

Abstract
Metabolic syndrome, as manifested by visceral obesity, hypertension, insulin resistance, and dyslipidemia, is reaching epidemic proportions in the Western World, specifically the United States. Epidemiologic studies suggest that the increased prevalence of metabolic syndrome directly correlates with an increase in the consumption of fructose, mainly in the form of high-fructose corn syrup. This inexpensive alternative to traditional sugar has been increasingly utilized by the food industry as a sweetener since the 1960s. While augmented caloric intake and sedentary lifestyles play important roles in the increasing prevalence of obesity, the pathogenesis of hypertension in metabolic syndrome remains controversial. One intriguing observation points to the role of salt in fructose-induced hypertension. Recent studies in rodents demonstrate that increased dietary fructose intake stimulates salt absorption in the small intestine and kidney tubules, resulting in a state of salt overload, thus setting in motion a cascade of events that will lead to hypertension. These studies point to a novel interaction between the fructose-absorbing transporter, Glut5, and the salt transporters, NHE3 and PAT1, in the intestine and kidney proximal tubule. This paper will focus on synergistic roles of fructose and salt in the pathogenesis of hypertension resulting from salt overload.
1. Metabolic Syndrome and Increased Fructose Consumption
The increased incidence of metabolic syndrome, as manifested by obesity, insulin resistance, hypertension, and cardiovascular ailment has brought the question of modern lifestyles and diets to the forefront of public debate. Numerous discussions and investigations have focused on whether sedentary lifestyles, contemporary changes in diet, or both are responsible for the rise in obesity found throughout Westernized cultures.
Proponents of the diet change argument point to the fact that our current diet is comprised of unbalanced nutrients, mainly too many carbohydrates, and has a total caloric intake that is well above our daily needs [1, 2]. Some anthropologists who subscribe to this view believe that early humans (hominids) consumed a significant amount of meat and possibly obtained most of their food from hunting [3]. Competing hypotheses within the same camp suggest that early humans may have consumed a plant-based diet in general [4], or that hunting and gathering possibly contributed equally to their diet [5]. The relative proportions of plant and animal foods in the diets of early humans (Paleolithic) probably varied between regions. For instance, hunter-gatherers in tropical regions such as Africa probably consumed a plant-based diet, while populations in colder regions such as Northern Europe most likely obtained most of their food from meat [6]. A plant-based diet contains a large amount of carbohydrates, whereas a meat-based diet is low in carbohydrates and high in protein.
Fructose is a monosaccharide and one of the three most important blood sugars, along with glucose and galactose [7]. It is predominantly metabolized in the liver and plays an essential role in vital metabolic functions in the body, including glycolysis and gluconeogenesis [8]. Fructose is naturally present in fruits such as apples, peaches, pears, and bananas, as well as in honey. Although it was present in the diets of early humans in the form of fruits, several studies indicate that the consumption of dietary fructose has skyrocketed during the last four decades. This is mostly due to the marked increase in the intake of high fructose corn syrup, a common sweetener used throughout the food industry. It has been shown that augmented absorption of fructose in the intestine disrupts glucose metabolism in the liver and leads to an enhanced rate of triglyceride synthesis.
Current data suggests that life originated in a saltwater environment billions of years ago and eventually migrated to dry land where both salt and water are not as plentiful. As a result, this migration required organisms to develop physiological adaptations and evolve organs to conserve and recover salt and water in their bodies. It is to be noted that the flora available on land are not only sodium deficient but are also potassium rich. Thus, human physiology evolved in a sodium deficient, but potassium rich paradigm. Unfortunately, enhanced salt consumption has been the hallmark of dietary changes in Westernized cultures. Not surprisingly, the changes from a low-sodium diet to a diet high in sodium resulted in a steep rise in hypertension, especially in populations where salt and water have been traditionally scarce. This is supported by studies showing that salt intake, as well as hypertension, was lowest in many rural and under-developed areas in Africa relative to the industrialized world [9–11].
2. Increased Dietary Fructose, High-Salt Diet, and Hypertension
A recent report by the American Heart Association underscored its concern over the shift in daily diet intake by indicating that Americans are consuming an average of 22 teaspoons of sugar each day [11]. More alarming is the dietary intakes in teens between the ages 14 and 18, who consume an astonishing 34 teaspoons of added sugar a day [11]. Most of the added sugar comes from soft drinks and candy-a staggering 355 calories, and is predominantly in the form of fructose.
In addition to fructose, Americans consume a significant amount of salt, well above what is needed for normal functioning of heart, kidney, and other organs. The National Academy of Sciences considers amounts of salt ranging from 1,100 to 3,300 milligrams per day safe and adequate for healthy adults. But the typical American consumes 6 to 8 times that amount, much of it “hidden” in the processed foods that constitute more than half of what Americans eat. The Center for Disease Control (CDC) warns that increased salt intake is significantly raising the risk of hypertension, cardiovascular disease, and kidney failure [11].
It is universally agreed that metabolic syndrome and the obesity epidemic are the result of increased caloric intake coupled with decreased physical activity. However, the pathogenesis of hypertension in metabolic syndrome remains less well understood. Possibilities such as enhanced insulin secretion, activation of the renin angiotensin system, elevated serum uric acid, obesity, and decreased nitric oxide generation by vascular endothelial cells have been proposed, and evidence in support of their role in the pathogenesis of hypertension has been presented [14–16]. Some studies point to sodium retention, impairment of endothelial nitric oxide production, or sympathetic activation as contributing factors to the generation of hypertension in metabolic syndrome [15, 16]. A recent report suggested that increased expression of angiotensin II type 1 receptor in mesangial cells plays an important role in the pathogenesis of hypertension in metabolic syndrome [17], while other studies have implicated stress response, with an underlying abnormality in the enzyme 11beta-hydroxysteroid dehydrogenase (HSD1), as a contributing factor [18].
