

Abstract
Learning and memory impairments occurring with Alzheimer's disease (AD) are associated with degeneration of the basal forebrain cholinergic neurons (BFCNs). BFCNs extend their axons to the hippocampus where they bind nerve growth factor (NGF) which is retrogradely transported to the cell body. While NGF is necessary for BFCN survival and function via binding to the high-affinity receptor TrkA, its uncleaved precursor, pro-NGF has been proposed to induce neurodegeneration via binding to the p75NTR and its coreceptor sortilin. Basal forebrain TrkA and NGF are downregulated with aging while pro-NGF is increased. Given these data, the focus of this paper was to determine a mechanism for how pro-NGF accumulation may induce BFCN degeneration. Twenty-four hours after a single injection of pro-NGF into hippocampus, we found increased hippocampal p75NTR levels, decreased hippocampal TrkA levels, and cholinergic degeneration. The data suggest that the increase in p75NTR with AD may be mediated by elevated pro-NGF levels as a result of decreased cleavage, and that pro-NGF may be partially responsible for age-related degenerative changes observed in the basal forebrain. This paper is the first in vivo evidence that pro-NGF can affect BFCNs and may do so by regulating expression of p75NTR neurotrophin receptors.
1. Introduction
Understanding the role of pro-nerve growth factor (pro-NGF) as an independent ligand is an important part of determining a mechanism for degeneration of the basal forebrain cholinergic neurons (BFCNs). BFCNs degenerate in Alzheimer's disease (AD) and Down syndrome (DS), and this phenomenon is believed to explain some of the cognitive deficits observed in both conditions [1]. In the United States alone, 5.3 million Americans of all ages have AD with one in eight people over the age of 65 diagnosed with AD (Alzheimer's Association, 2010). The hallmark of AD is progressive cognitive decline, defined by, but not limited to: profound memory loss, difficulty with planning and problem solving, visuospatial deficits, and difficulty with word use in speaking or writing (Alzheimer's Association, 2010). Ultimately, the progressive culmination of these symptoms leads to a loss of independent living. Current drug therapies for AD target the cholinergic system by increasing the amount of available acetylcholine without protecting cholinergic neurons from further degeneration and have been only mildly successful in slowing the progression of cognitive impairment in the clinic [2]. Because BFCN integrity is correlated with cognitive function, it would be most useful to target the cause of the degeneration rather than the outcome so that new therapeutics could serve to reverse or prevent further cholinergic degeneration. Thus, it is our interest to understand why BFCNs degenerate so that novel drug targets may be developed to prevent degeneration and sustain cognition.
Pro-NGF, the 32 kD precursor to 14 kD mature NGF, is the predominant form of NGF in the brain [3] and is elevated in AD [3–5]. Under pathological conditions such as seizures, pro-NGF is found in astrocytes, [6] and in the case of inflammatory neuropathology, astrocytic IL-1β is able to regulate NGF secretion [7]. Pro-NGF can be cleaved intracellularly by furin or proconvertases, or extracellularly by plasmin (for review see [8]). Briefly, there are essentially two routes for NGF or pro-NGF synthesis. The first pathway results in mature NGF production: (1) pro-NGF is cleaved by plasmin to yield mature NGF that will bind with high affinity to TrkA receptors on the distal axon in the hippocampus, followed by retrograde transport in signaling endosomes to the cell body in the basal forebrain. This pathway promotes survival of the BFCNs via PI3/Akt signaling. The second signaling pathway results in the production of un-cleaved pro-NGF; (2) pro-NGF in its native state binds with high affinity to p75 receptors (p75NTR) and the co-receptor sortilin to induce cell death via the JNK pathway [9]. These two separate pathways contribute to BFCN cell survival or cell degeneration, respectively (see Figure 1).
Figure 1.
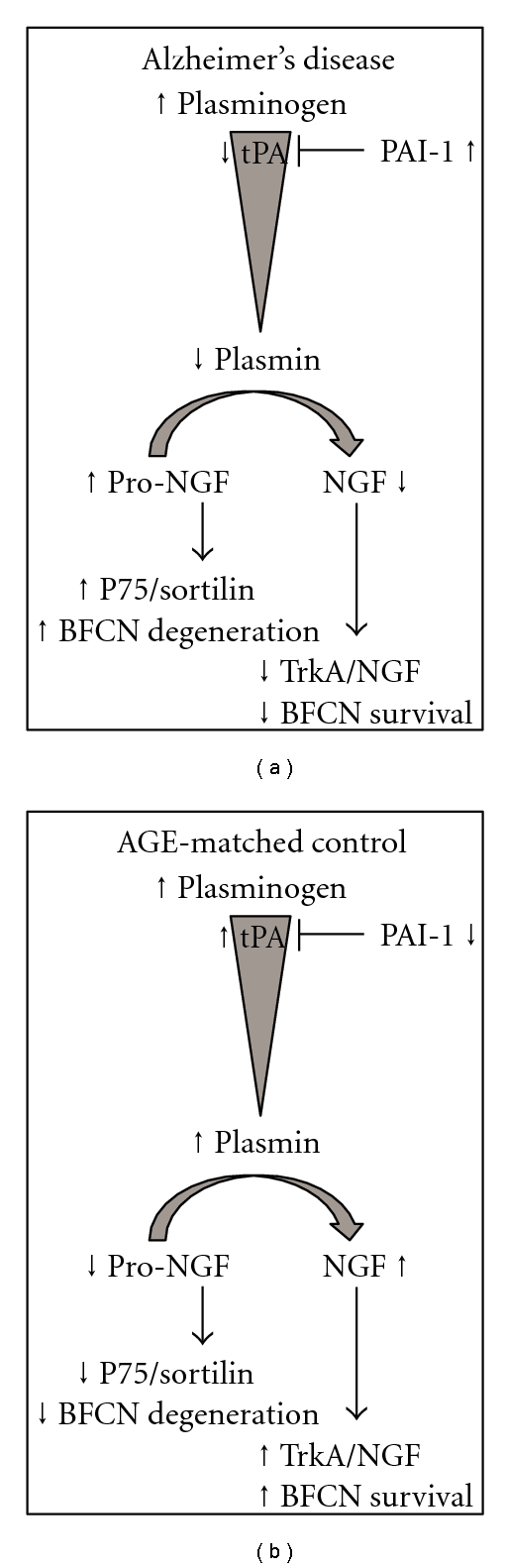
Proposed mechanism of pro-NGF induced neurodegeneration in Alzheimer's disease and age-matched controls. (1) The cleavage of plasminogen to plasmin is altered due to decreased tPA from increased PAI-1 activity, leading to an accumulation of pro-NGF in the hippocampus. (2) Increased pro-NGF leads to a preferential activation of the high affinity p75NTR/sortilin complex. (3) As NGF is down-regulated and pro-NGF is upregulated, high affinity NGF binding to TrkA receptors is compromised. (4) There is a shift in the balance of TrkA and p75NTR at the distal axon of the BFCN. (5) Pro-NGF/p75NTR/sortilin signaling increases JNK signaling and ultimately induces degeneration of the BFCNs. In age-matched controls (normal aging), balanced tPA activity allows for (1) cleavage of pro-NGF to yield NGF and (2) sustained NGF/TrkA signaling to promote cholinergic integrity. Abbreviations: tissue plasminogen activator (tPA), plasminogen activator inhibitor-1 (PAI-1), nerve growth factor (NGF), tropomyosin related kinase A (trkA = high affinity NGF receptor), pan neurotrophin receptor p75NTR (p75NTR = high affinity pro-NGF receptor).
