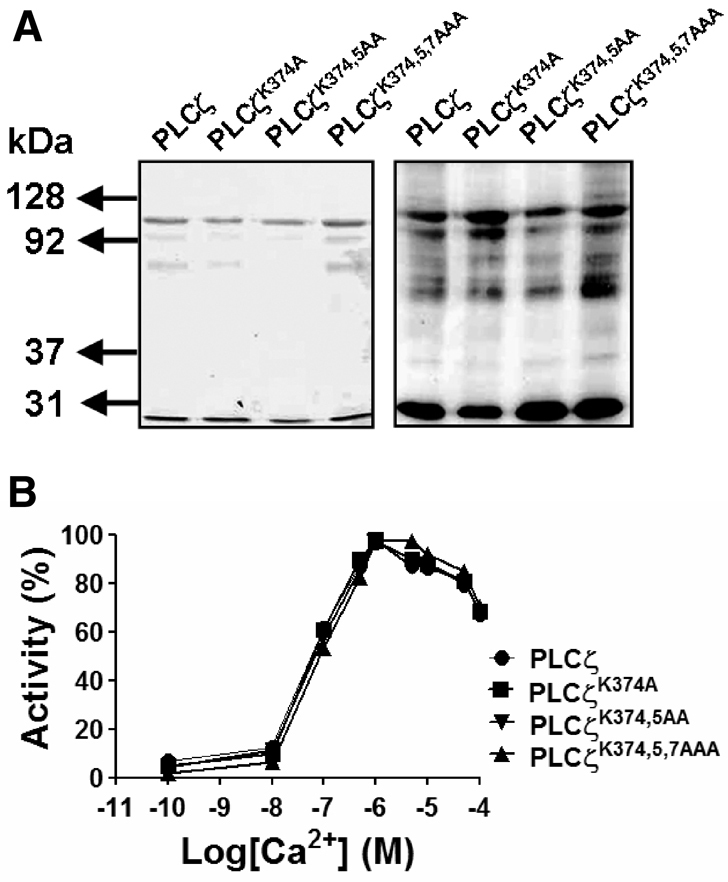
Ca2+-dependent enzyme activity of PLCζ XY-linker mutations. (A) Glutathione affinity-purified, wild-type and each of the K-to-A mutant GST–PLCζ fusion proteins (1 μg) were analyzed by 8% SDS-PAGE followed by either Coomassie Brilliant Blue staining (left panel) or immunoblot analysis using anti-GST antibody (right panel). Lanes 1–4 (left to right) show wild-type PLCζ and single, double and triple K-to-A PLCζ XY-linker mutants, respectively. (B) PtdIns(4,5)P2 hydrolysis activity of the purified wild-type and XY-linker mutant PLCζ proteins were determined in vitro with [3H]PtdIns(4,5)P2 at different Ca2+ concentrations. For these enzyme assays n=2, using two different batches of recombinant proteins and each experiment was performed in duplicate; values are means. In control experiments with GST, there was no specific PtdIns(4,5)P2 hydrolysis activity observed (data not shown).
To examine the impact of the XY-linker mutations on Ca2+ sensitivity of PLCζ enzyme activity, we assessed the ability of these GST–PLCζ fusion proteins to hydrolyze [3H]PtdIns(4,5)P2 at different Ca2+ concentrations, ranging from 0.1 nM to 0.1 mM. These experiments indicated that there was no significant difference in the Ca2+ sensitivity of PtdIns(4,5)P2 hydrolysis for the wild-type and the three XY-linker mutants (Fig. 4B), with a very similar EC50 value (81–90 nM) for all four recombinant PLCζ proteins (Table 2). To compare the enzyme kinetics of the wild-type and mutant PLCζs, the Michaelis–Menten constant, Km, was calculated from a Lineweaver–Burk reciprocal plot of the PtdIns(4,5)P2 hydrolysis activity of these proteins (Fig. 5; Table 2). The Km values obtained for wild-type PLCζ (80 μM) and PLCζK374,5A (88 μM) were very similar. By contrast, the Km value for PLCζK374,5AA and PLCζK374,5AAA was approximately 9-fold (752 μM) and 61-fold (4919 μM) higher than that of wild-type PLCζ, respectively. These results suggest that sequential neutralization of these three positively-charged residues within the XY-linker region reduces the in vitro affinity of PLCζ for PtdIns(4,5)P2 without affecting the Ca2+ sensitivity of this enzyme.
Table 2.
Summary of the EC50 of Ca2+ -dependent enzyme activity and the Km determined for the GST-tagged wild-type PLCζ and various PLCζ XY-linker K-to-A mutants (see Fig. 4B and Fig. 5)

Fig. 5.
