

Abstract
The psbD mRNA, which encodes the D2 reaction center polypeptide of photosystem II, is one of the most abundant chloroplast mRNAs. We have used genomic complementation to isolate the nuclear Nac2 gene, which is required for the stable accumulation of the psbD mRNA in Chlamydomonas reinhardtii. Nac2 encodes a hydrophilic polypeptide of 1385 amino acids with nine tetratricopeptide-like repeats (TPRs) in its C-terminal half. Cell fractionation studies indicate that the Nac2 protein is localized in the stromal compartment of the chloroplast. It is part of a high molecular weight complex that is associated with non-polysomal RNA. Change of a conserved alanine residue of the fourth TPR motif by site-directed mutagenesis leads to aggregation of Nac2 protein and completely abrogates its function, indicating that this TPR is important for proper folding of the protein and for psbD mRNA stability, processing and/or translation.
Keywords: Chlamydomonas/chloroplast/RNA stability/TPR protein
Introduction
Chloroplast biogenesis is a complex process that depends on the concerted action of the nuclear and chloroplast genomes. While the majority of chloroplast proteins are nucleus encoded, the chloroplast genome encodes a relatively small number of genes, the products of which are required mainly for photosynthesis and chloroplast gene expression. The level of expression of chloroplast genes varies considerably during plastid development and differentiation, and it is profoundly influenced by changes in light quantity and quality (for a review, see Goldschmidt-Clermont, 1998). In addition, some chloroplast genes are subjected to an endogenous circadian control. Genetic analysis of photosynthetic mutants of the unicellular green alga Chlamydomonas reinhardtii and of land plants has revealed a complex cross-talk between the nucleus and the chloroplast that is mediated by numerous nucleus-encoded factors either acting as constituents of the transcription/translation machinery or involved in several post-transcriptional steps of chloroplast gene expression, i.e. RNA stabilization, processing/maturation and translation (for reviews, see Rochaix 1992, 1996; Mayfield et al., 1995; Sugita and Sugiura, 1996; Goldschmidt-Clermont, 1998; Leon et al., 1998). Interestingly, in C.reinhardtii, most of these nuclear mutants are affected in the expression of specific chloroplast genes, whereas in land plants most nuclear mutations of this sort have pleiotropic effects (Goldschmidt-Clermont, 1998; Leon et al., 1998).
Several reports have provided evidence for post-transcriptional regulation of the abundance of chloroplast transcripts (Shiina et al., 1998; for reviews, see also Gruissem and Schuster, 1993; Mullet, 1993; Nickelsen, 1998). In alternating light–dark cycles, the level of individual mRNAs is regulated by differential rates of RNA synthesis and RNA stabilization (Salvador et al., 1993; Hwang et al., 1996; Shiina et al., 1998). The half-lives of many chloroplast transcripts are longer in the dark when transcription rates are at their lowest levels. In tobacco, the steady-state level of the chloroplast rbcL transcript is maintained by diurnal control. In the dark, the reduced transcription rate of the rbcL gene is compensated by an increase in the stability of its mRNA (Shiina et al., 1998). In C.reinhardtii, differences in the abundance of chloroplast transcripts were found to be light/dark dependent and/or to be under circadian control (Salvador et al., 1993; Hwang et al., 1996). The transcription rate of the chloroplast tufA gene is under the control of both circadian clock and diurnal rhythms, whereas its mRNA stability is under the control of diurnal rhythms only (Hwang et al., 1996).
Recent genetic analyses indicate that many nucleus-encoded factors are involved in RNA stabilization. Several C.reinhardtii mutants are affected in the stability of specific chloroplast transcripts such as those encoded by atpA, atpB (Drapier et al., 1998), petA, petB (Gumpel et al., 1995), petD (Drager et al., 1998), psbB (Sieburth et al., 1991; Monod et al., 1992), psbC (Sieburth et al., 1991) and psbD (Kuchka et al., 1989). In addition, the Arabidopsis thaliana nuclear mutant hcf109 exhibits defects in the stability of a distinct set of transcripts from four different plastid operons (Meurer et al., 1996).
Molecular and biochemical analyses have shown that both 5′- and 3′-untranslated regions (UTRs) can be implicated in RNA stabilization or degradation. Transformation experiments with chimeric genes in several of the C.reinhardtii stability mutants have revealed that the 5′-UTR conveys specific transcript instability in these mutants (Nickelsen et al., 1994; Drager et al., 1998; Vaistij et al., 2000). Furthermore, the affected transcripts are degraded by 5′–3′ exoribonucleolytic activities in these mutants (Drager et al., 1998, 1999; Nickelsen et al., 1999). The 5′-UTR of the tobacco rbcL mRNA also appears to be essential for the regulation of RNA stability (Shiina et al., 1998). Most chloroplast transcripts have stem–loop structures in their 3′-UTR, which are believed to be required for accurate 3′ end maturation and to protect the transcripts from 3′–5′ exonucleolytic degradation (Stern and Gruissem, 1987; Stern et al., 1989, 1991; Drager et al., 1996). Several nuclear proteins have been shown by in vitro RNA-binding experiments to interact with 3′-UTRs and to be involved in RNA metabolism. A 550 kDa protein complex from spinach that mediates petD RNA 3′ end processing in vitro contains a PNP-like exoribonuclease and an RNase E-like endonuclease (Hayes et al., 1996). Furthermore, a 41 kDa protein that also interacts with the petD 3′-UTR displays unspecific RNase activity (Yang et al., 1996), and a 54 kDa chloroplast protein from mustard mediates endonucleolytic 3′ end formation of some plastid transcripts (Nickelsen and Link, 1993).
Photosystem II (PSII) is a multisubunit complex embedded in the thylakoid membrane. The two reaction center polypeptides of PSII, D1 and D2, are translated on polysomes associated with the thylakoid membrane and are thought to be inserted co-translationally into the membrane. The stability of the psbD mRNA encoding D2 has been shown to be affected specifically in the nuclear photosynthetic mutant nac2-26 (Kuchka et al., 1989). This mRNA exists in two forms with 5′-UTRs of 74 and 47 nucleotides. The short form corresponds to the mature mRNA and is absent specifically in the nuclear nac2-26 mutant. Cis-acting elements have been localized within the psbD 5′-UTR which are required for psbD mRNA stabilization (Nickelsen et al., 1994, 1999). UV cross-linking experiments revealed the binding of a 47 kDa protein to the psbD 5′-UTR. This binding activity was altered in extracts from the nac2-26 mutant, thus revealing a correlation between the instability of psbD mRNA and loss of binding of the 47 kDa protein.
The molecular mechanisms that underlie the 5′ end formation of chloroplast mRNAs and its role in RNA stability are still poorly understood. As a first step towards this goal, we have isolated the Nac2 cDNA and found that it encodes a novel tetratricopeptide repeat (TPR)-containing protein. The Nac2 factor is a soluble chloroplast protein that is part of a large protein complex which is associated with non-polysomal RNA. This Nac2 protein complex is likely to play an important role in psbD RNA processing, stability and/or translation.
Results
Cloning of the Nac2 gene
To isolate the nuclear gene that is affected in the photosynthetic mutant nac2-26, the double mutant nac2-26 arg7 was first produced by appropriate genetic crosses. Cells of nac2-26 arg7 were transformed with DNA from a C.reinhardtii cosmid library in which the vector includes the arginino-succinate lyase gene (Purton and Rochaix, 1994, 1995). Transformants were selected for growth on minimal medium. After plating ∼109 cells, 12 arginine prototrophs were obtained that were able to grow photoautotrophically and displayed wild-type fluorescence transients (for details, see Materials and methods). This indicated that PSII activity had been restored in these transformants. Genomic DNA flanking the cosmid vector was recovered by plasmid rescue from the DNA of the transformants (see Materials and methods) and was used to isolate from a wild-type library the cosmid cosnac5, which was able to complement the nac2-26 mutation by nuclear transformation with an efficiency of 1.3 × 10–5 per cell. Cosnac5 DNA was then used to isolate a 2.7 kb cDNA, cnac1, and subsequently a longer 5.1 kb cDNA, cnac2. Both cDNAs were able to rescue nac2-26 although with a reduced efficiency when compared with cosnac5 (∼10–7/cell). Another PSII-deficient mutant lacking psbD mRNA, mø14 (S.Purton, unpublished results), could also be rescued by transformation with the genomic Nac2 clone.