Pro-NGF is the predominant form of NGF throughout adulthood [3], but in mouse models of AD [10] and human AD [5, 11], pro-NGF is significantly elevated compared to age-matched controls in the hippocampus and frontal cortex and entorhinal cortex, respectively [10, 11]. In a mouse model of DS, which displays the same neuropathology as AD, retrograde transport of the TrkA/NGF signaling endosomes is disrupted which leads to a decrease in NGF for the BFCNs [12]. The decrease in basal forebrain (BF) NGF coincides with a decrease in BF TrkA receptors [13–15] while p75NTR expression remains unchanged in the BF [4]. Research from our group in aged rats has demonstrated that lack of access to NGF in BFCNs is not due to reduced release of NGF from hippocampal neurons [16]; instead, the findings suggested a lack of ERK-mediated cell signaling in the septohippocampal pathway giving rise to increased levels of NGF in the hippocampus, on one hand, and reduced levels in the basal forebrain, on the other hand [17]. The inability to respond to injected exogenous NGF correlated strongly with performance on a spatial memory task, suggesting that the NGF signaling pathways were directly involved in performance in these working memory tasks [17]. Even though these findings point to altered downstream NGF problems with aging and in AD, recent findings suggest that upstream cleavage mechanisms for pro-NGF are also affected by aging and disease processes.
Upstream events include the metabolic events regulating pro-NGF expression. One key component of pro-NGF regulation is the plasminogen cascade (see Figure 1), which ultimately yields plasmin to convert pro-NGF to NGF. In human AD, activity of tissue plasminogen activator (tPA) is reduced as well as plasminogen and plasmin levels [10, 18], leaving pro-NGF unable to be converted to mature NGF. Similarly, in mouse models of AD, tPA levels are decreased [19] and the tPA inhibitor, PAI-1 is increased [20]. Current evidence in the field strongly suggests that these conditions favor accumulation of pro-NGF in AD [3, 4, 11].
Consequences of increased pro-NGF may be numerous, but at least one consequence supported by our data and others, is a compromise of TrkA signaling or expression. Using PC12 cells, Sobottka et al. [21] provided evidence that wildtype pro-NGF, when coincubated with NGF, decreased TrkA mediated MAPK activation due to pro-NGF outcompeting mature NGF. Additionally, it was recently shown that pro-NGF can suppress PI3K to inhibit TrkA signaling via PTEN in basal forebrain neurons in vitro [22]. Finally, Al-Shawi et al. [4] used superior cervical ganglia cultures in vitro and basal forebrain slices ex vivo, both of which responded with increased cell death when treated with pro-NGF, but these differences were not attributed to TrkA expression. While more evidence continues to implicate pro-NGF in cholinergic degeneration, causal studies understanding the sequence of events upstream and its consequences downstream in vivo have not been well developed. In an effort to understand BFCN degeneration, it is necessary to consider not only the ratio of pro-NGF to mature NGF but also the ratio of the high affinity receptors (p75NTR and TrkA, resp.) present at the distal axon that are communicating to the cholinergic cell body in the BF.
In line with studies conducted by Song et al. [22], we hypothesize that pro-NGF accumulation causes a subsequent decrease in TrkA signaling through preferential activation of the p75NTR receptor in vivo. Our finding that p75NTR receptor upregulation follows pro-NGF accumulation while simultaneously decreasing TrkA is an important finding to suggest that the receptor imbalance is a consequence of neurotrophin expression. Therefore, this is the first study to demonstrate that pro-NGF may directly relate to cholinergic degeneration in an intact animal.
2. Experimental Procedures
2.1. Animals
Fifteen male Fischer 344 rats were used: N = 9 aged (20 months of age) and N = 5 young (6 months of age). One aged saline animal was lost due to the presence of a pituitary tumor. In an additional control study, three aged rats were used to determine possible toxicity of pro-NGF in vivo. Rats were fed food and water ad libitum and housed on a 12 : 12 hour light cycle. All animal procedures were approved by the Institutional Animal Care and Use Committee at the Medical University of South Carolina and adhered to NIH guidelines.
2.2. Surgical Procedure
Young and aged rats were divided into three groups (n = 4-5 each): aged controls (20 months of age), young controls (6 months of age), and aged pro-NGF injected (20 months of age) rats. Under ketamine/xylazine anesthesia, all animals received bilateral hippocampal injections at coordinates targeted for dorsal hippocampus: AP-5.30, ML ± 4.5, DV-4.0 (modified from Williams et al. [23]). Animals received either saline (10 μL/hemisphere) or pro-NGF intracranial injections (5 μg in 10 μL/hemisphere; Scil Proteins, Halle, Germany). Saline or pro-NGF was delivered at a rate of 1 μL/minute over 10 minutes with a 5-minute wait period before and after injection. After surgery, rats were allowed to recover under careful supervision until normal locomotor activity returned. All animals were sacrificed 24 hours after injection by isoflurane overdose followed by decapitation, at which time the hippocampus and medial septal area of basal forebrain were dissected bilaterally with a dissecting microscope, placed on ice, flash frozen in separate tubes, and stored at −80°C until Western blot analyses. Brains collected from the toxicity study were used for immunohistochemistry and were placed in 4% paraformaldehyde in 0.1 M phosphate buffer (PB) for 48 hrs, and transferred to 30% sucrose in 0.1 M PB for a minimum of 48 hrs before being sectioned on a cryostat at 20 μm.
2.3. Verification of Injection Site
Preliminary studies were conducted in order to test the toxicity and/or physiological effects of pro-NGF injections into aged rats while simultaneously determining the success rate for bilateral injections into hippocampus. To this end, 6 animals were given either pro-NGF (n = 4) or saline (n = 2) and sacrificed 24 hours later to visualize injection sites within the hippocampus and examine ChAT immunostaining (see Section 2.6). Injection sites were verified with Cresyl violet staining (see Supplementary Figure 2 in supplementary Material available online at doi: 10.4061/2011/460543.).
2.4. Sample Preparation
The basal forebrain containing the medial septal area and bilateral hippocampi containing the injection site were microdissected, weighed, then snap frozen on dry ice. Before beginning the total protein bicinchoninic acid (BCA) assay (Pierce, Rockford, IL), all tissues were diluted 1 : 10 w/v in lysis buffer (in dH20: 1% NP40, 150.5 mM NaCl, 3.5 mM SDS, 5% Tris Base, pH 7.4) containing protease (Calbiochem, Los Angeles, CA) and phosphatase inhibitors (Roche, Indianapolis, IN), followed by approximately 15 triturations with a 20-gauge needle, stored on ice for 60 minutes, then centrifuged at 14000 rpm for 20 mins. After homogenization, the BCA assay was performed on the supernatant. The pellet and aliquots of the remaining supernatant were stored at −80°C for later use.