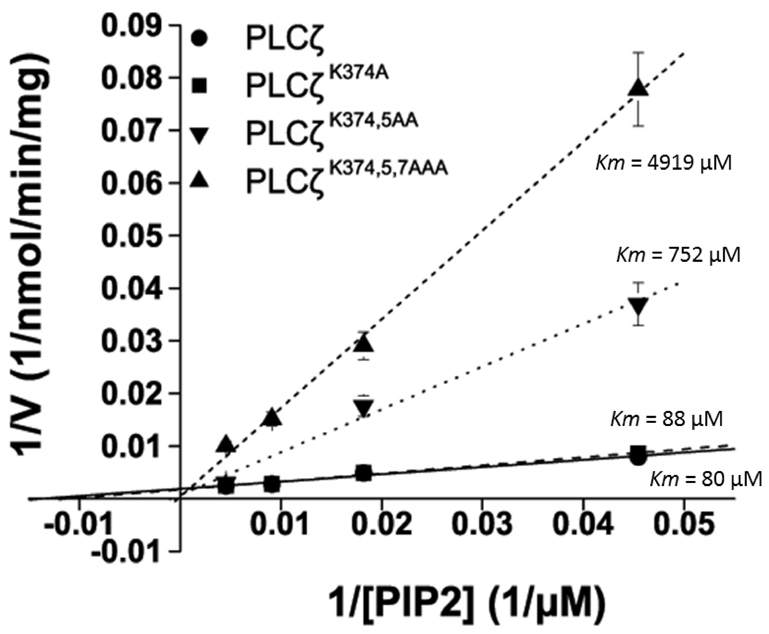
Determination of Km for the PLCζ XY-linker mutations, PLCζK374A, PLCζK374,5AA and PLCζK374,5,7AAA (see Table 2). Lineweaver–Burk reciprocal plots were prepared from hydrolysis activity assays for purified wild-type and mutant PLCζ GST fusion proteins in order to determine the Km for PtdIns(4,5)P2 (PIP2). Km values of 80 μM, 88 μM, 752 μM and 4919 μM were obtained for wild-type PLCζ, PLCζK374A, PLCζK374,5AA and PLCζK374,5,7AAA, respectively. For these enzyme assays n=2 using two different batches of recombinant proteins, and each experiment was performed in duplicate; values are means ± s.e.m.
Binding of wild-type PLCζ and the XY-linker K-to-A mutants to PtdIns(4,5)P2
To examine the PtdIns(4,5)P2 binding properties of wild-type PLCζ and the three XY-linker K-to-A mutants, we employed the liposome binding assay as described above (see Fig. 2B). In order to diminish any non-specific binding to highly charged lipids by this series of full-length PLC proteins, the liposome binding assays were performed in the presence of a near-physiological concentration of MgCl2 (0.5 mM). In addition to the wild-type PLCζ and PLCζ K-to-A mutants, the PtdIns(4,5)P2 binding of three other control PLC constructs (PLCδ1, PLCζΔC2 and PLCζD210R) was also assessed. The PLCδ1 served as positive control for these PtdIns(4,5)P2 binding assays, whereas PLCζΔC2, a deletion construct lacking the PLCζ C2 domain (Nomikos et al., 2005) was used to examine whether the C2 domain has any role in binding to PtdIns(4,5)P2. We also included PLCζD210R to investigate whether PtdIns(4,5)P2 binding requires the catalytically competent enzyme. Fig. 6 shows that PLCζ, PLCζD210R, PLCζΔC2, PLCζK374A and PLCζK374,5AA bind robustly only to the liposomes containing 1% PtdIns(4,5)P2, similar to that observed for the positive control, PLCδ1. There was no binding of proteins to liposomes without PtdIns(4,5)P2. By contrast, the majority (~60%) of the triple K-to-A mutant, PLCζK374,5,7AAA, was detected in the supernatant of liposomes containing 1% PtdIns(4,5)P2 (Fig. 6). These data indicate that although PLCζ lacks a PH domain, it remains capable of binding to PtdIns(4,5)P2. The binding observed with PLCζΔC2 and PLCζD210 reveals that PLCζ interaction with PtdIns(4,5)P2 in liposomes does not require the C2 domain, and there is no requirement for catalytic enzyme function, respectively. However, the replacement of three positively-charged residues within the PLCζ XY-linker region does dramatically diminish the interaction with PtdIns(4,5)P2.
Fig. 6.
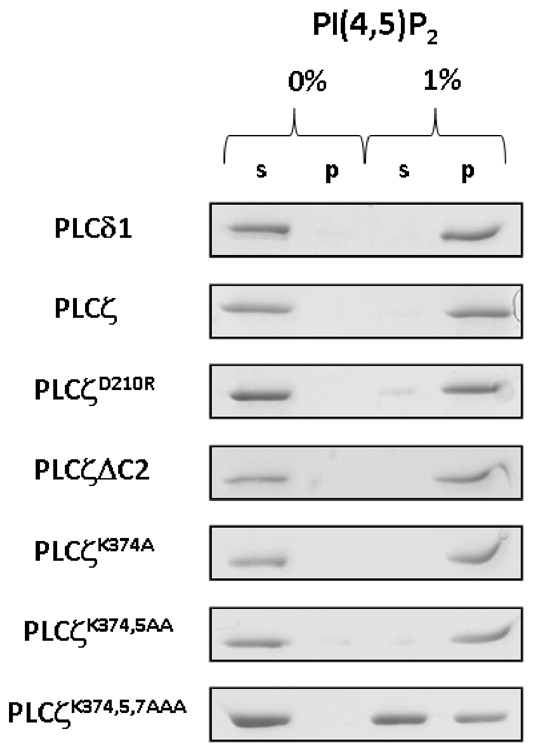
In vitro binding of wild-type PLCζ and PLCζ mutants to PtdIns(4,5)P2. Liposome ‘pull-down’ assay of wild-type and mutant PLCζ constructs. Unilamellar liposomes comprising PtdCho:CHOL:PtdEtn (4:2:1) with or without 1% PtdIns(4,5)P2 was incubated in the presence of 0.5 mM Mg2+ with the various purified recombinant PLCζ protein as described in Materials and Methods. Following centrifugation, both the supernatant (s) and liposome pellet (p) were subjected to SDS-PAGE and Coomassie Brilliant Blue staining.
