

Abstract
The ability to regulate any protein of interest in living systems with small molecules remains a challenge. We hypothesized that appending a hydrophobic moiety to the surface of a protein would mimic the partially denatured state of the protein, thus engaging the cellular quality control machinery to induce its proteasomal degradation. We designed and synthesized bifunctional small molecules that bind a bacterial dehalogenase (HaloTag protein) and present a hydrophobic group on its surface. Remarkably, hydrophobic tagging of the HaloTag protein with an adamantyl moiety induced the degradation of cytosolic, isoprenylated, and transmembrane fusion proteins in cell culture. We demonstrated the in vivo utility of hydrophobic tagging by degrading proteins expressed in zebrafish embryos and by inhibiting RasG12V-driven tumor progression in mice. Therefore, hydrophobic tagging of HaloTag fusion proteins affords small molecule control over any protein of interest, making it an ideal system for validating potential drug targets in disease models.
Introduction
One of the central challenges of chemical biology remains the ability to perturb the function of any intracellular protein using a small molecule. While significant strides have been made towards developing individual ligands to specific proteins, only approximately 300 molecular targets for approved drugs have been characterized1. Furthermore, the fraction of the proteome classified as “undruggable” by current methods is estimated to be about 80%2. It is likely that many appealing drug candidates have yet to be found and that future advances in drug development will be able to overcome the boundaries of what is thought to be an “undruggable” target3,4. Therefore, the challenge for biologists remains to identify those disease-causing drug targets. To this end, advances in deep sequencing, microarray technology and genome-wide RNAi screens have been employed successfully to identify promising new drug targets. For instance, genome-wide RNAi screens have been employed to identify synthetic lethal interactions with mutated oncogenes and to identify genes necessary for various pathogenic infections5-7.
While target identification is an obvious important first step in drug development, the in vivo validation of these potential targets remains a challenge. This is due in part to the unpredictable pharmacokinetics/pharmacodynamics of any inhibitory compound identified based on in vitro inhibition of protein function. In other words, is the failure of a small molecule inhibitor to give the desired in vivo result an unforeseen consequence of its in vivo metabolism or is its target protein simply a poor drug target? To address this question, general methods are needed to functionally validate whether modulation of a putative disease-relevant protein leads to the desired in vivo result. RNAi offered initial promise for organismal validation of putative drug targets, however, the delivery and stability of duplex RNA remain major hurdles in knocking down mRNA expression in a whole animal setting8. In the absence of a direct ligand for the target protein, there are currently three categories of small molecule-based methods to control the function of a protein of interest (POI)9. First, the plant hormone auxin can be employed to dimerize a plant E3 ubiquitin ligase (TIR1) with a domain from the AUX/IAA transcriptional repressor (Aid1), which when fused to a POI can be ubiquitinated by proximity to TIR110. This method requires fusing the POI to Aid1, along with an introduction of the plant E3 ligase TIR1 into cells. A second general method used to deregulate protein function involves dimerization of FKBP12 and the FKBP12-rapamycin binding (FRB) domain from mTOR. It has been shown that a POI can be recruited to the proteasome or to the mitochondrial outer membrane by this method11-13. Again, at least two fusion proteins must be introduced into the cell for this system to function9. Lastly, two destabilizing domains (DDs), one based on the FKBP12 protein and the other on E. coli DHFR protein14,15, have been developed to destabilize a DD-POI fusion protein. The degradation conferring DD can be stabilized by inclusion of derivatives of FK50616 (in the case of mutagenized FKBP12) or the E. coli DHFR inhibitor trimethoprim (in the case of DHFR), ultimately leading to increased levels of the fusion protein. While the DD method has been successfully used in several studies17-20, it requires the continued presence of the ligand for stable expression of the fusion protein. This requirement can be a concern when studying developing embryos, which might not receive sufficient stabilizing ligand, or when studying the long term effects of a POI, in which case the ligand would have to be injected into an animal for the duration of the study. Also, in the case of the long-term expression of the POI, one must bear in mind the possible fluctuations of the POI levels that are due to the intermittent injections of the stabilizing ligand.
To develop a general method to degrade any intracellular protein using a small molecule, we sought to enlist the cellular protein quality control machinery. The burial of internal hydrophobic residues within a protein's core is a major driving force behind protein folding, and, correspondingly, exposure of such hydrophobic regions is considered a hallmark of an unfolded protein21-23. For instance, the endoplasmic reticulum Hsp70-class chaperone BiP specifically binds hydrophobic amino acids and helps slow-folding proteins to fold22,24. Should the cell fail to fold the target protein correctly, the unfolded protein is eliminated by either the ubiquitin-proteasome system or autophagy25. We sought to mimic the partially denatured state of a protein by appending a hydrophobic tag on its surface in order to induce its degradation. To test this hypothesis, we selected the HaloTag dehalogenase system developed by Promega as the fusion protein component26. This system was chosen because HaloTag fusion proteins are commercially available in various formats and the haloalkane reactive linker binds to the HaloTag domain covalently, suggesting a high specificity of the ligand for HaloTag. Here, we demonstrate that hydrophobic tagging affords rapid and robust control of the abundance of numerous proteins, including transmembrane receptors, in cultured cells as well as in zebrafish and mouse models.