2.5. Western Blotting
Samples (20 μg for all blots except pro-NGF which required 75 μg) were loaded in duplicate and separated on 4–12% Bis Tris gels (Invitrogen, Carlsbad, CA) at 150 V for 45 minutes. After separation onto the gel, the gel was transferred via wet transfer onto a nitrocellulose membrane for one hour at 30 V. The membrane was then removed from the cassette and blocked for one hour in 5% nonfat milk in PBS-T (0.1% Tween-20) at room temperature and subsequently incubated overnight at room temperature in primary antibody. The following day blots were washed in PBS-T then incubated in secondary antibody in 5% nonfat milk in PBS-T for one hour at room temperature. Blots were then washed in PBS-T then imaged on a Kodak Image Station 4000 (4 exposures, 15 sec each) using Immobilon chemiluminescent reagent (Millipore, Bellerica, MA). For loading control, blots were incubated with mouse antiactin antibody for one hour at room temperature, then incubated in secondary antibody, then finally reimaged under the same settings. Primary antibodies used include rabbit anti-TrkA (1 : 1000, kindly provided by Dr. Louis Reichardt), mouse anti-β-Actin (1 : 10,000, Sigma, St. Louis, MO), mouse anti-p75NTR (1 : 1000, Millipore, Bellerica, MA), rabbit anti-caspase-3 (1 : 1,000, Stressgen, Ann Arbor, MI), rabbit antisortilin (1 : 1000, Abcam, Cambridge, MA), rabbit antiplasminogen (1 : 500, R&D Systems Inc., Minneapolis, MN), rabbit anti-Pro : NGF (1 : 200, Alomone Labs, Jerusalem, Israel) and secondary antibodies include donkey anti-rabbit HRP (1 : 5000, Jackson Immunoresearch, West Grove, PA), donkey antimouse HRP (1 : 5000, Jackson Immunoresearch, West Grove, PA). Rabbit anti-Pro-NGF blocking peptide was prepared in equal concentration to primary antibody and was incubated overnight at 4°C in 5% milk in PBS-T before use on membrane. Densitometry was performed using Fluorchem software (Alpha Innotech, San Leandro, CA); samples were normalized to protein standards on each blot then normalized to actin. Results are therefore reported as relative density.
2.6. ChAT Immunohistochemistry
Rats used to verify hippocampal injection sites were also used for ChAT immunohistochemistry; these brain sections also allowed us to preliminarily determine if pro-NGF would have a direct effect on BFCN morphology. To that end, brains were sectioned at 20 μm and every third section was collected for immunohistochemical analyses for a total of approximately 10 sections per brain area. Sections were mounted onto slides pretreated with etching solution for 10 minutes (70% TBS, 20% Methanol, 10% H202), then washed and blocked in 10% NDS in TBS-Triton for one hour. Slides were then incubated with rabbit anti-ChAT (1 : 500, Chemicon, Billerica, MA) overnight followed by donkey anti-rabbit secondary (1 : 200, Jackson Immunoresearch, West Grove, PA) for one hour. Finally, slides were incubated in ABC solution (Vector, Burlingame, CA) and developed with 3,3′-diaminobenzidine tetrahydrochloride (DAB, Sigma, St. Louis, MO). The slides were cover-slipped with Permount (Fisher Scientific, Fair Lawn, NJ) and allowed to dry.
2.7. Statistical Analyses
For each brain area (basal forebrain and hippocampus) and each marker, when all three groups were compared, a oneway ANOVA was used to assess for potential group differences, then Fischer's PLSD post hoc analyses were used to compare individual groups when group effects were present. In all cases, we were interested in comparing the young versus aged saline groups and the aged saline versus aged pro-NGF group. All statistics were performed using Statview version 5.0 (Abacus Corporation, Baltimore, MD), with a P ≤ .05 considered significant.
3. Results
3.1. Hippocampal and Basal Forebrain Levels of Pro-NGF
Intrahippocampal injections of pro-NGF produced a significant upregulation of pro-NGF protein in the hippocampus (Figure 2(a), F2,11 = 5.597, P = .0211). Pro-NGF injected rats had significantly more pro-NGF than aged saline injected rats (P = .05) and young saline injected rats (P = .0074) as indicated by Fisher's post-hoc analyses. Qualitatively, aged saline rats appeared to have more pro-NGF protein than young saline rats; however, this was not statistically significant (P = .3664). In the basal forebrain intra-hippocampal injections of pro-NGF had no effect on pro-NGF expression (Figure 2(b), F2,11 = 0.462, P = .6415), most likely suggesting that pro-NGF is not transported to the BFCN via retrograde transport.
Figure 2.
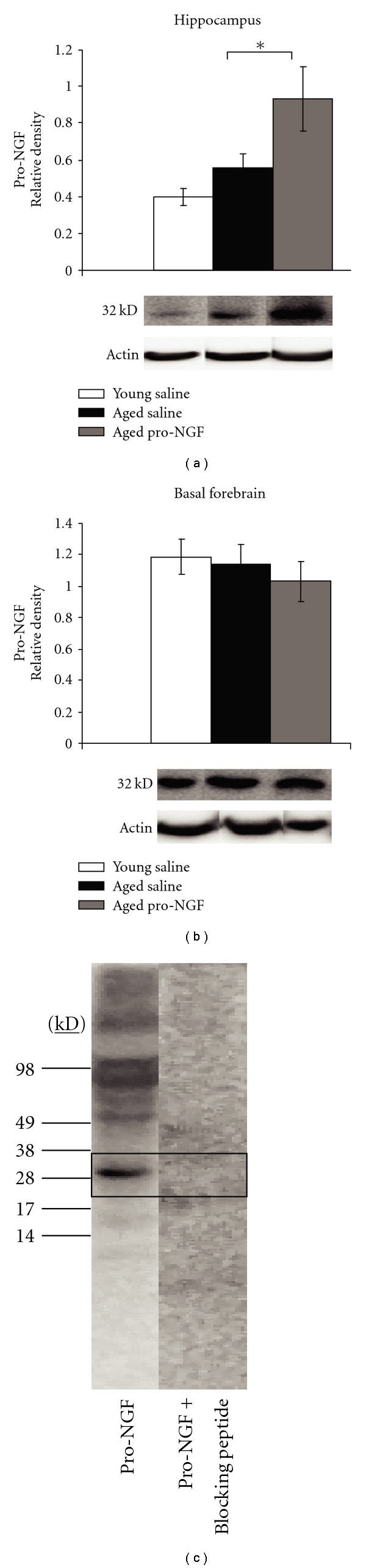
Intra-hippocampal injections of pro-NGF significantly increased hippocampal expression of pro-NGF (a), but did not alter basal forebrain pro-NGF levels (b). The pro-NGF antibody was specific for pro-NGF (~32 kD) and did not detect mature NGF protein levels as shown in a pro-NGF treated animal; when incubated with pro-NGF blocking peptide, no bands were present (c). Changes in protein levels were normalized to β-Actin; *P ≤ .05.