Molecular analysis of Nac2 mutants
To confirm that the isolated cDNA indeed corresponds to the NAC2 locus, DNA from the mutants nac2-26 and mø14 was examined for restriction fragment length polymorphism by Southern blot hybridization. The DNA from wild-type, nac2-26 and mø14 was digested with BglI and hybridized with the three different fragment probes A, B and C derived from the short cDNA cnac1 (Figure 1A). Hybridization of wild-type DNA with probes A and C revealed single bands of 900 and 300 bp, respectively (Figure 1A), indicating that Nac2 exists as a single copy gene. The region covered by the different cDNA probes appears to be deleted in the mø14 mutant since none of the probes hybridized to the mø14 DNA. Additional hybridizations of probes A and C to mø14 DNA digested with 10 different restriction endonucleases did not reveal any hybridization signal (data not shown), thus confirming that the 3′ end of the gene is missing in this strain. Differences in restriction fragment length between nac2-26 and wild-type were observed in the hybridization with probe B (Figure 1A). In wild-type, this probe hybridizes with BglI fragments of 1.5 and 0.6 kb, whereas in nac2-26 it hybridizes to fragments of 3.8, 1.3 and 0.6 kb. The observed difference in the hybridization pattern can be attributed to a rearrangement in the genomic region covered by the 660 bp BglI cDNA fragment. Analysis of the DNA from progeny of a cross between nac2-26 and the strain S1D2 revealed that the rearrangement co-segregated with the PSII-deficient phenotype (data not shown).

Fig. 1. Nac2 gene organization and expression. (A) Scheme of the 3′ region of the Nac2 cDNA with the coding region boxed. Total DNA from the wild-type (WT), nac2-26 and mø14 strains was digested with BglI restriction endonuclease, electrophoresed, transferred to nylon membranes and hybridized with the labeled probes A, B and C, the position of which is indicated on the cDNA map. The arrows represent the position of the primers used to synthesize probe A. The estimated sizes of the DNA fragments are indicated in kb. (B) RNA blot analysis of the wild-type, nac2-26 and mø14 strains. A 3 µg aliquot of poly(A) RNA from wild-type (WT), nac2-26 and mø14 was fractionated on a 1% agarose–formaldehyde gel, blotted to a nitrocellulose membrane and hybridized with the 32P-labeled Nac2 cDNA probes A and C. Equal loading of mRNA was checked by hybridization of the blots with a rbcS probe. The sizes of the RNA species are indicated in kb and were determined by comparison with size markers. (C) DNA blot analysis of four transformants T1, T2, T3 and T4 obtained by transformation of the nac2-26 strain with the Nac2 cDNA cnac1 encoding the 588 C-terminal residues of Nac2. Probe B was used for the hybridization.
Hybridization of wild-type RNA with the cDNA probes A and C revealed that Nac2 is transcribed as a single transcript of 5.3 kb (Figure 1B). As expected, these probes did not hybridize with any transcript from the mø14 mutant except for a transcript of 1.9 kb, which appears to be non-specific. In nac2-26, however, probe A, which is specific to the region upstream of the rearrangement, hybridized with a 3.8 kb RNA, whereas no specific transcript hybridized with probe C, which covers the region downstream of the rearrangement. Hence, in this mutant, the Nac2 gene is transcribed, but the transcript ends within the rearranged region resulting in a 3.8 kb truncated mRNA. Reprobing the blot with a probe specific for rbcS showed that equal amounts of mRNA had been loaded for the wild-type and nac2-26 strains and slightly less for mø14 (Figure 1B).
The 3′ region of the Nac2 gene is sufficient to convey psbD mRNA stability
The 2.7 kb cDNA clone cnac1, which contains only the C-terminal 588 codons of the Nac2 open reading frame (ORF; see below), is able to complement the nac2-26 mutation although at a much lower rate than the cosmid cosnac5. This suggests that this region of the Nac2 protein is sufficient to convey stability to the psbD transcript and that the N-terminal half of the protein is dispensable for this function. However, it is also possible that the Nac2 gene was restored in nac2-26 during transformation by homologous recombination. To distinguish between both possibilities, the DNAs from four different transformants rescued with the cnac1 DNA were examined by Southern blot analysis. The DNAs were digested with BglI and hybridized with the B probe from the cDNA (see Figure 1C). The three characteristic bands of 3.8, 1.3 and 0.6 kb from nac2-26 can be found in all four transformants, which contain, in addition, novel bands corresponding to the integrated cnac1 DNA. Thus, homologous recombination did not occur in any of the four transformants and we conclude that the truncated cDNA is able to rescue the mutant.
The Nac2 protein contains nine TPR-like motifs
Sequencing of the cnac2 cDNA revealed an ORF encoding a predicted polypeptide of 1385 amino acids corresponding to a molecular mass of 139.3 kDa (Figure 2A). The putative ATG start codon is preceded by three consecutive A residues (data not shown) characteristic of translation initiation sites of nuclear genes of C.reinhardtii (Silflow, 1998). In addition, a stop codon that is in-frame with the Nac2 ORF occurs upstream of this site (data not shown). The N-terminal region of the Nac2 protein is basic, and contains numerous hydroxylated amino acids and the Ala-Xxx-Ala motif at position 43, a characteristic feature of chloroplast transit sequences (Franzen et al., 1990). A BLAST search revealed that the C-terminal half of the protein is related to proteins containing TPRs. The Nac2 protein contains nine TPR-like motifs, each consisting of a 34 residue degenerate consensus sequence (Sikorski et al., 1990). TPR domains have been found in a variety of proteins with different functions (Goebl and Yanagida, 1991; Lamb et al., 1995). The TPR-like domains of the Nac2 protein are arranged tandemly, but not all of them are contiguous (Figure 2A and B) as found for other TPR proteins (Lamb et al., 1995). Each TPR domain is punctuated by proline-induced turns and consists of two α-helical domains A and B (Hirano et al., 1990; Sikorski et al., 1990; Das et al., 1998). Conserved hydrophobic residues in these domains at positions 4, 7, 8, 11, 20, 24 and 27 (see Figure 2C) have been proposed to interact with each other and thereby to mediate intra- or inter-protein interactions (Lamb et al., 1995). While the TPR domain B consensus is nearly fully conserved in the Nac2 TPR-like domains, domain A is not. In particular, a major difference is position 11, which is occupied in most cases by acidic amino acids, rather than by a bulky hydrophobic residue of the consensus TPR. The region containing the TPR-like motifs was the only domain of the Nac2 protein showing significant sequence similarity to known proteins. No other known protein motifs could be identified in the remaining part of the Nac2 protein except for the presence of a putative ATP/GTP-binding site (GXXXXGKST) at residues 401–409 (GVNGSGKSG). Another unusual feature of this protein is the presence of several long stretches of alanine, serine and aspartate residues.

Fig. 2. Nac2 polypeptide. (A) Sequence of the predicted Nac2 polypeptide. The nine TPR-like repeats are highlighted in gray; dots indicate boundaries between the repeats. The mutated A in the fourth TPR repeat is underlined. The stippled line marks the putative transit sequence. The GTP/ATP-binding site is double underlined. Arrowheads indicate the location of introns. (B) Schematic view of the Nac2 polypeptide. The nine TPR-like repeats are indicated by boxes; G, ATP/GTP-binding site; the upward arrow marks the end of the C-terminal part of the Nac2 protein which is able to complement the Nac2 deficiency; the asterisk marks the site of the mutation. (C) Alignment of the nine TPR motifs of the Nac2 protein. Residues which appear at least five times amongst the nine TPRs are highlighted. The conserved residues of the Nac2 TPRs are shown at the bottom together with the TPR consensus at positions 4, 7, 8, 11, 20, 24, 27 and 32 (Lamb et al., 1995); h, hydrophobic residue; A and B, TPR α-helical domains A and B (Das et al., 1998).
To characterize the Nac2 protein further, antibodies against a recombinant protein corresponding to the C-terminal 334 amino acids of Nac2 were raised in mouse (see Materials and methods). Whole-cell proteins from the wild-type strain cc137, the wild-type wall-less strain cw15, and the allelic mutants nac2-26 and mø14 were separated by polyacrylamide gel electrophoresis, transferred to nitrocellulose membranes and probed with the Nac2 polyclonal antibody. A 140 kDa protein was detected in the extracts of the two wild-type strains (Figure 3A). As expected, the Nac2 protein was not observed in the extracts from nac2-26 and mø14. Separation of wild-type cell extracts into soluble and membrane fractions revealed that the Nac2 protein is present exclusively in the soluble fraction (Figure 3A).