Discussion
During mammalian fertilization, egg activation leading to embryo development is believed to be triggered by entry of the sperm-specific PLCζ (Saunders et al., 2002; Cox et al., 2002; Kouchi et al., 2004). PLCζ stimulates repetitive Ca2+ oscillations in the egg upon fusion of the sperm and egg membranes by initiating Ca2+ release from intracellular stores via the activation of the Ins(1,4,5)P3 receptor signalling pathway. Recent clinical reports providing further evidence for the importance of PLCζ in mammalian fertilization have directly linked reduced expression levels and abnormal forms of PLCζ with documented cases of human male infertility (Yoon et al., 2008; Heytens et al., 2009). Despite the crucial significance of PLCζ in intracellular Ca2+ signalling at fertilization, the precise mechanism by which PLCζ locates and targets the substrate in eggs is still unclear. The potential role of positively charged PLCζ residues in targeting this enzyme to biological membranes via electrostatic interactions with the negatively charged PtdIns(4,5)P2 has been previously suggested (Nomikos et al., 2007). Fluorescence resonance energy transfer (FRET) experiments with a synthetic peptide corresponding to a portion of the XY linker have suggested that basic amino acids could help anchor PLCζ to the membrane and enhance local PtdIns(4,5)P2 concentration adjacent to the catalytic domain.
In this report, we have examined the ability of the mouse PLCζ XY-linker and C2 domain to interact with PtdIns(4,5)P2 by preparing recombinant protein constructs to enable expression and purification of these two structurally distinct PLCζ regions (Fig. 1). The protein-lipid overlay and liposome-binding assay results suggest that only the XY-linker, and not the C2 domain, binds strongly to PtdIns(4,5)P2 spotted onto nitrocellulose membrane (Fig. 2A) or to liposomes containing a near-physiological (1%) concentration of PtdIns(4,5)P2 (Fig. 2B). The lipid array analysis of interactions between the PLCζ C2 domain and other phosphoinositides detected binding only to the monophosphates, PtdIns3P and PtdIns5P (Fig. 2A). This result is consistent with previous observations using a similar protein-lipid overlay assay that showed the PLCζ C2 domain binds to PtdIns3P and with lower affinity to PtdIns5P (Kouchi et al., 2005). There is presently no evidence that C2 domain interaction with PtdIns3P or PtdIns5P plays a significant role in the subcellular targeting of PLCζ to biological membranes. However, a C2–PtdIns3P interaction might be involved in the regulation of PLCζ enzymatic activity, because it has been reported that PtdIns3P reduces the PtdIns(4,5)P2 hydrolytic activity of PLCζ (Kouchi et al., 2005).
In contrast to the C2 domain, the PLCζ XY-linker showed strong binding to all phosphoinositides, with highest preference for PtdIns(4,5)P2 and PtdIns(3,4,5)P3 (Fig. 2A). In agreement with the protein-lipid overlay approach, strong binding of the PLCζ XY-linker but not the C2 domain was also observed to liposomes containing PtdIns(4,5)P2 (Fig. 2B). Given that PLCζ lacks a PH domain, our results suggest that electrostatic interactions of the positively charged PLCζ XY-linker might provide the major contribution to the physiological interaction of PLCζ with PtdIns(4,5)P2. However, without further detailed kinetic analysis and high-resolution protein structure the role of additional PLCζ domains cannot be ruled out.
The importance of obtaining in vivo evidence led us to employ an additional approach to demonstrate the potential involvement of the PLCζ XY-linker in Ca2+-oscillation-inducing activity. Using site-directed mutagenesis to generate three cumulative mutations within the positively charged region of the mouse PLCζ XY linker (Fig. 3A), a cluster of three lysine residues (K374, K375 and K377) were sequentially replaced with the neutral amino acid alanine, to produce single (K374A), double (K374,375AA) and triple (K374,375,377AAA) mutant constructs of PLCζ. The lysine to alanine change was chosen as it is likely to bring about relatively small changes compared with charge-reversed amino acid substitution effects on PLCζ structure. Microinjection of the cRNA for luciferase-tagged PLCζ, PLCζK374A, PLCζK374,5AA or PLCζK374,5,7AAA into mouse eggs uniformly resulted in robust recombinant protein expression followed by the initiation of a series of Ca2+ oscillations that persisted for several hours (Fig. 3B). There was a significant reduction in the frequency of Ca2+ oscillations observed with the double and triple K-to-A mutants, PLCζK374,5AA (40% vs wild type) and PLCζK374,5,7AAA (14% vs wild type; Fig. 3B; Table 1). By contrast, oscillation frequency of the single PLCζK374A mutant was only slightly affected (90% vs wild type), suggesting that significantly altered in vivo PLCζ function was mediated by the loss of two or more positive charges in the XY-linker. Notably, for the double and triple K-to-A mutants there was an extended delay (~65 minutes) before the first Ca2+ spike appeared, compared with wild-type PLCζ (~30 minutes). In addition, a higher protein concentration was required of these two mutants (266% and 409% vs wild type) to initiate the first Ca2+ spike (Fig. 3B; Table 1). The requirement for substantially increased expression level of the double and triple K-to-A mutant proteins to generate the first Ca2+ spike, together with the lower Ca2+ oscillation frequency they produced, suggests that reduction of the net positive charge by two or more in the XY linker diminishes the affinity of PLCζ for its negatively charged, membrane-bound substrate, PtdIns(4,5)P2.