3.2. Hippocampal and Basal Forebrain Levels of p75NTR Receptors
After Intrahippocampal injection of pro-NGF, there was a significant effect of treatment on hippocampal p75NTR receptor levels using Western blots (Figure 3(a), F2,11 = 8.374, P = .0062). Following Fisher's post-hoc analyses, aged rats exhibited significantly higher p75NTR receptor protein levels in the hippocampus than young rats when comparing the two saline injected groups (P = .0427). In addition, aged rats treated with pro-NGF had significantly greater p75NTR protein expression than aged saline-injected rats, suggesting that pro-NGF administration further aggravated age-related alterations in hippocampal p75NTR levels (P = .0395). The observed increase in p75NTR levels in the hippocampus following pro-NGF injections was not replicated in the basal forebrain, even though an overall ANOVA revealed a main effect of group in the basal forebrain for p75NTR levels (Figure 3(b), F2,11 = 5.763, P = .0194). The main effect in the basal forebrain was largely due to an increase in p75NTR receptor levels in the aged saline group compared to the young saline group (P = .0084), rather than resulting from the pro-NGF treatment.
Figure 3.
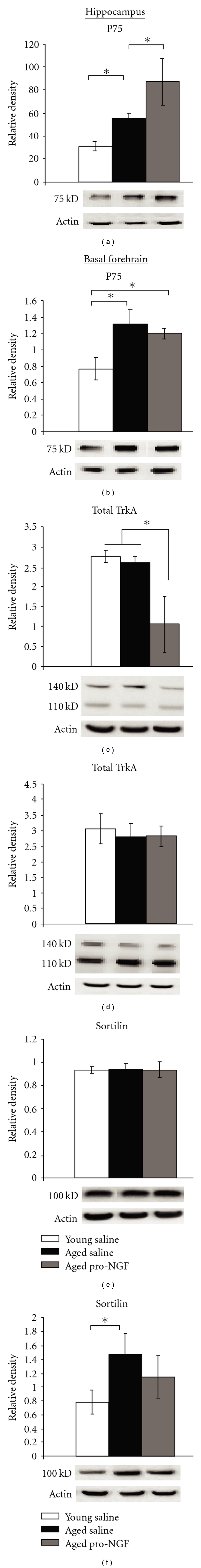
Aging resulted in an increase of p75NTR in the hippocampus (a) and basal forebrain (b), but only in the hippocampus was pro-NGF capable of significantly increasing p75NTR (a). Pro-NGF treatment decreased total hippocampal trkA (c) but did not significantly decrease total basal forebrain TrkA (d). Sortilin levels were not altered in the hippocampus (e) or basal forebrain (f). Changes in protein levels were normalized to β-Actin; *P ≤ .05.
3.3. Hippocampal and Basal Forebrain Levels of TrkA Receptors
In the hippocampus, total TrkA protein was significantly different across experimental groups (Figure 3(c), F2,11 = 4.481, P = .0377). This difference was due to aged pro-NGF rats having significantly less total TrkA protein compared to young (P = .0192) and aged saline rats (P = .0383). In an attempt to determine the cause of reduced total TrkA protein from pro-NGF treatment, the 110 kD and 140 kD forms of the TrkA receptor were individually analyzed. Because previous work has shown differences in NGF sensitivity for these two forms [24], we used western blotting to identify and quantify both forms. Intracellular, nonglycosylated 110 kD TrkA was significantly reduced in only pro-NGF treated rats (F2,11 = 5.867, P = .0184, Supplementary Figure 1C) but 140 kD TrkA levels were unaffected (F2,11 = .220, P = .8058, Supplementary Figure 1B). This was examined further by calculating the ratio of 140 kD versus 110 kD TrkA, where there was a main effect of group (F2,11 = 6.886, P = .0115; Supplementary Figure 1A), and Fisher's post-hoc analysis revealed that the differences were specifically due to the pro-NGF treated rats having significantly lower levels of the precursor (110 kD) TrkA form, compared to mature TrkA (140 kD) relative to both the aged (P = .0382) and young saline-treated rats (P = .0039).
Total TrkA was not significantly different between groups in the basal forebrain (Figure 3(d), F2,11 = .111, P = .8962). There was no difference between the groups for either the 110 kD (F2,11 = .008, P = .9918, Supplementary Figure 1H) or the 140 kD TrkA levels (F2,11 = 2.673, P = .1132, Supplementary Figure 1G) using a one-way ANOVA. Similar to the TrkA levels in the hippocampus described above, there was a significant difference in the ratio of 140 kD to 110 kD TrkA form in the basal forebrain across all groups (Supplementary Figure 1F, F2,11 = 4.143, P = .045). However, contrary to findings in the hippocampus, Fisher's post-hoc analysis demonstrated that basal forebrain levels were significantly different between the young and the aged saline group (P = .025) but not between the aged saline and pro-NGF groups (P = .0959), suggesting that TrkA levels in basal forebrain were not affected by pro-NGF treatment, at least at the 24-h postinjection time point examined here.
3.4. Hippocampal and Basal Forebrain Levels of Sortilin
Sortilin interacts with p75NTR to bind pro-NGF and induce cell death signaling [24]. In the hippocampus, total sortilin protein did not significantly differ between groups (Figure 3(e), F2,11 = .004, P = .9957). Total sortilin protein expression in the basal forebrain also did not differ between groups (Figure 3(f), F2,11 = 1.641, P = .2378), but an independent sample t-test revealed a trend for significantly less sortilin in the young saline group compared to the aged saline group (P = .0989). This suggests that pro-NGF treatment did not affect sortilin total protein expression in either the hippocampus or the basal forebrain.
3.5. BFCN Atrophy as a Result of Pro-NGF Injection
The effects of a single intrahippocampal injection of pro-NGF upon the morphology of BFCN neurons were examined using immunohistochemistry with an antibody directed against ChAT. Morphological assessment of sections from the pro-NGF treated group revealed cell body atrophy as well as reduced ChAT immunoreactivity within each neuron (Figure 4(b)) when compared to age matched saline-treated rats (Figure 4(a)). In addition, fewer neurites in the vicinity of cell bodies, as well as enlarged nuclei were observed following the pro-NGF injection (Figure 4(b)), indicative of a decrease in cholinergic integrity. Importantly, these are the first studies examining effects of pro-NGF injections in vivo in aged rats, demonstrating that pro-NGF may compromise cholinergic neuron integrity through a p75NTR-dependent mechanism.
Figure 4.