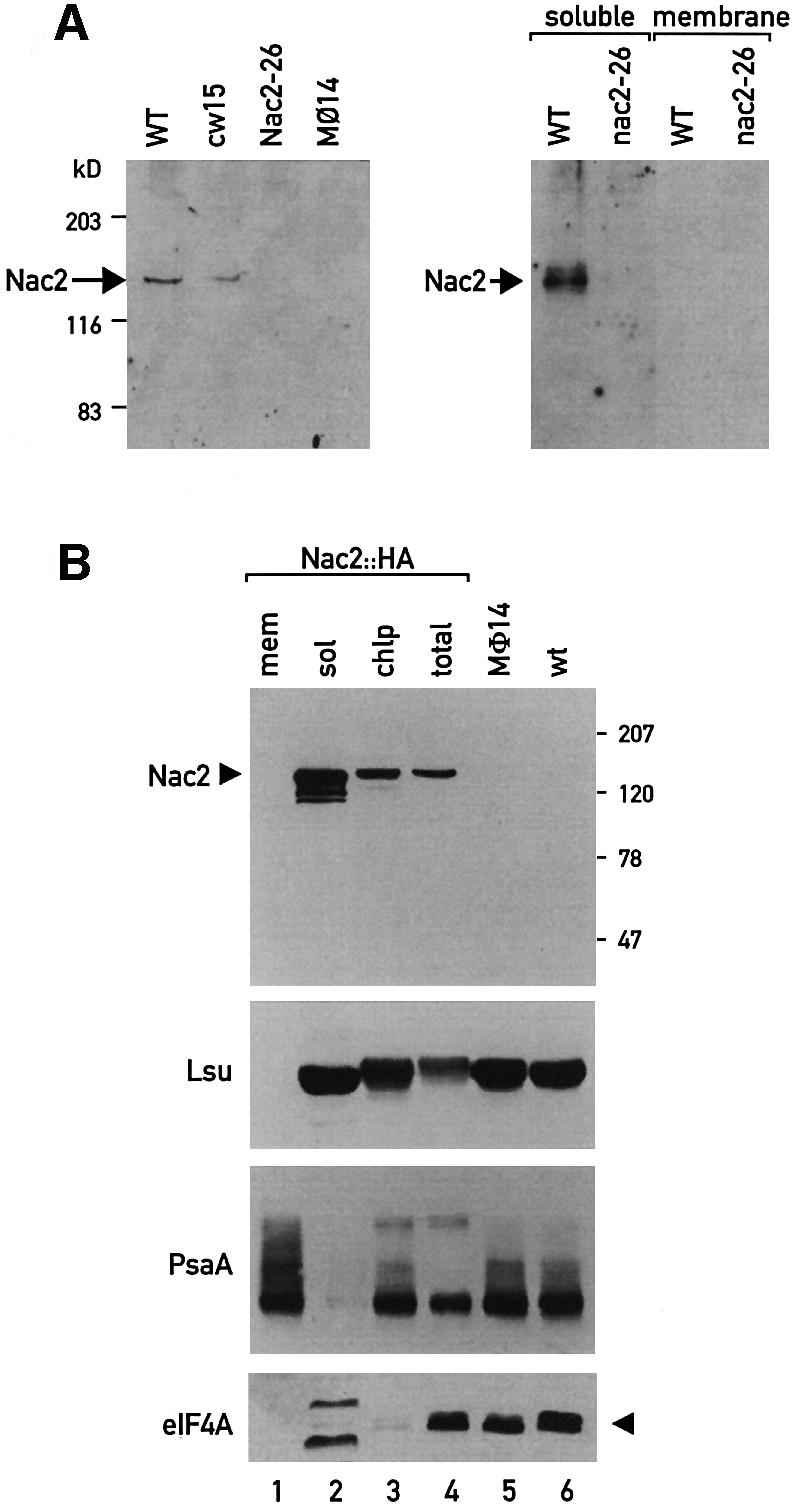
Fig. 3. Immunodetection of the Nac2 protein. (A) Immunoblot analysis of total cell proteins (75 µg) of the wild-type (WT), cw15, nac2-26 and mø14. Total soluble and membrane fractions were used. The blots were reacted with polyclonal antiserum against the 40 kDa C-terminal part of the Nac2 protein. Molecular mass markers are indicated in kDa. (B) Subcellular localization of the Nac2 protein. Immunoblot analysis of total cell proteins from the wild-type (WT), mø14, nac2::HA, chloroplasts from nac2::HA (chlp), soluble chloroplast fraction from nac2::HA (sol) and chloroplast membrane fraction from nac2::HA (mem). The blots were reacted with anti-HA monoclonal antibody and antibodies against the large subunit of Rubisco (RbcL), PsaA and eIF4A. The proteins reacting with the eIF4A antibody in the chloroplast lane could represent a plastid form of a factor related to eIF4A. Molecular mass markers are indicated in kDa.
To confirm that Nac2 encodes a 140 kDa protein, a chimeric genomic–cDNA gene containing a triple hemagglutinin (HA) epitope at the 3′ end of the coding sequence was constructed and used to rescue the mø14 null mutant by transformation. Whole-cell proteins from three transformants were probed by immunoblot analysis with the anti-HA monoclonal antibody. A 140 kDa protein could be detected readily in the extracts of the transformed strains (Figure 3B, lane 4), but not in the extracts from mø14 and the wild-type strains (Figure 3B, lanes 5 and 6). The Nac2::HA protein was found to be enriched ∼2-fold in isolated chloroplasts, as was also observed for the chloroplast protein PsaA and the large subunit of RubisCo (RbcL) (Figure 3B, lanes 3 and 4). The isolated chloroplasts were not contaminated significantly by cytosolic proteins since the cytosolic factor elF4A was only detected in whole-cell extracts (Figure 3B, lane 4), but not in the purified chloroplast fraction (Figure 3B, lane 3). Further fractionation of the chloroplasts into insoluble and soluble fractions confirmed that the Nac2::HA protein is a soluble chloroplast protein (Figure 3B, lanes 1 and 2).
The Nac2 protein is part of a large protein complex that is associated with RNA
The presence of TPR-like domains raises the possibility that Nac2 interacts with other proteins and that it might be part of a multiprotein complex. To test this hypothesis, the soluble fraction from whole cells was either subjected to sedimentation analysis by ultracentrifugation on a continuous 0.1–1.3 M sucrose gradient (Figure 4A) or fractionated by size exclusion chromatography (Figure 4B). In both cases, the bulk of the Nac2::HA protein was detected in fractions corresponding to a size of 500 kDa. However, a significant part of Nac2::HA protein was also found in the heavier fractions (Figure 4B). Nac2 thus appears to be part of a high molecular weight complex. When this complex was analyzed by size exclusion chromatography in the presence of MgCl2 and heparin, the complex was found to peak in the 600 kDa fractions, with substantial amounts observed in fractions corresponding to a larger size (Figure 4B). In the presence of EDTA, however, the elution profile was changed considerably, with most of the Nac2::HA protein in the 600 kDa region and with much less protein in the heavier fractions. In the presence of RNase and Mg2+ ions, the protein complex was found in the 500 kDa fraction. Under similar conditions, Rubisco, which is known to form a complex of 560 kDa, was found in the same fractions whether RNase was present or not in the buffer. Because the Nac2 protein could also be involved in translation (Nickelsen et al., 1999), we tested whether the Nac2 complex might include other known chloroplast proteins involved in translation. One factor of this sort, RB60, has been shown to be part of a protein complex binding to the 5′-UTR of psbA mRNA, which encodes the PSII reaction center D1 polypeptide from C.reinhardtii (Danon and Mayfield, 1991). The RNA-binding activity of this complex correlates with the activation of translation of D1 by light (Danon and Mayfield, 1991). When the fractions of the column were probed with an antibody against RB60, the peak fractions in the presence of MgCl2 and heparin, or MgCl2 and RNase, did not coincide with those obtained with the Nac2 protein (Figure 4B), thus indicating that Nac2 and RB60 are not part of the same complex. In addition, since the size of the RB60 complex, known to be associated with RNA, is lowered after RNase treatment, this experiment provides a control for the RNase treatments performed with the Nac2 complex. To test whether the Nac2 protein is associated with polysomes, a chloroplast extract was centrifuged through a two-step 0.5 M/1.75 M sucrose cushion and the polysomes were recovered at the bottom. Immunoblot analysis revealed that under these conditions, the Nac2 protein does not co-fractionate with polysomes, but remains in the supernatant fraction (Figure 5). In addition, RNAs were isolated from the fractions collected after size exclusion chromatography of extracts from the nac2::HA strain (Figure 4C). rRNAs were found in heavier fractions than the Nac2 complex (Figure 4B, rows 1–3), thus confirming that the Nac2 protein does not co-fractionate with polysomes. To determine the location of psbD mRNA in these fractions, RNA from each fraction was slot-blotted and hybridized to a psbD probe. While most of psbD mRNA co-fractionated with the polysomes (Figure 4C, fractions 5–13), a smaller portion of this RNA was found in the same fractions of the Nac2 complex (Figure 4C, fractions 15–17). This result is compatible with an association of psbD mRNA with the Nac2 complex.