The steep increase in fructose consumption directly correlates with the increased incidence of metabolic syndrome and prevalence of hypertension in developed countries [19]. In rats, mice, and dogs, increased dietary fructose intake for several weeks has been shown to recapitulate many parameters of metabolic syndrome, including hypertension, insulin resistance, and hyperlipidemia [13, 19–27]. Indeed, increased dietary fructose intake has been shown to cause hypertension in rats, as early as 2 to 4 weeks after the start of the experiment [19–23]. There are notable species differences with regard to the rapidity of the onset of hypertension in mammalians: While dogs develop hypertension as early as 2 weeks, it takes more than 10 weeks for mice to develop hypertension when fed increased dietary fructose [13, 25–27].
Hypertension is a complex, multifactorial disorder, and attributing its etiology to a single factor is too simplistic. One major factor, however, which is essential to the understanding of blood pressure regulation and hypertension, is altered salt absorption in the kidney [28–31]. Several studies have implicated enhanced salt intake and absorption in the kidney, along with high insulin levels, as important factors in the pathophysiology of hypertension in metabolic syndrome [32–34]. It is worth mentioning that most of the high salt-containing (e.g., processed foods) diets are also deficient in potassium and adequate potassium intake is shown to improve hypertension control [35].
Given the independent roles of increased dietary fructose and salt in causing hypertension and given the increased consumption of both fructose and salt in our daily diet, the question needs be asked as to whether the increased dietary fructose and salt intake have any synergistic effect on blood pressure elevation. Recent studies have shed new light on the role of dietary fructose on salt absorption in the intestine or kidney, which will be discussed below.
Given the important role of fructose and salt in the pathogenesis of hypertension, a review of the literature on the pathways and molecules mediating their absorption in the intestine and kidney provides a timely discourse. Further, the newly identified role of fructose in activating salt absorption in the intestine and kidney will be discussed.
3. Fructose Absorption in the Small Intestine and Kidney Proximal Tubule: Glut2 versus Glut5
Fructose is absorbed in the small intestine and kidney proximal tubule, predominantly via Glut5 and Glut2, which are members of facilitative carbohydrate transporting family [36–41]. Glut5 is exclusively transporting fructose while Glut2 is capable of transporting both glucose and fructose [36–41]. Glut5 is expressed on the apical membrane of enterocytes; whereas, Glut2 is located intracellularly and on the basolateral membrane at basal state. Both Glut5 and Glut2 are upregulated in response to high levels of dietary fructose intake; however, in the presence of elevated fructose or glucose intake it is Glut2 that is recruited from the basolateral to the apical membrane while Glut5 remains in its original apical position [39–41]. Figures 1(a) and 1(b) depicts the localization and role of Glut2 and Glut5 in fructose absorption in jejunum at basal state (a) and in response to increased dietary fructose intake (b).
Figure 1.
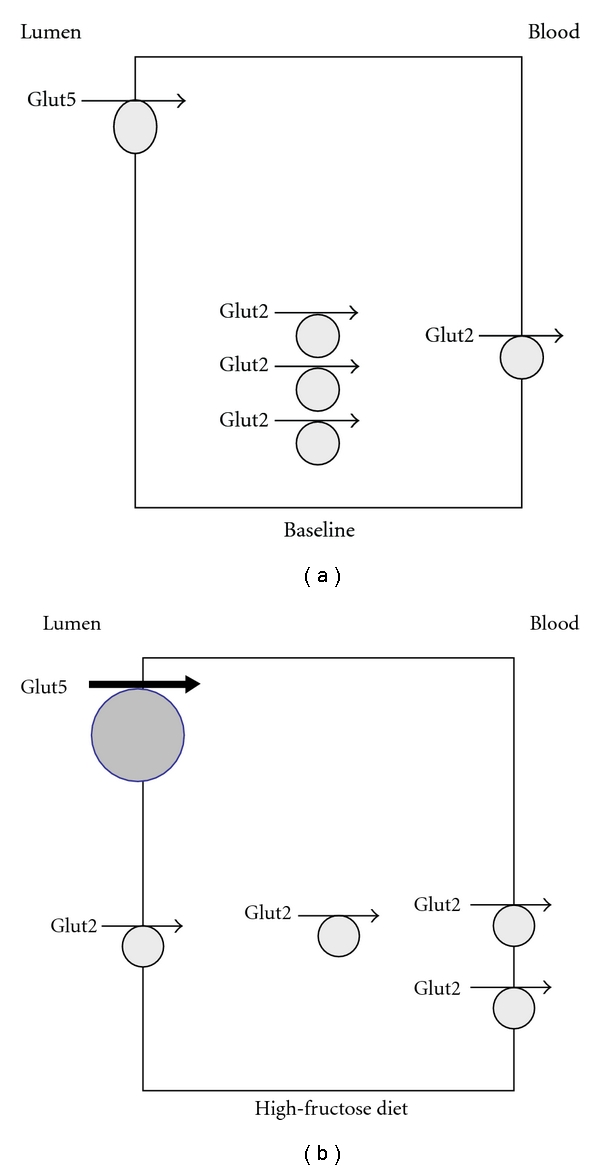
Localization of Glut2 and Glut5 in jejunum and their regulation in response to increased dietary fructose intake. Glut5 which only transports fructose but not glucose resides in the apical membrane at basal state (a) and is activated in response to increased dietary fructose intake (b). Glut2 which can transport both glucose and fructose is expressed in the cytoplasm and on basolateral membrane at basal state (a) and is recruited to the apical membrane in the presence of increased dietary fructose or glucose intake.
While it has been proposed that Glut2 is a major glucose- and fructose-absorbing transporter [36, 37], there is little evidence in support of fructose malabsorption in Glut2 deficient mice. On the other hand, Glut5 deletion abolishes the absorption of fructose in the small-intestine and causes a profound fructose malabsorption in Glut5 knock-out mice [42]. In addition to the small_intestine, Glut5 is abundantly expressed in the proximal tubule, specifically in the S3 segment [41].