Results
Hydrophobic tagging destabilizes HaloTag fusion proteins
We designed 21 structurally distinct scaffolds as the basis for our Hydrophobic Tags (HyTs), and synthesized and tested 30 compounds across these scaffolds composed of hydrophobic moieties linked to the HaloTag haloalkane reactive linker (Supplementary Table 1). In designing the hydrophobic portion of these bifunctional molecules, we used the compound library available in the Yale University Small Molecule Discovery Center as an informal resource to identify compounds that (1) maximized hydrophobicity, (2) minimized molecular weight, and (3) incorporated chemically diverse and commercially available scaffolds. To determine their biological activity, we generated a stable HEK 293T cell line expressing a luciferase-HaloTag fusion protein and treated these cells with the HyT compounds at 1 μM for 24 hours. Remarkably, several non-toxic compounds appeared to reduce luciferase activity and we characterized the five most potent compounds further (Fig. 1a). All five HyTs exhibited high hydrophobicity scores (logP ranging from +3 to +5) and were active in a concentration-dependent manner, whereas the HyT5 control compound with two PEG groups did not decrease the luciferase activity (Fig. 1b). Based on these initial data, we continued our investigation of hydrophobic tagging-induced degradation with hydrophobic containing HyT13 because of the reported high stability and cell permeability of compounds bearing adamantyl groups27,28.
Figure 1. Hydrophobic tagging strategy using the HaloTag fusion protein system.
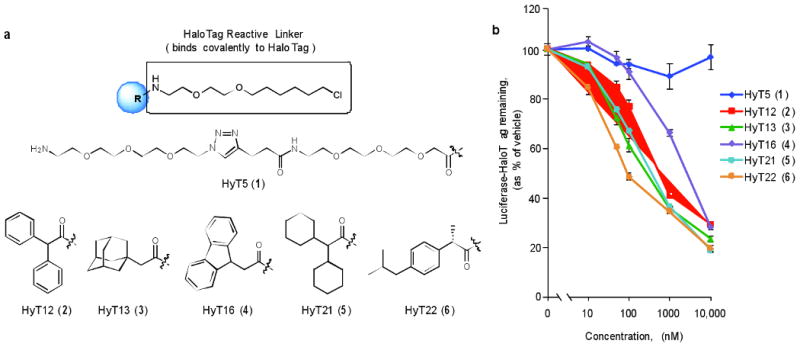
(a) Chemical structures of the representative HaloTag Ligands: HyT5, HyT12, HyT13, HyT16, HyT21 and HyT22. (b) HEK 293T cells expressing HA-HaloTag-luciferase were treated with indicated compounds at 1 μM for 24 hours, at which point luciferase assays were performed.
As our luciferase assay relied on the loss-of-activity of the luciferase-HaloTag fusion protein, we wanted to determine whether the decrease in luciferase activity resulted from the degradation of the entire fusion protein or perhaps simply inhibition of luciferase activity. We generated a stable Flp-In 293 cell line with a single integration site containing HA-EGFP-HaloTag fusion protein, and employed this cell line to perform kinetic studies with HyT13. Immunoblotting showed that HyT13 efficiently degraded the fusion protein, with a maximal effect achieved at 100 nM (Fig. 2a). The IC50 of HyT13 was determined to be 21 nM (Supplementary Results, Supplementary Fig. 1). A time course experiment revealed that the full effect is reached within 8 hours, with 50% degradation observed by 1.5 hours (Fig. 2b and Supplementary Fig. 2). When cells were treated with 1 μM HyT13 for 24 hours, and then the HyT13 was removed for 24 hours, the protein level recovered to half the starting levels. No cellular toxicity was observed at 20 μM of HyT13, a dose of 1000-fold over the IC50 value (Supplementary Fig. 3). Consistent with our hypothesis that hydrophobic tagging mimics a partially denatured protein state and that the protein is ultimately delivered to the proteasome for degradation, inclusion of proteasome inhibitors MG132 and YU10129 blocked HyT13 mediated degradation (Fig. 2c). To verify that the observed decrease in HA-EGFP-HaloTag levels does not result from masking of the HA epitope during immunoblotting, we generated a HeLa cell line stably expressing EGFP-HaloTag and analyzed the intracellular fluorescence by flow cytometry. Consistent with our previous observations, treatment of these cells with 1 μM of HyT13 for 24 hours reduced the mean fluorescence intensity of cells almost 7-fold (Fig. 2d). Together, these findings provide the first experimental evidence that hydrophobic tagging represents a viable strategy for the control of protein levels.
Figure 2. HyT13 leads to degradation of HaloTag fusion proteins.

(a) Flp-In 293 cells expressing HA-EGFP-HaloTag were treated with indicated concentrations of HyT13 for 24 hours. The lysates were probed with anti-HA and anti-β-actin antibodies. (b) The same cell line as in (a) was treated for the indicated times with 1 μM HyT13. The rightmost sample was treated with HyT13 for 24 hours, after which HyT13-free media was provided for 24 hours. (c) The same cell line as in (a) was pretreated with proteasome inhibitors MG132 (10 μM) and YU101 (10 μM) for 1 hour prior to addition of 1 μM HyT13. The lysates were prepared from cells 6 hours after HyT13 addition. (d) HeLa cells stably expressing EGFP-HaloTag were treated with vehicle or 1 μM HyT13 for 24 hours, whereupon the intracellular GFP fluorescence was quantified by flow cytometry. MFI = mean fluorescence intensity. (e) HEK 293T cells stably expressing indicated transmembrane HA-HaloTag fusion proteins were treated with 1 μM HyT13 for 24 hours. Shown are representative images from at least three experiments; bands were quantified and mean degradation ± SEM is shown. (f) One-cell stage zebrafish embryos were injected with 100 ng of HA-HaloTag-Smad5 cRNA, grown to 256-cell stage and then treated with 10 μM HyT13 for 24 hours. Shown are representative images from at least three experiments; bands were quantified and mean degradation ± SEM is shown. Full gels are available in Supplementary Results.