Fig. 4. The Nac2 protein is part of a high molecular weight complex. Soluble cell extracts were prepared as described in Materials and methods. (A) A soluble cell extract from mø14 transformed with Nac2::HA was centrifuged on a 0.3–1.3 M linear sucrose gradient for 21 h at 230 000 g. Sedimentation was from left to right. Aliquots of the fractions were electrophoresed on a SDS–6% polyacrylamide gel, immunoblotted and reacted with anti-HA monoclonal antibody. (B) The soluble cell extracts from mø14 transformed with wild-type Nac2::HA (rows1–3) or mutant Nac2 A1038E::HA (rows 4–6) were concentrated to a final concentration of 20 µg/ml and fractionated on a Superose 6 PC3.2/30 column using the SMART system (Pharmacia Biotech, Sweden). Twenty-four fractions were collected and aliquots were electrophoresed on a 6% SDS–polyacrylamide gel and reacted with anti-HA monoclonal antibody (rows 1–6) and antibodies against RB60 (rows 7 and 8) and RbcL (rows 9 and 10). Sizes in each fraction of (A) and (B) were determined by comparison with size standards included in the HMW Gel Filtration Calibration kit (Pharmacia Biotech, Sweden). (C) Total RNA was extracted from 28 fractions collected after size exclusion chromatography with extracts from the nac2::HA strain. RNAs were stained with ethidium bromide and electrophoresed through an agarose–formaldehyde denaturing gel and visualized under UV light. (D) RNA from each fraction was slot-blotted and hybridized with a 32P-labeled psbD probe.
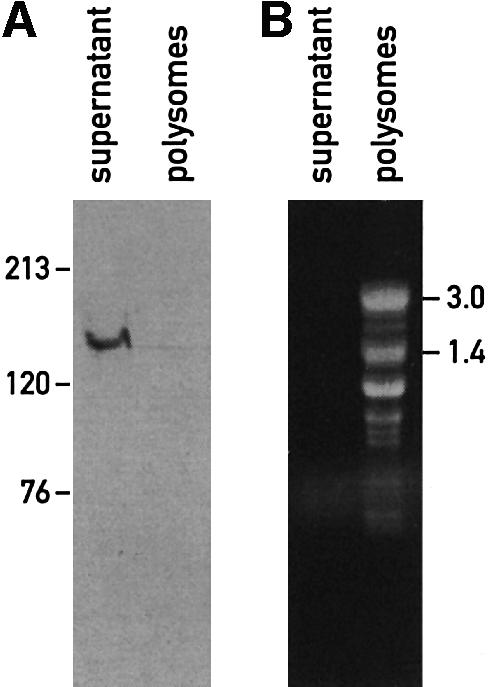
Fig. 5. The Nac2 protein is not associated with polysomes. Polysomes were prepared as described in Materials and methods. (A) Immunoblot analysis of the supernatant and polysome fractions from the nac2::HA strain. Samples were loaded on a 6% SDS–polyacrylamide gel and reacted with anti-HA monoclonal antibody. Molecular mass markers are indicated in kDa. (B) RNA content of the supernatant and polysome fractions from the nac2::HA strain. Total RNA was isolated by phenol/chloroform extraction followed by ethanol precipitation. The samples were stained with ethidium bromide and electrophoresed on an agarose–formaldehyde denaturing gel. RNA sizes are indicated in kb.
To determine whether the TPR domains are essential for Nac2 function, the conserved alanine (A1038) at position 20 of TPR domain 4 was changed to glutamate in the Nac2::HA gene construct. This mutated gene was introduced into the mø14 strain by nuclear co-transformation using plasmids containing the cry (which confers cryptopleurine and emetine resistance, Nelson et al., 1994) or the ble gene (which confers phleomycin and zeomycin resistance, Stevens et al., 1996). In a control co-transformation experiment with the wild-type Nac2::HA construct and the PSP115 plasmid (containing the ble gene), 20% of the zeomycin-resistant transformants displayed wild-type fluorescence transients. However, when the mutant Nac2(A1038E)::HA construct was used, none of the drug-resistant transformants could grow photoautotrophically. Amongst 100 transformants tested by immunoblotting for the presence of the Nac2 protein, only one accumulated detectable amounts of Nac2 protein representing 10% of the level found in the nac2::HA strain (Figure 6A). This transformant, nac2 A1038E::HA, was completely deficient in PSII activity as assayed by fluorescence transients (Figure 6B) and unable to grow photoautotrophically. This suggests that the mutation in the fourth TPR repeat inactivates and partially destabilizes the protein and that only in rare cases do limited amounts of mutant protein accumulate when the transgene is highly expressed, presumably because of its insertion near a strong promoter. Analysis of this transformant revealed that the altered Nac2 protein forms a high molecular weight aggregate that is fully resistant to both EDTA treatment and RNase digestion (Figure 4B, rows 4–6).

Fig. 6. The Nac2 A1038E protein is expressed but does not rescue the mø14 null mutant. (A) Immunoblot analysis of total cell proteins from the nac2::HA, mø14 and nac2 A1038E::HA strains. Serial dilutions of the nac2::HA extracts were performed with total cell extracts from the mø14 strain in order to maintain the protein amount (100 µg) constant in each lane. Samples were loaded on a 6% SDS–polyacrylamide gel and reacted with anti-HA monoclonal antibody. (B) Fluorescence transients of dark-adapted cells of the wild-type (WT), mø14, nac2::HA and nac2 A1038E::HA strains. Cells were grown in liquid TAP medium under dim light and dark-adapted for 1 min before measurements.
Discussion
We have used a genomic rescue approach to isolate and characterize the nucleus-encoded Nac2 factor that is required for the stabilization of the mature chloroplast psbD RNA. The isolated Nac2 cDNA contains the entire Nac2 ORF based on the following observations. (i) It is nearly equal in size to the 5.3 kb Nac2 mRNA. (ii) It encodes a polypeptide of 1385 amino acids with a predicted molecular mass of 140 kDa. This size agrees with that of the protein observed by immunodetection with the anti-Nac2 and the anti-HA antibodies. (iii) A stop codon is present in-frame upstream of the ATG initiation codon.
We have used cell fractionation and immunological means to show that the Nac2 protein is localized within the chloroplast stromal compartment. The N-terminal end of the protein has features that are compatible with those of a chloroplast transit peptide; in particular, it contains numerous basic and hydroxylated residues. A surprising finding is that the Nac2-deficient strain can be rescued with a truncated cDNA in which more than half of the Nac2 coding sequence is missing from the 5′ end. However, the efficiency of this rescue is considerably lower, at least by two orders of magnitude, as compared with the genomic Nac2 DNA. This suggests that upon integration into the nuclear genome, the cDNA is in rare cases fused in-frame to a coding sequence which could act as a transit sequence. It is well documented that signal sequences specific for a given membrane transport system lack a strict consensus and that up to 25% of randomly produced peptides can act as signal sequences for the endoplasmic reticulum, mitochondria or the bacterial plasma membrane (Schatz and Dobberstein, 1996). The same situation is likely to hold for chloroplast transit sequences. Immunoblot analysis of the transformants probed with the Nac2 polyclonal antibody revealed proteins of aberrant sizes (data not shown). This is consistent with the fact that in order to be imported into the chloroplast, the cDNA had to recruit transit-like sequences in these transformants.