4. Salt Absorption in the Small Intestine and Kidney Proximal Tubule
The absorption of salt (NaCl) in the small intestine is mediated via the Na+/H+ exchanger NHE3 (Slc9a3) working in parallel with Cl−/HCO3− exchangers DRA (Slc26a3) and PAT1 (Slc26a6) [43–57]. Figure 2 depicts the localization of salt absorbing transporters in the small intestine (a) and kidney proximal tubule (b). As shown, SLC26A6 (human)/Slc26a6 (mouse), also known as PAT1 (Putative Anion Transporter 1) is expressed on the apical membranes of upper villous epithelium in the small intestine and the kidney proximal tubule [46–54]. PAT1, which can function in vivo as both a Cl−/HCO3− and Cl−/oxalate exchanger, plays important roles in the absorption of salt and secretion of bicarbonate in the small intestine and in chloride absorption in the kidney proximal tubule [46–54]. DRA (Slc26a3) is expressed on the apical membrane of mid to lower villous epithelium in the small intestine and the upper villous epithelium of large intestine [55, 56] where it plays an essential role in chloride absorption in both locations. Human mutations rendering DRA inactive results in chloride-losing diarrhea [55]. Recent studies indicate that genetic deletion of DRA recapitulates the phenotype of chloride losing diarrhea in mice as well [56].
Figure 2.
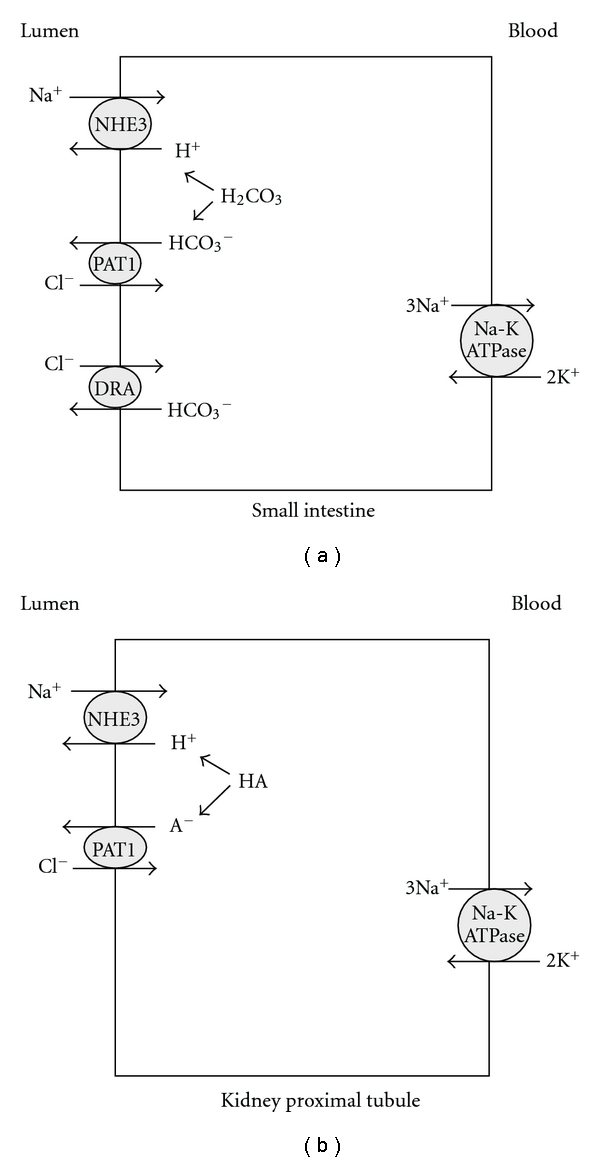
Localization of salt absorbing transporters in the small intestine (a) and kidney proximal tubule (b). NHE3: sodium hydrogen exchanger3: SLC9A3; PAT1: putative anion transporter 1: SLC26A6; DRA: down regulated in adenoma: SLC26A3.
5. Fructose Stimulates Salt Absorption in Perfused Jejunum: Interaction of Glut5 with PAT1
Recent studies have unmasked a remarkable interaction between fructose and salt absorption in the small intestine. These studies demonstrated that in vivo perfusion of the proximal jejunum in WT mice resulted in a significant increase in fluid absorption in the jejunum [13]. However, the stimulatory effect of fructose on salt was significantly blunted in mice deficient in PAT1−/− [13]. Contrary to the blunted response seen in the jejunum of PAT1 ko mice, luminal fructose elicited a very robust stimulatory effect on salt absorption in the jejunum of DRA ko mice (Seidler and Soleimani, unpublished data), indicating that DRA is not activated by fructose. The fructose-stimulated salt absorption in jejunum was completely dependent on luminal chloride or sodium and was abrogated in NHE3 ko mice (Seidler and Soleimani, unpublished data). Taken together, these studies strongly suggest that fructose-stimulated salt absorption is mediated via PAT1 and NHE3.
Recent studies have unmasked the minimum molecular machinery that mediates fructose-stimulated salt absorption in the small intestine. In vivo perfusion of the jejunum from Glut 5−/− mice indicated that the stimulatory effect of fructose on salt absorption was completely blocked [42], indicating that the presence of Glut5 is essential for fructose-stimulated salt absorption in the intestine. Given the essential roles of NHE3, and PAT1 in enhanced salt absorption in the intestine (above) it is speculated that Glut5, NHE3 and PAT1 are the major players in fructose-stimulated salt absorption. Figure 3 depicts the role of fructose and Glut5 in fructose absorption and fructose-stimulated salt absorption in the small intestine.
Figure 3.