Degradation of transmembrane and zebrafish proteins
One limitation of existing technologies for small molecule control of protein levels has been the difficulty of degrading transmembrane proteins9. To determine if hydrophobic tagging shares this limitation, we constructed several transmembrane-HA-HaloTag fusion proteins, such that the HaloTag portion would be intracellular. Ror2 is a single-pass receptor tyrosine kinase-like orphan receptor, which functions in Wnt ligand signaling30. Likewise, CD3E is a single-pass cell surface glycoprotein involved in antigen recognition31. CD9 is a 4-pass transmembrane protein from the tetraspanin family and it functions in integrin signaling32. Finally, G-protein coupled receptors GPR40 and Frizzled-4 are 7-pass transmembrane receptors for long-chain free fatty acids and Wnt proteins, respectively33,34. Treatment of HEK 293T cell lines stably expressing these transmembrane HaloTag fusion proteins with HyT13 efficiently induced their degradation (Fig. 2e), demonstrating the potential of our hydrophobic tagging system to degrade transmembrane proteins. These experiments show that fusions to either the amino or carboxy terminus of the HaloTag protein are susceptible to this small molecule-induced degradation strategy and that transmembrane proteins can be degraded by HyT13.
We also explored the possibility of employing the hydrophobic tagging system in the zebrafish Danio rerio. We injected HA-HaloTag-Smad5 cRNA into zebrafish embryos and then treated the embryos with either vehicle or HyT13. Immunoblotting of injected embryo lysates revealed that the fusion protein is very efficiently degraded, demonstrating that HyT13 is able to penetrate the chorion and can direct the HaloTag fusion proteins for degradation in zebrafish (Fig. 2f). These experiments show that HyT13 is capable of degrading fusion proteins in various cell lines, as well as in zebrafish embryos.
HyT13 suppresses HaloTag-RasG12V tumor burden in mice
We next explored the functional utility of HaloTag-based degradation of an oncogene by HyT13 both in cell culture and in mice. The small GTPase H-Ras is one of the most commonly mutated genes in cancer, with up to 90% of cancers harboring activating mutations in this gene35. Activating mutations, such as the H-RasG12V allele, lead to decreased dependence on extracellular mitogenic signals. Ectopic expression of H-RasG12V in mouse fibroblast cell line NIH-3T3 can lead to a transformed phenotype, as demonstrated by assays in cell culture and in mice. When H-RasG12V expressing cells are grown in culture under low serum conditions they lose cell-to-cell contact inhibition and form distinct foci instead of growing as a cellular monolayer. Furthermore, these transformed cells are capable of tumor formation when injected into immuno-compromised nude mice36,37. We investigated whether (1) HaloTag-H-RasG12V driven focus formation can be suppressed in NIH-3T3 cells and (2) HaloTag-H-RasG12V driven tumor burden in mice can be reduced by administration of HyT13. First, NIH-3T3 cells were stably infected with a HA-HaloTag-H-RasG12V retroviral construct. The encoded fusion protein was readily degraded with HyT13 (Fig. 3a). To test the HaloTag receptor specificity for HyT13, we generated a point mutation in the HaloTag protein (HaloTagD106A) that is unable to form a covalent bond with the reactive chloroalkane in HyT1326. Unlike HA-HaloTag-H-RasG12V, HA-HaloTag(D106A)-H-RasG12V fusion protein was unaffected by HyT13 (Fig. 3a). Next, we plated both cell lines sparsely (105 cells/10-cm plate) in 10% FBS containing media. The next day, the media was replaced with 1% FBS containing media and the cultures were treated with either vehicle or HyT13. By day 6, both vehicle-treated cell lines and HyT13-treated HA-HaloTag(D106A)-H-RasG12V expressing cells had formed many foci, whereas HA-HaloTag-H-RasG12V expressing cells treated with HyT13 had grown a normal monolayer of cells, much like the parental NIH-3T3 cells (Fig. 3b-c). In the absence of HyT13, HA-HaloTag-H-RasG12V expressing cells exhibited slightly higher number of colonies than HA-HaloTag(D106A)-H-RasG12V cells. However, we attribute this observation to slight differences in retroviral infection efficiencies, since we have observed instances where the HaloTag(D106A)-H-RasG12V cells exhibit more colonies than the HA-HaloTag-H-RasG12V cells as well (data not shown). These results demonstrate that hydrophobic tagging can be used to reduce protein activity in the context of in vitro cell culture.
Figure 3. Functional validation of HaloTag degradation by HyT13.