The cDNA rescue experiments indicate that the N-terminal half of the Nac2 protein is not essential for its function. A striking feature of the C-terminal half of the protein is the presence of nine TPR-like domains, eight of which are tandemly arranged. TPR proteins have been found in a wide range of organisms and in different cellular locations including the nucleus, the cytosol, mitochondria and chloroplasts (Lamb et al., 1995; Boudreau et al., 1997). The crystal structure of the three TPR domains of a protein phosphatase has revealed that these domains consist of a pair of antiparallel α-helices with adjacent TPR motifs packed together in a parallel arrangement (Das et al., 1998). TPR proteins are involved in many different biological processes. Furthermore, proteins containing multiple copies of TPR motifs appear to function as scaffolding proteins and, in some cases, to coordinate the assembly of proteins into multisubunit complexes. Several multimolecular complexes involving TPR proteins have indeed been identified, such as the mitochondrial import receptor complex, the peroxisomal import receptor complex, a transcription repression complex and the anaphase-promoting complex (for a review, see Lamb et al., 1995). Interestingly, the Nac2 protein also appears to be part of a high molecular weight complex that is associated with RNA. Although EDTA treatment of the complex shifts it to a smaller size, the Nac2 protein is not stably associated with polysomes. It is not yet clear whether the high molecular weight complex is a homo- or hetero-multimer. At least one TPR motif appears to be essential for the proper folding of the Nac2 protein in a functional form based on the observation that the Ala1038→Glu mutation in the fourth TPR domain partially destabilizes the protein and leads to the formation of a high molecular weight aggregate which is fully inactive.
Previous work has clearly indicated that the target site for the Nac2 function is the psbD 5′-UTR. The specificity of this interaction is rather high as other chloroplast mRNAs are unaffected by the absence of the Nac2 factor and, reciprocally, mutations affecting the stability of other chloroplast mRNAs do not destabilize the psbD RNA, e.g. in the 222E nuclear mutant, which specifically lacks the psbB mRNA (Monod et al., 1992). The gene required for psbB mRNA accumulation, Mbb1, has recently been isolated and characterized (F.Vaistij, E.Boudreau, S.D. Lemaire, M.Goldschmidt-Clermont and J.-D.Rochaix, unpublished results). Interestingly, like Nac2, Mbb1 interacts with the 5′-UTR of its target mRNA, contains 10 tandemly arranged TPR-like motifs and is part of a high molecular weight complex, although of different size from the Nac2 complex. Thus two nucleus-encoded factors of C.reinhardtii share similar structural motifs, but are involved in the metabolism of two different chloroplast mRNAs. This raises the possibility that each of these complexes specifically recognizes one chloroplast 5′-UTR and recruits, through its TPRs, a common factor involved in this process, which could be a nuclease, a processing enzyme or a translation factor.
Attempts to demonstrate RNA binding of the Nac2 protein have been inconclusive (J.Nickelsen and J.D.Rochaix, unpublished results). It is thus likely that the Nac2 multiprotein complex recognizes the psbD 5′-UTR through another subunit of the complex or through an interaction with a specific RNA-binding protein. A possible candidate is a 47 kDa protein, the binding of which to the psbD 5′-UTR was strongly diminished in the absence of the Nac2 protein (Nickelsen et al., 1994). Other candidates include three independent nuclear gene products that have been identified genetically as suppressors of chloroplast psbD 5′-UTR mutations that destabilize psbD RNA (Nickelsen, 2000). The Nac2 protein is involved, directly or indirectly, in the processing of the psbD precursor RNA into its mature form (Nickelsen et al., 1999). In this respect, it is interesting to note that the maize Crp1 protein required for chloroplast RNA processing and translation is also part of a high molecular weight complex and contains pentatricopeptide repeats (PPR) of 35 amino acids that have been found in several plant organellar proteins and that are related to the TPR motifs (Fisk et al., 1999; Small and Peeters, 2000). Recent studies have revealed the existence of a 5′–3′ chloroplast RNA exonuclease (Drager et al., 1999; Nickelsen et al., 1999). One possibilty is that the Nac2 complex binds near the psbD RNA maturation site and thereby protects the psbD RNA from further digestion. Alternatively, it could catalyze an endonucleolytic process coupled with the protection of the 5′ end of the mature psbD mRNA. It is also possible that the Nac2 complex plays a role in the initiation of translation and that the observed instability of the psbD RNA in the mutant results from its inability to perform this process. However, in several psbD 5′-UTR mutants deficient in the initiation of translation, RNA accumulation and 5′ processing occur at almost wild-type levels (Nickelsen et al., 1999).
Materials and methods
Strains and media
The nac2-26 and the cw15 mutant strains have been described previously (Harris, 1989; Kuchka et al., 1989); mø14 was kindly provided by S.Purton. Tris acetate medium (TAP), high salt medium (HSM) and TAP minus arginine plates were prepared as described by Rochaix et al. (1988).
Isolation of nucleic acids and hybridizations
Total DNA from the wild-type, nac2-26, mø14 strains and the transformants were extracted as previously described (Boudreau et al., 1997; Rochaix et al., 1988). DNA preparations were digested with BglI restriction endonuclease. The resulting fragments were separated by agarose gel electrophoresis, transferred onto Hybond-N+ membranes (Amersham, Arlington Heights, IL) and hybridized with 32P-labeled Nac2-specific probes: probe A, a 615 bp PCR-amplified fragment synthesized from bases 2371–2986 relative to the start of the ORF; probe B, a 666 bp BglI cDNA fragment from bases 2952–3618; and probe C, a 303 bp BglI fragment from bases 3618–3921.
Total cellular RNA was isolated from cells grown under constant low light (40 µE/m2/s) of the C.reinhardtii strains 137c, nac2-26 and mø14 using Tri Reagent (Sigma Chemical Co, St Louis, MO). The mRNA was isolated using the PolyA Tract mRNA Isolation System IV (Promega Corporation, Madison, WI). Aliquots of mRNA (3 µg) were electrophoresed and transferred to Nitrocellulose Hybond-C nylon membranes (Amersham, Arlington Heights, IL) and hybridized with probes A and C. The blots were also hybridized with a 32P-labeled fragment specific to the rbcS gene.
Nuclear transformation of C.reinhardtii and plasmid rescue
Transformation of nac2-26 arg7 cw15 cells with total DNA from the C.reinhardtii genomic cosmid library containing the arginino-succinate lyase gene in the vector pPR691 was essentially as described (Purton and Rochaix, 1994, 1995). Twelve transformants selected for growth on HSM medium were obtained after plating 2 × 109 cells. A 20 µg aliquot of total DNA from four of these transformants was partially digested with HpaII, and electrophoretically separated on a 0.5% low melting point agarose gel. DNA fragments in the range of 10–25 kb were cut out and religated within the agarose as described (Sambrook et al., 1989). Samples were phenol/chloroform extracted twice and DNA was used for transformation of Escherichia coli using an electroporation apparatus (Bio-Rad). Bacterial clones containing the cosmid-derived bla gene were selected on ampicillin (100 µg/ml) LB plates. Plasmid DNA from the E.coli transformants was digested with NcoI and the resulting fragments were isolated after electrophoretic separation. These fragments were used to screen the same cosmid library that had been used to complement nac2-26 arg7. A 1 kb NcoI fragment was used to identify a cosmid called cosnac5 that complemented the nac2-26 mutant after glass bead-mediated transformation. Further subcloning, after partial digestion of cosnac5 with Sau3A, yielded a genomic fragment of 6 kb that was still able to complement nac2-26.
cDNA isolation and sequencing
A wild-type cDNA library of C.reinhardtii constructed by H.Sommer was screened with cosnac5 DNA (Sambrook et al., 1989). Five different phages, out of 300 000 screened, hybridized to cosnac5. Only one of these, called cnac1, also hybridized with the genomic 6 kb subclone able to rescue the nac2-26 mutant. Cnac1 contains a 2.7 kb cDNA. Nested deletion subclones were generated (Henikoff, 1987) and sequenced using the USB sequencing kit in combination with a terminal transferase treatment after termination reactions following the manufacturer’s instructions. The longer 5.1 kb cDNA clone, called cnac2, was isolated after rescreening the cDNA library (500 000 p.f.u.) with a 0.9 kb BamHI–SmaI fragment located upstream of the genomic region corresponding to cnac1. Automated sequencing of subcloned cnac2 DNA was carried out at MWG Biotech, and homology searches were performed using the BLAST service (National Center for Biotechnology Information, Bethesda, MD). Sequence analysis was performed using PC/Gene software (University of Geneva). These sequence data have been submitted to the DDBJ/EMBL/GenBank databases under accession No. AJ271460.
For complementation analysis, the 2.7 kb cDNA was excised with XbaI and SalI and inserted into the likewise restricted nuclear expression vector pSP105 (Stevens et al., 1996). This gave rise to a transcriptional fusion with the rbcS leader.