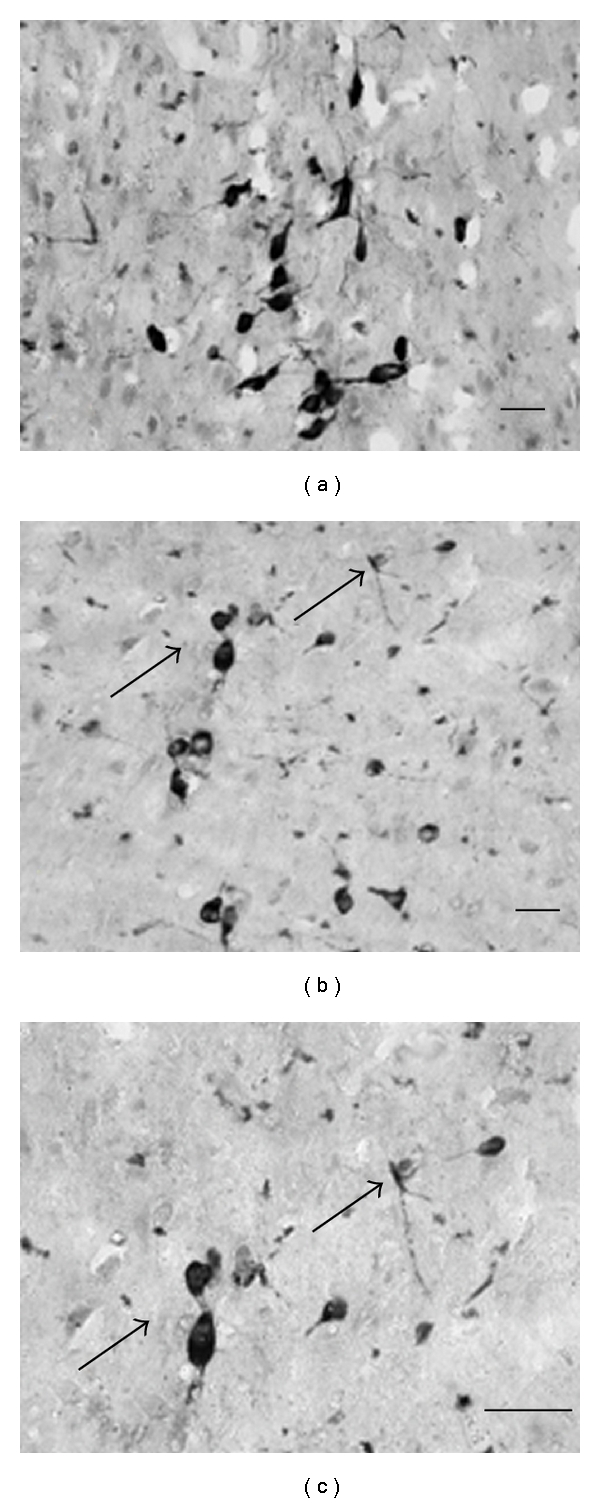
Pro-NGF treatment resulted in observable degeneration of the cholinergic neurons stained for Choline Acetyl Transferase (ChAT; (b), (c)) when compared to age matched saline animals (a). Arrows in (b) point to neurons represented in (c) under higher magnification. Scale bar = 50 μm, 20x ((a) and (b)) and 40x (c).
4. Discussion
Pro-NGF is elevated in the hippocampus in AD and may be result in an altered TrkA/p75 ratio as well as BFCN degeneration. In an attempt to model the elevated pro-NGF levels observed in patients with AD, we injected pro-NGF into the hippocampus of aged rats. We report that injecting pro-NGF directly into the hippocampus of aged rats significantly increased p75NTR receptor expression while decreasing total TrkA receptor protein. Until now, the role of pro-NGF levels for TrkA/p75NTR expression had not been examined in vivo. Our data confirm and extend previous in vitro studies [22] that suggest that p75NTR receptor activation can down-regulate TrkA expression, except our findings are in the intact animal. We also have preliminary data to suggest that injection of pro-NGF into the hippocampus affects cholinergic neuron morphology in the basal forebrain, previously only shown in vitro [4, 9]. Understanding the precise modulation of the NGF trophic factor system in the AD disease process is essential for developing novel drug compounds that could rescue cholinergic degeneration and enhance cognition in AD. Combined, the present data strongly suggest that elevation of pro-NGF levels, at least in the aged brain, leads to a switch from TrkA to p75NTR expression and that such a receptor switch may have implications for cholinergic degeneration.
4.1. Alterations in Pro-NGF Levels
Pro-NGF is the predominant form of NGF in the brain [3] and is elevated in AD [3, 4, 11]. In the present study, aged rats were injected with pro-NGF to determine in vivo effects of these injections on the TrkA/p75NTR receptor expression and on cholinergic degeneration. There was a slight increase in pro-NGF with aging alone, but when aged animals were injected with pro-NGF, there was significantly more pro-NGF protein expressed in the hippocampus but not in the basal forebrain, suggesting that the injected pro-NGF was not transported via retrograde transport to the BFCN. We also observed that there was not a significant increase in pro-NGF with aging alone in the basal forebrain. While the lack of increased pro-NGF in injected rats was not a surprise, we expected that pro-NGF levels would be increased with normal aging based on the evidence for increased pro-NGF in the hippocampus [3, 4], decreased mature NGF in the basal forebrain [12], and evidence that pro-NGF applied to the basal forebrain directly is capable of inducing neurodegeneration [4, 9]. We believe that the failure to see any changes in the basal forebrain does not imply that the ratio of pro-NGF to mature NGF at the distal axon is irrelevant but instead warrants further investigation as our studies were performed in the intact animal.
4.2. Alterations in p75NTR Levels
Interactions with the p75NTR neurotrophin receptor have been implicated in synapse elimination [25], cell death following seizures [22], and retinal degeneration [26]. However, evidence regarding altered p75NTR receptor levels in the basal forebrain with aging or disease has been controversial. Mufson et al. [27] reported that there is a loss of basal forebrain/medial septal p75NTR positive neurons in humans with AD while Al-Shawi et al. [4] reported no change in p75NTR-positive cells in aged mice by using mean gray value density analyses. By analyzing tissue homogenates from specific brain regions, we found a significant increase in p75NTR levels in both the basal forebrain and the hippocampus in the aged rat in the current study. Interestingly, treatment with pro-NGF exacerbated the p75NTR upregulation in the hippocampus, but not in the basal forebrain. This finding could be a result of the p75NTR remaining at the location of the distal axon in the hippocampus so that it may then be shed and recycled for reinsertion without ever being fully transported back to the basal forebrain. Further studies are necessary to investigate this potential phenomenon. The changes in p75NTR expression as a result of aging alone may be explained by a long term, chronic process that may be regulated by inherent pro-NGF production. Future studies will need to determine the membrane bound versus cytosolic p75NTR expression at various time points in order to fully appreciate the role that p75NTR may have in dictating the pro-NGF response.
4.3. TrkA Expression in the Hippocampus
Expression of the TrkA receptor in the hippocampus is a controversial topic. While we acknowledge that the majority of the TrkA in the septohippocampal pathway is expressed in the basal forebrain, TrkA is also present in cholinergic neurites within the hippocampus, and it is clear from a multitude of studies that the TrkA protein [28–30], while produced in the BFCN, is transported via axonal transport to the terminal field in the hippocampus, where it exerts the majority of its effects at the synaptic cleft following activity-dependent release of NGF from the postsynaptic hippocampal neuron. TrkA receptors are also highly expressed on the cell body surface in the basal forebrain, and we suggest that these receptors may be produced locally from mRNA in the cell body, or regulated by the TrkA/NGF signal from the distal axon via signaling pathways. In summary, the presence of TrkA in the hippocampus (on the axon terminal) may play an essential role in regulating the interaction with p75NTR that may convey the death/survival signal to the basal forebrain cholinergic neurons.