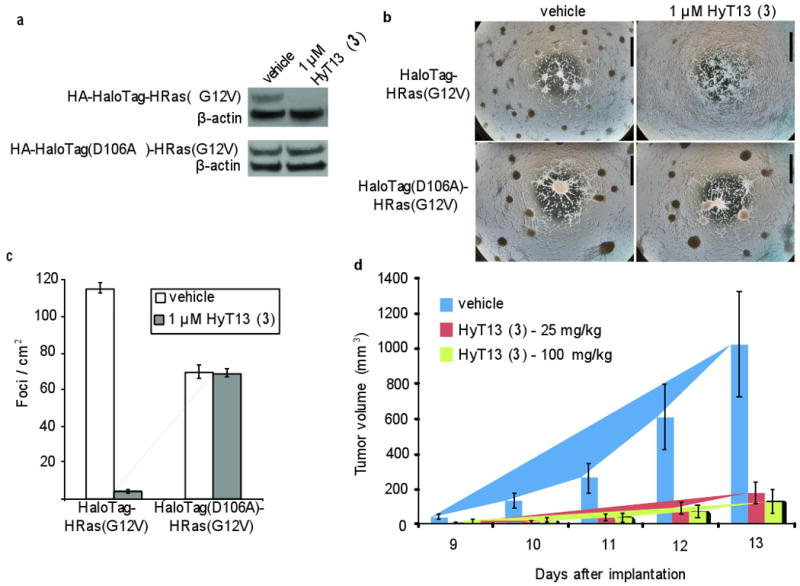
(a) NIH-3T3 cells were retrovirally infected with a construct expressing either HA-HaloTag-HRas(G12V) or HA-HaloTag(D106A)-HRas(G12V). The cells were then treated with vehicle or 1 μM HyT13 for 24 hours. The lysates were prepared for immunoblotting and the blots were probed with anti-HA and anti-β-actin antibodies. Full gels are available in Supplementary Results. (b) One hundred thousand NIH-3T3 cells infected with HA-HaloTag-HRas(G12V) or HA-HaloTag(D106A)-HRas(G12V) were plated in 10% FBS containing medium onto 10-cm plates. The next day, the medium was replaced with 1% FBS containing medium, along with vehicle or 1 μM HyT13. The media was refreshed every 2 days, and the plates were pictured on day 6. Bar, 5 mm. (c) Quantification of foci as described in (b). The number of foci/cm2 was counted from three separate plates, with error bars representing SEM. (d) One hundred thousand HA-HaloTag-HRasG12V-expressing NIH-3T3 cells were injected into the flank of nude mice on day 0. The mice were administered IP injections of vehicle or HyT13 daily from day 0. Tumor size was measured daily, and the tumor volume was calculated. Each treatment group employed 7 mice. Error bars represent SEM.
To examine whether the HaloTag:HyT13 based system could be used in mouse models to relieve the H-RasG12V-driven tumor burden, we first evaluated the pharmacokinetics of HyT13. We performed a maximum tolerated dose experiment with HyT13 in nude mice at doses up to 100 mg/kg over a 14-day treatment regimen. No obvious phenotype was observed even at the highest dose (Supplementary Fig. 4). Next, we sought to determine the serum bioavailability of HyT13 following injections. HyT13 was administered at 25 mg/kg by intraperitoneal (IP) injection into Swiss Webster mice and the serum was collected at 1 and 24 hours post-injection. At 1 hour post HyT13 administration the blood serum concentration was approximately 2 μM, and by 24 hours the HyT13 concentration had dropped to about 500 nM (Supplementary Fig. 5). Based on our previous experiments in a cell culture setting, we speculated that these serum HyT13 concentrations would be sufficient to suppress H-RasG12V tumor formation in mice. To test this, we injected NIH-3T3 cells expressing HA-HaloTag-H-RasG12V into the flank of nude mice and on the same day started a daily treatment regimen of vehicle, 25 mg/kg HyT13 or 100 mg/kg HyT13. Obvious solid tumor masses were observed on day 9 in vehicle-treated mice and the tumor volume grew exponentially until day 13, when the animals were sacrificed. The tumors in HyT13 mice were on average 6 times smaller than in vehicle treated mice, suggesting that HyT13 was able to reduce H-RasG12V tumor formation (Fig. 3d). These data clearly demonstrate the utility of the HaloTag:HyT13 system in perturbing protein function in live animals.
Discussion
Here we describe a novel hydrophobic tagging technology to systematically degrade levels of a specific protein upon addition of a small molecule (Fig. 4). This strategy has several benefits over the existing technologies. First, protein degradation is achieved upon compound administration as opposed to following ligand withdrawal. This aspect is particularly relevant when a protein needs to be expressed for long periods before the study, as there is no continuous ligand treatment necessary to maintain expression of the POI. In contrast, DD-based methods (see Introduction) of controlling protein abundance require constant drug administration, which can be both time-consuming and expensive. Also, there are likely fluctuations in the concentration of the fusion protein between ligand administrations using the DD-based system, whereas the expression of the HaloTag fusion protein is stable in the absence of the degradation signal. Therefore, depending on the application, it can be desirable to have a system where the small molecule induces degradation, rather than stabilization, of the POI. Second, our HaloTag:HyT13 method relies on the single introduction of a fusion domain to the POI. This feature contrasts with the auxin system, where an exogenous plant E3 ligase must be expressed in addition to the fusion protein. Third, almost all human and mouse genes are commercially available as both N- and C-terminal HaloTag fusions in transient and lentiviral expression vectors. These protein fusions with the 34 kDa HaloTag receptor are proving useful in many studies of protein function since they can be readily labeled in vivo and purified using fluorescent or biotinylated HaloTag reagents. The ability to degrade these fusion proteins with the hydrophobic tag HyT13 only adds to the repertoire of possible HaloTag applications. Although HyT13 is not yet commercially available, this small molecule can be obtained using standard synthetic methods in four steps from commercially available starting materials with an overall yield of 63% (see Supplementary Methods – Scheme 2).