To determine the effect of the single, double and triple K-to-A mutations on the enzymatic properties of PLCζ, we purified each of them as GST fusion proteins (Fig. 4A) to enable in vitro analysis of their ability to hydrolyze PtdIns(4,5)P2. Previous characterization of PLCζ activity has shown a steep Ca2+ dependence, with a nanomolar EC50 (82 nM) for the hydrolysis of PtdIns(4,5)P2, in contrast to a micromolar EC50 (6 μM) for PLCδ1 (Nomikos et al., 2005). We found no evidence of a role for these XY-linker lysines in Ca2+ regulation of PLCζ activity because all three K-to-A mutants exhibited an EC50 similar to that of the wild-type enzyme (81–90 nM vs 88 nM; Table 2). By contrast, the Km values for PLCζK374,5AA and PLCζK374,5AAA mutants were approximately 9-fold and 61-fold higher than wild-type PLCζ (Fig. 5; Table 2). This suggests that the sequential cumulative neutralization of these basic residues within the XY-linker region substantially reduces the in vitro affinity of PLCζ for PtdIns(4,5)P2 without affecting the Ca2+ sensitivity of this enzyme (Fig. 4B; Table 2).
Further evidence of a specific role for the polybasic region within the PLCζ XY-linker in interacting with PtdIns(4,5)P2-enriched biological membranes was obtained from liposome binding assays performed with expressed wild-type and mutant PLCζs in the presence of near-physiological Mg2+ (Fig. 6). We observed strong PtdIns(4,5)P2 binding of both the wild-type PLCζ and the catalytically inactive mutant PLCζD210R, indicating that substrate interaction and hydrolytic activity are distinct, separable steps in the mechanism of enzyme action. Interestingly, both single and double K-to-A mutants also displayed robust PtdIns(4,5)P2 binding, whereas binding of PLCζK374,5,7AAA was dramatically reduced, by over 60%, suggesting that the cumulative reduction of XY-linker positive residues in the PLCζ triple mutant had crossed below a crucial charge-density threshold resulting in diminution of PtdIns(4,5)P2 binding (Fig. 6). Furthermore, the PLCζ deletion construct lacking the C2 domain showed binding to liposomes containing PtdIns(4,5)P2 equivalent to that of wild-type PLCζ, suggesting that the C2 domain does not play a direct role in the interaction with PtdIns(4,5)P2.
Notably, ClustalW sequence alignment analysis of PLCζ from various species shows that the XY-linker region, which connects the two highly conserved halves of the catalytic barrel (the X and Y domains) is the most non-conserved region of PLCζ (Saunders et al., 2007). However, although the primary structure is poorly conserved, the XY-linker region of all mammalian PLCζ species thus far sequenced retains a net positive charge. The significance of this XY-linker structure diversity is unclear, but it might explain the different rates of PtdIns(4,5)P2 hydrolysis and relative potency in inducing Ca2+ oscillations between PLCζ isoforms of different species. For example, the deduced isoelectric points of human (9.1), monkey (7.2) and mouse (5.4), appears to correlate with the ability of the PLCζs of these different species to trigger Ca2+ oscillations in mouse eggs; increased potency is observed with higher isoelectric point (Saunders et al., 2002; Cox et al., 2002; Saunders et al., 2007).
Significantly, none of the other mammalian PLC isozymes possesses a highly positively charged XY-linker region. In contrast to PLCζ, the XY-linker of PLCβ, δ and ε is highly negatively charged, mostly disordered and it has been suggested that these XY linkers might mediate auto-inhibition of PLC activity (Hicks et al., 2008). It has been proposed that the negatively charged XY-linkers of these PLCs prevent PtdIns(4,5)P2 gaining access to the active site, by a combination of steric exclusion and electrostatic repulsion of negatively charged membranes (Hicks et al., 2008). The PLCγ XY-linker possesses additional regulatory domains: a PH domain, two SH2 domains and an SH3 domain, and this enzyme is activated by tyrosine phosphorylation within the XY-linker region (Rodriguez et al., 2001; Ozdener et al., 2002; Sekiya et al., 2004). A recent study suggested that the general mechanism of PLC auto-inhibition mediated by the XY-linker region also applies to PLCγ isozymes and the crucial determinant for the auto-inhibition is the C-terminal SH2 domain (Gresset et al., 2010). Interestingly, the XY-linkers of PLCη isozymes are not negatively charged overall but contain clusters of acidic residues near their ends. However, it is unclear whether there is a role of this region in the regulation of PLCη activity (Fukami et al., 2010).
PLCζ has the simplest domain organization and could be considered a prototypic mammalian PLC (Saunders et al., 2002; Swann et al., 2006; Saunders et al., 2007). This is consistent with the pivotal physiological role it plays in reproduction by triggering the primary Ca2+ signalling event at mammalian fertilization to initiate egg activation and embryo development. However, our observations suggest that the PLCζ regulatory mechanism and the role of its XY-linker region might be distinct and not follow the pattern observed for other mammalian somatic cell PLCs. Further understanding of the potentially unique molecular mechanism of PLCζ action will be aided by additional kinetic analysis and structural definition of the mammalian PLCζ. High resolution three-dimensional structure analysis by X-ray crystallography could help to reveal the crucial ion, lipid and protein binding sites within PLCζ. This would provide a useful base for further kinetic investigation of the putative interactions of PLCζ with the ions, lipids or proteins that might play an important role in its function and mode of regulation. Unravelling the complex subcellular processes mediated by PLCζ should provide a molecular explanation for the fundamental Ca2+ signalling event that initiates early embryo development.