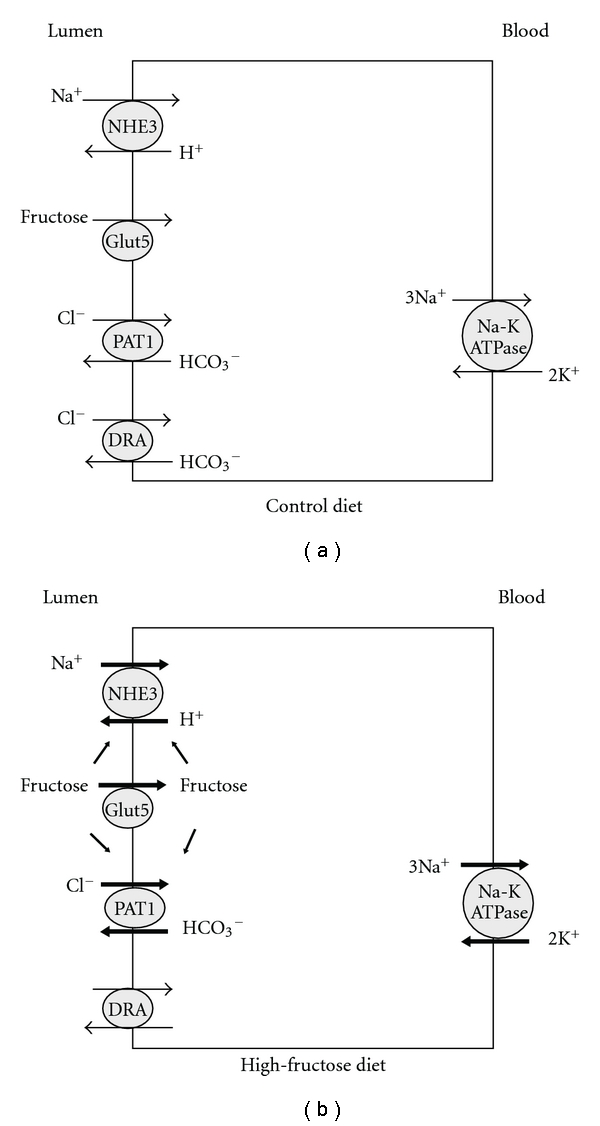
The role of fructose and Glut5 in fructose absorption and fructose-stimulated salt absorption in the small intestine. Luminal fructose is predominantly absorbed via Glut5 action and stimulates PAT1 and NHE3, therefore enhancing salt absorption in the jejunum. The stimulatory effect of luminal fructose on salt absorption in jejunum was completely abrogated in Glut5 null mice (see [12]).
6. Effect of Increased Dietary Fructose on the Expression of Glut5, PAT1, and NHE3 in the Small Intestine
In addition to the acute stimulatory effect of fructose on salt absorption in perfused jejunum, balanced studies demonstrated additional mechanisms through which fructose stimulates salt absorption. These studies indicated that animals that were fed 60% fructose diet for two weeks enhanced the mRNA expression and protein abundance of Glut5, PAT1, and NHE3 in their jejunum versus control diet (60% starch) [27].
The presence of a functional Glut5 is essential for fructose absorption and fructose-stimulated salt absorption in the small intestine. These conclusions are based on studies performed in Glut5 deficient animals that were subjected to a high-fructose diet. Results from these studies revealed that increased dietary fructose (60%) caused severe small intestinal malabsorption, as illustrated by massive dilation of bowel loops, specifically the cecum [42]. Furthermore, Glut5+/+ mice showed a sixfold increase in their blood fructose levels, whereas Glut5−/− mice had no noticeable increase in their blood fructose level even after five days on a high-fructose diet [42]. Lastly, Glut5−/− mice developed hypotension after 5 days of being fed a high fructose diet, due to volume depletion [42]. Taken together, these results strongly suggest that Glut5 is essential for the absorption of fructose and in conjunction with NHE3 and PAT1 play the central role in fructose-stimulated salt absorption in the intestine.
7. Effect of Increased Dietary Fructose Intake on Urinary Salt Excretion in Rats and Mice
The fructose concentration in the lumen of the small intestine directly reflects the amount of fructose present in the diet and can range from almost zero to more than 30–40 mM in humans or animals on a high-fructose diet. The fructose concentration in the blood and kidney tubule filtrate is ~2 orders of magnitude lower than the blood, with values ranging between 50 and 500 μM/liter in subjects on normal- and high-fructose diets, respectively. Despite a much lower fructose concentration in the kidney, there is clear evidence which supports an important role for fructose on salt absorption in the kidney proximal tubule. These results demonstrated that a high-fructose diet significantly decreased the daily excretion of chloride and sodium in the rat kidney while kidney function, including BUN and serum creatinine and urine osmolality, remained unchanged [13]. The stimulatory effect of a high-fructose diet on salt absorption in the kidney was also evident in wild-type mice.
In addition to its pivotal role in fructose-stimulated salt absorption in jejunum, PAT1 also plays an important role in enhanced kidney salt absorption in mice fed increased dietary fructose. Recent observation in our laboratory indicate that while sodium and chloride excretion rates in PAT1 wild and null mice are comparable on control diet, they are significantly increased in PAT1 null mice on high-fructose diet (unpublished data). Kidney function and serum uric acid remained normal and comparable in PAT+/+ and PAT−/− mice on high-fructose diet. Figure 4 depicts the effect of increased dietary fructose intake on Glut5 and salt absorbing transporters in the kidney proximal tubule.
Figure 4.