Figure 4. Schematic of HyT13 mediated degradation of HaloTag fusion proteins.
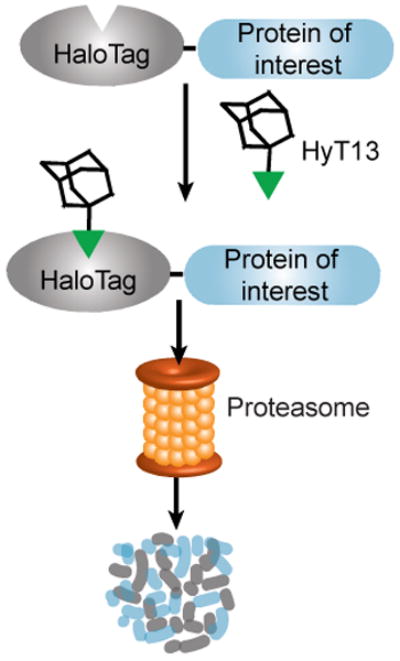
A fusion protein composed of a protein of interest and the HaloTag protein is degraded upon HyT13 treatment by the proteasome.
One of the criticisms that surround the several FKBP12 based degradation systems is their reliance on either rapamycin, FK506 or their derivatives to cause protein perturbation. Since these are bioactive small molecules, they could induce biological effects unrelated to perturbing the POI. In contrast, HaloTag dehalogenase is a bacterial gene and covalent binding of HyT13 to HaloTag affords this system a high degree of specificity. This bioorthogonality may explain the lack of noticeable HyT13 cytotoxicity even upon 1,000-fold administration over its IC50 value of 21 nM in cell culture. Moreover, mice injected daily with HyT13 at 100 mg/kg for 14 days gained weight normally, suggesting that HyT13 possesses no in vivo toxicity even at this high dose.
Like several other systematic degradation methods, the HaloTag:HyT13 methodology is not able to degrade endogenous proteins unless the HaloTag gene is fused with the gene of interest. However, there are two viable strategies to overcome this limitation and subject endogenous proteins to Halotag:HyT13-mediated regulation in culture or live animals. First, it is possible to generate HaloTag fusion constructs via targeted genome engineering. Recent advances in zinc finger nucleases20,38,39 and homologous recombination40 technologies open the possibility of systematically tagging endogenous proteins in rodents in a manner similar to yeast. The second approach would be to inactivate the endogenous gene by knockdown or knockout techniques and introduce the corresponding HaloTag fusion gene into the animal. Both approaches should be amenable to bypassing an early requirement of an essential gene, thus allowing the study of its function later during organogenesis or disease development.
In summary, herein we describe a chemical biology approach to systematically degrade any POI in either cell culture or whole animals. The system requires construction of a single fusion protein, which is specifically degraded by the addition of a non-toxic, low-molecular weight hydrophobic tag. We believe this system is particularly amenable to animal studies, as we have shown here with experiments in zebrafish and mice. Additionally, our findings suggest that hydrophobic tagging represents a novel approach to promote targeted degradation of endogenous proteins independent of the HaloTag:HyT13 system.
Methods
Synthesis and characterization of HyT compounds is described in Supplementary Methods
Cell culture and materials
Indicated cells were grown at 37°C in DMEM, supplemented with 10% fetal bovine serum and penicillin/streptomycin. The HaloTag protein was obtained from pHT2 vector (Promega). The luciferase sequence was obtained from pGL3-Basic vector (Promega), mouse Ror2 was kindly provided by Sigmar Stricker (Max Planck-Institute for Molecular Genetics), Danio rerio Smad5 was cloned from a zebrafish cDNA library and H-RasG12V was obtained from Addgene plasmid 9051, contributed by Robert Weinberg (MIT). The remaining transmembrane proteins were cloned from a human spleen cDNA library (Invitrogen). A D106A point mutation was introduced into the HaloTag gene by the QuikChange Site Directed mutagenesis kit (Stratagene). Flp-In 293 cells were purchased from Invitrogen. HA-HaloTag-Smad5 and EGFP-HaloTag were cloned into the pCS2+ vector, while the rest of the constructs were cloned into a retroviral pEYK3.1 vector (kindly provided by George Daley, MIT) by excising GFP41. Retrovirus was generated in GP2-293 cells (Clontech) with a pVSV-G and a corresponding pEYK plasmid, and the indicated cells were infected as described41. Anti-HA antibody was purchased from Covance (clone 16B12) and anti-β-actin antibody was purchased from Sigma (clone AC-74). HyT compounds were stored and aliquoted in DMSO as 1000× stock solutions.
Luciferase assay
Ten thousand stable HEK 293T cells infected with HA-luciferase-HaloTag were plated into each well in a 96-well plate. The next day, indicated HyT compounds were added in triplicate and the cells were cultured for another 24 hours. The cells were washed once with cold PBS and lysed in Passive Lysis Buffer (Promega). The luciferase activity was performed by Steady-Glo Luciferase Assay System (Promega) on a Wallac Victor 2 Plate Reader (Perkin Elmer) and the luciferase activity was normalized by protein concentration, as determined by the Bradford assay.