Materials and Methods
Cloning of XY-linkerζ, C2ζ and PHδ1 constructs
The XY-linker [amino acids (aa) 308–385] and the C2 domain (aa521–647) of mouse PLCζ were amplified by PCR from the original cDNA clone (GenBank™ accession number AF435950) (Saunders et al., 2002), using Phusion polymerase (Finnzymes) and the appropriate primers to incorporate a 5′-EcoRI site and a 3′-SalI site. PCR products were cloned into the pGEX-6P1 vector (GE Healthcare). The following primers were used for each construct: for XY-linker ζ, 5′-GTCGAATTCAAAGTGGGAACCTTATCTGA-3′ (forward) and 5′-GTAGTCGACTCAGGCCATGGCTATTTTCAT-3′ (reverse), and for C2ζ, 5′-GCTGAATTCACCCTCACAATCCGAATCAT-3′ (forward) and 5′-TAACGTCGACTCACTCTCTGAAGTACCAAAC-3′ (reverse). PHδ1 (aa1–136) was amplified by PCR from the rat PLCδ1 clone (GenBank™ accession number M20637) that was kindly provided by M. Katan (Cancer Research UK, Centre for Cell and Molecular Biology, London, UK). We used the appropriate primers to incorporate a 5′-EcoRI site and a 3′-SalI site and the PCR product was cloned into the pGEX-6P1 vector. The forward primer was 5′-ACATGAATTCATGGACTCGGGTAGGGACTTC-3′ and the reverse 5′-GCCAGTCGACTCAGTCCATGGAGCCGGAGTG-3′. Each of the above expression vector constructs was confirmed by dideoxynucleotide sequencing (Prism Big Dye kit; ABI Prism® 3100 Genetic Analyzer, Applied Biosystems).
Protein expression and purification
For bacterial expression of glutathione S-transferase (GST) fusion proteins, E. coli [Rosetta (DE3) cells; Novagen] was transformed with the appropriate pGEX construct and cultured at 37°C until the A600 reached 0.6, and then protein expression was induced for 18 hours at 16°C with 0.1 mM isopropyl β-D-thiogalactopyranoside (IPTG; Promega). Cells were harvested by centrifugation at 6000 g for 10 minutes, resuspended in phosphate-buffered saline (PBS; 137 mM NaCl, 2.7 mM KCl, 4.3 mM Na2HPO4·7H2O, 1.4 mM KH2PO4, pH 7.4) containing 2 mM dithiothreitol and protease inhibitor mixture (Roche), and then sonicated four times for 15 seconds on ice. After 15 minutes of centrifugation at 15,000 g at 4°C, soluble GST fusion proteins were purified by affinity chromatography using glutathione–Sepharose™ 4B beads following standard procedures (GE Healthcare). Eluted proteins were dialyzed overnight (Pierce; SnakeSkin 10,000 molecular weight cut-off) at 4°C against 4 litres of PBS, and concentrated with centrifugal concentrators (Sartorius; 10,000 molecular weight cut-off).
Protein–lipid overlay assay
PIP Array membranes (Molecular Probes) were blocked for 2 hours with binding buffer [TBS-T (20 mM Tris, 137 mM NaCl, 0.1% Tween 20, pH 7.4] containing 3% bovine serum albumin (lipid-free), and incubated with 25 pmoles of each GST–PLCζ fusion protein for 1 hour at room temperature. After washing three times in TBS-T, GST–PLCζ fusion protein interaction with the phosphatidylinositols was detected by first incubating the PIP Array membranes with rabbit anti-GST polyclonal antibody (T103; 1:10,000 dilution in 5 ml of binding buffer) (Nomikos et al., 2005; Nomikos et al., 2007) overnight at 4°C, followed by 3′ 15-minute washes. This was followed by incubation with horseradish peroxidase-conjugated anti-rabbit antibody in the same binding buffer for 1 hour at room temperature, followed by 3′ 15-minute washes with TBS-T. Super Signal West Dura (Pierce) was used to visualise the HRP-coupled antibodies followed by a Bio-Rad Gel Doc system for image capture.
Liposome preparation and binding assay
Unilamellar liposomes were prepared by the extrusion method using a laboratory extruder (LiposoFast-Pneumatic, Avestin Inc., Ottawa, ON, Canada) with lipids purchased from Avanti Polar Lipids Inc. (Alabaster, AL). In a typical experiment requiring a 2 ml dispersion of liposomes, 0.038 mmol (19′10−3 M) 1,2-dipalmitoyl-sn-glycero-3-phosphocholine (PtdCho), 0.019 mmol (9.5′10−3 M) of cholesterol (CHOL; molar ratio of PtdCho:CHOL 2:1), 0.0095 mmol (4.8′10−3 M) 1,2-dimyristoyl-sn-glycero-3-phosphoethanolamine (PtdEtn; molar ratio of PtdCho:PtdEtn 4:1) and 1% of total lipids (16:50:38) phosphatidylcholine and phosphatidylinositol (PtdIns) were dissolved in chloroform:methanol (2:1 v/v) for the formation of lipid films. The film was hydrated with 2 ml of PBS and the resultant suspension was extruded through two-stacked polycarbonate filters of 100 nm pore size. Twenty-five cycles of extrusion were applied at 50°C. For the control experiments, unilamellar liposomes without PtdIns were prepared in an analogous manner. Dynamic light scattering (DLS) was employed to determine the size of the liposomes, which used a light scattering apparatus (AXIOS–150/EX, Triton Hellas, Thessaloniki, Greece) with a 30 mW laser source and an Avalanche photodiode detector set at a 90° angle. DLS measurements of the extruded lipid preparation showed a narrow monomodal size distribution with average liposome diameter of 100 nm and a polydispersity index of 0.20–0.25. For protein-binding studies the liposomes (100 μg of 4:2:1 PtdCho:CHOL:PtdEtn) containing 0–1% PtdIns(4,5)P2 were incubated with 1 μg of recombinant protein for 20 minutes at room temperature and centrifuged for 5 hours at 4°C. The supernatant and pellet were then analyzed by SDS-PAGE and Coomassie Blue staining.
