

Abstract
A chemo- and regioselective α-hydroxylation reaction of carbonyl compounds with molecular oxygen as oxidant is reported. The hydroxylation reaction is catalyzed by a dinuclear Pd(II) complex, which functions as an oxygen transfer catalyst, reminiscent of an oxygenase. The development of this oxidation reaction was inspired by discovery and mechanism evaluation of previously unknown Pd(III)–Pd(III) complexes.

Bimetallic catalysis, as we defined it in our previous work, is catalysis with synergistic redox cooperation between two metal centers.1 Several dinuclear complexes are known to catalyze redox transformations in organic chemistry and Nature,2 but the design of new dinuclear complexes for bimetallic redox catalysis is rare.3 While in monometallic redox transformations only one metal participates in redox chemistry, two metals share the redox work in bimetallic catalysis and can therefore lower activation barriers compared to mononuclear catalysts.1 In 2009, we reported the first reactions from organometallic Pd(III) compounds and implicated dinuclear Pd(III) complexes with metal–metal bonds in bimetallic directed oxidation catalysis.4 Subsequently, we sought to evaluate dinuclear Pd catalysts with potential for metal-metal redox cooperation for other challenging redox transformations. Here we report a chemo- and regioselective α-hydroxylation reaction of carbonyl compounds with molecular oxygen or air as oxidant, catalyzed by the dinuclear Pd(II) complex 1 (eq 1).
 |
(1) |
Dioxygen (O2) is a readily available, inexpensive oxidant. Catalysts that use O2 to re-oxidize a reduced catalyst but do not transfer either oxygen atom from O2 to the product are called oxidases. Pd complexes with oxidase reactivity have a successful history in synthesis, as exemplified by the Wacker oxidation and work by Sigman, Stahl, and Stoltz.5 Oxygenases incorporate one (monooxygenase) or both oxygen atoms (dioxygenase) from O2 into a substrate and are less common in organic synthesis.6 Here we report the first dinuclear Pd(II) oxygen transfer catalyst, which functions as a dioxygenase. We identified the dipalladium paddlewheel complex 1, originally developed by Cotton,7 which features four bridging hexahydro-2H-pyrimido[1,2-a]pyrimidine ligands (hppH, 2), and its use for chemo- and regioselective α-hydroxylation of carbonyl compounds to form tertiary alcohols with O2 as the only oxidant (eq 1).
Oxidation of α-methyl-β-tetralone (3) under one atmosphere of O2 in the presence of 5 mol% 1 produced α-hydroxy-α-methyl-β-tetralone (4) in 77% isolated yield in the absence of any other reagent or additive except solvent. α-Hydroxylation reactions of carbonyl compounds are typically performed by oxidation of the corresponding enolates or silyl enol ethers with an oxygen transfer reagent, such as m-CPBA, dimethyldioxirane (DMDO), or N-sulfonyl oxaziridines.8 During deprotonation, silyl enol ether formation, and oxidation, a stoichiometric amount of waste is generated in each operation. Other metal-mediated α-hydroxylation reactions of carbonyls with dioxygen typically afford hydroperoxides and require subsequent reduction, or have significantly smaller demonstrated substrate scope.9