Immunoblotting
The indicated cells were washed twice with cold PBS and the cells were lysed in lysis buffer (1× PBS, 1% NP-40, 1 mM EDTA, 40 mM HEPES) with protease inhibitors. The lysates were cleared by centrifugation at 10,000 g for 5 min. The total protein concentration was determined by Bradford assay and 50 μg of protein was loaded onto an 8% Bis-Tris gel. To solubilize polyubiquitinated and aggregated proteins upon proteasome inhibition42 samples generated for Fig. 2e were lysed with a SDS lysis buffer (1× PBS, 1% NP-40, 1% SDS, 1% sodium deoxycholate, 1 mM EDTA, 40mM HEPES) with protease inhibitors. The blots were processed by standard procedures with indicated antibodies, and the band intensities were quantified by ImageJ.
Flow cytometry analysis
Stable HeLa cells were raised by cotransfection of pCS2/EGFP-HaloTag and p-Puro containing the puromycin resistance gene. A clonal population of cells expressing EGFP-HaloTag was isolated. These cells were treated with vehicle or 1 μM HyT13 for 24 hours, washed with PBS and trypsinized. The cells were resuspended in FBS-free DMEM and the intracellular GFP level was measured by FACSCalibur (BD Biosciences).
Zebrafish Danio rerio experiments
The wild-type fish line TLF was used for this study. The HA-HaloTag-Smad5 in pCS2+ plasmid was in vitro transcribed with the SP6 transcription kit (Ambion). The mRNA was injected at 100 ng/μL at the one cell stage and embryos were raised to the 256-cell stage, when they were moved to glass depression slides (10-per-well) and put in 1 ml E2 media with or without HyT13 (10 μM). Embryos were cultured at 28.6°C for 24 hours and then dechorionated and de-yolked as described43. Approximately 60 embryos per condition were collected for immunoblot analysis, as described above.
Focus formation assay
One hundred thousand NIH-3T3 cells infected with HA-HaloTag-H-RasG12V and HA-HaloTag(D106A)-H-RasG12V were plated onto 10-cm cell culture plates in 10% FBS with DMEM. The next day, the media was replaced with 1% FBS media and the cells were administered either vehicle or 1 μM HyT13. The media and the drug were replaced every two days. On day 6, the foci were photographed and counted as the number of distinct foci per 1-cm2 area.
Tumor formation assay
One hundred thousand NIH-3T3 cells expressing HA-HaloTag-H-RasG12V were injected into the flank of anesthetized 6-week old female nu/nu nude mice (Charles River Laboratories). Two hours later, the mice were IP injected with either vehicle (10 μL volume, with 5 μL DMSO and 5 μL of Cremophor EL), 25 mg/kg HyT13 or 100 mg/kg HyT13. The drug injections continued daily until the end of the experiment. Upon the appearance of tumors on day 7, the tumors were measured daily with calipers, and their volumes were calculated using the formula: a(b)2 / 2, where a and b represent the longest and shortest diameters of the tumor, respectively.
Animal experiments
All experimental protocols involving zebrafish and mouse work were performed under the auspices of Yale University's Institutional Animal Care and Use Committee (IACUC).
Supplementary Material
Acknowledgments
We wish to acknowledge financial support from the NIH (R01AI084140) and to thank the members of the Crews lab for critical reading of the manuscript. TWC was the Canadian Institutes of Health Research Jean-François St-Denis Fellow in Cancer Research and a Bisby Fellow. TBS is a recipient of the American Cancer Society fellowship.
Footnotes
Author contributions: T.K.N., H.S.T., A.R.S. and C.M.C. designed the research. T.K.N., H.S.T., A.R.S., M.J.S., T.W.C., T.B.S. and K.R. performed the experiments. T.K.N., H.S.T., S.A.H. and C.M.C. analyzed the data. T.K.N., H.S.T. and C.M.C. wrote and edited the manuscript.
Competing financial interests: The authors declare no competing financial interests.