We hypothesize that increased pro-NGF binding to the P75 receptor may lead to a subsequent disruption in the maturation process of the functional TrkA receptor. This is illustrated by significantly lower levels of the nonglycosylated 110 kD precursor form of the TrkA protein in the hippocampus of pro-NGF treated rats compared to either young saline or aged saline-treated rats and a trend for decreased mature 140 kD hippocampal TrkA in pro-NGF treated rats. An earlier time point may have been necessary to capture a more absolute effect of pro-NGF treatment on TrkA because we previously demonstrated an upregulation of TrkA as early as 15 minutes after exogenous NGF injection into the hippocampus [17]. Also, TrkA has a short half life of approximately 138 minutes [24], and any significant changes that may have occurred would be missed by a 24-hour time point. Future studies will be conducted involving the administration of pro-NGF with an immediate and long-term time course to further characterize the timeline for the TrkA : p75NTR shift observed herein.
4.4. TrkA Expression in the Basal Forebrain
Previous work from our group and others has shown reduced basal forebrain TrkA with aging [12, 17, 27]. Results in the current study only reflect a trend for an age-related decrease in TrkA that is likely due to low sample size. It is interesting, however, that pro-NGF injections did not affect the basal forebrain in the same way as the hippocampus was affected. This may be due to a time-dependent process whereby first the TrkA is downregulated at the distal axon (which we observed at 24 hours), followed by decreased TrkA mRNA and protein in the cell body in the basal forebrain. It is also possible that increased p75NTR signaling may suppress the anterograde transport of TrkA receptors to the distal axon to bind to (decreased) NGF as an early process in p75NTR suppression of TrkA signaling. We have previously demonstrated that administration of mature NGF can rapidly and significantly increase the expression level of TrkA receptors in the aged rat [17, 31], showing a direct relationship between access to the growth factor and expression of its high-affinity receptor. Elevated pro-NGF levels would therefore function in the same manner, limiting the access of mature NGF to cholinergic neurites in the hippocampus.
4.5. Alterations in Sortilin
Sortilin interacts with p75NTR to bind pro-NGF at the distal axon of the BFCNs which then promotes cell death signaling to the BFCNs [32]. In both the hippocampus and the basal forebrain, sortilin levels were unchanged. However, in the basal forebrain, we found that there was a trend for increased sortilin with aging, which has been observed by others [4, 33]. We hypothesized that an increase in p75NTR expression with pro-NGF treatment and aging would be associated with an increase in sortilin because sortilin is required for p75NTR interactions with pro-NGF [32]. However, our lack of findings are interesting considering that sortilin is also essential for anterograde transport of TrkA receptors [34], which would suggest that sortilin levels may be unchanged because of the ubiquitous presence of sortilin in the CNS, and suggests distribution rather than overall level changes.
4.6. Alterations in Plasminogen
The plasminogen cascade has gained much attention due to its role in proteolytic cleavage processes. Plasminogen is converted to plasmin by the enzyme tissue plasminogen activator (tPA), and plasmin cleaves pro-NGF to mature NGF where tPA is regulated by plasminogen activator inhibitor-1 (PAI-1). One mechanism that may explain BFCN degeneration in AD is that the processing enzymes for the conversion of pro-NGF to NGF are disrupted; therefore, we explored plasminogen levels as a result of aging and pro-NGF treatment. Individuals with AD have decreased plasmin [18] and tissue plasminogen activator (tPA) activity [19]. Additionally, the inhibitor of tPA, PAI-1, is elevated in AD [19]. All of these processes may lead to increased pro-NGF expression. Our results replicate the idea of decreased plasminogen in the presence of excess pro-NGF, suggesting a disruption in the proteolytic cleavage process of pro-NGF.
4.7. Effects of Pro-NGF on Cell Morphology
We also observed qualitative changes in ChAT staining after pro-NGF treatment in the basal forebrain; pro-NGF treatment resulted in atrophy of cell bodies and retraction of neurites which we believe represents the early stages of degeneration. This decrease in cholinergic integrity parallels the increase in pro-NGF protein (see Figure 2). Cleaved caspase-3 levels are increased in basal forebrain cultures after treatment with pro-NGF and are associated with decreased neuronal survival [9]. In an attempt to replicate these findings in vivo and explore a mechanism by which the pro-NGF treatment could result in p75NTR upregulation, trkA downregulation and cholinergic compromise, we examined caspase-3 levels (Supplementary Figure 1). We found no difference in either cleaved or uncleaved caspase-3 protein in either the hippocampus or basal forebrain. While we were hopeful to replicate these findings, we believe our lack of results may be due to the prolonged timepoint used in our study compared to the acute timepoint of 45 minutes used by Volosin et al. [9] or may also be due to differences in in vitro versus in vivo conditions.
4.8. Comments Regarding a Possible Mechanism
Until this report, there have been no studies regarding the in vivo effects of pro-NGF treatment. These experiments are essential to understanding how upstream proteolytic cleavage of pro-NGF may affect the TrkA/p75NTR ratio known to play an important role in BFCN survival and function. It is understood that (1) retrograde signaling mechanisms of NGF are disrupted in AD, (2) proteolytic processing is disrupted in AD, and (3) pro-NGF interacts with p75NTR and sortilin. However, the sequence of events is only starting to be developed. This study provides the first insight to how increasing pro-NGF levels may alter or further exacerbate the TrkA/p75NTR balance in the aged rat. We hypothesize that upstream mechanisms (plasminogen cleavage to plasmin) regulating pro-NGF cleavage are altered first, followed by a shift in the TrkA/p75NTR balance in the distal axon that leads to a down-regulation in retrograde NGF/TrkA signaling and an increase in p75NTR/sortilin/pro-NGF signaling. This is illustrated in Figure 1. Future studies will aim to further characterize the time course, the sequence of events, and signaling pathways involved in pro-NGF induced BFCN degeneration.
4.9. Conclusion
In conclusion, this is the first report to demonstrate that pro-NGF administration into the hippocampus in the aged rat gives rise to an increase in p75NTR signaling while subsequently decreasing TrkA signaling. We propose that this altered TrkA/p75NTR balance leads to BFCN degeneration that is associated with cognitive impairment in AD [1]. These important findings serve as a guide for future work related to novel drug design in the field of age-related cholinergic degeneration and memory loss with Alzheimer's disease.
Supplementary Material
Intra-hippocampal injections of pro-NGF altered the expression of 110kD immature TrkA and 140kD mature TrkA in the hippocampus and basal forebrain, but no differences were observed in Caspase-3 activation. Plasminogen, the precursor to plasmin, was increased with aging but not as a result of pro-NGF injections into the hippocampus. Intra-hippocampal injections of pro-NGF into the dorsal hippocampus were verified using Cresyl Violet staining.