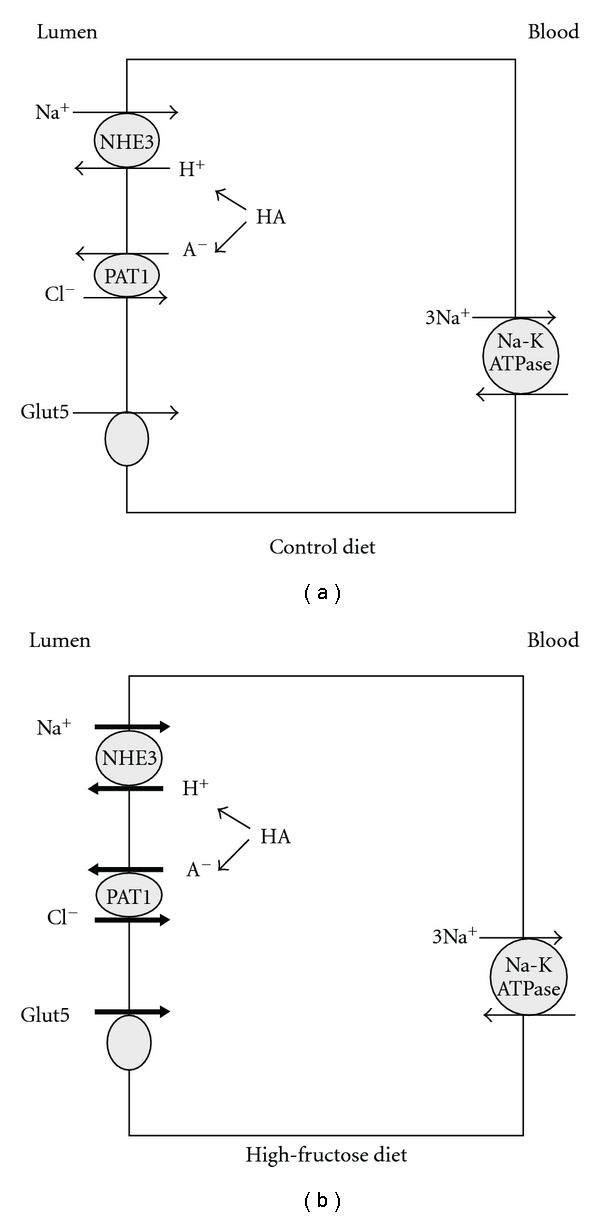
Effect of increased dietary fructose on Glut5 and salt-absorbing transporters in kidney proximal tubule. Increased dietary fructose intake enhances the expression of Glut5 and increases the absorption of salt via NHE3 and PAT1 in the kidney proximal tubule (see [13] and personal observation).
8. Fructose-Induced Hypertension: Role of NHE3, PAT1, and Glut5
Recent studies indicated that the development of fructose-induced hypertension requires functional PAT1 and Glut5 in the intestine and kidney. These studies demonstrated that PAT1+/+ mice on a high-fructose diet for 12 weeks developed significant increase in their blood pressure [13]. In contrast, PAT1−/− mice failed to develop hypertension on a high-fructose diet [13]. PAT1−/− mice displayed normal food intake and weight gain on either control or high-fructose diet versus PAT1+/+ mice. Both PAT1+/+ and PAT1−/− mice showed comparable increase in plasma fructose on increased dietary fructose regiment [13]. The role of intestinal Glut5 in fructose-stimulated salt absorption was dramatic: while Glut5+/+ mice tolerated the high-fructose diet and developed hypertension after 12 weeks, Glut5−/− mice developed massive fructose malabsorption and became hypotensive after only 5 days of augmented dietary fructose intake [12]. Taken together, these results indicate that PAT1 and Glut5 are essential for the generation of fructose-induced hypertension.
9. Role of Gender in Fructose-Induced Hypertension
Recent findings in our laboratory point to the role of gender in the severity of fructose-induced hypertension. When fed a high-fructose diet, male mice displayed a more severe hypertension relative to female mice (personal observation). It is intriguing to speculate that this effect might be in part related to the role of gender on the expression of salt absorbing transporters in the kidney proximal tubule [12].
10. Role of Salt Intake in Fructose-Induced Hypertension
If fructose stimulates salt absorption in the intestine and kidney, then it is reasonable to ask whether varying the amount of dietary salt has any impact on fructose-induced hypertension. Studies in rats have confirmed this speculation by showing that high salt intake enhances and low salt intake blunts the magnitude of fructose-induced hypertension [20]. These studies are in agreement with observations that fructose stimulates salt absorption in the intestine and kidney [13], and alterations in the amount of dietary salt will affect the magnitude of salt absorption from the intestine and kidney which in turn can impact the state of salt overload and hypertension.
11. Role of Various Monosaccharides in Salt Absorption in the Intestine and Kidney
It has been known for a very long time that glucose enhances sodium absorption in the small intestine via activation of Na-Glucose transporter (SGLT1), which actually formed the basis of the “oral rehydration solutions” adapted by the World Health Organization. In humans, oral glucose intake induces a slight and transient increase of blood pressure [58]. However, that effect is secondary to sympathetic activation [58] and not due to salt overload. It has been suggested that the consumption of excess sugar (glucose)-sweetened beverages is associated with the development and maintenance of obesity, which in turn can increase blood pressure via multiple factors [59]. However, there is no data indicating that increased dietary glucose intake increases blood pressure by increasing salt absorption in both the intestine and kidney.
A major difference between glucose and fructose metabolism is the fact that glucose absorption from the intestine triggers insulin release whereas fructose absorption does not. As a result of insulin release, the glucose concentration in the blood remains normal. It is therefore unlikely that increased glucose intake increases salt absorption in the kidney tubules since the glucose concentration in the glomerular filtrate is normal. However, increased dietary fructose intake does not stimulate insulin release, and fructose concentration in the blood and glomerular filtrate reaches levels 5–7 times above normal in rodents on increased dietary fructose intake [13, 42]. These differences might explain the increased salt absorption in the kidney tubules of rats or mice on increased dietary fructose intake [13].
12. Hyperuricimia Does Not Play a Major Role in the Generation of Fructose-Induced Hypertension
The possible contribution of hyperuricemia in fructose-induced hypertension has been the subject of several studies and many speculations. Our studies in rats on high-fructose diet for five weeks or in mice on high-fructose diet for 12 weeks did not demonstrate any elevation in serum uric acid; although, they did show enhanced excretion of uric acid in the urine [13]. These results clearly demonstrate that serum uric acid is not causally linked to the generation of hypertension in fructose-induced hypertension. Furthermore, it indicates that as long as kidney function remains stable elevated dietary fructose intake does not cause hyperuricemia. We speculate that the hyperuricemia that has been reported in rodents on high-fructose diet (reviewed in [22]) is a secondary phenomenon and results from decreased kidney function (GFR) subsequent to hypertension [13].