References
- 1.Overington JP, Al-Lazikani B, Hopkins AL. How many drug targets are there? Nat Rev Drug Discov. 2006;5:993–996. doi: 10.1038/nrd2199. doi:nrd2199 [pii] [DOI] [PubMed] [Google Scholar]
- 2.Russ AP, Lampel S. The druggable genome: an update. Drug Discov Today. 2005;10:1607–1610. doi: 10.1016/S1359-6446(05)03666-4. doi:S1359-6446(05)03666-4 [pii] [DOI] [PubMed] [Google Scholar]
- 3.Dixon SJ, Stockwell BR. Identifying druggable disease-modifying gene products. Curr Opin Chem Biol. 2009;13:549–555. doi: 10.1016/j.cbpa.2009.08.003. doi:S1367-5931(09)00107-0 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Crews CM. Targeting the undruggable proteome: the small molecules of my dreams. Chem Biol. 2010;17:551–555. doi: 10.1016/j.chembiol.2010.05.011. doi:S1074-5521(10)00196-1 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Luo J, et al. A genome-wide RNAi screen identifies multiple synthetic lethal interactions with the Ras oncogene. Cell. 2009;137:835–848. doi: 10.1016/j.cell.2009.05.006. doi:S0092-8674(09)00529-7 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Krishnan MN, et al. RNA interference screen for human genes associated with West Nile virus infection. Nature. 2008;455:242–245. doi: 10.1038/nature07207. doi:nature07207 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Karlas A, et al. Genome-wide RNAi screen identifies human host factors crucial for influenza virus replication. Nature. 2010;463:818–822. doi: 10.1038/nature08760. doi:nature08760 [pii] [DOI] [PubMed] [Google Scholar]
- 8.Whitehead KA, Langer R, Anderson DG. Knocking down barriers: advances in siRNA delivery. Nat Rev Drug Discov. 2009;8:129–138. doi: 10.1038/nrd2742. doi:nrd2742 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schrader EK, Wilmington SR, Matouschek A. Making it easier to regulate protein stability. Chem Biol. 2010;17:917–918. doi: 10.1016/j.chembiol.2010.09.004. doi:S1074-5521(10)00316-9 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nishimura K, Fukagawa T, Takisawa H, Kakimoto T, Kanemaki M. An auxin-based degron system for the rapid depletion of proteins in nonplant cells. Nat Methods. 2009;6:917–922. doi: 10.1038/nmeth.1401. doi:nmeth.1401 [pii] [DOI] [PubMed] [Google Scholar]
- 11.Schneekloth JS, Jr, et al. Chemical genetic control of protein levels: selective in vivo targeted degradation. J Am Chem Soc. 2004;126:3748–3754. doi: 10.1021/ja039025z. doi:10.1021/ja039025z. [DOI] [PubMed] [Google Scholar]
- 12.Sakamoto KM, et al. Protacs: chimeric molecules that target proteins to the Skp1-Cullin-F box complex for ubiquitination and degradation. Proc Natl Acad Sci U S A. 2001;98:8554–8559. doi: 10.1073/pnas.141230798. doi:10.1073/pnas.141230798141230798[pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Robinson MS, Sahlender DA, Foster SD. Rapid inactivation of proteins by rapamycin-induced rerouting to mitochondria. Dev Cell. 2010;18:324–331. doi: 10.1016/j.devcel.2009.12.015. doi:S1534-5807(10)00013-4 [pii]10.1016/j.devcel.2009.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Iwamoto M, Bjorklund T, Lundberg C, Kirik D, Wandless TJ. A general chemical method to regulate protein stability in the mammalian central nervous system. Chem Biol. 2010;17:981–988. doi: 10.1016/j.chembiol.2010.07.009. doi:S1074-5521(10)00305-4 [pii]10.1016/j.chembiol.2010.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Banaszynski LA, Chen LC, Maynard-Smith LA, Ooi AG, Wandless TJ. A rapid, reversible, and tunable method to regulate protein function in living cells using synthetic small molecules. Cell. 2006;126:995–1004. doi: 10.1016/j.cell.2006.07.025. doi:S0092-8674(06)01013-0 [pii]10.1016/j.cell.2006.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Clackson T, et al. Redesigning an FKBP-ligand interface to generate chemical dimerizers with novel specificity. Proc Natl Acad Sci U S A. 1998;95:10437–10442. doi: 10.1073/pnas.95.18.10437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Herm-Gotz A, et al. Rapid control of protein level in the apicomplexan Toxoplasma gondii. Nat Methods. 2007;4:1003–1005. doi: 10.1038/nmeth1134. doi:nmeth1134 [pii]10.1038/nmeth1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Banaszynski LA, Sellmyer MA, Contag CH, Wandless TJ, Thorne SH. Chemical control of protein stability and function in living mice. Nat Med. 2008;14:1123–1127. doi: 10.1038/nm.1754. doi:nm.1754 [pii]10.1038/nm.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dvorin JD, et al. A plant-like kinase in Plasmodium falciparum regulates parasite egress from erythrocytes. Science. 2010;328:910–912. doi: 10.1126/science.1188191. doi:328/5980/910 [pii]10.1126/science.1188191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pruett-Miller SM, Reading DW, Porter SN, Porteus MH. Attenuation of zinc finger nuclease toxicity by small-molecule regulation of protein levels. PLoS Genet. 2009;5:e1000376. doi: 10.1371/journal.pgen.1000376. doi:10.1371/journal.pgen.1000376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Agashe VR, Shastry MC, Udgaonkar JB. Initial hydrophobic collapse in the folding of barstar. Nature. 1995;377:754–757. doi: 10.1038/377754a0. doi:10.1038/377754a0. [DOI] [PubMed] [Google Scholar]