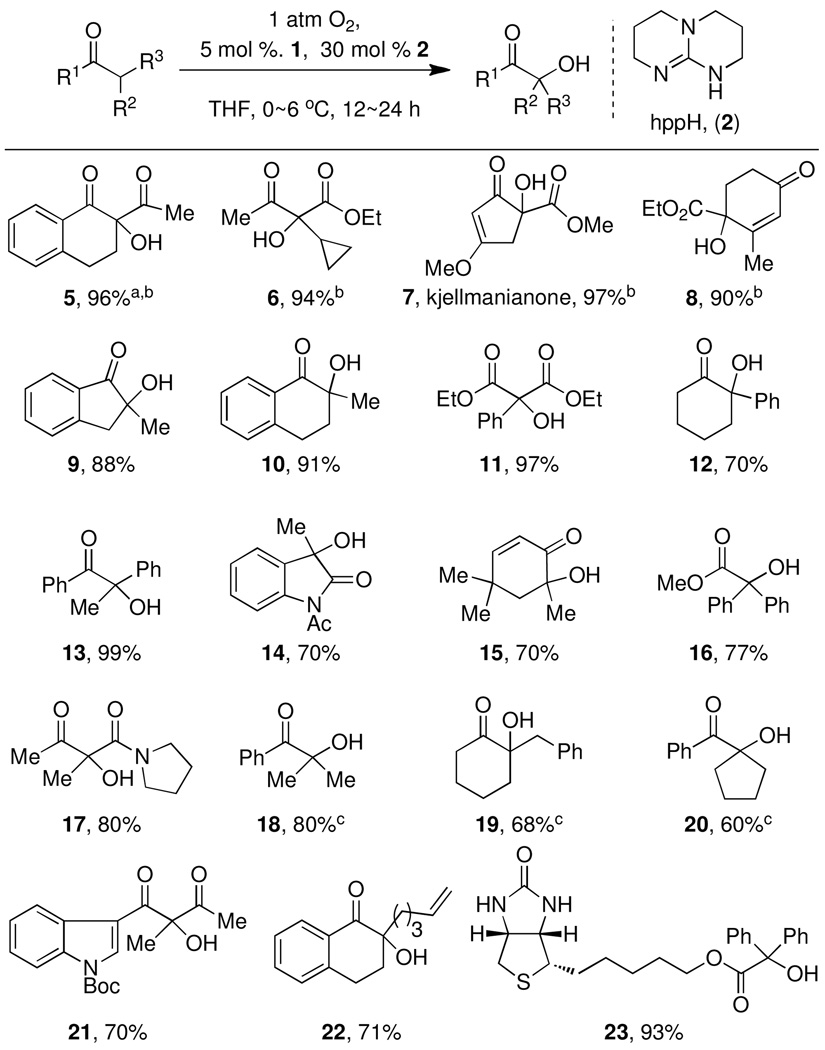
Hydroxylation proceeded smoothly between 0 °C and 6 °C in THF or toluene (for results in toluene and benzene, see Supporting Information). When the reaction was performed under one atmosphere of air instead of O2, isolated yields of hydroxylated products were 20–30% lower. The reaction could be applied to a variety of carbonyl compounds, similar or higher in acidity than tetralone, such as 5, with as little as 2.5 mol% catalyst 1 as shown in Table 1. Addition of substoichiometric amounts of the commercially available guanidine derivative hppH (2) enabled hydroxylation of carbonyl compounds less acidic than tetralone (see for example reaction to 19). The additive hppH likely does not function solely as a base for enolate formation; other strong amine bases such as hppMe and DBU did not have a beneficial effect. Hydroxylation of α-allyl cyclohexanone proceeded in less than 40% yield, even with 10 mol% catalyst, likely due to lower acidity, with starting material being the remainder of the isolated material. Carbonyl substitutions that favor enol formation by either hydrogen bonding (11) or conjugation (10) are advantageous for the hydroxylation reaction.
Table 1.
α-Hydroxylation reaction of carbonyl compounds with O2.
 |
2.5 mol% of 1 was used.
no 2 was added.
10 mol% of 1, and 60 mol% of 2 were used.
The hydroxylation reaction is regioselective and forms tertiary alcohols, even when more than one acidic C–H group is present such as in α-benzylcyclohexanone (reaction to 19). Oxidation of C–H acidic methylene groups to afford secondary alcohols was not observed due in part to overoxidation to the corresponding α-diketo compounds (see Supporting Information). The hydroxylation reaction catalyzed by 1 is chemoselective; double bonds (7, 15, 21, and 22) and sulfides (23), known to react with other electrophilic and nucleophilic oxygen transfer reagents,10 were compatible with the Pd-catalyzed oxidation presented here.
Our results indicate that the dinuclear Pd(II) catalyst 1 behaves as a dioxygenase: Oxygen transfer from O2 to the substrate was established by hydroxylation in an 18O2 atmosphere, which afforded the 18O-isotopologue as hydroxylation product in 97% isotopic purity as determined by mass spectrometry (eq 2). Based on an oxygen uptake experiment, both oxygen atoms from O2 were incorporated into the product. Hence, the oxidation reported here proceeds without generation of stoichiometric amounts of waste except solvent.