Hippocampal and basal forebrain levels of plasminogen: Because of its role in regulating plasmin, the enzyme responsible for pro-NGF cleavage, we examined the expression level of plasminogen in the hippocampus and the basal forebrain. Plasminogen is the precursor to plasmin and it is postulated that an increase in plasminogen may represent failure of plasminogen to be converted to plasmin and therefore decrease pro-NGF cleavage to mature NGF. In the hippocampus, there was a main effect of treatment for plasminogen (Supplemental Figure 1E, F2,11 = 4.888, P = .0331). This was due to significantly higher plasminogen in aged saline rats compared to young saline rats (P = .0173) and aged pro-NGF treated rats (P = .0266). In the basal forebrain, plasminogen levels were also significantly elevated (Supplemental Figure 1J, F2,11 = 5.688, P = .0203). Plasminogen expression was higher in the aged saline group versus the young saline group (P = .0063) but the pro-NGF injected group demonstrated a trend to being upregulated compared to young saline rats (P = .0809).
Hippocampal and basal forebrain levels of caspase-3: It was previously reported that basal forebrain cultures treated with pro-NGF results in caspase-3 regulated cell death [9]. Because cleaved caspase-3 is the active form for neurodegeneration, we examined caspase-3 activation by using the ratio of uncleaved (pro-caspase-3) to cleaved caspase-3. In the hippocampus, there was not a significant difference in the ratio of uncleaved to cleaved caspase-3 (Supplemental Figure 1D, F2,11 = 1.534, P = .2585). Similarly, in the basal forebrain, the ratio of caspase-3 activation was also unaltered (Supplemental Figure 1I, F2,11 = .287, P = .7561).
Conflict of Interests
The authors report no actual or potential conflicts of interest.
Acknowledgment
This paper was made possible by a Grant from National Institutes on Aging (AG10755).
References
- 1.Bierer LM, Haroutunian V, Gabriel S, et al. Neurochemical correlates of dementia severity in Alzheimer’s disease: relative importance of the cholinergic deficits. Journal of Neurochemistry. 1995;64(2):749–760. doi: 10.1046/j.1471-4159.1995.64020749.x. [DOI] [PubMed] [Google Scholar]
- 2.Schneider LS, Insel PS, Weiner MW. Treatment with cholinesterase inhibitors and memantine of patients in the Alzheimer's disease neuroimaging initiative. Archives of Neurology. 2011;68(1):58–66. doi: 10.1001/archneurol.2010.343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fahnestock M, Michalski B, Xu B, Coughlin MD. The precursor pro-nerve growth factor is the predominant form of nerve growth factor in brain and is increased in Alzheimer’s disease. Molecular and Cellular Neuroscience. 2001;18(2):210–220. doi: 10.1006/mcne.2001.1016. [DOI] [PubMed] [Google Scholar]
- 4.Al-Shawi R, Hafner A, Olson J, et al. Neurotoxic and neurotrophic roles of proNGF and the receptor sortilin in the adult and ageing nervous system. European Journal of Neuroscience. 2008;27(8):2103–2114. doi: 10.1111/j.1460-9568.2008.06152.x. [DOI] [PubMed] [Google Scholar]
- 5.Peng S, Wuu J, Mufson EJ, Fahnestock M. Increased proNGF levels in subjects with mild cognitive impairment and mild Alzheimer disease. Journal of Neuropathology and Experimental Neurology. 2004;63(6):641–649. doi: 10.1093/jnen/63.6.641. [DOI] [PubMed] [Google Scholar]
- 6.Volosin M, Trotter C, Cragnolini A, et al. Induction of proneurotrophins and activation of p75-mediated apoptosis via neurotrophin receptor-interacting factor in hippocampal neurons after seizures. Journal of Neuroscience. 2008;28(39):9870–9879. doi: 10.1523/JNEUROSCI.2841-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jurič DM, Čarman-Kržan M. Interleukin-1β, but not IL-1α, mediates nerve growth factor secretion from rat astrocytes via type I IL-1 receptor. International Journal of Developmental Neuroscience. 2001;19(7):675–683. doi: 10.1016/s0736-5748(01)00044-2. [DOI] [PubMed] [Google Scholar]
- 8.Friedman WJ. Proneurotrophins, seizures, and neuronal apoptosis. Neuroscientist. 2010;16(3):244–252. doi: 10.1177/1073858409349903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Volosin M, Song W, Almeida RD, Kaplan DR, Hempstead BL, Friedman WJ. Interaction of survival and death signaling in basal forebrain neurons: roles of neurotrophins and proneurotrophins. Journal of Neuroscience. 2006;26(29):7756–7766. doi: 10.1523/JNEUROSCI.1560-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bruno MA, Leon WC, Fragoso G, Mushynski WE, Almazan G, Cuello AC. Amyloid β-induced nerve growth factor dysmetabolism in Alzheimer disease. Journal of Neuropathology and Experimental Neurology. 2009;68(8):857–869. doi: 10.1097/NEN.0b013e3181aed9e6. [DOI] [PubMed] [Google Scholar]
- 11.Pedraza CE, Podlesniy P, Vidal N, et al. Pro-NGF isolated from the human brain affected by Alzheimer’s disease induces neuronal apoptosis mediated by p75NTR. American Journal of Pathology. 2005;166(2):533–543. doi: 10.1016/S0002-9440(10)62275-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cooper JD, Salehi A, Delcroix JD, et al. Failed retrograde transport of NGF in a mouse model of Down’s syndrome: reversal of cholinergic neurodegenerative phenotypes following NGF infusion. Proceedings of the National Academy of Sciences of the United States of America. 2001;98(18):10439–10444. doi: 10.1073/pnas.181219298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kelly A, Maguire C, Lynch MA. Deficits in nerve growth factor release and tyrosine receptor kinase phosphorylation are associated with age-related impairment in long-term potentiation in the dentate gyrus. Neuroscience. 1999;95(2):359–365. doi: 10.1016/s0306-4522(99)00460-1. [DOI] [PubMed] [Google Scholar]
- 14.Hunter CL, Bimonte HA, Granholm ACE. Behavioral comparison of 4 and 6 month-old Ts65Dn mice: age-related impairments in working and reference memory. Behavioural Brain Research. 2003;138(2):121–131. doi: 10.1016/s0166-4328(02)00275-9. [DOI] [PubMed] [Google Scholar]
- 15.Salehi A, Delcroix JD, Swaab DF. Alzheimer’s disease and NGF signaling. Journal of Neural Transmission. 2004;111(3):323–345. doi: 10.1007/s00702-003-0091-x. [DOI] [PubMed] [Google Scholar]
- 16.Williams BJ, Eriksdotter-Jonhagen M, Granholm AC. Nerve growth factor in treatment and pathogenesis of Alzheimer’s disease. Progress in Neurobiology. 2006;80(3):114–128. doi: 10.1016/j.pneurobio.2006.09.001. [DOI] [PubMed] [Google Scholar]