- 22.Gething MJ. Role and regulation of the ER chaperone BiP. Semin Cell Dev Biol. 1999;10:465–472. doi: 10.1006/scdb.1999.0318. doi:S1084-9521(99)90318-X [pii] [DOI] [PubMed] [Google Scholar]
- 23.Lins L, Brasseur R. The hydrophobic effect in protein folding. FASEB J. 1995;9:535–540. doi: 10.1096/fasebj.9.7.7737462. [DOI] [PubMed] [Google Scholar]
- 24.Blond-Elguindi S, et al. Affinity panning of a library of peptides displayed on bacteriophages reveals the binding specificity of BiP. Cell. 1993;75:717–728. doi: 10.1016/0092-8674(93)90492-9. doi:0092-8674(93)90492-9 [pii] [DOI] [PubMed] [Google Scholar]
- 25.Kubota H. Quality control against misfolded proteins in the cytosol: a network for cell survival. J Biochem. 2009;146:609–616. doi: 10.1093/jb/mvp139. doi:mvp139 [pii] [DOI] [PubMed] [Google Scholar]
- 26.Los GV, et al. HaloTag: a novel protein labeling technology for cell imaging and protein analysis. ACS Chem Biol. 2008;3:373–382. doi: 10.1021/cb800025k. doi:10.1021/cb800025k. [DOI] [PubMed] [Google Scholar]
- 27.Mathias LJ, Jensen JJ, Reichert VT, Lewis CM, Tullos GL. Adamantane-containing polymers. Acs Sym Ser. 1996;624:197–207. [Google Scholar]
- 28.Tsuzuki N, et al. Adamantane as a brain-directed drug carrier for poorly absorbed drug. 2. AZT derivatives conjugated with the 1-adamantane moiety. J Pharm Sci. 1994;83:481–484. doi: 10.1002/jps.2600830407. [DOI] [PubMed] [Google Scholar]
- 29.Elofsson M, Splittgerber U, Myung J, Mohan R, Crews CM. Towards subunit-specific proteasome inhibitors: synthesis and evaluation of peptide alpha',beta'-epoxyketones. Chem Biol. 1999;6:811–822. doi: 10.1016/s1074-5521(99)80128-8. doi:S1074-5521(99)80128-8 [pii] [DOI] [PubMed] [Google Scholar]
- 30.Oishi I, et al. The receptor tyrosine kinase Ror2 is involved in non-canonical Wnt5a/JNK signalling pathway. Genes Cells. 2003;8:645–654. doi: 10.1046/j.1365-2443.2003.00662.x. doi:662 [pii] [DOI] [PubMed] [Google Scholar]
- 31.DeJarnette JB, et al. Specific requirement for CD3epsilon in T cell development. Proc Natl Acad Sci U S A. 1998;95:14909–14914. doi: 10.1073/pnas.95.25.14909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Masellis-Smith A, Shaw AR. CD9-regulated adhesion. Anti-CD9 monoclonal antibody induce pre-B cell adhesion to bone marrow fibroblasts through de novo recognition of fibronectin. J Immunol. 1994;152:2768–2777. [PubMed] [Google Scholar]
- 33.Briscoe CP, et al. The orphan G protein-coupled receptor GPR40 is activated by medium and long chain fatty acids. J Biol Chem. 2003;278:11303–11311. doi: 10.1074/jbc.M211495200. doi:10.1074/jbc.M211495200M211495200[pii] [DOI] [PubMed] [Google Scholar]
- 34.Kirikoshi H, et al. Molecular cloning and characterization of human Frizzled-4 on chromosome 11q14-q21. Biochem Biophys Res Commun. 1999;264:955–961. doi: 10.1006/bbrc.1999.1612. doi:10.1006/bbrc.1999.1612S0006-291X(99)91612-1[pii] [DOI] [PubMed] [Google Scholar]
- 35.Bos JL. ras oncogenes in human cancer: a review. Cancer Res. 1989;49:4682–4689. [PubMed] [Google Scholar]
- 36.Parada LF, Tabin CJ, Shih C, Weinberg RA. Human EJ bladder carcinoma oncogene is homologue of Harvey sarcoma virus ras gene. Nature. 1982;297:474–478. doi: 10.1038/297474a0. [DOI] [PubMed] [Google Scholar]
- 37.Shih C, Weinberg RA. Isolation of a transforming sequence from a human bladder carcinoma cell line. Cell. 1982;29:161–169. doi: 10.1016/0092-8674(82)90100-3. doi:0092-8674(82)90100-3 [pii] [DOI] [PubMed] [Google Scholar]
- 38.Porteus M. Design and testing of zinc finger nucleases for use in mammalian cells. Methods Mol Biol. 2008;435:47–61. doi: 10.1007/978-1-59745-232-8_4. doi:10.1007/978-1-59745-232-8_4. [DOI] [PubMed] [Google Scholar]
- 39.Ostrand-Rosenberg S. Animal models of tumor immunity, immunotherapy and cancer vaccines. Curr Opin Immunol. 2004;16:143–150. doi: 10.1016/j.coi.2004.01.003. doi:10.1016/j.coi.2004.01.003S0952791504000068[pii] [DOI] [PubMed] [Google Scholar]
- 40.Rago C, Vogelstein B, Bunz F. Genetic knockouts and knockins in human somatic cells. Nat Protoc. 2007;2:2734–2746. doi: 10.1038/nprot.2007.408. doi:nprot.2007.408 [pii]10.1038/nprot.2007.408. [DOI] [PubMed] [Google Scholar]
- 41.Koh EY, Chen T, Daley GQ. Novel retroviral vectors to facilitate expression screens in mammalian cells. Nucleic Acids Res. 2002;30:e142. doi: 10.1093/nar/gnf142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gies E, et al. Niclosamide prevents the formation of large ubiquitin-containing aggregates caused by proteasome inhibition. PLoS One. 2010;5:e14410. doi: 10.1371/journal.pone.0014410. doi:10.1371/journal.pone.0014410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Link V, Shevchenko A, Heisenberg CP. Proteomics of early zebrafish embryos. BMC Dev Biol. 2006;6:1. doi: 10.1186/1471-213X-6-1. doi:1471-213X-6-1 [pii]10.1186/1471-213X-6-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.