Antiserum production
For antibody production, a 1.6 kb MseI–EcoRI fragment containing the 3′-terminal part of the nac2-26 coding region was cloned into the NdeI–EcoRI sites of the expression vector pET-15b (Novagen Inc., Madison, WI). Constructs were introduced by transformation in the expression host E.coli strain BL21 and expression was induced with 1 mM isopropyl-β-d-thiogalactopyranoside. The expected 40 kDa N-terminal His-tagged polypeptide was found in inclusion bodies. These were purified further on Ni-NTA–agarose columns (Qiagen) and eluates were dialyzed overnight against a buffer containing 50 mM ammonium carbonate and 0.1% SDS. Samples were extracted using the procedure of Wessel and Flügge (1984) to remove SDS. Proteins were resuspended in 50 mM Tris–HCl pH 7.6, 150 mM NaCl and dispersed by sonication prior to injection into mice.
DNA constructs
In order to integrate the triple HA epitope sequence at the end of the Nac2 ORF, an EcoRV site was created upstream of the Nac2 stop codon by PCR mutagenesis. A KpnI–XhoI fragment containing the stop codon was subcloned in the Bluescript KS vector. A first PCR was performed with the universal primer and the antisense Nac2-RV oligonucleotide (5′-TCCTCAGATATCCATGTCGTCGGCATCGG-3′). The resulting PCR fragment was digested with KpnI and EcoRV and cloned into KS, giving rise to pNACKRV. A second PCR was made with the reverse primer and the sense NAC2-RV primer (5′-GATATGGATATCTGAGGAGGTGCCAGGCT-3′). The resulting fragment was digested with EcoRV and XhoI and cloned in the respective sites of pNACKRV, thus yielding a Nac2 subclone with an EcoRV site just upstream of the stop codon. A His6-Myc cassette (gift of M.Goldschmidt-Clermont) was inserted in-frame with the Nac2 ORF at the EcoRV restriction site. This subclone was recloned into the nac2 cDNA using the KpnI–XhoI restriction sites. A chimeric genomic::cDNA plasmid was created by replacing the 3.0 kb genomic SrfI–XhoI with the corresponding 1.96 kb SrfI–XhoI cDNA His-Myc sequence. The 3HA epitope was introduced in-frame in the BstEI site of the His-Myc sequence. The resulting clones were sequenced prior to transformation.
Cellular fractionation
Whole-cell samples were prepared by resuspending sedimented cells in protein inhibitor mix (20 mM EDTA, 10 mM ε-amino caproic acid, 50 µg/ml pepstatin A, 20 µg/ml leupeptin, 2 mM benzamidine HCl) and lysed in an equal volume of cell lysis buffer (100 mM Tris–HCl pH 6.8, 4% SDS) at 37°C for 30 min. Samples were centrifuged at 10 000 g for 10 min and the supernatant was transferred to a new tube. To prepare whole-cell soluble and insoluble fractions, resuspended cells were sonicated on ice for 1 min. Broken cells were centrifuged at 100 000 g for 1 h. The supernatant was transferred to a new tube whereas membranes were resuspended in equal volumes of protein inhibitor mix and cell lysis buffer.
Chloroplasts from cell wall-deficient strains were isolated according to Zerges and Rochaix (1998). Intact chloroplasts were isolated from a continuous Percoll gradient (10%/85%). To prepare chloroplast fractions, isolated intact chloroplasts were osmotically lysed in chloroplast lysis buffer [50 mM HEPES–KOH pH 7.8, 5 mM MgCl2, 5 mM β-mercaptoethanol, 5 mM ε-amino caproic acid, 25 µg/ml pepstatin A, 10 µg/ml leupeptin, 1 mM benzamidine HCl and 1 mM phenylmethylsulfonyl fluoride (PMSF)] by repeated pipetting followed by freezing at –70°C and thawing on ice. Ammonium sulfate was added to broken chloroplasts at a concentration of 0.5 M and incubated on ice for 20 min, then centrifuged at 100 000 g for 30 min. Pelleted membranes were washed once with 0.5 M ammonium sulfate, then centrifuged for 30 min at 100 000 g. Soluble proteins were precipitated from the supernatant by slowly adding ammonium sulfate (0.31 g/ml) on ice and centrifuging at 35 000 g. Membranes and pelleted soluble proteins were resuspended in equal volumes of protein inhibitor mix and cell lysis buffer. Extracts were standardized for concentration with the bicinchoninic acid protein assay (Smith et al., 1985).
The soluble fraction of the mø14 strain transformed with the Nac2::HA or the Nac2 A1038E::HA construct that was used for sucrose gradient centrifugation or size exclusion chromatography was prepared as follows. A 250 ml culture at 2 × 106 cells/ml was centrifuged and resuspended in 4 ml of breaking buffer (20 mM HEPES–KOH pH 7.8, 50 mM KCl, 5 mM ε-amino caproic acid, 25 µg/ml pepstatin A, 10 µg/ml leupeptin, 1 mM benzamidine HCl and 1 mM PMSF). For the size exclusion chromatography, the breaking buffer also contained, depending on the experiment, 10 µg/ml of RNase A with 10 mM MgCl2, or 0.5 mg/ml heparin with either 10 mM MgCl2 or 2.5 mM EDTA. The cells were broken with a French press at 1380 p.s.i.. Broken cells were centrifuged at 35 000 g for 45 min. The supernatant was centrifuged at 100 000 g for 1 h. For sucrose gradients, the supernatant was loaded on a 0.3–1.3 M linear sucrose gradient and centrifuged for 21 h at 230 000 g in an SW40 rotor (Beckman Instruments Inc., Fullerton, CA). One-tenth of each 500 µl fraction was loaded on a 6% SDS–polyacrylamide gel. For size exclusion chromatography, the supernatant was concentrated in a Centricon YM-30 (Millipore Corporation, Belford, MA) to a final concentration of 20 µg/µl. Fractionations were done on a Superose 6 PC3.2/30 column (Pharmacia Biotech, Upsala, Sweden) using the SMART System (Pharmacia Biotech). A 50 µl (1000 µg) aliquot of the concentrated proteins was injected in the column and elution was performed at 4°C with a buffer containing 20 mM HEPES–KOH pH 7.8, 50 mM KCl and 10 mM MgCl2 at a rate of 40 µl/min. Twenty-four 50 µl fractions, eluted 600 and 1800 µl after injection, were collected. For nac2 A1038E::HA extracts, three consecutive fractionations were performed and each 150 µl fraction was precipitated by addition of 8 vols of ethanol. Proteins were pelleted by centrifugation (15 min, 15 000 g), washed with 70% ethanol and resuspended in 10 µl of loading buffer. The fractions were loaded on a 6% SDS–polyacrylamide gel. The molecular weight range of each fraction from the sucrose gradient and the size exclusion chromatography was estimated by comparison with size standards included in the HMW Gel Filtration Calibration Kit (Pharmacia Biothech, Sweden).
Total RNA was prepared from 28 fractions of 150 µl eluted between 600 and 2000 µl after injection and obtained by three consecutive runs. After phenol/chloroform extraction and ethanol precipitation, the pellets were resuspended in 4 µl of sterile water, stained with ethidium bromide and electrophoresed through a 1.5% agarose–formaldehyde denaturing gel. The RNAs were either stained with ethidium bromide and electrophoresed through a 1.5% agarose–formaldehyde denaturing gel or denatured and slot-blotted onto a Hybond-N+ membrane (Amersham, Arlington Heights, IL). The blots were hybridized with a 32P-labeled EcoRI–PvuII fragment specific for the psbD gene.
Immunoblot analysis
A 75 µg aliquot of proteins from various extracts was incubated at 37°C at a final concentration of 10% glycerol, 1.4% SDS, 100 mM dithiothreitol, 30 mM Tris–HCl pH 6.8. Proteins were fractionated by electrophoresis in 6% SDS–polyacrylamide gels (Sambrook et al., 1989). Proteins were blotted onto nitrocellulose filters (Protran, Schleicher and Schuell, Inc., Keene, NH) blocked in phosphate-buffered saline containing 4% non-fat dry milk and 0.01% Tween-20, incubated with primary antisera or HA monoclonal antibody (Eurogentec, Belgium) at 4°C overnight, and reacted with peroxidase-linked anti-mouse Ig or anti-rabbit Ig for 1 h. Signals were visualized by enhanced chemiluminescence (ECL) (Durrant, 1990).