Mutagenesis and cRNA synthesis
A pCR3 mouse PLCζ-luciferase construct (Nomikos et al., 2005) was subjected to site-directed mutagenesis (QuikChange II, Stratagene) to sequentially generate the three substitutions of alanine for lysine at K374, K375 and K377, thus producing the PLCζK374A, PLCζK374,5AA and PLCζK374,5,7AAA mutants. Following linearization of wild-type and mutated PLCζ constructs, cRNA was synthesized using the mMessage Machine T7 kit (Ambion) and then polyadenylated using the poly(A) tailing kit (Ambion), as per the manufacturer's instructions.
Preparation and handling of gametes
Experiments were carried out with mouse eggs in Hepes-buffered medium (H-KSOM) as described previously (Summers et al., 2000; Nomikos et al., 2005). All compounds were from Sigma unless stated otherwise. Female mice were superovulated and eggs were collected 13.5–14.5 hours after injection of human chorionic gonadotrophin and maintained in droplets of M2 medium (Sigma) or H-KSOM under mineral oil at 37°C. Microinjection of the eggs was carried out 14.5–15.5 hours after the hormone injection (Nomikos et al., 2005).
Microinjection and measurement of intracellular Ca2+ and luciferase expression
Mouse eggs were washed in M2 and microinjected with cRNA diluted in injection buffer (120 mM KCl, 20 mM Hepes, pH 7.4) (Nomikos et al., 2005). The volume injected was estimated from the diameter of cytoplasmic displacement caused by the bolus injection. All injections were 3–5% of the egg volume. Eggs were microinjected with the appropriate cRNA, mixed with an equal volume of 1 mM Oregon Green BAPTA dextran (Molecular Probes) in the injection buffer. Eggs were then maintained in H-KSOM with 100 μM luciferin and imaged on a Nikon TE2000 or Zeiss Axiovert 100 microscope equipped with a cooled intensified CCD camera (Photek Ltd., St Leonards on Sea, UK). Ca2+ was monitored in these eggs for 4 hours after injection by measuring the Oregon Green BAPTA dextran fluorescence with low level excitation light from a halogen lamp. At the end of Ca2+ measurements, the same set of eggs was monitored for luminescence by integrating light emission (in the absence of fluorescence excitation) for 20 minutes using the same intensified CCD camera. The fluorescence signals were typically 10–100 times greater than the luminescence signals. Ca2+ measurements for an egg were further analysed only if the same egg was luminescent. The luminescence from eggs was converted into an amount of luciferase using a standard curve that was generated by placing eggs in a luminometer that had been previously calibrated by microinjection with known amounts of luciferase protein (Sigma) (Nomikos et al., 2005; Swann et al., 2009).
Assay of PLC activity
PtdIns(4,5)P2 hydrolytic activity of recombinant wild-type PLCζ and the various mutant constructs was assayed as described previously (Nomikos et al., 2005). The final volume of the assay mixture was 50 μl containing 100 mM NaCl, 0.4% sodium cholate (w/v), 2 mM CaCl2, 4 mM EGTA, 20 μg of bovine serum albumin, 5 mM 2-mercaptoethanol and 20 mM Tris-HCl buffer, pH 6.8. The final concentration of PtdIns(4,5)P2 in the reaction mixture was 220 μM, containing 0.05 μCi of [3H]PtdIns(4,5)P2. The assay conditions were optimized for linearity, requiring a 10-minute incubation of 20 pmol GST–PLCζ protein sample at 25°C. Reactions were stopped by the addition of 0.25 ml chloroform:methanol:concentrated HCl (100:100:0.6 v/v) followed by 0.075 ml of concentrated HCl. The mixture was vortexed and centrifuged at 2000 g for 2 minutes, and then 0.2 ml of the upper aqueous phase was removed and added to 10 ml Optiphase ‘Hisafe 3’ scintillation mixture (Wallac), and the radioactivity was determined by liquid scintillation spectrofluorimetry (Packard Tri-Carb 2100TR). In assays to determine dependence on PtdIns(4,5)P2 concentration, 0.05 μCi [3H]PtdIns(4,5)P2 was mixed with nonradioactive PtdIns(4,5)P2 to give the appropriate final concentration. In assays examining the Ca2+ sensitivity, Ca2+ buffers were prepared by mixing EGTA and CaCl2, as described previously (Nomikos et al., 2005).