13. Summary and Conclusions
Based on recent published reports and observations in our laboratory we suggest that fructose-induced hypertension is generated in large part by a state of salt overload resulting from enhanced salt absorption in intestine and kidney. The increase in dietary fructose intake stimulates salt absorption in the small intestine and kidney proximal tubule through coordinated activation of PAT1, NHE3, and Glut5. We propose that reducing dietary intake of fructose and salt, as well as maneuvers aimed at inhibiting fructose absorption in the intestine and kidney tubules, could have profound beneficial effect on controlling blood pressure in patients with metabolic syndrome.
Acknowledgments
The authors acknowledge support from the Center on Genetics of Transport at University of Cincinnati and research funds from US Renal Care (to M. Soleimani).
References
- 1.Brand-Miller J, McMillan-Price J, Steinbeck K, Caterson I. Dietary glycemic index: health implications. Journal of the American College of Nutrition. 2009;28(4 SUPPL.):446S–449S. doi: 10.1080/07315724.2009.10718110. [DOI] [PubMed] [Google Scholar]
- 2.Acheson KJ. Carbohydrate for weight and metabolic control: where do we stand? Nutrition. 2010;26(2):141–145. doi: 10.1016/j.nut.2009.07.002. [DOI] [PubMed] [Google Scholar]
- 3.Cordain L. Implications of plio-pleistocene hominin diets for modern human. In: Ungar P, editor. Early Hominin Diets: The Known, the Unknown, and the Unknowable. Oxford, UK: Oxford University Press; 2006. pp. 363–383. [Google Scholar]
- 4.Jenkins DJA, Kendall CWC. The garden of Eden: plant-based diets, the genetic drive to store fat and conserve cholesterol, and implications for epidemiology in the 21st century. Epidemiology. 2006;17(2):128–130. doi: 10.1097/01.ede.0000199527.67506.1e. [DOI] [PubMed] [Google Scholar]
- 5.Corning P. Nature's Magic: Synergy in Evolution and the Fate of Humankind. Cambridge University Press; 2003. [Google Scholar]
- 6.Gowlett JAJ. What Actually was the Stone Age Diet? Journal of Nutritional and Environmental Medicine. 2003;13(3):143–147. [Google Scholar]
- 7.Sigman-Grant M, Morita J. Defining and interpreting intakes of sugars. The American journal of clinical nutrition. 2003;78(4):815S–826S. doi: 10.1093/ajcn/78.4.815S. [DOI] [PubMed] [Google Scholar]
- 8.Wolf G. The effect of fasting and fructose and glucose infusion on gluconeogenesis and triose phosphate flux in rats in vivo. Nutrition Reviews. 1995;53(10):299–301. doi: 10.1111/j.1753-4887.1995.tb01481.x. [DOI] [PubMed] [Google Scholar]
- 9.Carvalho JJM, Baruzzi RG, Howard PF, et al. Blood pressure in four remote populations in the INTERSALT study. Hypertension. 1989;14(3):238–246. doi: 10.1161/01.hyp.14.3.238. [DOI] [PubMed] [Google Scholar]
- 10.Forrester T. Historic and early life origins of hypertension in Africans. Journal of Nutrition. 2004;134(1):211–216. doi: 10.1093/jn/134.1.211. [DOI] [PubMed] [Google Scholar]
- 11.Johnson RK, Appel LJ, Brands M, et al. Dietary sugars intake and cardiovascular health a scientific statement from the american heart association. Circulation. 2009;120(11):1011–1020. doi: 10.1161/CIRCULATIONAHA.109.192627. [DOI] [PubMed] [Google Scholar]
- 12.Quigley R. Androgens stimulate proximal tubule transport. Gender Medicine. 2008;5(supplement 1):S114–S120. doi: 10.1016/j.genm.2008.03.011. [DOI] [PubMed] [Google Scholar]
- 13.Singh AK, Amlal H, Haas PJ, et al. Fructose-induced hypertension: essential role of chloride and fructose absorbing transporters PAT1 and Glut5. Kidney International. 2008;74(4):438–447. doi: 10.1038/ki.2008.184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Reusch JEB. Current concepts in insulin resistance, type 2 diabetes mellitus, and the metabolic syndrome. American Journal of Cardiology. 2002;90(5, supplement):19G–26G. doi: 10.1016/s0002-9149(02)02555-9. [DOI] [PubMed] [Google Scholar]
- 15.Kagota S, Yamaguchi YU, Tanaka N, et al. Disturbances in nitric oxide/cyclic guanosine monophosphate system in SHR/NDmcr-cp rats, a model of metabolic syndrome. Life Sciences. 2006;78(11):1187–1196. doi: 10.1016/j.lfs.2005.06.029. [DOI] [PubMed] [Google Scholar]
- 16.Grassi G, Seravalle G, Quarti-Trevano F, et al. Excessive sympathetic activation in heart failure with obesity and metabolic syndrome: characteristics and mechanisms. Hypertension. 2007;49(3):535–541. doi: 10.1161/01.HYP.0000255983.32896.b9. [DOI] [PubMed] [Google Scholar]
- 17.Packer CS. Angiotensin II as a multifunctional hormone: a regulator of haemodynamics and metabolism with potential roles in the etiologies of hypertension and the metabolic syndrome. Journal of Hypertension. 2006;24(1):37–38. doi: 10.1097/01.hjh.0000198044.69999.31. [DOI] [PubMed] [Google Scholar]
- 18.Sukhija R, Kakar P, Mehta V, Mehta JL. Enhanced 11β-hydroxysteroid dehydrogenase activity, the metabolic syndrome, and systemic hypertension. American Journal of Cardiology. 2006;98(4):544–548. doi: 10.1016/j.amjcard.2006.03.028. [DOI] [PubMed] [Google Scholar]