Nac2 mutagenesis
Mutagenesis of the Ala1038 residue into Glu was performed by PCR amplification with oligonucleotides F03 (5′-AAAAGGGCTACTGCCAGC) and F04 (5′-AAGCTGGGTACCTGCC). After digestion with KpnI, the resulting PCR fragment was exchanged with the BalI–KpnI fragment of cnac1, thus yielding the mutated cDNA cnac1Ala 1038Glu. The correct mutation was verified by sequencing. The mutated plasmid was digested with SrfI and Eco72I and introduced into the corresponding sites of the Nac2::HA gene. The resulting Nac2 A1038E::HA plasmid was introduced into the nuclear genome of the mø14 strain by co-transformation as previously described with the cry (Nelson et al., 1994) or the ble gene (Stevens et al., 1996). Transformants were selected on TAP plates supplemented with zeomycin (25 µg/ml) or emetine (75 µg/ml) under dim light. Strains expressing detectable levels of proteins of the Nac2 A1038E::HA protein were screened by immunoblot analysis.
Polysome extraction
The procedure for preparation of polysomes is a modification of the methods described by Barkan (1988) and Yohn et al. (1996). A pellet of 7.4 × 108 cells was resuspended in 2 ml of polysome extraction buffer (200 mM Tris–HCl pH 8.0, 50 mM KCl, 35 mM MgCl2, 25 mM EGTA, 0.2 M sucrose, 1% Triton X-100 and 2% polyethelene-10-tridecyl-ether) supplemented with inhibitors [0.5 mg/ml heparin, 100 mM β-mercaptoethanol, 100 µg/ml chloramphenicol, 1 mM PMSF, 1 mM 1,10-phenanthroline and 0.5% (v/v) protease inhibitor cocktail (Sigma, P8849)]. The cells were broken with a French press at 1380 p.s.i. and centrifuged immediately at 4°C for 15 min at 10 000 g. The supernatant was supplemented with sodium deoxycholate to a final concentration of 0.5% and layered onto cushions of 1.5 ml of 1.75 M sucrose overlaid with 1 ml of 0.5 M sucrose, both prepared in cushion buffer (40 mM Tris–HCl pH 8.0, 20 mM KCl, 30 mM MgCl2, 5 mM EGTA). The sucrose cushions were centrifuged at 4°C with a fixed angle rotor (Beckman TLA 100.3) for 3 h at 100 000 g. Polysomes were recovered in the pellet and resuspended in 50 µl of cushion buffer supplemented with inhibitors as above. The green supernatant (1 ml) was also recovered for further analysis. RNA extractions were performed with half of the polysome extract (25 µl) and half of the supernatant (500 µl) by phenol/chloroform extraction and ethanol precipitation. The pellets were resuspended in 8 µl of sterile water and 4 µl were stained with ethidium bromide and electrophoresed through a 1.5% agarose–formaldehyde denaturing gel.
Fluorescence transients
Fluorescence transients of cells adapted in the dark for 1 min were measured with a Plant Efficiency Analyzer (PEA, Hansatech Instruments, UK). Cells were grown in TAP liquid medium with continuous shaking under dim light
Acknowledgments
Acknowledgements
We thank M.Goldschmidt-Clermont for helpful comments, N.Roggli for preparing the figures, B.Schwencke for skilled technical assistance, and U.Kück for providing laboratory space to J.N. in the latter phase of the work. E.B. was supported by a postgraduate scholarship from the Natural Science and Engineering Research Council of Canada. S.D.L. was supported by a long-term EMBO fellowship ALTF 111-1999. This work was supported by grant 3100-050895.97 from the Swiss National Fund to J.-D.R. and grant Ni390/2-1 from the Deutsche Forschungsgemeinschaft to J.N.
References
- Barkan A. (1988) Proteins encoded by a complex chloroplast transcription unit are each translated from both monocistronic and polycistonic mRNAs. EMBO J., 7, 2637–2644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boudreau E., Takahashi,Y., Lemieux,C., Turmet,M. and Rochaix,J.-D. (1997) The chloroplast ycf3 and ycf4 open reading frames of Chlamydomonas reinhardtii are required for the accumulation of the photosystem I complex. EMBO J., 16, 6095–6104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danon A. and Mayfield,S. (1991) Light-regulated translational activators: identification of chloroplast gene specific mRNA binding proteins. EMBO J., 10, 3993–4001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das A.K., Cohen,P.W. and Barford,D. (1998) The structure of the tetratricopeptide repeats of protein phosphatase 5: implications for TPR-mediated protein–protein interactions. EMBO J., 17, 1192–1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drager R.G., Zeidler,M., Simpson,C.L. and Stern,D.B. (1996) A chloroplast transcript lacking the 3′ inverted repeat is degraded by 3′–5′ exoribonuclease activity. RNA, 2, 652–663. [PMC free article] [PubMed] [Google Scholar]
- Drager R.G., Girard-Bascou,J., Choquet,Y., Kindle,K.L. and Stern,D.B. (1998) In vivo evidence for 5′–3′ exoribonuclease degradation of an unstable chloroplast mRNA. Plant J., 13, 85–96. [DOI] [PubMed] [Google Scholar]
- Drager R.G., Higgs,D., Kindle,K.L. and Stern,D.B. (1999) 5′ to 3′ exoribonucleolytic activity is a normal component of chloroplast mRNA decay pathways. Plant J., 19, 521–531. [DOI] [PubMed] [Google Scholar]
- Drapier D., Suzuki,H., Levy,H., Rimbault,B., Kindle,K.L., Stern,D.B and Wollman,F.A. (1998) The chloroplast atpA gene cluster in Chlamydomonas reinhardtii. Functional analysis of a polycistronic transcription unit. Plant Physiol., 117, 629–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durrant I. (1990) Light-based detection of biomolecules. Nature, 346, 297–298. [DOI] [PubMed] [Google Scholar]
- Fisk D.G., Walker,M.B. and Barkan,A. (1999) Molecular cloing of the maize gene crp1 reveals similarity between regulators of mitochondrial and chloroplast geen expression. EMBO J., 18, 2621–2630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franzen L.G., Rochaix,J.-D. and von Heijne,G. (1990) Chloroplast transit peptides from the green alga Chlamydomonas reinhardtii share features with both mitochondrial and higher plant chloroplast presequences. FEBS Lett., 260, 165–168. [DOI] [PubMed] [Google Scholar]
- Goebl M. and Yanagida,M. (1991) The TPR snap helix: a novel protein repeat motif from mitosis to transcription. Trends Biochem. Sci., 16, 173–177. [DOI] [PubMed] [Google Scholar]
- Goldschmidt-Clermont M. (1998) Coordination of nuclear chloroplast expression in plants cells. Int. Rev. Cytol., 177, 115–180. [DOI] [PubMed] [Google Scholar]
- Gruissem W. and Schuster,G. (1993) Control of mRNA degradation in organelles. In Brawerman,G. and Belasco,J. (eds), Control of Messenger RNA Stability. Academic Press, Orlando, FL, pp. 329–365. [Google Scholar]
- Gumpel N.J., Ralley,L., Girard-Bascou,J., Wollman,F.A., Nugent,J.H. and Purton,S. (1995) Nuclear mutants of Chlamydomonas reinhardtii defective in the biogenesis of the cytochrome b6f complex. Plant Mol. Biol., 29, 921–932. [DOI] [PubMed] [Google Scholar]
- Harris E.H. (1989) The Chlamydomonas Sourcebook. Academic Press, San Diego, CA. [Google Scholar]
- Hayes R., Kudla,J., Schuster,G., Gabay,L., Maliga,P. and Gruissem,W. (1996) Chloroplast mRNA 3′ end processing by high molecular weight complex regulated by nuclear encoded RNA binding proteins. EMBO J., 15, 1132–1141. [PMC free article] [PubMed] [Google Scholar]
- Henikoff S. (1987) Unidirectional digestion with exonuclease III in DNA sequence analysis. Methods Enzymol., 155, 156–165. [DOI] [PubMed] [Google Scholar]
- Hirano T., Kinoshita,N., Morikawa,K. and Yanagida,M. (1990) Snap helix with knob and hole: essential repeats in S.pombe nuclear protein nuc2+. Cell, 60, 319–328. [DOI] [PubMed] [Google Scholar]
- Hwang S., Kawazoe,R. and Herrin,D.L. (1996) Transcription of tufA and other chloroplast-encoded genes is controlled by a circadian clock in Chlamydomonas. Proc. Natl Acad. Sci. USA, 93, 996–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuchka M., Goldschmidt-Clermont,M., van Dillewijn,J. and Rochaix, J.-D. (1989) Mutation at the Chlamydomonas nuclear NAC2 locus specifically affects stability of the chloroplast psbD transcript encoding polypeptide D2 of PSII. Cell, 58, 869–876. [DOI] [PubMed] [Google Scholar]