SDS-PAGE and western blotting
Recombinant proteins were separated by SDS-PAGE as described previously (Nomikos et al., 2005). Separated proteins were transferred onto polyvinylidene difluoride membrane, incubated overnight at 4°C in Tris-buffered saline, 0.1% Tween 20 (TBS-T) containing 5% nonfat milk powder, and probed with anti-GST polyclonal antibody (1:10,000 dilution). Detection of horseradish peroxidase-coupled secondary antibody was achieved using Super Signal West Dura (Pierce) and a Bio-Rad ChemiDoc gel documentation system for image capture.
Acknowledgments
This work was supported by a grant from the Wellcome Trust (080701 to F.A.L. and K.S.). K.E. and M.T. are the recipients of PhD studentships from the Libyan Ministry of Education and NCSR Demokritos, respectively. We thank Zili Sideratou for assistance with preparation of liposomes and Matilda Katan for the gift of PLCδ1. Deposited in PMC for immediate release.
References
- Bae Y. S., Cantley L. G., Chen C. S., Kim S. R., Kwon K. S., Rhee S. G. (1998). Activation of phospholipase C-gamma by phosphatidylinositol 3,4,5-trisphosphate. J. Biol. Chem. 273, 4465-4469 [DOI] [PubMed] [Google Scholar]
- Cox L. J., Larman M. G., Saunders C. M., Hashimoto K., Swann K., Lai F. A. (2002). Sperm phospholipase Czeta from humans and cynomolgus monkeys triggers Ca2+ oscillations, activation and development of mouse oocytes. Reproduction 124, 611-623 [DOI] [PubMed] [Google Scholar]
- Falasca M., Logan S. K., Lehto V. P., Baccante G., Lemmon M. A., Schlessinger J. (1998). Activation of phospholipase C gamma by PI 3-kinase-induced PH domain-mediated membrane targeting. EMBO J. 17, 414-422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukami K., Inanobe S., Kanemaru K., Nakamura Y. (2010). Phospholipase C is a key enzyme regulating intracellular calcium and modulating the phosphoinositide balance. Prog. Lipid Res. 49, 429-437 [DOI] [PubMed] [Google Scholar]
- Gresset A., Hicks S. N., Harden T. K., Sondek J. (2010). Mechanism of phosphorylation-induced activation of phospholipase C (PLC)-{gamma} isozymes. J. Biol. Chem. 285, 35836-35847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heytens E., Parrington J., Coward K., Young C., Lambrecht S., Yoon S. Y., Fissore R. A., Hamer R., Deane C. M., Ruas M., et al. (2009). Reduced amounts and abnormal forms of phospholipase C zeta (PLCzeta) in spermatozoa from infertile men. Hum. Reprod. 24, 2417-2428 [DOI] [PubMed] [Google Scholar]
- Hicks S. N., Jezyk M. R., Gershburg S., Seifert J. P., Harden T. K., Sondek J. (2008). General and versatile autoinhibition of PLC isozymes. Mol. Cell 31, 383-394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knott J. G., Kurokawa M., Fissore R. A., Schultz R. M., Williams C. J. (2005). Transgenic RNA interference reveals role for mouse sperm phospholipase Czeta in triggering Ca2+ oscillations during fertilization. Biol. Reprod. 72, 992-996 [DOI] [PubMed] [Google Scholar]
- Kouchi Z., Fukami K., Shikano T., Oda S., Nakamura Y., Takenawa T., Miyazaki S. (2004). Recombinant phospholipase Czeta has high Ca2+ sensitivity and induces Ca2+ oscillations in mouse eggs. J. Biol. Chem. 279, 10408-10412 [DOI] [PubMed] [Google Scholar]
- Kouchi Z., Shikano T., Nakamura Y., Shirakawa H., Fukami K., Miyazaki S. (2005). The role of EF-hand domains and C2 domain in regulation of enzymatic activity of phospholipase Czeta. J. Biol. Chem. 280, 21015-21021 [DOI] [PubMed] [Google Scholar]
- Lomasney J. W., Cheng H. F., Wang L. P., Kuan Y., Liu S., Fesik S. W., King K. (1996). Phosphatidylinositol 4,5-bisphosphate binding to the pleckstrin homology domain of phospholipase C-delta1 enhances enzyme activity. J. Biol. Chem. 271, 25316-25326 [DOI] [PubMed] [Google Scholar]
- Lomasney J. W., Cheng H. F., Roffler S. R., King K. (1999). Activation of phospholipase C delta1 through C2 domain by a Ca(2+)-enzyme-phosphatidylserine ternary complex. J. Biol. Chem. 274, 21995-22001 [DOI] [PubMed] [Google Scholar]
- Malcuit C., Kurokawa M., Fissore R. A. (2006). Calcium oscillations and mammalian egg activation. J. Cell. Physiol. 206, 565-573 [DOI] [PubMed] [Google Scholar]
- Medkova M., Cho W. (1999). Interplay of C1 and C2 domains of protein kinase C-alpha in its membrane binding and activation. J. Biol. Chem. 274, 19852-19861 [DOI] [PubMed] [Google Scholar]
- Nalefski E. A., Falke J. J. (1996). The C2 domain calcium-binding motif: structural and functional diversity. Protein Sci. 5, 2375-2390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nomikos M., Blayney L. M., Larman M. G., Campbell K., Rossbach A., Saunders C. M., Swann K., Lai F. A. (2005). Role of phospholipase C-ζ domains in Ca2+-dependent phosphatidylinositol 4,5-bisphosphate hydrolysis and cytoplasmic Ca2+ oscillations. J. Biol. Chem. 280, 31011-31018 [DOI] [PubMed] [Google Scholar]