- 19.Elliott SS, Keim NL, Stern JS, Teff K, Havel PJ. Fructose, weight gain, and the insulin resistance syndrome. American Journal of Clinical Nutrition. 2002;76(5):911–922. doi: 10.1093/ajcn/76.5.911. [DOI] [PubMed] [Google Scholar]
- 20.Giacchetti G, Sechi LA, Griffin CA, Don BR, Mantero F, Schambelan M. The tissue renin-angiotensin system in rats with fructose-induced hypertension: overexpression of type 1 angiotensin II receptor in adipose tissue. Journal of Hypertension. 2000;18(6):695–702. doi: 10.1097/00004872-200018060-00006. [DOI] [PubMed] [Google Scholar]
- 21.Lee DH, Lee JU, Kang DG, Paek YW, Chung DJ, Chung MY. Increased vascular endothelin-1 gene expression with unaltered nitric oxide synthase levels in fructose-induced hypertensive rats. Metabolism. 2001;50(1):74–78. doi: 10.1053/meta.2001.19527. [DOI] [PubMed] [Google Scholar]
- 22.Johnson RJ, Sanchez-Lozada LG, Nakagawa T. The effect of fructose on renal biology and disease. Journal of the American Society of Nephrology. 2010;21(12):2036–2039. doi: 10.1681/ASN.2010050506. [DOI] [PubMed] [Google Scholar]
- 23.Hsieh P-S, Tai Y-H, Loh C-H, Shih K-C, Cheng W-T, Chu C-H. Functional interaction of AT1 and AT2 receptors in fructose-induced insulin resistance and hypertension in rats. Metabolism. 2005;54(2):157–164. doi: 10.1016/j.metabol.2004.07.016. [DOI] [PubMed] [Google Scholar]
- 24.Farah V, Elased KM, Morris M. Genetic and dietary interactions: role of angiotensin AT receptors in response to a high-fructose diet. American Journal of Physiology. 2007;293(2):H1083–H1089. doi: 10.1152/ajpheart.00106.2006. [DOI] [PubMed] [Google Scholar]
- 25.Sanchez-Lozada LG, Tapia E, Jiménez A, et al. Fructose-induced metabolic syndrome is associated with glomerular hypertension and renal microvascular damage in rats. American Journal of Physiology. 2007;292(1):F423–F429. doi: 10.1152/ajprenal.00124.2006. [DOI] [PubMed] [Google Scholar]
- 26.Martinez FJ, Rizza RA, Romero JC. High-fructose feeding elicits insulin resistance, hyperinsulinism, and hypertension in normal mongrel dogs. Hypertension. 1994;23(4):456–463. doi: 10.1161/01.hyp.23.4.456. [DOI] [PubMed] [Google Scholar]
- 27.Farah V, Elased KM, Morris M. Genetic and dietary interactions: role of angiotensin AT receptors in response to a high-fructose diet. American Journal of Physiology. 2007;293(2):H1083–H1089. doi: 10.1152/ajpheart.00106.2006. [DOI] [PubMed] [Google Scholar]
- 28.O'Shaughnessy KM, Karet FE. Salt handling and hypertension. Journal of Clinical Investigation. 2004;113(8):1075–1081. doi: 10.1172/JCI21560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Meneton P, Jeunemaitre X, De Wardener HE, Macgregor GA. Links between dietary salt intake, renal salt handling, blood pressure, and cardiovascular diseases. Physiological Reviews. 2005;85(2):679–715. doi: 10.1152/physrev.00056.2003. [DOI] [PubMed] [Google Scholar]
- 30.Karppanen H, Mervaala E. Sodium Intake and Hypertension. Progress in Cardiovascular Diseases. 2006;49(2):59–75. doi: 10.1016/j.pcad.2006.07.001. [DOI] [PubMed] [Google Scholar]
- 31.Haddy FJ. Role of dietary salt in hypertension. Life Sciences. 2006;79(17):1585–1592. doi: 10.1016/j.lfs.2006.05.017. [DOI] [PubMed] [Google Scholar]
- 32.Hall JE, Brands MW, Hildebrandt DA, Mizelle HL. Obesity-associated hypertension: hyperinsulinemia and renal mechanisms. Hypertension. 1992;19(1, supplement):I45–I55. doi: 10.1161/01.hyp.19.1_suppl.i45. [DOI] [PubMed] [Google Scholar]
- 33.Björntorp P, Holm G, Rosmond R, Folkow B. Hypertension and the metabolic syndrome: closely related central origin? Blood Pressure. 2000;9(2-3):71–82. doi: 10.1080/08037050050151762. [DOI] [PubMed] [Google Scholar]
- 34.Ogihara T, Asano T, Fujita T. Contribution of salt intake to insulin resistance associated with hypertension. Life Sciences. 2003;73(5):509–523. doi: 10.1016/s0024-3205(03)00315-1. [DOI] [PubMed] [Google Scholar]
- 35.Adrogué HJ, Madias NE. Sodium and potassium in the pathogenesis of hypertension. New England Journal of Medicine. 2007;356(19):1966–1978+1914. doi: 10.1056/NEJMra064486. [DOI] [PubMed] [Google Scholar]
- 36.Ferraris RP. Dietary and developmental regulation of intestinal sugar transport. Biochemical Journal. 2001;360(2):265–276. doi: 10.1042/0264-6021:3600265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gouyon F, Caillaud L, Carrière V, et al. Simple-sugar meals target GLUT2 at enterocyte apical membranes to improve sugar absorption: a study in GLUT2-null mice. Journal of Physiology. 2003;552(3):823–832. doi: 10.1113/jphysiol.2003.049247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kayano T, Burant CF, Fukumoto H, et al. Human facilitative glucose transporters. Isolation, functional characterization, and gene localization of cDNAs encoding an isoform (GLUT5) expressed in small intestine, kidney, muscle, and adipose tissue and an unusual glucose transporter pseudogene-like sequence (GLUT6) Journal of Biological Chemistry. 1990;265(22):13276–13282. [PubMed] [Google Scholar]
- 39.Shu R, David ES, Ferraris RP. Dietary fructose enhances intestinal fructose transport and GLUT5 expression in weaning rats. American Journal of Physiology. 1997;272(3):G446–G453. doi: 10.1152/ajpgi.1997.272.3.G446. [DOI] [PubMed] [Google Scholar]