 |
(2) |
The mechanism of the hydroxylation reaction, formally an oxygen insertion into a C–H bond, is currently unknown. Because more acidic carbonyl compounds afford higher yields, we speculate that enol formation is important for hydroxylation. One role of hppH (2) may be to facilitate enolization. Several C–H autooxidation reactions by O2 and metal-catalyzed oxidation reactions with O2 proceed by radical mechanisms via hydrogen abstraction and often afford peroxides or hydroperoxides as products.11 No hydroperoxide was observed in the O2 oxidation reactions catalyzed by 1 (see Supporting Information). Successful hydroxylation via a potential hydrogen abstraction mechanism to afford 6 would require that radical recombination after hydrogen abstraction were faster than cyclopropane ring opening. Moreover, triphenylmethane (HCPh3), with a small C–H bond dissociation energy, was not oxidized under the reaction conditions. We therefore think that a hydrogen abstraction mechanism is less likely, and that the mechanism of the hydroxylation reaction presented here is distinct from the mechanism of radical metal-mediated and C–H autoxidation reactions by O2.
Our original design targeted high-valent Pd oxygen species, accessible by bimetallic synergistic redox participation, for oxidation chemistry. When the hydroxylation reaction shown in eq 1 was analyzed by in situ UV-vis spectroscopy, an absorption centered at 650 nm was observed spectroscopically (Figure 1). Upon discontinued agitation, this absorption disappeared within a minute, and remaining coloration was observed at the interface between reaction solution and dioxygen atmosphere. Previously, we have reported a variety of dinuclear Pd(III) complexes, all of which display a UV-vis absorption maximum around 600 nm, which was not observed for the corresponding Pd(II) complexes.1,4 For comparison, we prepared the dinuclear Pd(III) benzoate complex 24, featuring oxygen ligands on both Pd centers. As shown in Figure 1, 24 displays a similar UV-vis absorption centered at 650 nm as observed during the reaction. Computational analysis predicted that the absorption at 650 nm is due to a transition from a metal–metal bonding to a metal–metal antibonding orbital.12 Catalyst 1, substrate 3, and a combination thereof do not display a similar absorption, and no mononuclear Pd catalyst that was investigated afforded hydroxylation comparable to 1 (see Supporting Information for details). While the intermediacy of a dinuclear Pd(III) complex is an enticing possibility, consistent with our original reaction design, substantiated discussion of mechanism is premature at this stage and requires additional experimentation.
Figure 1.

In situ UV-vis spectroscopy of reaction intermediate and dinuclear Pd(III) benzoate complex 24.
In conclusion, we report a chemo- and regioselective α-hydroxylation reaction of carbonyl compounds by oxygen transfer from O2, catalyzed by a dinuclear Pd(II) complex. While the hydroxylation reaction presented here is a first example of reaction development inspired by bimetallic Pd(III) catalysis, we anticipate that bimetallic Pd(III) catalysis may serve as a concept for synthetically useful reaction discovery.
Supplementary Material
Acknowledgement
We thank NSF for financial support (CHE-0952753), Theodore Betley and David Powers for discussions, and NSC of Taiwan for a post graduate fellowship for GJC (NSC-972917I564145). DFT computation was performed using the computer facilities at the Odyssey cluster at Harvard University.
Footnotes
Supporting Information Available: Detailed experimental procedures and spectroscopic data for all new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- 1.Powers DC, Benitez D, Tkatchouk E, Goddard WA, III, Ritter T. J. Am. Chem. Soc. 2010;132:14092. doi: 10.1021/ja1036644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.For examples, see: van der Beuken EK, Feringa BL. Tetrahedron. 1998;54:12985. Espino C, Du Bois J. Angew Chem Int Ed. 2001;40:598. doi: 10.1002/1521-3773(20010202)40:3<598::AID-ANIE598>3.0.CO;2-9. Bugg TDH. Tetrahedron. 2003;59:7075. Doyle M. J. Org. Chem. 2006;71:9253. doi: 10.1021/jo061411m.
- 3.A bimetallic reaction is characterized by synergistic metal-metal redox participation.1 For a leading reference on non-redox catalysis with multinuclear catalysts, see: Shibasaki M, Yamamoto Y, editors. Multimetallic Catalysts in Organic Synthesis. Wiley-VCH; 2004.