- Nomikos M., Mulgrew-Nesbitt A., Pallavi P., Mihalyne G., Zaitseva I., Swann K., Lai F. A., Murray D., McLaughlin S. (2007). Binding of phosphoinositide-specific phospholipase C-zeta (PLC-zeta) to phospholipid membranes: potential role of an unstructured cluster of basic residues. J. Biol. Chem. 282, 16644-16653 [DOI] [PubMed] [Google Scholar]
- Nomikos M., Elgmati K., Theodoridou M., Calver B. L., Cumbes B., Nounesis G., Swann K., Lai F. (2011). Male infertility-linked point mutation disrupts the Ca2+ oscillation-inducing and PIP2 hydrolysis activity of sperm PLCzeta. Biochem. J. 434, 211-217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozdener F., Dangelmaier C., Ashby B., Kunapuli S. P., Daniel J. L. (2002). Activation of phospholipase Cgamma2 by tyrosine phosphorylation. Mol. Pharmacol. 62, 672-679 [DOI] [PubMed] [Google Scholar]
- Pawelczyk T., Lowenstein J. M. (1993). Binding of phospholipase C delta 1 to phospholipid vesicles. Biochem. J. 291, 693-696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rebecchi M. J., Pentyala S. N. (2000). Structure, function, and control of phosphoinositide-specific phospholipase C. Physiol. Rev. 80, 1291-1335 [DOI] [PubMed] [Google Scholar]
- Rodriguez R., Matsuda M., Perisic O., Bravo J., Paul A., Jones N. P., Light Y., Swann K., Williams R. L., Katan M. (2001). Tyrosine residues in phospholipase Cgamma 2 essential for the enzyme function in B-cell signaling. J. Biol. Chem. 276, 47982-47992 [DOI] [PubMed] [Google Scholar]
- Saunders C. M., Larman M. G., Parrington J., Cox L. J., Royse J., Blayney L. M., Swann K., Lai F. A. (2002). PLC zeta: a sperm-specific trigger of Ca(2+) oscillations in eggs and embryo development. Development 129, 3533-3544 [DOI] [PubMed] [Google Scholar]
- Saunders C. M., Swann K., Lai F. A. (2007). PLCzeta, a sperm-specific PLC and its potential role in fertilization. Biochem. Soc. Symp. 74, 23-36 [DOI] [PubMed] [Google Scholar]
- Sekiya F., Poulin B., Kim Y. J., Rhee S. G. (2004). Mechanism of tyrosine phosphorylation and activation of phospholipase C-gamma 1. Tyrosine 783 phosphorylation is not sufficient for lipase activation. J. Biol. Chem. 279, 32181-32190 [DOI] [PubMed] [Google Scholar]
- Singh S. M., Murray D. (2003). Molecular modeling of the membrane targeting of phospholipase C pleckstrin homology domains. Protein Sci. 12, 1934-1953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stricker S. A. (1999). Comparative biology of calcium signaling during fertilization and egg activation in animals. Dev. Biol. 211, 157-176 [DOI] [PubMed] [Google Scholar]
- Suh P. G., Park J. I., Manzoli L., Cocco L., Peak J. C., Katan M., Fukami K., Kataoka T., Yun S., Ryu S. H. (2008). Multiple roles of phosphoinositide-specific phospholipase C isozymes. BMB Rep. 41, 415-434 [DOI] [PubMed] [Google Scholar]
- Summers M. C., McGinnis L. K., Lawitts J. A., Raffin M., Biggers J. D. (2000). IVF of mouse ova in a simplex optimized medium supplemented with amino acids. Hum. Reprod. 15, 1791-1801 [DOI] [PubMed] [Google Scholar]
- Swann K. (1994). Ca2+ oscillations and sensitization of Ca2+ release in unfertilized mouse eggs injected with a sperm factor. Cell Calcium 15, 331-339 [DOI] [PubMed] [Google Scholar]
- Swann K., Saunders C. M., Rogers N. T., Lai F. A. (2006). PLCzeta(zeta): a sperm protein that triggers Ca2+ oscillations and egg activation in mammals. Semin. Cell Dev. Biol. 17, 264-273 [DOI] [PubMed] [Google Scholar]
- Swann K., Campbell K., Yu Y., Saunders C., Lai F. A. (2009). Use of luciferase chimaera to monitor PLCzeta expression in mouse eggs. Methods Mol. Biol. 518, 17-29 [DOI] [PubMed] [Google Scholar]
- Yoda A., Oda S., Shikano T., Kouchi Z., Awaji T., Shirakawa H., Kinoshita K., Miyazaki S. (2004). Ca2+ oscillation-inducing phospholipase C zeta expressed in mouse eggs is accumulated to the pronucleus during egg activation. Dev. Biol. 268, 245-257 [DOI] [PubMed] [Google Scholar]
- Yoon S. Y., Jellerette T., Salicioni A. M., Lee H. C., Yoo M. S., Coward K., Parrington J., Grow D., Cibelli J. B., Visconti P. E., et al. (2008). Human sperm devoid of PLC, zeta 1 fail to induce Ca(2+) release and are unable to initiate the first step of embryo development. J. Clin. Invest. 118, 3671-3681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng L., Krishnamoorthi R., Zolkiewski M., Wang X. (2000). Distinct Ca2+ binding properties of novel C2 domains of plant phospholipase dalpha and beta. J. Biol. Chem. 275, 19700-19706 [DOI] [PubMed] [Google Scholar]