- Lamb J.R., Tugendreich,S. and Hieter,P. (1995) Tetratricopeptide repeat interactions: to TPR or not to TPR? Trends Biochem. Sci., 20, 257–259. [DOI] [PubMed] [Google Scholar]
- Leon P., Arroyo,A. and Mackenzie,S. (1998) Nuclear control of plastid and mitochondrial development in higher plants. Annu. Rev. Plant Physiol. Plant Mol. Biol., 49, 453–480. [DOI] [PubMed] [Google Scholar]
- Mayfield S.P., Christopher,B.Y., Cohen,A. and Danon,A. (1995) Regulation of chloroplast gene expression. Annu. Rev. Physiol. Plant. Mol. Biol., 46, 147–166. [Google Scholar]
- Meurer J., Berger,A. and Westhoff,P. (1996) A nuclear mutant of Arabidopsis with impaired stability on distinct transcripts of the plastid psbB, psbD/psbC, ndhH and ndhC operons. Plant Cell, 8, 1193–1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monod C., Goldschmidt-Clermont,M. and Rochaix,J.-D. (1992) Accumulation of chloroplast psbB RNA requires a nuclear factor in Chlamydomonas reinhardtii. Mol. Gen. Genet., 231, 449–459. [DOI] [PubMed] [Google Scholar]
- Mullet J.E. (1993) Dynamic regulation of chloroplast transcription. Plant Physiol., 103, 309–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson J.A.E., Savereide,P.B. and Lefebvre,P.A. (1994) The CRY1 gene in Chlamydomonas reinhardtii: structure and use as a dominant selectable marker for nuclear transformation. Mol. Cell. Biol., 14, 4011–4019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nickelsen J. (1998) Chloroplast RNA stability. In Rochaix,J.-D., Goldschmidt-Clermont,M. and Merchant,S. (eds), The Molecular Biology of Chloroplasts and Mitochondria in Chlamydomonas. Kluwer Academic Publishers, Dordrecht, The Netherlands, pp. 151–163. [Google Scholar]
- Nickelsen J. (2000) Mutations at three different nuclear loci of Chlamydomonas suppress a defect in chloroplast psbD mRNA accumulation. Curr. Genet., 37, 136–142. [DOI] [PubMed] [Google Scholar]
- Nickelsen J. and Link,G. (1993) The 54 kDa RNA-binding protein from mustard chloroplasts mediates endonucleolytic transcript 3′ end formation in vitro. Plant J., 3, 537–544. [DOI] [PubMed] [Google Scholar]
- Nickelsen J., van Dillewijn,J., Rahire,M. and Rochaix,J.-D. (1994) Determinants for stability of the chloroplast psbD RNA are located within its short leader region in Chlamydomonas reinhardtii. EMBO J., 13, 3182–3191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nickelsen J., Fleischmann,M., Boudreau,E., Rahire,M. and Rochaix,J.-D. (1999) Identification of cis-acting RNA leader elements required for chloroplast psbD gene expression in Chlamydomonas. Plant Cell, 11, 957–970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purton S. and Rochaix,J.-D. (1994) Complementation of a Chlamydomonas reinhardtii mutant using a genomic cosmid library. Plant. Mol. Biol., 24, 533–537. [DOI] [PubMed] [Google Scholar]
- Purton S. and Rochaix,J.-D. (1995) Characterization of the ARG7 gene of Chlamydomonas reinhardtii and its applications to nuclear transformation. Eur. J. Phycol., 30, 141–148. [Google Scholar]
- Rochaix J.-D. (1992) Post-transcriptional steps in the expression of chloroplast genes. Annu. Rev. Cell Biol., 8, 1–28. [DOI] [PubMed] [Google Scholar]
- Rochaix J.-D. (1996) Post-transcriptional regulation of chloroplast gene expression in Chlamydomonas reinhardtii.Plant Mol. Biol., 32, 327–341. [DOI] [PubMed] [Google Scholar]
- Rochaix J.-D., Mayfield,S., Goldschmidt-Clermont,M. and Erickson,J. (1988) Molecular biology of Chlamydomonas. In Shaw,C.H. (ed.), Plant Molecular Biology: A Practical Approach. IRL Press, Oxford, UK, pp. 253–275. [Google Scholar]
- Salvador M.L., Klein,U. and Bogorad,L. (1993) Light-regulated and endogenous fluctuations of chloroplast transcript levels in Chlamydomonas. Regulation by transcription and RNA degradation. Plant J., 3, 213–219. [DOI] [PubMed] [Google Scholar]
- Sambrook J., Fritsch,E.F. and Maniatis,T. (1989) Molecular Cloning: A Laboratory Manual. 2nd edn. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- Schatz G. and Dobberstein,B. (1996) Common principles of protein translocation across membranes. Science, 271, 1519–1526. [DOI] [PubMed] [Google Scholar]
- Shiina T., Allison,L. and Maliga,P. (1998) rbcL transcript levels in tobacco plastids are independent of light: reduced dark transcription rate is compensated by increased mRNA stability. Plant Cell, 10, 1713–1722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sieburth L.E., Berry-Lowe,S. and Schmidt,G.W. (1991) Chloroplast RNA stability in Chlamydomonas reinhardtii: rapid degradation of psbB and psbC transcripts in two nuclear mutants. Plant Cell, 3, 175–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sikorski R.S., Boguski,M.S., Goebl,M. and Hieter,P. (1990) A repeating amino acid motif in CDC23 defines a family of proteins and a new relationship among genes required for mitosis and RNA synthesis. Cell, 60, 307–317. [DOI] [PubMed] [Google Scholar]
- Silflow C. (1998) Organization of the nuclear genome. In Rochaix,J.-D., Goldschmidt-Clermont,M. and Merchant,S. (eds), The Molecular Biology of Chloroplasts and Mitochondria in Chlamydomonas., Kluwer Academic Publishers, Dordrecht, The Netherlands, pp. 25–40. [Google Scholar]
- Small I.D. and Peeters,N. (2000) The PPR motif—a TPR-related motif prevalent in plant organellar proteins. Trends Biochem. Sci., 25, 46–47. [DOI] [PubMed] [Google Scholar]
- Smith P.K. et al. (1985) Measurement of protein using bicinchoninic acid. Anal. Biochem., 150, 76–85. [DOI] [PubMed] [Google Scholar]
- Stern D.B. and Gruissem,W. (1987) Control of plastid gene expression: 3′ inverted repeats act as mRNA processing and stabilizing elements, but do not terminate transcription. Cell, 51, 1145–1157. [DOI] [PubMed] [Google Scholar]
- Stern D.B., Jones,H. and Gruissem,W. (1989) Function of plastid mRNA 3′ inverted repeats. J. Biol. Chem., 264, 18742–18750. [PubMed] [Google Scholar]
- Stern D.B., Radwanski,E.R. and Kindle,K.L. (1991) A 3′ stem/loop structure of the Chlamydomonas chloroplast atpB gene regulates mRNA accumulation in vivo. Plant Cell, 3, 285–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens D.R., Rochaix,J.-D. and Purton,S. (1996) The bacterial phleomycin resistance gene ble as a domonant selectable marker in Chlamydomonas. Mol. Gen. Genet., 251, 23–30. [DOI] [PubMed] [Google Scholar]
- Sugita M. and Sugiura,M. (1996) Regulation of gene expression in chloroplasts of higher plants. Plant Mol. Biol., 32, 315–326. [DOI] [PubMed] [Google Scholar]
- Vaistij F., Goldschmidt-Clermont,M. and Rochaix,J.-D. (2000) Stability determinants of the chloroplast psbB/T/H mRNAs of Chlamydomonas reinhardtii. Plant J., 21, 469–482. [DOI] [PubMed] [Google Scholar]
- Wessel D. and Flügge,U. (1984) A method for the quantitative recovery of protein in dilute solution in the presence of detergents and lipids. Anal. Biochem., 138, 141–143. [DOI] [PubMed] [Google Scholar]
- Yang J., Schuster,G. and Stern,D.B. (1996) CSP41, a sequence-specific chloroplast mRNA binding protein is an endoribonuclease. Plant Cell, 8, 1409–1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yohn C.B., Cohen,A., Dannon,A. and Mayfield,S.P. (1996) Altered mRNA binding and decreased translation initiation in a nuclear mutant lacking translation of the chloroplast psbA mRNA. Mol. Cell. Biol., 16, 3560–3566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zerges W. and Rochaix,J.-D. (1998) Low density membranes are associated with RNA-binding proteins and thylakoids in the chloroplast of Chlamydomonas reinhardtii.J. Cell Biol., 140, 101–110. [DOI] [PMC free article] [PubMed] [Google Scholar]