- 17.Williams B, Granholm AC, Sambamurti K. Age-dependent loss of NGF signaling in the rat basal forebrain is due to disrupted MAPK activation. Neuroscience Letters. 2007;413(2):110–114. doi: 10.1016/j.neulet.2006.11.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ledesma MD, Abad-Rodriguez J, Galvan C, et al. Raft disorganization leads to reduced plasmin activity in Alzheimer’s disease brains. EMBO Reports. 2003;4(12):1190–1196. doi: 10.1038/sj.embor.7400021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Melchor JP, Pawlak R, Strickland S. The tissue plasminogen activator-plasminogen proteolytic cascade accelerates amyloid-β (Aβ) degradation and inhibits Aβ-induced neurodegeneration. Journal of Neuroscience. 2003;23(26):8867–8871. doi: 10.1523/JNEUROSCI.23-26-08867.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu RM, van Groen T, Katre A, et al. Knockout of plasminogen activator inhibitor 1 gene reduces amyloid beta peptide burden in a mouse model of Alzheimer’s disease. Neurobiology of Aging. 2011;32(6):1079–1089. doi: 10.1016/j.neurobiolaging.2009.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sobottka B, Reinhardt D, Brockhaus M, Jacobsen H, Metzger F. ProNGF inhibits NGF-mediated TrkA activation in PC12 cells. Journal of Neurochemistry. 2008;107(5):1294–1303. doi: 10.1111/j.1471-4159.2008.05690.x. [DOI] [PubMed] [Google Scholar]
- 22.Song W, Volosin M, Cragnolini AB, Hempstead BL, Friedman WJ. ProNGF induces PTEN via p75NTR to suppress Trk-mediated survival signaling in brain neurons. Journal of Neuroscience. 2010;30(46):15608–15615. doi: 10.1523/JNEUROSCI.2581-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Williams B, Nelson M, Granholm AC, Coultrap S, Browning M, Curtis M. Altered NGF response but not release in the aged septo-hippocampal cholinergic system. Experimental Neurology. 2005;196(1):30–40. doi: 10.1016/j.expneurol.2005.07.001. [DOI] [PubMed] [Google Scholar]
- 24.Jullien J, Guili V, Reichardt LF, Rudkin BB. Molecular kinetics of nerve growth factor receptor trafficking and activation. Journal of Biological Chemistry. 2002;277(41):38700–38708. doi: 10.1074/jbc.M202348200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Deppmann CD, Mihalas S, Sharma N, Lonze BE, Niebur E, Ginty DD. A model for neuronal competition during development. Science. 2008;320(5874):369–373. doi: 10.1126/science.1152677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bai Y, Dergham P, Nedev H, et al. Chronic and acute models of retinal neurodegeneration TrkA activity are neuroprotective whereas p75NTR activity is neurotoxic through a paracrine mechanism. Journal of Biological Chemistry. 2010;285(50):39392–39400. doi: 10.1074/jbc.M110.147801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mufson EJ, Lavine N, Jaffar S, Kordower JH, Quirion R, Saragovi HU. Reduction in p140-trkA receptor protein within the nucleus basalis and cortex in Alzheimer’s disease. Experimental Neurology. 1997;146(1):91–103. doi: 10.1006/exnr.1997.6504. [DOI] [PubMed] [Google Scholar]
- 28.Roskoden T, Otten U, Schwegler H. Early postnatal corticosterone administration regulates neurotrophins and their receptors in septum and hippocampus of the rat. Experimental Brain Research. 2004;154(2):183–191. doi: 10.1007/s00221-003-1656-5. [DOI] [PubMed] [Google Scholar]
- 29.Fiore M, Amendola T, Triaca V, Alleva E, Aloe L. Fighting in the aged male mouse increases the expression of TrkA and TrkB in the subventricular zone and in the hippocampus. Behavioural Brain Research. 2005;157(2):351–362. doi: 10.1016/j.bbr.2004.08.024. [DOI] [PubMed] [Google Scholar]
- 30.Bulbarelli A, Lonati E, Cazzaniga E, et al. TrkA pathway activation induced by amyloid-beta (Abeta) Molecular and Cellular Neuroscience. 2009;40(3):365–373. doi: 10.1016/j.mcn.2008.12.006. [DOI] [PubMed] [Google Scholar]
- 31.Bäckman C, Rose GM, Hoffer BJ, et al. Systemic administration of a nerve growth factor conjugate reverses age-related cognitive dysfunction and prevents cholinergic neuron atrophy. Journal of Neuroscience. 1996;16(17):5437–5442. doi: 10.1523/JNEUROSCI.16-17-05437.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nykjaer A, Lee R, Teng KK, et al. Sortilin is essential for proNGF-induced neuronal cell death. Nature. 2004;427(6977):843–848. doi: 10.1038/nature02319. [DOI] [PubMed] [Google Scholar]
- 33.Terry AV, Jr., Kutiyanawalla A, Pillai A. Age-dependent alterations in nerve growth factor (NGF)-related proteins, sortilin, and learning and memory in rats. Physiology & Behavior. 2011;102(2):149–157. doi: 10.1016/j.physbeh.2010.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vaegter CB, Jansen P, Fjorback AW, et al. Sortilin associates with Trk receptors to enhance anterograde transport and neurotrophin signaling. Nature Neuroscience. 2011;14(1):54–63. doi: 10.1038/nn.2689. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Intra-hippocampal injections of pro-NGF altered the expression of 110kD immature TrkA and 140kD mature TrkA in the hippocampus and basal forebrain, but no differences were observed in Caspase-3 activation. Plasminogen, the precursor to plasmin, was increased with aging but not as a result of pro-NGF injections into the hippocampus. Intra-hippocampal injections of pro-NGF into the dorsal hippocampus were verified using Cresyl Violet staining.
Hippocampal and basal forebrain levels of plasminogen: Because of its role in regulating plasmin, the enzyme responsible for pro-NGF cleavage, we examined the expression level of plasminogen in the hippocampus and the basal forebrain. Plasminogen is the precursor to plasmin and it is postulated that an increase in plasminogen may represent failure of plasminogen to be converted to plasmin and therefore decrease pro-NGF cleavage to mature NGF. In the hippocampus, there was a main effect of treatment for plasminogen (Supplemental Figure 1E, F2,11 = 4.888, P = .0331). This was due to significantly higher plasminogen in aged saline rats compared to young saline rats (P = .0173) and aged pro-NGF treated rats (P = .0266). In the basal forebrain, plasminogen levels were also significantly elevated (Supplemental Figure 1J, F2,11 = 5.688, P = .0203). Plasminogen expression was higher in the aged saline group versus the young saline group (P = .0063) but the pro-NGF injected group demonstrated a trend to being upregulated compared to young saline rats (P = .0809).
Hippocampal and basal forebrain levels of caspase-3: It was previously reported that basal forebrain cultures treated with pro-NGF results in caspase-3 regulated cell death [9]. Because cleaved caspase-3 is the active form for neurodegeneration, we examined caspase-3 activation by using the ratio of uncleaved (pro-caspase-3) to cleaved caspase-3. In the hippocampus, there was not a significant difference in the ratio of uncleaved to cleaved caspase-3 (Supplemental Figure 1D, F2,11 = 1.534, P = .2585). Similarly, in the basal forebrain, the ratio of caspase-3 activation was also unaltered (Supplemental Figure 1I, F2,11 = .287, P = .7561).