- 40.Corpe CP, Basaleh MM, Affleck J, Gould G, Jess TJ, Kellett GL. The regulation of GLUT5 and GLUT2 activity in the adaptation of intestinal brush-border fructose transport in diabetes. Pflugers Archiv European Journal of Physiology. 1996;432(2):192–201. doi: 10.1007/s004240050124. [DOI] [PubMed] [Google Scholar]
- 41.Helliwell PA, Richardson M, Affleck J, Kellett GL. Stimulation of fructose transport across the intestinal brush-border membrane by PMA is mediated by GLUT2 and dynamically regulated by protein kinase C. Biochemical Journal. 2000;350(1):149–154. [PMC free article] [PubMed] [Google Scholar]
- 42.Barone S, Fussell SL, Singh AK, et al. Slc2a5 (Glut5) is essential for the absorption of fructose in the intestine and generation of fructose-induced hypertension. Journal of Biological Chemistry. 2009;284(8):5056–5066. doi: 10.1074/jbc.M808128200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schultheis PJ, Clarke LL, Meneton P, et al. Renal and intestinal absorptive defects in mice lacking the NHE3 Na+/H+ exchanger. Nature Genetics. 1998;19(3):282–285. doi: 10.1038/969. [DOI] [PubMed] [Google Scholar]
- 44.Zachos NC, Tse M, Donowitz M. Molecular physiology of intestinal Na+/H+ exchange. Annual Review of Physiology. 2005;67:411–443. doi: 10.1146/annurev.physiol.67.031103.153004. [DOI] [PubMed] [Google Scholar]
- 45.Bookstein C, DePaoli AM, Xie Y, et al. Na+/H+ exchangers, NHE-1 and NHE-3, of rat intestine. Expression and localization. Journal of Clinical Investigation. 1994;93(1):106–113. doi: 10.1172/JCI116933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang Z, Petrovic S, Mann E, Soleimani M. Identification of an apical Cl−/HCO exchanger in the small intestine. American Journal of Physiology. 2002;282(3):G573–G579. doi: 10.1152/ajpgi.00338.2001. [DOI] [PubMed] [Google Scholar]
- 47.Wang Z, Wang T, Petrovic S, et al. Kidney and intestine transport defects in Slc26a6 null mice. American Journal of Physiology. 2005;288:C957–C965. doi: 10.1152/ajpcell.00505.2004. [DOI] [PubMed] [Google Scholar]
- 48.Simpson JE, Schweinfest C, Shull GE, et al. PAT-1 (Slc26a6) is the predominant apical membrane Cl−/HCO3− exchanger in the upper villous epithelium of murine duodenum. American Journal of Physiology. 2006;292(4):G1079–G1088. doi: 10.1152/ajpgi.00354.2006. [DOI] [PubMed] [Google Scholar]
- 49.Seidler U, Rottinghaus I, Hillesheim J, et al. Sodium and chloride absorptive defects in the small intestine in Slc26a6 null mice. Pflugers Archiv European Journal of Physiology. 2008;455(4):757–766. doi: 10.1007/s00424-007-0318-z. [DOI] [PubMed] [Google Scholar]
- 50.Mount DB, Romero MF. The SLC26 gene family of multifunctional anion exchangers. Pflugers Archiv European Journal of Physiology. 2004;447(5):710–721. doi: 10.1007/s00424-003-1090-3. [DOI] [PubMed] [Google Scholar]
- 51.Soleimani M. Expression, regulation and the role of SLC26 Cl−/HCO3− exchangers in kidney and gastrointestinal tract. Novartis Foundation Symposium. 2006;273:91–102. [PubMed] [Google Scholar]
- 52.Xie Q, Welch R, Mercado A, Romero MF, Mount DB. Molecular characterization of the murine Slc26a6 anion exchanger: functional comparison with Slc26a1. American Journal of Physiology. 2002;283(4 52-4):F826–F838. doi: 10.1152/ajprenal.00079.2002. [DOI] [PubMed] [Google Scholar]
- 53.Knauf F, Yang CL, Thomson RB, Mentone SA, Giebisch G, Aronson PS. Identification of a chloride-formate exchanger expressed on the brush border membrane of renal proximal tubule cells. Proceedings of the National Academy of Sciences of the United States of America. 2001;98(16):9425–9430. doi: 10.1073/pnas.141241098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Petrovic S, Ma L, Wang Z, Soleimani M. Identification of an apical Cl−/HCO3− exchanger in rat kidney proximal tubule. American Journal of Physiology. 2003;285(3 54-3):C608–C617. doi: 10.1152/ajpcell.00084.2003. [DOI] [PubMed] [Google Scholar]
- 55.Höglund P, Haila S, Socha J, et al. Mutations of the down-regulated in adenoma (DRA) gene cause congenital chloride diarrhoea. Nature genetics. 1996;14(3):316–319. doi: 10.1038/ng1196-316. [DOI] [PubMed] [Google Scholar]
- 56.Schweinfest CW, Spyropoulos DD, Henderson KW, et al. slc26a3 (dra)-deficient mice display chloride-losing diarrhea, enhanced colonic proliferation, and distinct up-regulation of ion transporters in the colon. Journal of Biological Chemistry. 2006;281(49):37962–37971. doi: 10.1074/jbc.M607527200. [DOI] [PubMed] [Google Scholar]
- 57.Wang T, Yang CL, Abbiati T, et al. Mechanism of proximal tubule bicarbonate absorption in NHE3 null mice. American Journal of Physiology. 1999;277(2):F298–F302. doi: 10.1152/ajprenal.1999.277.2.F298. [DOI] [PubMed] [Google Scholar]
- 58.Valensi P. Hypertension, single sugars and fatty acids. Journal of Human Hypertension. 2005;19(supplement 3):S5–S9. doi: 10.1038/sj.jhh.1001954. [DOI] [PubMed] [Google Scholar]
- 59.Feig DI. Sugar-sweetened beverages and hypertension. Future Cardiology. 2010;6(6):773–776. doi: 10.2217/fca.10.92. [DOI] [PubMed] [Google Scholar]