- 4.(a) Powers DC, Ritter T. Nat. Chem. 2009;1:302. doi: 10.1038/nchem.246. [DOI] [PubMed] [Google Scholar]; (b) Powers DC, Geibel MAL, Klein JEMN, Ritter T. J. Am. Chem. Soc. 2009;131:17050. doi: 10.1021/ja906935c. [DOI] [PubMed] [Google Scholar]; (c) Powers DC, Xiao DY, Geibel MAL, Ritter T. J. Am. Chem. Soc. 2010;132:14530. doi: 10.1021/ja1054274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.(a) Smidt J, Hafner W, Jira R, Sieber R, Sedlmeier J, Sabel A. Angew. Chem. 1962;74:93. [Google Scholar]; (b) Stahl SS. Angew. Chem., Int. Ed. 2004;43:3400. doi: 10.1002/anie.200300630. [DOI] [PubMed] [Google Scholar]; (c) Sigman MS, Jensen DR. Acc. Chem. Res. 2006;39:221. doi: 10.1021/ar040243m. [DOI] [PubMed] [Google Scholar]; (d) Ebner DC, Bagdanoff JT, Ferreira EM, McFadden RM, Caspi DD, Trend RM, Stoltz BM. Chem. Eur. J. 2009;15:12978. doi: 10.1002/chem.200902172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.For recent examples of O2 fixation, see: Zhang J, Khaskin E, Anderson NP, Zavalij PY, Vedernikov AN. Chem. Commun. 2008:3625. doi: 10.1039/b803156h. Zhang Y-H, Yu J-Q. J. Am. Chem. Soc. 2009;131:14654. doi: 10.1021/ja907198n. Zhang C, Jiao N. J. Am. Chem. Soc. 2010;132:28. doi: 10.1021/ja908911n. Zhu M-K, Zhao J-F, Loh T-P. J. Am. Chem. Soc. 2010;132:6284. doi: 10.1021/ja100716x. Chiba S, Zhang L, Lee J-Y. J. Am. Chem. Soc. 2010;132:7266. doi: 10.1021/ja1027327. Schmidt VA, Alexanian EJ. Angew. Chem., Int. Ed. 2010;49:4491. doi: 10.1002/anie.201000843.
- 7.Cotton FA, Gu J, Murillo CA, Timmons DJ. J. Am. Chem. Soc. 1998;120:13280. [Google Scholar]
- 8.(a) Rubottom GM, Vazquez MA, Pelegrina DR. Tetrahedron Lett. 1974;15:4319. [Google Scholar]; (b) Adam W, Prechtl F. Chem. Ber. 1991;124:2369. [Google Scholar]; (c) Davis FA, Chen B-C. Chem. Rev. 1992;92:919. [Google Scholar]; (d) Davis FA, Clark C, Kumar A, Chen B-C. J. Org. Chem. 1994;59:1184. [Google Scholar]; (e) Davis FA, Liu H, Chen B-C, Zhou P. Tetrahedron. 1998;54:10481. [Google Scholar]
- 9.(a) Conover LH, Butler K, Johnston JD, Korst JJ, Woodward RB. J. Am. Chem. Soc. 1962;84:3222. [Google Scholar]; (b) Christoffers J. J. Org. Chem. 1999;64:7668. [Google Scholar]; (c) Baucherel X, Levoirier E, Uziel J, Juge S. Tetrahedron Lett. 2000;41:1385. [Google Scholar]; (d) Arai T, Sato T, Noguchi H, Kanoh H, Kaneko K, Yanagisawa A. Chem. Lett. 2006;35:1094. [Google Scholar]; (e) Monguchi Y, Takahashi T, Iida Y, Fujiwara Y, Inagaki Y, Maegawa T, Sajiki H. Synlett. 2008;15:2291. [Google Scholar]
- 10.(a) Davis FA, La1 SG. J. Org. Chem. 1988;53:5004. [Google Scholar]; (b) Zhang X, Foote CS. J. Am. Chem. Soc. 1993;115:8867. [Google Scholar]; (c) Aldea R, Alper H. J. Org. Chem. 1995;60:8365. [Google Scholar]
- 11.Frank CE. Chem. Rev. 1950;46:155. doi: 10.1021/cr60143a003. [DOI] [PubMed] [Google Scholar]
- 12.For computational details, see Supporting Information. For a related computational study of the UV-vis absorption spectrum of a Pd(III)–Pd(III) complex, see Cotton FA, Koshevoy IO, Lahuerta P, Murillo CA, Sanaú M, Ubeda MA, Zhao QL. J. Am. Chem. Soc. 2006;128:13674. doi: 10.1021/ja0656595.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.